
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Explorations in a galaxy of sialic acids: a review of sensing horizons, motivated by emerging biomedical and nutritional relevance
Saurav K.
Guin
 *abc,
Trinidad
Velasco-Torrijos
ad and
Eithne
Dempsey
*ad
*abc,
Trinidad
Velasco-Torrijos
ad and
Eithne
Dempsey
*ad
aDepartment of Chemistry, Maynooth University, Maynooth, Co. Kildare, Ireland. E-mail: SauravKumar.Guin@mu.ie; sauravkrguin@yahoo.co.in; Eithne.Dempsey@mu.ie
bFuel Chemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai – 400085, India
cHomi Bhabha National Institute, Anushaktinagar, Mumbai – 400094, India
dKathleen Lonsdale Institute for Human Health, Maynooth University, Maynooth, Co. Kildare, Ireland
First published on 25th November 2021
Abstract
Sialic acid (Sia) is widely distributed in free, polymeric and conjugated forms in living creatures and humans. It serves various roles in human physiology including brain development, function of central nervous system, the immune system, lactation and infant cognition. Sia is over- or under-expressed in human cells, tissues and fluids during physiological disorders and the onset of cancers. Furthermore, the exploitation of Sia by several bacteria and viruses including SARS-CoV-2 to cause human infections has been recently revealed. A special chemical form of Sia and 3′- and 6′-sialyllactose have recently received regulatory approval for use in commercial food products as nutrition additives. Therefore, Sia production and assay have immense clinical and industrial interests. However, sensing or analytical approaches to Sia measurement in complex clinical and food samples are less developed due to lack of a comprehensive review covering both fundamental and perspectives to general readers working at the periphery of Sia. This review provides a carefully balanced and comprehensive collation of key aspects of the monosaccharide family of Sia, covering their sources, synthesis, biological significance, role in disease and human health and nutrition, culminating in a robust account of their analysis in complex matrices.
 Saurav K. Guin | Dr. Saurav K. Guin is an early-career Scientist working in the domain of electrosynthesis, electrocatalysis and electroanalysis for more than 14 years in the Fuel Chemistry Division, Bhabha Atomic Research Centre, Department of Atomic Energy, Government of India. He is an Assistant Professor of Chemical Sciences in Homi Bhabha National Institute, Mumbai, India. At present time, he is working as a Marie Skłodowska-Curie Fellow and Career Fit PLUS Fellow at the Department of Chemistry, National University of Ireland Maynooth (Maynooth University) in Ireland on the development of biosensors for the determination of sialic acids in complex clinical samples, commercial foods and drinks. He worked in the Ulm University, Germany (2016) and National University of Singapore (2009) as visiting research fellow availing international fellowships. He is (co)author of 19 peer-reviewed articles published in international peer-reviewed journals and 44 other scientific publications. He is recipient of several competitive international research-fellowships, travel-grants and presentation-awards in international conferences. He has published a peer-reviewed Special Issue as a Guest Editor in ‘Electroanalysis (Wiley)’. He served Indian Society for ElectroAnalytical Chemistry as the Secretary for 6 years. He is also member of reputed academic societies. |
 Trinidad Velasco-Torrijos | Dr. Trinidad Velasco-Torrijos is currently an Associated Professor and coordinator of BSc in Pharmaceutical and Biomedical Chemistry program at the Department of Chemistry, Maynooth University (Ireland). Trinidad received her PhD in from the University of Bristol (UK) under the supervision of Prof. Tony Davis in the field of carbohydrate recognition and supramolecular Chemistry. She pursued post-doctoral research at Ghent University (Belgium) with Prof. Annemieke Madder. She then joined the group of Prof. Paul Murphy at University College Dublin (Ireland) as a Marie Curie Postdoctoral Fellow (2003–2005). Trinidad's current research interests include the synthesis of glycomimetics and carbohydrate-based soft materials for biomedical applications. |
 Eithne Dempsey | Dr. Eithne Dempsey received a PhD in Electroanalytical Chemistry from Dublin City University followed by postdoctoral research at St. Vincent's Hospital, Dublin. She then took up a position as Lecturer in Chemistry at Technological University Dublin – Tallaght Campus where she managed the Centre for Research in Electroanalytical Technologies. She was appointed visiting Professor of Chemistry at the University of the Western Cape, Capetown, South Africa in 2012 and recently (2017) took up a position at Maynooth University, currently as Associate Professor (since 2020) in Dept. Chemistry. Her core research objective is to address electroanalytical challenges using bespoke (nano)materials integrated with biosensing systems suitable for onsite deployment in multiple application scenarios. This includes nanoassembly and characterisation with use of modern electrochemical and surface based techniques, utilisation of electrocatalytic materials in biomedical and environmental science and the design and fabrication of integrated fluidics/sensor microsystems which exploit electrocatalytic/nanomaterials for biomedical diagnostics. |
1. Introduction
Sialic acid (Sia) having basic D-glycero-D-galacto conformational unit belongs to a sub-set of nonulosonic acid monosaccharides with 9-carbon (C) backbone molecular structure. Sia is endogenously produced in many living organisms and generally occupies the terminal position of cell surface glycans, glycoconjugates (i.e., glycoproteins and glycolipids), oligosaccharides, lipo-oligosaccharides and polysaccharides. To the best of our knowledge, 94 types of Sia have been identified and separated to date from various biological sources. Sia has the anomeric position at C-2 atom of the backbone structure and different combinations of the functional groups at C-4, 5, 7, 8 and 9 atoms (Fig. 1[A]). D-Neuraminic acid, N-acetyl-D-neuraminic acid (Neu5Ac), N-glycolyl-D-neuraminic acid (Neu5Gc) and 2-keto-3-deoxy-D-glycero-D-galacto-nononic acid (KDN) are common Sia present in many biological sources (Fig. 1[B]). The pKa of Neu5Ac is reported as 2.6 and thus in the physiological conditions (i.e., at pH 7.4), it exists as an anionic pyranose form (Fig. 1[C]).1 About 92–95% of free Sia at equilibrium exists in the thermodynamically stable β-pyranose form. The anomeric carbon (C-2) is involved in the formation of glycosidic linkages and thus bound Sia predominantly exists in the α-pyranose form (Fig. 1[C]). Sia is generally part of oligosaccharides bonded to glycoproteins and glycolipids through either α2,3- or α2,6-glycosidic bond. They can also be connected through either α2,9- or α2,8-bond to other Sia units i.e. oligo-(2–7 units of Sia) and poly-(≥8 units of Sia) sialic acid (polySia) as a linear polysaccharide chain.2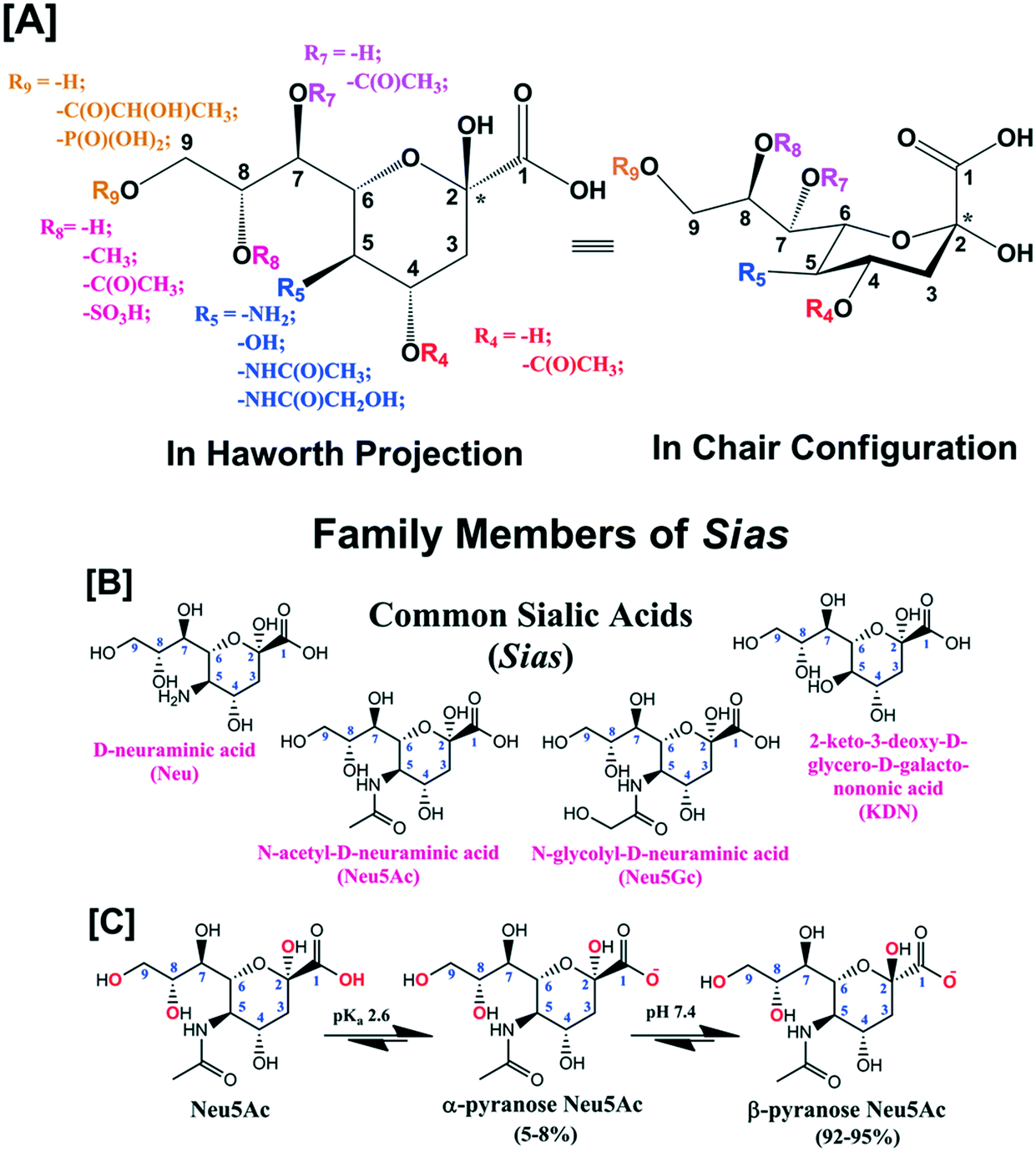 | ||
| Fig. 1 [A] Generic molecular structure of Sia. [B] Molecular structures of common Sia i.e.D-neuraminic acid, N-acetyl-D-neuraminic acid (Neu5Ac), N-glycolyl-D-neuraminic acid (Neu5Gc) and 2-keto-3-deoxy-D-glycero-D-galacto-nononic acid (KDN). [C] Anomeric forms of free Neu5Ac at physiological pH (7.4). | ||
It is important to mention that several research groups around the world independently identified Sia in a range of biological sources and unintentionally named them differently. In 1935, Klenk's research group in Germany extracted and identified a new monosaccharide from human brain glycolipid and named it “neuraminic acid”.3–5 In the following year, Blix's research group in Sweden extracted a similar compound from the mucin of submaxillary gland of bovine and named that as “kohlenhydrat I”.6,7 Yamakawa's research group in Japan extracted a similar compound from equine blood stroma glycolipid in 1951 and designated it as “hemataminic acid”.8 In the same year, Gottschalk in Australia extracted a similar compound from homogeneous mucoproteins by using an influenza virus enzyme and named as “N-substituted-isoglucosamine”.9 György's research group, in USA in 1953, extracted a similar type of monosaccharide from the nondialyzable fraction of skimmed human milk and called it “gynaminic acid”.10,11 A similar type of compound was extracted by Richard et al. in Germany in 1954 from the mucoprotein fraction of bovine colostrum and was identified as “lactaminic acid”.12 In 1955, Gottschalk identified the structural similarity among the above-mentioned compounds.13 In order too avoid the global confusion in the identification and reporting of this group of monosaccharides, he in association with Blix and Klenk, provided a unique nomenclature “sialic acid” that was unanimously accepted in 1957.14
Sia is a “shy” sugar unlike glucose and fructose and is thus relatively less known. Sia is endogenously synthesised de novo via an anabolic mechanism in the cytosol of eukaryotic cells. The uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc), which is a nucleotide sugar, successively undergoes enzymatic reactions with UPD-GlcNAc-2-epimerase (EC: 5.1.3.14), N-Acetyl-D-mannosamine (ManNAc) kinase (EC: 2.7.1.60), Neu5Ac-g-phosphate synthetase (EC: 2.5.1.57) and Neu5Ac-9-phosphatase (EC: 3.1.3.29) to produce Neu5Ac inside the nucleus where it is activated by cytidine-5′-monophospho (CMP)-Neu5Ac synthetase (EC: 2.7.7.43) as CMP-Neu5Ac. The CMP-Neu5Ac transporter transports CMP-Neu5Ac from the nucleus back to the cytosol for the following catabolic cycle. The bifunctional GNE enzyme (i.e., UPD-GlcNAc-2-epimerase and ManNAc kinase) regulates the anabolic enzymatic reaction through feedback action.15,16 CMP-N-acetylneuraminic acid hydroxylase (EC: 1.14.18.2) hydroxylates CMP-Neu5Ac and produces CMP-Neu5Gc for the catabolic cycle.17 This enzyme is absent in humans, ferrets and new world monkeys as a result of an independent inactivating mutation and thus Neu5Gc is not endogenously synthesised in their body.18–20 Recently, it was experimentally observed that the distribution and concentration of Neu5Gc in other mammals depend on the type of species and tissues. For example, the skeletal muscle of goat was found enriched with Neu5Gc (166 ± 48.70 μg g−1 of protein), while it was found to be absent in the skeletal muscles of dog and kangaroo. The lung, heart, liver, kidney and spleen of female deer had negligible (in the range 0.31–0.61 μg g−1 of protein) amount of Neu5Gc, but it was considerably (36.89 ± 42.60 μg g−1 of protein) present in skeletal muscle. Similarly, the large- (851.50 ± 314.20 μg bound Neu5Gc per g of protein; 79.76 ± 57.11 μg free Neu5Gc per g of protein) and small-intestines of sheep (760.52 ± 295.08 μg bound Neu5Gc per g of protein; 65.96 ± 49.34 μg free Neu5Gc per g of protein) had the highest concentration of Neu5Gc amongst all organs. The concentration of bound Neu5Gc in the commercial cuts of lamb/mutton varied in the range ∼31–77 μg g−1 of protein.21 Although KDN is highly abundant in bacteria and lower vertebrates, it is highly expressed on ovarian tumour tissue and in the foetal haemoglobin of humans.22Sia is also found in some viruses, bacteria, fungi, protozoa, insects, mollusca, sea urchin, and different organs and cells of humans.23 The edible bird's nest (EBN) of Asian swiftlet showed significantly high concentration of Sia due to the involvement of Sia rich saliva secreted from the sublingual glands of Asian swiftlets during nest formation.24–26 The presence of Sia in plants and fruits is usually rare and mainly originated from extraneous contaminants.27–29 However, recently Sun et al. reported the presence of a considerable amount of Neu5Ac in pea seedlings.30 Furthermore, Xia et al. extracted Sia from the sialylated glycoprotein from the root of a Tibetan plant Potentilla anserina L.31 Hence, the present state of art is not able to confirm that Sia is absent in all parts of the plants.
It would be a mistake to ignore the biological importance of Sia. Schauer and Kamerling published a book chapter in 2018 exploring the world of Sia, covering their discovery, structural analysis and some biological roles.32 In 2021, Yang et al. has published a review article emphasising the biosynthesis, metabolism and various means of production of Sia.33 Furthermore, a couple of interesting reviews have been published in 2020–2021 covering the progress and promises of free Sia storage disorder,34 development of the entry inhibitors for Sia targeting viruses,35 the role of Sia as a receptor of various pathogens,36 role in human immune regulation and therapeutic potentials37 and utilisation of Sia for identifying tumours.38 In 2010, Lacomba et al. published a review emphasising analytical methodologies for Sia and gangliosides in the biological and dairy products.39 In 2021, Cheeseman et al. published another review on the identification and quantification of Sia covering the traditional colorimetric, fluorometric, enzymatic and chromatographic methods.40 It is important to mention here, that the European Commission has recently approved inclusion of Sia as a supplement in infant and follow-up formula and other food products under regulation EC/258/97.41 However, the beneficial or inhibiting role of Sia in the present pandemic caused by the COVID-19 disease is yet an open question.42–44 Hence, this serendipitous review has the intriguing ambition to concisely outline the universe of Sia to both general and expert readers covering Sia's involvement in human biochemistry; its role in human health, the immune system, pathogenicity and disease; dietary regulations and commercial production and most importantly the evolution of analytical methods from traditional to the latest emerging techniques for assays of Sia in complex biological and food matrices.
2. Biological relevance of Sia
The CMP-sialic acid transporter (viz. CMP-Neu5Ac transporter) transports CMP-Sia to the lumen of the Golgi body, where Sia becomes associated with glycoproteins, glycolipids and oligosaccharides by various silyl transferases. The sialylated glycoproteins/glycolipids/oligosaccharides and poly- or oligo- sialic acids are distributed in the lysosome, cytosol, outer-plasma membrane and intra-cellular cell compartment membrane, where exo-α-sialidase (EC: 3.2.1.18) releases the terminal Sia unit in the cell. Sialic acid aldolase (EC: 4.1.3.3) then splits Sia into pyruvate and ManNAc in the cell. In this catabolic cycle both free Sia and ManNAc may be salvaged to the appropriate stage of the anabolic cycle.45 The absence of (CMP)-Neu5Ac synthetase and sialic acid aldolase in human milk helps in the accumulation of free Neu5Ac in human milk.2.1 Diverse biological functions of Sia in healthy humans
Sia is generally present on the human cell glycocalyx, which is a sugar-coat or a thick (∼500 nm) network of glycans on the surface of human cells. The glycocalyx not only protects the cells, but it also interacts with other cells and outer-cellular environment. Neu5Ac is a major occupant of stroma of human erythrocytes i.e., red blood cell (RBC) and it eventually helps in the dispersion of RBC in blood owing to electrostatic repulsions as shown in Fig. 2[A].46 Furthermore, the podocalyxin epithelial polyanions consist of negatively charged Sia (as shown in Fig. 2[B]), sulphated oligosaccharides and glycopeptides control the glomerular filtration in the kidneys in association with phosphorylated ezrin and Na+/H+ exchanger regulatory factor 2.47 On the other hand, glycans are covalent linear and branched oligosaccharide chains, whose monosaccharide component includes Neu5Ac, glucose, galactose, mannose, fucose, N-acetyl-glucosamine (GlcNAc) and N-acetyl-galactosamine (GalNAc). Glycans are attached to glycoproteins (by either N- or O-linkage) and glycolipids on the glycocalyx and extracellular fluids and matrices.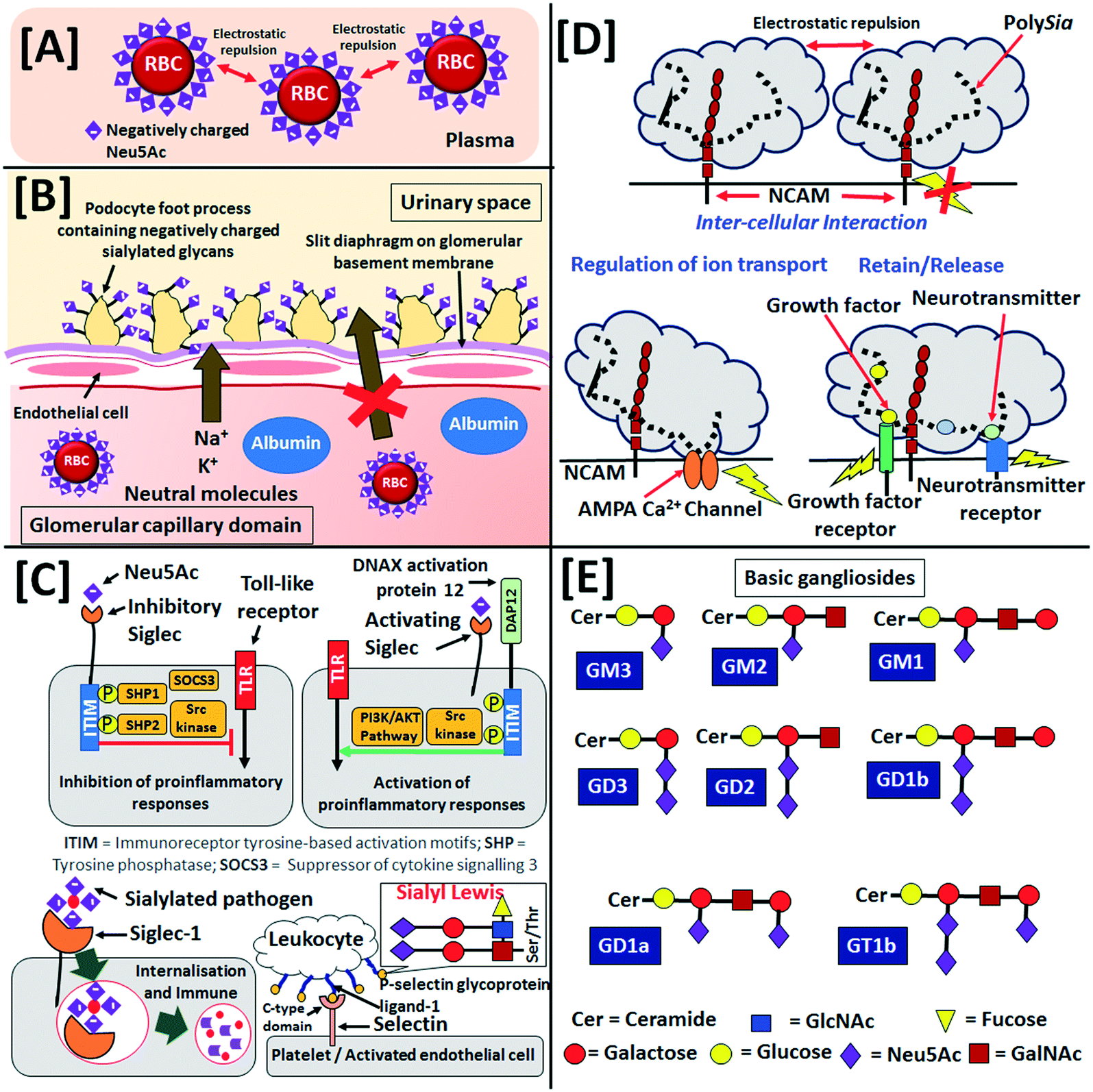 | ||
| Fig. 2 Negative charge of Neu5Ac in [A] dispersion of RBC in blood and [B] glomerular filtration in kidney. [C] The inhibitory (top left) and activating (top right) siglec actions in immune system, internalisation and immune response of Siglec-1 (bottom left), interaction of C-type domain of selectin with sialyl Lewis of P-selectin glycoprotein ligand-1 of leukocyte (bottom right). [D] Intercellular repulsive interaction of polySia-NCAM, activation of ion channels and growth factor receptors through attractive interactions. [E] Sequence of saccharides in some of the basic gangliosides (the relative scale of objects of the schemes are not in actual dimensions). | ||
The sialoglycans on the glycocalyx of healthy cells usually form specific self-associated molecular pattern (SAMP).48 The sialoglycan-SAMP is recognised by sialic acid-binding immunoglobulin type lectin (Siglec) protein receptors on the surface of immune cells (such as monocytes, macrophages, dendritic cells, natural killer cells, B lymphocytes, neutrophils and eosinophils), myelin, myeloid progenitors, trophoblasts and osteoclasts. Siglec discriminates the body's own and foreign antigens and can interact with Sia of the same as well as different cells. A total number of 15 human Siglecs (1–12, 14–16) have been identified so far and it is believed that Siglec-13 and -17 genes have been inactivated during the course of human evolution.49 Siglec-1 (also known as sialoadhesin), -2 (also known as CD22), -4 (also known as myelin-associated glycoprotein) and -15 are found in all mammals and thus are known as conserved classic Siglecs. The other Siglecs have rapidly evolved in humans and are categorised as Siglec-3 (also known as CD-33) related Siglecs. On the other hand, Siglecs are often classified as per their functionality such as inhibitory (2, 3, 5–12), activating (14–16) and non-signalling (1, 4) Siglecs.
The binding of Sia by the inhibitory-type Siglecs induces the Src family kinases by the immunoreceptor tyrosine-based inhibitory motif or its alike tyrosine present in the cytoplasmic domain and eventually it down-regulates the pro-inflammation caused by toll-like receptors (Fig. 2[C]).50 This phenomenon prevents the pro-inflammatory damage of the self-antigens and healthy cells. On the other hand, the transmembrane DNAX activation protein 12 of the activating-type Siglec electrostatically binds with negatively charged Sia and stimulates pro-inflammatory responses through mitogen-activated protein kinase and AKT pathways to destroy the pathogens having specific sialoglycan-SAMPs to circumvent inhibitory Siglecs (Fig. 2[C]).51 The Siglec-2, -6, -14 and -15 bind α2,6-Neu5Ac, whereas Siglec-5 and -11 bind α2,3-Neu5Ac and α2,8-Neu5Ac, respectively. The Siglec-9 and -10 do not show any preference to α2,3- and α2,6-Neu5Ac; but Siglec-1, -4 and -8 preferentially bind α2,3-Neu5Ac over α2,6-Neu5Ac and the reverse is true for Siglec-3. On the other hand, Siglec-7 preferentially binds to α2,8- over α2,6- and α2,3-Neu5Ac.52 The α2,3-Neu5Ac-SAMP is recognised by the serum complement factor H of the alternate pathway of innate immune system to identify and protect “self” cells.53,54
The selectins are a family of calcium-dependent (C-type) lectins also involved in immune cell adhesion to endothelial cells. They mediate access to lymphoid organs and sites of inflammation. Sialylated tetrasaccharides, i.e., sialyl Lewisx (sLex, Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAc) and sialyl Lewisa (sLea, Neu5Acα2-3Galβ1-3(Fucα1-4)GlcNAc) are among the best described ligands for the selectins but they can also have high-affinity interactions with sulfatides and non-sialylated glycosphingolipids (Fig. 2[C]).55 The selectin family can be subdivided in three main groups: P-selectins, expressed on platelets; E-selectins, expressed on endothelial cells and L-selectin, expressed on leukocytes. The biological function and ligand selectivity of these three selectins sub-families are also different. L-selectin is involved in leukocyte trafficking. E-selectins are constitutively expressed on endothelial cells in the bone marrow and skin, but exposure to inflammatory cytokines is required for their expression in other organs. P-selectins are found in activated platelets. Sołkiewicz et al. recently reported that the specific reactivity of α2,3-Neu5Ac recognising selectin Maackia amurensis agglutinin with sialylated serum immunoglobulin G as well as quantitative evaluation of sialylation and agalactosylation factor could be useful to diagnose inflammatory endometriosis disease.56
NCAM participates in cytoplasmic signalling, intercellular and extracellular matrix interactions in the central nervous system. The long negatively charged hydrated polySia on NCAM are responsible for ∼20–30 nm intercellular space (Fig. 2[D]). However, polySia on NCAM attract neuro- and bio-logically active molecules viz. brain-derived neurotrophic factor, pro-brain-derived neurotrophic factor, neurotrophic factors-3, fibroblast growth factor-2, norepinephrine, dopamine, epinephrine, histone H1, chemokine ligand 21, etc. (Fig. 2[D]).73 It also shows attractive interaction to Ca2+ permeable AMPA channel (Fig. 2[D]). PolySia helps in the migration of neuronal progenitor cells from the subventricular zone of lateral ventricle and rostral migratory stream to the olfactory bulb in the embryonic nervous system. The polySia on the progenitors in the subventricular zone influences the timing of the differentiation process into interneurons in the olfactory bulb.74 During the embryonic and neonatal stages, polySia expressed on the growing axons helps to avoid interference of ectopic synapses projected towards specific targets; then, its under-expression on the developing axons and oligodendrocyte precursors starts at the onset of myelination. Furthermore, during the critical neonatal stage polySia provides insulation and controls synaptogenesis helping maturation of the central nervous system of the infant. However, the expression of polySia in the central nervous system is gradually decreased from the embryonic stage to mature infant (∼10 years age) and mostly located at very low concentration in adults up to 80 years old in subventricular zone and granule cell layer of the hippocampus to maintain neurogenesis and neural plasticity. Furthermore, trace amounts of polySia present in the adult thalamus (paratenial, periventricular and anteroventral anterodorsal nuclei), hypothalamus (supraoptic and suprachiasmatic nuclei), habenular nuclei, entorhinal–hippocampal complex, lateral geniculate nucleus, dorsal spinal laminae and mesencephalic central grey control the physiological plasticity. PolySia is also expressed on some sensory neurons in the spinal cord, optic nerve, tectum (axons of ganglion cells) and piriform cortex. The reintroduction of external polySia in adults has shown natural neural tissue repair.75–77 Weisgerber et al. reported 15.0 ± 1.5 μg mL−1 polyNeu5Ac in the cerebrospinal fluid of new-born and premature infants and its concentration decreased to 0.2 ± 0.02 μg mL−1 in children ≥1 year old.58 Simon et al. have recently reported the expression of polySia in human semen owing to the post-translational modification of NCAM and α2,8-polysialyltransferases ST8SiaII.78 The expression of polySia to CD36 ratio in human breast milk is initially increased from 0.572 ± 0.225 during 7 days of colostrum to 3.424 ± 0.938 at 1 month and then again decreased to 1.491 ± 0.044 and 1.018 ± 0.478 after 3 and 6 months, respectively, from birth.65
Owing to the above-mentioned physiological roles of the polySia to humans, it is usually recognised as a harmless molecule by the human immune system. This phenomenon is often exploited by some bacteria such as Escherichia coli, Listeria monocytogenes, Neisseria meningitidis, Haemophilus influenzae b, Streptococcus agalactiae and Streptococcus pneumoniae having their own polySia glycocalyx causing invasive meningococcal diseases viz. meningitis, fulminant septicaemia, etc.79 However, Siglec-11 preferentially binds to polySia and inhibits inflammatory neurotoxicity of phagocytosis.80 The expression of polySia on several cancer cells is highly invasive and proliferative, being recognised as onco-developmental antigens. Thus, polySia is sometimes considered a potential biomarker to diagnose lung carcinoma, neuroblastoma, glioblastoma and pituitary tumours. Cancer therapy sometimes utilises antibodies and inhibitors specific to polySia-NCAM.81 Furthermore, the abnormal expression and impairment of polySia are associated with mental disorders viz. schizophrenia, bipolar disorder, autism spectrum disorder.82 Relapsing–remitting multiple sclerosis is caused by the inflammatory attacks on myelin and nerve fibers. The high activity of immunoglobulin G towards exogenous Neu5Gc and Neu5Ac showed some potential use in immunotherapy of relapsing–remitting multiple sclerosis.83
In 1978, Svennerholm et al. reported the concentration of gangliosides in different human organs.85,86 The highest concentration (i.e., 3500–3000 nmol g−1 of tissue weight) of Neu5Ac was reported for the human brain's cerebral cortex followed by the cells in the cerebral white matter (1250–1000 nmol g−1 of tissue weight). The spleen (300–200 nmol g−1 of tissue weight), placenta (200–100 nmol g−1 of tissue weight) and retina (150–100 nmol g−1 of tissue weight) share almost similar concentration of gangliosides. Other organs viz. liver (100–50 nmol g−1 of tissue weight), thyroid (100–60 nmol g−1 of tissue weight), peripheral nerve (80–35 nmol g−1 of tissue weight), skeletal muscle and muscular layer of small intestine (80–50 nmol g−1 of tissue weight), kidney (60–30 nmol g−1 of tissue weight), skin (30–35 nmol g−1 of tissue weight), fat tissue (10–15 nmol g−1 of tissue weight) and mucosa (5–8 nmol g−1 of tissue weight) also showed moderate concentration of gangliosides. In the nomenclature of gangliosides, ‘G’ refers to the ganglio series of glycosphingolipids, while ‘M’, ‘D’, ‘T’ and ‘Q’ define ‘mono’, ‘di’, ‘tri’ and ‘tetra’ Neu5Ac units, respectively. The migration order of gangliosides in thin-layer chromatography is designated by the number 1, 2, 3, etc., whereas ‘a’, ‘b’ and ‘c’ refer to ‘one’, ‘two’ and ‘three’ units of Neu5Ac, respectively, linked to the innermost galactose unit of the glycan chain.87Fig. 2[E] shows the arrangement of saccharides in some of the basic gangliosides. Ganglioside distribution and composition in human brain can vary greatly depending on age. The simple gangliosides viz. GM3, GD3 and 9OAc-GD3 are predominant in human embryonic brains, but as age increases, comparatively complex gangliosides viz. GM1, GD1a, GD1b and GT1b are upregulated with concomitant down-regulation of embryonic gangliosides.88,89 Ando et al. were able to determine the composition of adult white- and grey-matter of human brain by densitometry. They found the ganglioside bound Neu5Ac concentration in the human grey- and white-matter as 875 and 275 μg g−1 of the wet tissue, respectively. They also reported that GD1a (21.7%), GD1b (18.2%), GT1b (16.3%) and GM1 (14.9%) are the major components of the grey matter, while GM1 (21.6%), GD1b (16.9%), GT1b (11.1%) and GD1a (8.8%) are major components of white matter. The other gangliosides such as GM2, GM3, GM4, GD2, GD3, GT1a, GQ1b and GD1a-GalNAc are also found in both the grey- and white- matter of the human brain.90 Sarbu et al. recently utilised electrospray-ionization-ion-mobility-mass-spectrometry to study the ganglioside profile of purified normal foetal frontal lobe in the 37th gestation week. They identified a total number of 143 ganglioside species having different glycan structures and ceramide constitutions including 47, 37 and 25 structures of GT, GD and GM, respectively. They also reported the existence of a minimum 12 GQ structures and glycans having Neu5Ac up to 8 units (which was earlier believed to be a maximum 5 units).91 The same research group later identified more than 140 ganglioside species in the adult human hippocampus by using an ion-mobility-mass-spectrometry hyphenated with the collision-induced-dissociation fragmentation method.92 They also compared the frontal and the occipital lobes of the brains of the 20 and 82 year-old males by nano-electron-spray-ionisation orbitrap-mass-spectrometry hyphenated with collision-induced-dissociation fragmentation method and reported that the content of sialylated, fucosylated and acetylated gangliosides in the overall human brain could decrease with age. Interestingly, the gangliosides are over-expressed in the frontal lobe compared to the occipital lobe in young ages; while the opposite becomes true in old ages.93 Through ion-mobility-mass-spectrometry they have recently identified 113 gangliosides including GD2 and GD3 in the human cerebrospinal fluid and confirmed that some of the brain gangliosides could be passed to the cerebrospinal fluid due to immediate proximity to brain.94
The gangliosides are expressed along with phospholipids, cholesterol and glycosphingolipids on the plasma membrane's lipid rafts and participate in signal transduction (inhibition, activation, promotion, etc.) in cancer cells. This over-/under- expression of gangliosides is often used to diagnose several cancers and other diseases.95 The malignant brain tumour known as glioblastoma multiforme can be diagnosed from the overexpression of both GD3 and GT1 gangliosides.96 The high incidence of GD3 and GM3 gangliosides is a signature of human melanoma, a skin cancer.97 GM1 activates tropomyosin receptor kinase-A and promotes differentiation of the neuroblastoma cells by formation of receptor-GM1 complex on the cell surface.98 The GM1's expression in the skin increases with aging and senescence. It induces human aortic endothelial cells' insulin resistance.99 The exogeneous GM1 showed a neuroprotective effect for neurodegeneration, neuronal injury; as well as a beneficial role to patients having Huntington, Alzheimer, Parkinson diseases, epilepsy, stroke and multiple sclerosis.100 Furthermore, GM1 binds with the platelet-derived growth factor receptors, inhibits the signal to the glycolipid-enriched microdomain and suppresses the cell growth signal.101 On the contrary, platelet-derived growth factor B induces GD3 to enhance platelet-derived growth factor signal and promotes the cell proliferation and invasion in the gliomas in the glial cells of brain or spine.102 GD3 blocks the activation of nuclear factor kappa-light-chain-enhancer of activated B cells in the human hepatoblastoma HepG2 cell-line after ionizing radiation or daunorubicin therapy. Hence, GD3 pre-treatment promotes apoptotic cell death during radiation or chemo therapy in patients having hepatocellular carcinoma.103 Furthermore, GD3 activates CD95 induced apoptosis of lymphoid and myeloid tumour cells for leukaemia.104 On the other hand, the overexpression of GD3 on the malignant melanoma and human osteosarcomas cells activates p130Cas and promotes focal adhesion kinase and paxillin pathway.105–107 GD3 helps in the signalling of epidermal growth factor receptor and its activated form in the breast cancer stem cells and cell line.108 GM3 showed inhibition in the epidermal growth factor signalling pathway for the human neuroblastoma cell line NBL-W (for neural and brain cancer);109 human epidermoid carcinoma A431 cell (for skin cancer);110 PC-3 and LNCaP cell (for prostate cancer);111 YTS-1, T24, 5637, and KK47 cell (for human bladder cancer);112 HSC-2 and SAS cell (for squamous carcinoma).113 GM3 inhibits PI-3K/AKT/MDM2 signalling as well as Wnt/β-catenin signalling pathways,114,115 but promotes oxidative stress-mediated mitochondrial pathway in HCT116 cell for human colorectal cancer.116 GM2 has potential in the diagnosis and therapy of pancreatic ductal adenocarcinoma as it is overexpressed in MIA-PaCa-2 cell line and promotes TFG-β1 signalling for invasion.117 GD2 is expressed on the stem-like cells of triple-negative breast cancer118 and it is involved in the proliferation of MDA-MB-231 cells by promoting signalling of c-Met receptor.119 The GD3 and GD2 are overexpressed on glioma cell line U-251MG and promote the signalling of ERK1/2 and AKT pathway.120 GD2, GD1b, and GT1b are generally expressed on the small cell lung cancer cells121 and GD2 promotes the activation of focal adhesion kinase pathway for integrin-mediated signal transduction.122 GD1a targets c-Met receptor and suppresses the metastasis of hepatocellular carcinoma in human hepatoma HepG2 cell by inhibiting the signalling of hepatocyte growth factor.123 Both exogenous and endogenous GD1b regulate breast cancer apoptosis as evidenced in the MCF-7 human breast cancer cells.124 The treatment with GT1b induces an early apoptosis in A549 lung cancer cells by modulating both caveolin-1 and p53 to inhibit fibronectin-a5b1-integrin-ERK signalling.125
2.2 Involvement of Sia in human bacterial and viral infections
Bacteria and viruses often evolve to utilise and exploit Sia for evading the human's natural immune system. Hence, Sia is responsible for many bacterial and viral infections and eventually a greater number of viruses exploit Sia compared to bacterial infections.The first group would include bacteria which can de novo biosynthesise polySia or sialylated capsular polysaccharides, lipopolysaccharides, lipooligosaccharides and peptidoglycans by themselves on their own cell surface to mask the host's immune surveillance. Escherichia coli bacterial phage receptor displays an endogenous polySia network similar to polySia structures of brain microglia.142 This molecular mimicry helps E. coli in the cell-internalisation process as well as conferring protection from the innate immune network by engaging inhibitory Siglec-11. On the other hand, uropathogenic E. coli non-specifically binds to Sia on the urinary bladder epithelium.143 However, the interaction of E. coli with the activating Siglec-16 enhances proinflammatory cytokine expression. Thus, fortunately the impaired dynamics of Siglecs-11 and -16 leads to the elimination of this pathogen from the human body.144 Han et al. recently used N-acetyl-9-azido-9-deoxy-neuraminic acid label in flow cytometry, metagenomic and whole-genome sequencing to identify a new strain of E. coli in complex human faecal microbiome.145Neisseria meningitidis bacteria, which cause meningitis disease, also can synthesise α2,8-(serogroup B) and O-acetylated or non-acetylated α2,9-(serogroup C) polySia on the lipooligosaccharide cell surface.146 Hence, it inhibits the activation of an alternative pathway of the immune system.147 However, the preferential binding of N. meningitidis with Siglecs-1 and -5 leads to its efficient phagocytosis.148Campylobacter jejuni bacteria can synthesise α2,3- and α2,8-Neu5Ac themselves on their own lipooligosaccharide cell structures and thus they can easily mimic the human peripheral nerve gangliosides, causing autoimmune neuropathy known as Guillain–Barré syndrome.149 The sialylated lipooligosaccharide of C. jejuni enhances dendritic cell activation and promotes B-cell responses by producing IFN-β and TNF-α cytokines.150 Furthermore, α2,3- and α2,8- sialylated lipooligosaccharides interact with Siglecs-1 and -7, respectively, on the dendritic cells and polarize naïve T-cells differently to T-helper-2 and -1 cells, respectively.151Staphylococcus agalactiae bacteria, also known as group B Streptococcus, can produce α2,3-Neu5Ac linked capsular polysaccharide themselves, to mimic the human glycocalyx and can cause meningitis in human infant and sepsis. Group B Streptococcus blocks the antimicrobial factor derived from the platelets and engages the inhibitory receptor Siglec-9 to block platelet activation.152 However, genetically or biochemically mediated O-acetylation of α2,3-Neu5Ac capsular polysaccharide of group B Streptococcus minimises the virulence and neutrophil suppression.153
A second group of bacteria cannot synthesise Sia by themselves but can exploit host cell's Sia as a source of carbon for their own catabolism, colonization and biofilm formation. Haemophilus influenzae bacteria use dietary Neu5Gc as well as Neu5Ac from the host cell to sialylate outer membrane glycolipids and lipooligosaccharide and form biofilms.154Streptococcus pneumonia bacteria, acquire the terminal Sia of human nasopharynx sialoglycans as a carbon source for airway colonisation.155Pseudomonas aeruginosa bacteria which cause chronic infections of lungs, absorb Sia on their own surface from the sialylated glycoproteins present in human body fluids and engage the inhibitory Siglec-9 to attenuate the inflammatory responses of neutrophils.156,157Neisseria gonorrhoeae bacteria cause the gonococcal infection known as gonorrhea. The bacterial sialyltransferase transfers Neu5Ac from host cell's CMP-Neu5Ac to gonococcal lipooligosaccharide and downregulates the alternative complement pathway of innate immune surveillance. However, the use of either a fusion protein (consisting of complement inhibition factor H and fragment crystallizable region of immunoglobulin G) or un-protective analogue of Neu5Ac to interact with the bacterial lipooligosaccharide can be utilised for the immunotherapy of gonorrhoea.158Haemophilus ducreyi bacteria, responsible for the chancroid disease, and Pasteurella multocida pathogen, causing soft-tissue infection, also utilise the host's sialylated glycans to evade human immune surveillance.159,160
The third group of bacteria use their own Sia or host cell's Sia as either receptor or ligand to mediate infection. The erythrocyte binding antigen 175 of Plasmodium falciparum pathogen (responsible for malaria) preferentially binds to Neu5Ac available on the RBC membrane.161 GM1 ganglioside is a known receptor of cholera toxin. The Vibrio cholerae neuraminidase enzyme released from Vibrio cholerae bacteria cleaves Sia from higher order gangliosides to unmask GM1 on the host cell surface.162 On the other hand, the cluster of genes in the vibrio pathogenicity island 2 of the Vibrio cholerae strain indicates that it can utilise the released Sia as the sole source of carbon and nitrogen for their own catabolism reactions.163 The Sia-binding adhesin of Helicobacter pylori bacteria targets the gangliosides of the host cell surface, stimulating neutrophils to produce reactive oxygen species, which often damage the gastric epithelium.164 On the other hand, Sia-binding adhesin of Streptococcus gordonii binds with Veillonella atypica; this process is necessary for intergeneric coaggregation, which causes dental biofilm.165
The human influenza virus A, B, C have two surface glycoproteins viz. hemagglutinin and neuraminidase, which interact with the host cells. The globular domain of hemagglutinin trimer in the influenza A and B viruses preferentially binds with α2,6- (in human) and α2,3- (in avian) sialylated glycans, but influenza C virus binds to 9OAc-Neu5Ac. On the other hand, the neuraminidase cleaves the post-infection glycoside bonds and releases the progeny in the cytosol.180 The human upper respiratory epithelial cells in the nasal mucosa, paranasal sinuses, pharynx, bronchi, etc., are highly susceptible to the influenza infection owing to higher availability of cell surface α2,6-Neu5Ac glycoconjugates in the human upper airway.181 Human saliva and serum contain a minimum of 116 and 144 glycan binding proteins, which can block the terminal α2,6-Neu5Ac glycoconjugates in competition to the hemagglutinin protein of influenza virus, thus helping to inhibit influenza infection.182,183 However, the expression of α2,3-Neu5Ac in the salivary glycoprotein MUC5B and immunoglobulin-A of pregnant women decreased drastically, resulting in less screening of avian influenza virus specially in the third trimester.184 Antiviral agents such as zanamivir, oseltamivir, peramivir, laninamivir are known commercially available neuraminidase inhibitor molecules, while tert-butyl hydroquinone, arbidol and their derivatives could restrict the hemagglutinin attachment to the sialylated glycoconjugates of host cells.185 Human mumps virus and parainfluenza virus use viral hemagglutinin-neuraminidase proteins to target unbranched α2,3-Neu5Ac glycoconjugates, in contrast to the α2,6-Neu5Ac glycoconjugates preferred by the influenza virus.186–188 The hemagglutinin protein of adeno-associated virus serotype-1, -4, -5 and -6 recognises both α2,3-Neu5Ac and α2,6-Neu5Ac on N-linked glycoproteins.189
The VP1 of the human capsid protein viruses viz. BK polyomavirus and Merkel cell polyomavirus preferentially bind to α2,3-Neu5Ac glycoconjugates of urogenital tissues, astrocytes, oligodendrocytes, lymphocytes, renal cells, etc.,190,191 whereas human JC polyomavirus prefers α2,6-Neu5Ac glycoconjugates of the glial cells.192 The σ1 protein of reovirus binds GM2 (for type 1 serotype) and α2,3/6/8-Neu5Ac glycans (for type 3 serotype).193,194 The polypeptide VP8* of the spike protein VP4 of rotavirus interacts with subterminal Neu5Ac residue of the gangliosides GD1a and GM1 for key binding of the virus to host cells.195 The human adenovirus 37, causing severe epidemic keratoconjunctivitis, uses α2,3-Neu5Ac-glycans on GD1a ganglioside for the host cell binding and infectivity.196,197 On the other hand, the short fibre knob of human adenovirus 52 causing human gastroenteritis, preferentially recognizes polySia sequence for causing infection.198
The glycocalyx of human cells is important in the mechanism of infection of the human corona viruses (CoVs) viz. CoV-229E, CoV-NL63, OC43-CoV, HKU1-CoV, severe acute respiratory syndrome coronavirus (SARS-CoV), middle east respiratory syndrome coronavirus (MERS-CoV) and novel corona virus (SARS-CoV-2).199 Upon attachment of CoV, the transmembrane protease serine-2 of the host cell splits the spike protein S of CoV at site S2′ (in between fusion protein and internal fusion protein of S2) into two subunits viz. S1 and S2, responsible for recognition and membrane fusion, respectively (Fig. 3[A]). The N-terminal domain (NTD) of the S1 unit i.e., S1A identifies the sialylated glycans on the cell surface and facilitates the initial binding of CoV to the host cell, while the C-terminal domain (CTD) of S1 i.e., S1B binds to the specific host receptors to assist fusion and cell internalization.200 Among the human CoVs, the CoV-229E and CoV-NL63 do not require Sia for host cell adhesion, rather they directly interact with aminopeptidase N receptor and angiotensin-converting enzyme 2 (ACE2), respectively.201,202 The CTD of SARS-CoV binds to ACE2 cell receptor for recognition and host cell entry. On the other hand, OC43-CoV and HKU1-CoV viruses, causing common cold, pneumonia and bronchiolitis, have homodimeric hemagglutinin-esterase in S1A and that recognises 9OAc-Neu5Ac glycans as host cell receptors.203 The S1A of MERS-CoV preferentially interacts with α2,3-Neu5Ac glycans compared to α2,6-Neu5Ac glycans, while S1B is involved in adherence to the specific cell surface entry receptor dipeptidyl peptidase 4.204
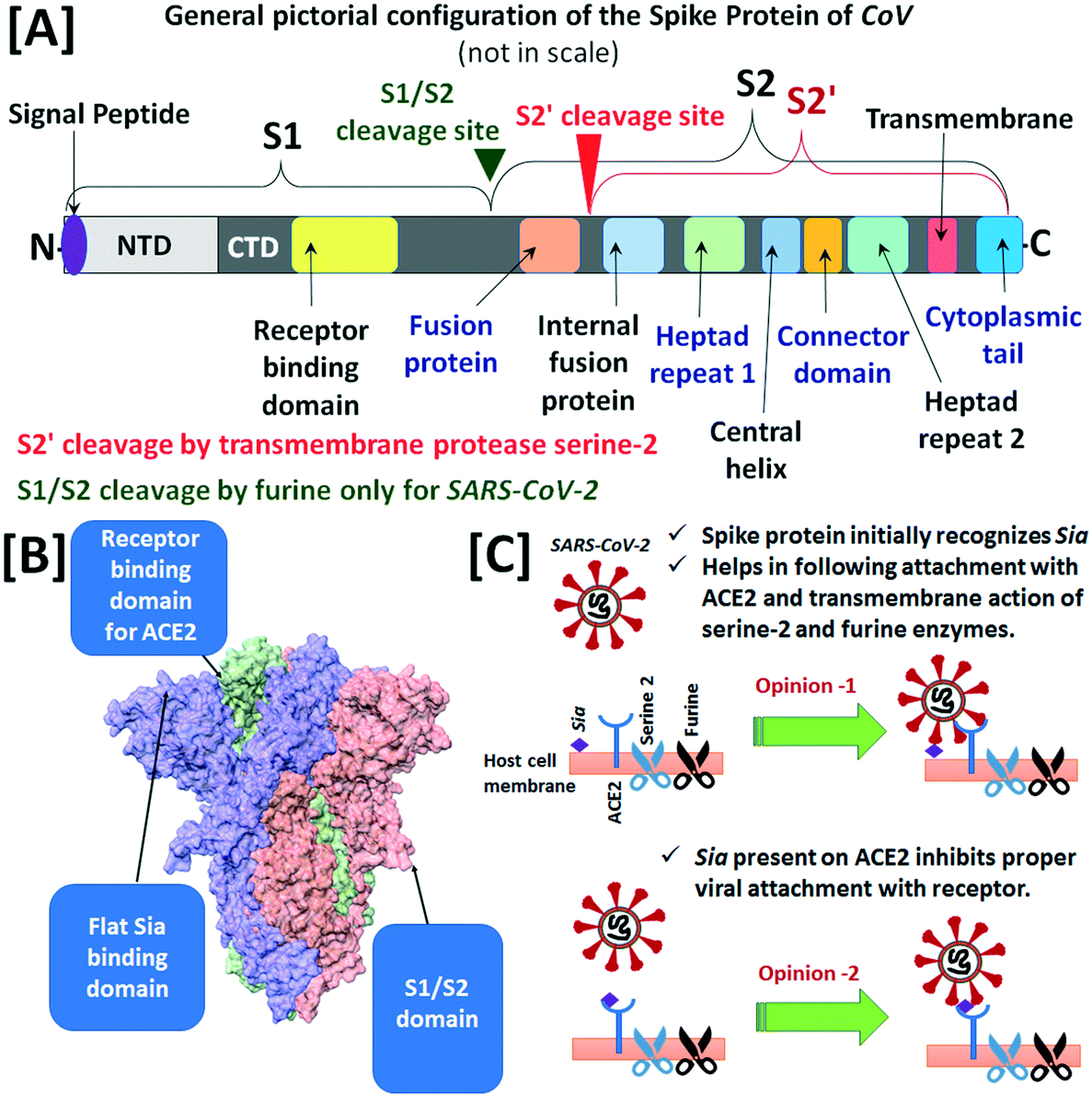 | ||
| Fig. 3 [A] A pictorial configuration of the main constituents of the spike protein of SARS-CoV-2 indicating the cleavage sites viz. S1/S2 and S2′. [B] The model of unique feature of the spike protein of SARS-CoV-2 (main figure used with permission from ref. 206) Copyright 2020 John Wiley and Sons. [C] The contradictory opinions about the role of Sia in the attachment to human cells (figures are not to scale). | ||
The novel corona virus SARS-CoV-2 which is responsible for the COVID 19 disease, is now considered most infectious among the group of human CoVs. The spike protein of SARS-CoV-2 has another cleavage motif at the boundary of S1–S2 subunits for furin-like enzymes of the host cell in addition to the transmembrane protease serine-2 unit, which is less expressed in the human lungs (Fig. 3[A]). The specific amino-acid sequence of the furin-like cleavage site is not present in other types of human CoVs. Hence, more efficient cleavage of the spike protein sub-units by both transmembrane protease serine-2 and furin of the host cell may be one of the possible reasons for higher pathogenicity of SARS-CoV-2 compared to SARS-CoV and MERS-CoV.205 On the other hand, the presence of a flat and non-sunken sialic acid-binding unit at NTD of S1 subunit of the spike protein has theoretically been proposed in a recent article (Fig. 3[B]).206 The recent in silico (performed through molecular docking simulations and electron density mapping) and computational studies, in line with the cryogenic microscopic evidence, have suggested the involvement of a flat Sia binding domain in the NTD of the spike protein in the establishment of initial contact with the sialylated glycoconjugates and GM1 of the host epithelium. This initial interaction is proposed to provide higher affinity binding between the spike protein and ACE2 receptor (opinion 1 in Fig. 3[C]).42,207,208 Furthermore, the in silico computational representation of the molecular isoelectronic density surface in terms of 2D Zernike polynomials also suggested the interaction of spike protein of SARS-CoV-2 with Sia receptors of the cells in the upper airways.209 Recently, a computational study in synchronization with mass-spectrometric and surface plasmon resonance (SPR) experiments on the interaction between Neu5Ac and SARS-CoV-2 spike protein reported that both up and down states of receptor-bonding-domain (known as RBD) of the spike protein moderately bind with Neu5Ac.210 Furthermore, an exploration in the Human Protein Atlas repository revealed high expression of Neu5Ac biosynthesis enzymes viz. glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase, Neu5Ac-9-phosphate synthase and Neu5Ac-9-phosphate phosphatase in the salivary glands and hence, proposed about a possible role of Neu5Ac in SARS-CoV-2 infection.211 On the other hand, the participation of other co-receptors viz. CD147,212 heparan sulfate,213etc. is also proposed for the initial recognition of the virus by the host cell. Moreover, complex sialylated N- and O-glycans have recently been evident on human ACE2,214 but those glycans have shown indication of resistance in the viral recognition by the host cells.215 Furthermore, the studies of SARS-CoV-2 infection on human epithelial lung cells and ex vivo human lung tissues revealed that heparan sulfate is an important co-receptor of this virus, while the sialylated glycans present on ACE2 can prevent protein spike and ACE2 interaction, which is most needed for viral attachment to the host cell (opinion 2 in Fig. 3[C]).216 A study of the binding of soluble form of SARS-CoV-2 spike protein (in which entire external domain was stabilised by six proline residues and secondary antibody StrepMAB Classic, anti-twin-Strep-tag MoAb) to a sialoglycan microarray presenting 139 glycans representative of common terminal structures on vertebrate glycans showed the preferential affinity order as α2,3-Neu5Ac > 4OAc-Neu5Ac > α2,6-Neu5Ac ∼ 9OAc-Neu5Ac ∼ 9NAc-Neu5Ac > α2,3-Neu5Gc ∼ α2,6-Neu5Gc. This multivalent Sia-affinity of the spike protein may be responsible for the formation of small and long-lived aerosol particle production from human upper airways.217 Recently published work by Nguyen et al. (during the proof-reading period of this review and included here for completion), revealed that the receptor-binding domain of the spike protein of SARS-CoV-2 has good binding affinity towards the gangliosides (GM1 and GM2), acidic (sialylated) HMOs and ABH blood group antigens (particularly A and H types 2, 3, 4).218 The authors used catch-and-release ESI-MS and hydrophilic-interaction-ultra HPLC to scan the affinity of both receptor-binding domain and S protein of SARS-CoV-2 over 132 mammalian and 7 non-mammalian glycans. The work provides an insight into aspects of COVID 19 disease 1) glycolipids of the host cell, especially gangliosides GM1 and GM2 in competition with cell surface heparan sulfate, bind to a common site of receptor-binding domain and S protein of SARS-CoV-2 and facilitate viral entry by the action of ACE2. The human brain and central nervous system share a higher fraction of gangliosides compared to other human organs (see Section 2.1.2), and this work aids understanding of the spread of SARS-CoV-2 infection to the central nervous system.219 2) Human milk has the lowest fraction (∼12–14%) of acidic (sialylated) HMOs which can bind to the receptor-binding domain and the S protein of SARS-CoV-2. In this study, none of the 72 neonates breastfed for 14 days by COVID 19 positive mothers tested COVID 19 positive.220 3) Antigen A of human blood cells predominantly stimulates (compared to other antigens B and AB) the formation of sialoside clusters in target cells through cis-carbohydrate–carbohydrate interactions. Therefore, humans having blood group A are more susceptible to SARS-CoV-2 infection.221 Furthermore, Nguyen et al. showed that decreasing cell surface Sia by either sialyltransferase inhibition, genetic knockout of biosynthesis of Sia or neuraminidase treatment decreased the SARS-CoV-2 infectivity by 30–40% and this information is now very useful for the development of COVID 19 therapy.218 However, several questions on Sia-SARS-CoV-2 interactions including the influence on the human immune response and change of viral affinity with new variants of SARS-CoV-2 are yet to be explored.222 Overall, the role of Sia for SARS-CoV-2 infection is still an open question.44
The infection of SARS-CoV, MERS-CoV and SARS-CoV-2 to human organs leads to the release of high levels of pro-inflammatory cytokines and chemokines, which is known as a “cytokine storm”. It is proposed that the cross reactivity of Sia with the host immune lectins might be responsible for this cytokine storm.43 Gas and water phase quantum mechanical calculations revealed that the monoprotonated form of anti-malarial drugs chloroquine and hydroxychloroquine, having anti-inflammatory properties, can form stable complexes with Neu5Ac. This may interfere with the ACE2 receptor and spike protein interaction and subsequently inhibit SARS-CoV-2 infection.223 Thus, in the uncertain therapeutic trials against COVID-19, these two drugs are being administrated to COVID-19 patients to fight against SARS-CoV-2 infection.224–226 However, more clinical studies are recommended for the prolonged use of chloroquine and hydroxychloroquine solely or in combination with other drugs for COVID-19 treatment.227 Another drug viz. remdesivir, which showed reliable clinical performance for MERS-CoV and SARS-CoV in the past pandemics, is also being used for clinical treatment of COVID-19.228,229 Interestingly, owing to the preferential affinity of Neu5Ac towards the spike protein glycoprotein of SARS-CoV-2 compared to SARS-CoV, a paper-based lateral flow point-of-care diagnostic sensor is prepared with Neu5Ac modified poly(N-hydroxyethyl acrylamide) coated gold nanoparticles to detect SARS-CoV-2 spike protein down to 5 mg L−1 concentration within 30 min through a biolayer interferometry analysis.230
2.3 Sia as potential biomarker for human diseases
| Human organ/system affected | Patients having addiction/cancer/disease and healthy controls | Type of samples | Number of individuals (age group) | Concentration of Sia [probability (P) value] | Ref. | ||
|---|---|---|---|---|---|---|---|
| Total Sia | Bound Sia | Free Sia | |||||
| Addictions | |||||||
| Oral | Smokeless tobacco (Paan) consumers | Saliva | 44 (27.52 ± 9.9 years) | 39.57 ± 26.58 mg L−1 (P 0.63) | NR | 23.21 ± 18.98 mg L−1 (P 0.001) | 239 |
| Non Paan consumers | 50 (29.5 ± 10.3 years) | 38.39 ± 28.55 mg L−1 | NR | 18.11 ± 16.5 mg L−1 | |||
| Smokers | 33 (23.80 ± 3.43 years) | 62.60 ± 3.91 mg L−1 (P < 0.001) | NR | NR | 240 | ||
| Maras powder users | 37 (29.03 ± 6.46 years) | 75.52 ± 6.86 mg L−1 (P < 0.001) | NR | NR | |||
| Healthy control | 30 (22.23 ± 2.45 years) | 51.60 ± 3.51 mg L−1 | NR | NR | |||
| Liver | Alcoholics | Serum | 51 (mean 49 years) | 1.449 ± 0.3019 mM (NR) | NR | NR | 242 |
| Alcoholics with liver disease | 32 | 1.421 ± 0.3082 mM (NR) | NR | NR | |||
| Alcoholic without liver disease | 19 | 1.497 ± 0.2927 mM (NR) | NR | NR | |||
| Non-alcoholics | 20 (mean 51 years) | 1.154 ± 0.1702 mM | NR | NR | |||
| Alcoholics | 106 (20–71 years) | 76.8 ± 14.9 mg dL−1 (P < 0.001) | 13.67 ± 3.55 mg dL−1 (lipid bound) (P 0.003) | NR | 245 | ||
| Non-alcoholics | 50 (21–76 years) | 63.1 ± 8.2 mg dL−1 | 11.67 ± 2.08 mg dL−1 (lipid bound) | NR | |||
| Alcoholics | 49 (24–70 years) | 82 ± 13.5 mg dL−1 (female); 85 ± 19.8 mg dL−1 (male); P < 0.001 | NR | NR | 246 | ||
| Non-alcoholics | 38 (40 years) | 60 ± 10.4 mg dL−1 (female); 64 ± 16.0 mg dL−1 (male) | NR | NR | |||
| Diseases | |||||||
|---|---|---|---|---|---|---|---|
| Central nervous system and brain | Pyogenic meningitis | Cerebrospinal fluid | 5 (NR) | NR | 38–1450 μM (NR) | 80–620 μM (NR) | 247 |
| Tuberculous meningitis | 5 (NR) | NR | 32–220 μM (NR) | 13–26 μM (NR) | |||
| Healthy control | 9 (NR) | NR | 12–40 μM (NR) | 6–32 μM (NR) | |||
| Alzheimer | 39 (47–86 years) | NR | NR | 16.75 ± 3.89 μM (P = 0.001) | 248 | ||
| Idiopathic normal pressure hydrocephalus | 19 (70–86 years) | NR | NR | 13.35 ± 3.2 μM (P = 0.001) | |||
| Patients having cerebellar ataxia with free sialic acid | 5 (19–37 years) | NR | NR | 43.4 ± 11.0 μM | 250 | ||
| Patients having neurological symptoms including psychomotor retardation, cerebellar ataxia, paraplegia, parkinsonism or other extrapyramidal manifestations, neuropathy, psychiatric symptoms and leukodystrophy | 144 (1–80 years) | NR | NR | 8.2 ± 4.2 μM (P < 0.001) | |||
| Patients having other well-defined metabolic diseases | 88 (1–80 years) | NR | NR | 9.9 ± 5.6 μM (P < 0.001) | |||
| Salla | 3 (1–80 years) | NR | NR | 31.3 ± 4.9 μM | |||
| Alzheimer | Plasma | 60 (74.13 ± 1.683 years) | 0.7133 ± 0.0201 ng L−1 (P < 0.0001) | NR | NR | 249 | |
| Healthy control | 60 (70.00 ± 1.255 years) | 0.5972 ± 0.01866 ng L−1 | NR | NR | |||
| Autism spectrum disorder | 82 (4.22 ± 1.01 years) | 3.27 ± 0.90 mM (P < 0.01) | NR | NR | 252 | ||
| Healthy control | 60 (4.52 ± 0.84 years) | 5.32 ± 1.85 mM | NR | NR | |||
| Autism spectrum disorder with medication (risperidone, 0.32 ± 0.013 mg per day; 2–6 months) | Saliva | 28 (5.50 ± 2.05 years) | 0.102 ± 0.062 mM (P 0.027) | NR | NR | 251 | |
| Autism spectrum disorder without medication | 18 (5.50 ± 2.05 years) | 0.100 ± 0.099 mM (P 0.027) | NR | NR | |||
| Healthy control | 30 (5.35 ± 2.15 years) | 0.160 ± 0.097 mM | NR | NR | |||
| Blood and related organs (diabetes mellitus and associated diseases) | Type 1 diabetes mellitus | Serum | 20 (22–79 years) | 2.00 ± 0.37 mM (P < 0.02) | NR | NR | 253 |
| Healthy non-diabetes | 20 (19–66 years) | 1.98 ± 0.67 mM | NR | NR | |||
| Type 2 diabetes mellitus (T2DM) | 16 (36–67 years) | 2.32 ± 0.41 mM | NR | NR | |||
| Type 1 diabetes mellitus | 16 (34–71 years) | 1.84 ± 0.24 mM (P < 0.001) | NR | NR | |||
| Healthy non-diabetes | 16 (22–64 years) | 1.92 ± 0.35 mM (P < 0.001) | NR | NR | |||
| Type 1 diabetes mellitus with retinopathy | 6 (NR) | 2.38 ± 0.33 mM (P < 0.01) | NR | NR | |||
| Type 1 diabetes mellitus without retinopathy | 6 (NR) | 1.85 ± 0.26 mM | NR | NR | |||
| T2DM normoalbuminuria (albumin excretion rate 16.5 ± 6.6 μg min−1) | Serum (urine) | 26 (54.2 ± 9.9 years) | 1.9 ± 0.4 mM (0.08 ± 0.01 mol% Sia/creatinine) (P < 0.001) | 0.5 ± 0.2 mM (lipid bound) | NR | 254 | |
| T2DM microalbuminuria (albumin excretion rate 78.1 ± 23.2 μg min−1) | 52 (54.2 ± 9.9 years) | 2.0 ± 0.4 mM (0.08 ± 0.01 mol% Sia/creatinine) (P < 0.001) | 0.6 ± 0.2 mM (lipid bound) | NR | |||
| Healthy nondiabetic control (albumin excretion rate 1.1 ± 0.8 μg min−1) | 28 (NR) | 1.6 ± 0.1 mM (0.04 ± 0.01 mol% Sia/creatinine) | 0.5 ± 0.1 mM (lipid bound) | NR | |||
| T2DM and nephropathy comorbidity | 190 (26–79 years) | 93.2 ± 3.6 mg dL−1 (17.7 ± 1.4 mg dL−1) (P < 0.05) | NR | NR | 255 | ||
| Nephropathy | 36 (30–73 years) | 84.0 ± 2.7 mg dL−1 (14.3 ± 0.7 mg dL−1) (P < 0.05) | NR | NR | |||
| T2DM | 30 (38–70 years) | 82.6 ± 1.3 mg dL−1 (12.5 ± 0.9 mg dL−1) (P < 0.05) | NR | NR | |||
| Healthy control without T2DM and nephropathy | 240 (20–79 years) | 59.1 ± 3.6 mg dL−1 (9.2 ± 1.0 mg dL−1) | NR | NR | |||
| T2DM | Plasma (RBC membrane) | 146 (22–80 years) | 175.00 ± 68.89 μM (4.20 ± 2.14 mmol per mg protein) (P < 0.05) | NR | NR | 256 | |
| T2DM and nephropathy comorbidity | 174.13 ± 23.20 μM (4.00 ± 2.51 mmol per mg protein) (P < 0.05) | NR | NR | ||||
| T2DM and hypertension comorbidity | 176.00 ± 54.82 μM (4.14 ± 2.00 mmol per mg protein) (P < 0.05) | NR | NR | ||||
| T2DM and hyperlipidemia comorbidity | 160.80 ± 48.61 μM (3.93 ± 2.10 mmol per mg protein) (P < 0.05) | NR | NR | ||||
| Healthy control | 15 (22–80 years) | 149.9 ± 47.30 μM (5.84 ± 1.43 mmol per mg protein) | NR | NR | |||
| Gestational diabetes mellitus | RBC membrane | 16 (29 ± 6 years) | 81.29 ± 41.80 mg per g protein (P < 0.001) | NR | NR | 129 | |
| Healthy pregnant women | 25 (28 ± 5 years) | 96.30 ± 34.20 mg per g protein (P < 0.001) | NR | NR | |||
| Healthy nonpregnant women | 10 (30 ± 4 years) | 42.33 ± 15.85 mg per g protein | NR | NR | |||
| High in 50 g glucose challenge test (1 h; glucose > 140 mg dL−1) and 100 g oral glucose tolerance test (3 h; glucose > 176 mg dL−1) | Serum | 8 (20–38 years) | 3.05 (2.67–3.49) mM (P < 0.001) | NR | NR | 258 | |
| High in 50 g glucose challenge test (1 h; glucose > 140 mg dL−1); but low in 100 g oral glucose tolerance test (3 h; glucose ≤ 176 mg dL−1) | 36 (19–38 years) | 3.22 (2.34–5.04) mM (P < 0.001) | NR | NR | |||
| Normal in 50 g glucose challenge test (1 h; glucose ≤ 140 mg dL−1) | 61 (18–38 years) | 2.66 (1.2–4.59) mM | NR | NR | |||
| Behcet's syndrome | 16 (NR) | 95.6 ± 15.3 (76–129) mg dL−1 (P < 0.01) | 29.8 ± 7.4 (17.2–45.6) mg dL−1 (P > 0.05) | NR | 259 | ||
| Uveitis disease | 12 (NR) | 75.8 ± 8.1 (64–91.8) mg dL−1 (P < 0.01) | 26.6 ± 3.0 (21.4–32.4) mg dL−1 (P > 0.05) | NR | |||
| Healthy control | 22 (NR) | 59.5 ± 7.5 (47–75.5) mg dL−1 | 19.2 ± 3.2 (15–24.5) mg dL−1 | NR | |||
| Gum | Periodontitis | Saliva | 5 (NR) | NR | NR | 0.0040% (whole saliva) | 260 |
| 0.0019% (parotis saliva) | |||||||
| Rheumatoid arthritis | 5 (NR) | NR | NR | 0.0053% (whole saliva) | |||
| 0.0035% (parotis saliva) | |||||||
| Healthy control | 8 (NR) | NR | NR | 0.0047% (whole saliva) | |||
| 0.0024% (parotis saliva) | |||||||
| Gingivitis | 30 (46.70 ± 13.59 years) | 12.98 ± 18.06 mg dL−1 | NR | NR | 262 | ||
| Periodontitis | 33 (45.18 ± 8.98 years) | 19.81 ± 16.45 mg dL−1 | NR | NR | |||
| Healthy control | 30 (44.67 ± 9.51 years) | 5.79 ± 2.49 mg dL−1 | NR | NR | |||
| Chronic gingivitis | Saliva (serum) | 30 (27.73 ± 7.51 years) | 68.23 ± 2.71 mg dL−1 (65.65 ± 3.56 mg dL−1) P < 0.0001 | NR | NR | 261 | |
| Chronic periodontitis | 30 (31.23 ± 6.74 years) | 81.33 ± 3.94 mg dL−1 (75.98 ± 3.58 mg dL−1) P < 0.0001 | NR | NR | |||
| Healthy control | 30 (27.37 ± 9.69 years) | 39.05 ± 6.35 mg dL−1 (49.75 ± 4.87 mg dL−1) | NR | NR | |||
| Aggressive periodontitis | 10 (35.16 ± 6.81 years) | 5.83 ± 0.74 mg dL−1 (parotis); 10.01 ± 1.92 mg dL−1 (whole) (134.28 ± 27.84 mg dL−1); P < 0.001 | NR | NR | 263 | ||
| Chronic periodontitis | 10 (35.16 ± 6.81 years) | 4.16 ± 1.10 mg dL−1 (parotis); 8.87 ± 1.57 mg dL−1 (whole) (108.65 ± 17.71 mg dL−1); P < 0.001 | NR | NR | |||
| Chronic gingivitis | 10 (35.16 ± 6.81 years) | 3.72 ± 0.84 mg dL−1 (parotis); 4.72 ± 1.08 mg dL−1 (whole) (80.69 ± 9.09 mg dL−1); P < 0.001 | NR | NR | |||
| Healthy control | 10 (22.20 ± 1.55 years) | 3.01 ± 0.57 mg dL−1 (parotis); 3.54 ± 0.93 mg dL−1 (whole) (60.03 ± 10.62 mg dL−1) | NR | NR | |||
| Heart and arteries | Patient had myocardial infarction before 1 day | Plasma | 19 (64.8 ± 11 years) | 0.88 ± 0.29 g L−1 (P < 0.05) | 0.28 ± 0.13 g L−1 (P < 0.05) | NR | 264 |
| Patient had myocardial infarction before 2 days | 0.89 ± 0.30 g L−1 (P < 0.05) | 0.20 ± 0.12 g L−1 (P < 0.05) | NR | ||||
| Patient had myocardial infarction before 5 days | 0.75 ± 0.19 g L−1 (P < 0.05) | 0.27 ± 0.13 g L−1 (P < 0.05) | NR | ||||
| Healthy control | 19 (50.6 ± 18.6 years) | 0.62 ± 0.13 g L−1 | 0.24 ± 0.08 g L−1 | NR | |||
| Myocardial infarction | Serum | 100 (63.44 ± 10.51 years) | 297 (220–374) μg L−1 (P < 0.001) | NR | NR | 265 | |
| Unstable angina pectoris | 537 (64.96 ± 10.13 years) | 227 (114–312) μg L−1 (P < 0.001) | NR | NR | |||
| Healthy control | 129 (57.43 ± 9.67 years) | 207 (114–276) μg L−1 | NR | NR | |||
| Normal coronary artery | Plasma | 235 (58.6 ± 9.0 years) | 147.20 (123.42–186.79) μg L−1 | NR | NR | 267 | |
| Nonobstructive coronary atherosclerosis | 496 (62.5 ± 8.8 years) | 148.43 (123.88–181.57) μg L−1 | NR | NR | |||
| Stable angina | 135 (64.3 ± 8.1 years) | 143.70 (128.30–177.97) μg L−1 | NR | NR | |||
| Unstable angina | 587 (64.9 ± 9.0 years) | 178.25 (135.46–212.38) | NR | NR | |||
| Acute myocardial infarction | 566 (63.1 ± 10.8 years) | 182.34 (147.16–234.70) μg L−1 | NR | NR | |||
| Coronary artery disease | Serum | 16 (63.6 ± 10.3 years) | 71.91 (68.33–75.60) mg dL−1 | NR | NR | 268 | |
| Healthy control | 25 (56.9 ± 13.3 years) | 60.76 (57.04–65.92) mg dL−1 | NR | NR | |||
| Periodontitis in comorbidity with cardiovascular disease | Serum (saliva) | 26 (40–85 years) | 86.87 ± 15.61 mg dL−1 (9.48 ± 4.55 mg dL−1) (P < 0.0001) | NR | NR | 269 | |
| Periodontitis only | 26 (45–75 years) | 53.2 ± 7.89 mg dL−1 (5.05 ± 1.49 mg dL−1) | NR | NR | |||
| Carotid atherosclerosis | Serum | 322 (59.9 ± 5.2 years) | 75.0 ± 9.7 mg dL−1 (P < 0.0001) | NR | NR | 270 | |
| Healthy control | 323 (58.7 ± 5.4 years) | 70.7 ± 8.9 mg dL−1 | NR | NR | |||
| Hypertriglyceridaemia | 15 (59 ± 9 years) | 84.9 ± 21.5 mg dL−1 (P < 0.03) | 23.0 ± 4.3 mg dL−1 (P < 0.001) | NR | 271 | ||
| Cholesterolemia | 15 (53 ± 11 years) | 58.4 ± 11.7 mg dL−1 (P < 0.03) | 14.9 ± 4.7 mg dL−1 (P < 0.001) | NR | |||
| Healthy control | 15 (52 ± 14 years) | 64.9 ± 20.8 mg dL−1 | 12.0 ± 3.2 mg dL−1 | NR | |||
| Eye | Sympathetic ophthalmia | 16 (13–43 years) | 120.2 ± 12.6 (99–142) mg dL−1 (P < 0.001) | NR | NR | 272 | |
| Traumatic uveitis | 36 (12–46 years) | 71.3 ± 8.4 (60–86) mg dL−1 (P > 0.1) | NR | NR | |||
| Healthy control | 40 (16–40 years) | 67.8 ± 7.9 (56–83) mg dL−1 | NR | NR | |||
| Thyroid | Sub-clinical hypothyroidism | 35 (34.4 ± 10.3 years) | 37.1 ± 15.5 mg L−1 (P 0.775) | NR | NR | 273 | |
| Overt hypothyroidism | 25 (33 ± 9.3 years) | 58.2 ± 18.9 mg L−1 (P < 0.001) | NR | NR | |||
| Healthy control | 30 (32.5 ± 7.5 years) | 38.1 ± 12.0 mg L−1 | NR | NR | |||
| Bowl | Active ulcerations | 6 (24.3 ± 3.8 years) | 69.7 ± 9.4 mg dL−1 (P < 0.01) | NR | NR | 274 | |
| No active ulcerations (post-surgery) | 6 (35.5 ± 12.8 years) | 54.7 ± 5.5 mg dL−1 (P < 0.01) | NR | NR | |||
| Healthy control | 6 (32.3 ± 4.7 years) | 50.3 ± 4.4 mg dL−1 | NR | NR | |||
| Liver | Hepatitis B | 50 (19–71 years) | 1.32 (0.06–1.84) mM (P 0.047) | NR | NR | 275 | |
| Hepatitis C | 40 (19–67 years) | 1.67 (1.18–2.96) mM (P 0.025) | NR | NR | |||
| Healthy control | 30 (21–54 years) | 1.43 (0.96–2.16 mM) | NR | NR | |||
| Malignant jaundice | 55 (35–87 years) | 7.95 mg L−1 (median); Sia/protein 1.20 | NR | NR | 276 | ||
| Jaundice | 30 (35–87 years) | 6.86 mg L−1 (median); Sia/protein 1.03 (P < 0.001) | NR | NR | |||
| Healthy control | 24 (24–56 years) | 5.97 mg L−1 (median); Sia/protein 0.79 (P 0.041) | NR | NR | |||
| Alcoholic patients having cirrhosis | 31 (27–52 years) | 5.81 (3.89–10.06) mg L−1 (P < 0.001) | NR | 0.55 (0.34–0.94) mg L−1 (P 0.017) | 277 | ||
| Non-alcoholic patients having cirrhosis | 24 (27–52 years) | 5.43 (3.79–7.88) mg L−1 (P < 0.001) | NR | 0.49 (0.34–0.82) mg L−1 (P 0.035) | |||
| Patients having chronic viral hepatitis C | 24 (27–52 years) | 5.76 (4.42–8.11) mg L−1 (P < 0.001) | NR | 0.48 (0.34–0.63) mg L−1 (P 0.016) | |||
| Healthy control | 49 (21–79 years) | 6.22 (5.4–8.0) mg L−1 | NR | 0.49 (0.36–0.61) mg L−1 | |||
| Kidney | Chronic glomerulonephritis | Serum (urine) | 35 (25–60 years) | 82.3 ± 6.5 mg dL−1 (0.150 ± 0.03 mg mL−1) (P < 0.001) | 19.7 ± 0.9 mg dL−1 lipid bound (NR) (P < 0.001) | NR | 278 |
| Chronic renal failure | 27 (19–54 years) | 70.7 ± 5.5 mg dL−1 (pre-dialysis) | 17.7 ± 0.8 mg dL−1 lipid bound (pre-dialysis) | NR | |||
| 71.2 ± 6.8 mg dL−1 (post-dialysis) (0.147 ± 0.04 mg mL−1) (P < 0.001) | 20.6 ± 1.1 mg dL−1 lipid bound (post-dialysis) (NR) (P < 0.001) | ||||||
| Healthy control | 60 (16–64 years) | 59.8 ± 7.9 mg dL−1 (0.098 ± 0.04 mg mL−1) | 15.9 ± 0.8 mg dL−1 lipid bound | NR | |||
| Low glomerular filtration rate (8–82 mL min−1 per 1.73 m2) | Plasma | 16 (27–74 years) | NR | NR | 310–1260 μg L−1 (NR) | 279 | |
| GNE myopathy (glomerular filtration rate 85–155 mL min−1 per 1.73 m2) | 19 (30–65 years) | NR | NR | 151 ± 29 (109–206) μg L−1 (NR) | |||
| Healthy control (glomerular filtration rate > 90 mL min−1 per 1.73 m2) | 6 (NR) | NR | NR | 100–200 μg L−1 (NR) | |||
| Multiorgans | Hyperemesis gravidarum | Serum | 30 (27.17 ± 5.63 years) | 28.370 ± 8.69 mg L−1 (P < 0.05) | NR | NR | 280 |
| Healthy control | 30 (28.63 ± 6.21 years) | 20.978 ± 9.96 mg L−1 | NR | NR | |||
| Preeclampsia | Plasma | 47 (27.1 ± 5.5 years) | 103.9 (91.3–111.0) mg dL−1 (P < 0.05) | NR | NR | 281 | |
| Transient hypertension in pregnancy | 50 (24.7 ± 7.1 years) | 102.2 (96.7–108.9) mg dL−1 (P < 0.05) | NR | NR | |||
| Uncomplicated pregnant women | 40 (23.8 ± 5.8 years) | 104.4 (91.2–106.9) mg dL−1 (P < 0.05) | NR | NR | |||
| Non pregnant women | 20 (22.2 ± 4.0 years) | 72.0 (62.7–78.8) mg dL−1 | NR | NR | |||
| Brucellosis | Serum | 29 (NR) | 2386 ± 82 μM (P < 0.01) | NR | NR | 282 | |
| Healthy control | 64 (NR) | 1821 ± 44 μM | NR | NR | |||
| Cancers | |||||||
|---|---|---|---|---|---|---|---|
| NR: not reported. | |||||||
| Oral | Oral squamous cell carcinoma | Saliva | 30 (26–73 years) | NR | 3.02 ± 0.01 mg dL−1 (P < 0.001) | 4.30 ± 0.01 mg dL−1 (P < 0.001) | 295 |
| Healthy control | 30 (26–46 years) | NR | 2.06 ± 0.01 mg dL−1 | 3.40 ± 0.02 mg dL−1 | |||
| Oral cancer | Serum | 26 (23–50 years) | 94.69 ± 7.26 mg dL−1 (P < 0.001) | NR | NR | 296 | |
| Leukoplakia (pre-cancer) | 27 (23–50 years) | 60.30 ± 7.96 mg dL−1 (P < 0.001) | NR | NR | |||
| Oral sub mucous fibrosis (pre-cancer) | 27 (23–50 years) | 59.97 ± 6.03 mg dL−1 (P < 0.001) | NR | NR | |||
| Healthy tobacco consumers | 40 (23–50 years) | 48.52 ± 6.49 mg dL−1 (P > 0.05) | NR | NR | |||
| Healthy tobacco non-consumers | 40 (23–50 years) | 48.77 ± 5.88 mg dL−1 | NR | NR | |||
| Gingival cancer, palatal cancer, buccal cancer | 37 (16–80 years) | 69.46 ± 11.23 mg dL−1 (P < 0.01) | NR | NR | 297 | ||
| Salivary gland tumours | 23 (16–80 years) | 70.58 ± 12.07 mg dL−1 (P < 0.01) | NR | NR | |||
| Lip and skin cancer | 22 (16–80 years) | 59.24 ± 12.10 mg dL−1 (P < 0.01) | NR | NR | |||
| Tongue and floor cancer | 20 (16–80 years) | 75.01 ± 12.00 mg dL−1 (P < 0.01) | NR | NR | |||
| Malignant fibrous histiocytoma | 4 (16–80 years) | 85.63 ± 5.45 mg dL−1 (P < 0.01) | NR | NR | |||
| Sarcoma | 3 (16–80 years) | 64.06 ± 5.01 mg dL−1 (P < 0.01) | NR | NR | |||
| Mandibular cancer | 1 (NR) | 69.72 mg dL−1 (P < 0.01) | NR | NR | |||
| Benign tumours in oral and maxillofacial region | 60 (23–54 years) | 58.28 ± 8.34 mg dL−1 (P > 0.05) | NR | NR | |||
| Healthy control | 80 (22–44 years) | 55.02 ± 7.2 mg dL−1 | NR | NR | |||
| Bile duct | Cholangiocarcinoma | 89 (55 ± 10 years) | 2.75 ± 0.67 mM (P < 0.001) | NR | NR | 298 | |
| Benign hepatobiliary diseases | 38 (55 ± 14 years) | 2.33 ± 0.69 mM (P < 0.002) | NR | NR | |||
| Healthy control | 43 (44 ± 14 years) | 1.89 ± 0.46 mM | NR | NR | |||
| Colon/rectum | Colorectal cancer | 123 (34–88 years) | 3.27 ± 1.12 mM; 49.01 ± 16.52 μmol g−1 of total protein (P < 0.001) | 3.24 ± 1.11 mM; 48.59 ± 16.43 μmol g−1 of total protein (P < 0.001) | 0.03 ± 0.01 mM | 299 | |
| Healthy control | 72 (34–88 years) | 2.14 ± 0.41 mM; 23.90 ± 4.81 μmol g−1 of total protein | 2.12 ± 0.41 mM; 23.59 ± 4.78 μmol g−1 of total protein | 0.03 ± 0.01 mM | |||
| Ovary | Endometrial neoplasia | 45 (37–81 years) | 2.38 ± 0.72 mM; 27.91 ± 8.00 μmol g−1 of protein (P < 0.001) | NR | NR | 300 | |
| Other neoplasia | 7 (37–81 years) | 2.53 ± 1.29 mM; 28.45 ± 11.80 μmol g−1 of protein (P 0.0086) | NR | NR | |||
| Healthy women | 20 (20–36 years) | 1.52 ± 0.25 mM; 19.28 ± 2.24 μmol g−1 of protein | NR | NR | |||
| Ascites cells | Tissue | 6 (adults), 7 (cords) | 1.46 μg g−1 (KDN); 91.2 μg g−1 (Neu5Ac) (NR) | 0.16 μg g−1 (KDN); 82 μg g−1 (Neu5Ac) (NR) | 1.3 μg g−1 (KDN); 9.2 μg g−1 (Neu5Ac) (NR) | 301 | |
| Tumour ovary | 0.87 μg g−1 (KDN); 244 μg g−1 (Neu5Ac) | 0.10 μg g−1 (KDN); 230 μg g−1 (Neu5Ac) | 0.77 μg g−1 (KDN); 14 μg g−1 (Neu5Ac) | ||||
| Healthy ovarian tissue | 0.56 μg g−1 (KDN); 170 μg g−1 (Neu5Ac) | 0.11 μg g−1 (KDN); 135 μg g−1 (Neu5Ac) | 0.45 μg g−1 (KDN); 35 μg g−1 (Neu5Ac) | ||||
| Throat | Tumour | 49 (mean 62.2) | 1.8 ± 0.2 μg g−1 (KDN); 85.2 ± 7.6 μg g−1 (Neu5Ac); 0.03 ± 0.01 μg g−1 (Neu5Gc) (P < 0.01) | 0.1 ± 0.01 μg g−1 (KDN); 79.7 ± 7.5 μg g−1 (Neu5Ac); 0.02 ± 0.01 μg g−1 (Neu5Gc) (P < 0.01) | 1.7 ± 0.2 μg g−1 (KDN); 5.5 ± 0.7 μg g−1 (Neu5Ac); 0.01 ± 0.003 μg g−1 (Neu5Gc) (P < 0.01) | 302 | |
| Regional lymph nodes | 10 (mean 62.2) | 1.2 ± 0.2 μg g−1 (KDN); 70.8 ± 7.9 μg g−1 (Neu5Ac); 0.015 ± 0.005 μg g−1 (Neu5Gc) | 0.1 ± 0.01 μg g−1 (KDN); 67.2 ± 7.8 μg g−1 (Neu5Ac); 0.01 ± 0.004 μg g−1 (Neu5Gc) | 1.1 ± 0.2 μg g−1 (KDN); 3.6 ± 0.5 μg g−1 (Neu5Ac); 0.005 ± 0.003 μg g−1 (Neu5Gc) | |||
| Lung | Advanced level of lung cancer | Serum | 3 (NR) | 4839 ± 443 μM | NR | NR | 282 |
| Healthy control | 64 (NR) | 1821 ± 44 μM | NR | NR | |||
| Breast | Breast cancer with secondary growth | 36 (NR) | 1773 ± 58 μM (not differentiable) | NR | NR | ||
| Breast cancer | 6 (NR) | 2034 ± 135 μM (not differentiable) | NR | NR | |||
| Healthy women | 64 (NR) | 1821 ± 44 μM | NR | NR | |||
| Blood forming tissue | Leukemia | Serum (plasma) | 16 (NR) (9 (NR)) | 2780 ± 110 μM (2769 ± 142 μM) (P < 0.01) | NR | NR | |
| Healthy women | 64 (NR) (13 (NR)) | 1821 ± 44 μM (1715 ± 42 μM) | NR | NR | |||
| Bladder | Freshly diagnosed tumour at bladder | Serum (urine) | 5 (NR) | 2400 ± 141 μM (P < 0.01) (327 ± 6 μM; 0.05 > P > 0.02) | NR | NR | |
| Healthy control | 64 (NR, serum) 18 (NR, urine) | 1821 ± 44 μM (176 ± 17 μM) | NR | NR | |||
| Bladder tumour | Urine | 60 (50 ± 15 years) | 99.80 ± 35.80 μg g−1 (Sia/creatinine) (P < 0.001) | NR | NR | 303 | |
| Healthy control | 34 (15 ± 8 years) | 52.57±15.60 μg g−1 (Sia/creatinine) | NR | NR | |||
| Bladder tumour | 73 (NR) | 92.37 ± 42.55 μg g−1 (P < 0.001) | NR | NR | 304 | ||
| Healthy control | 34 (NR) | 52.57 ± 15.6 μg g−1 | NR | NR | |||
2.3.2.1. Sia as biomarker of harmful addictions. The tobacco specific nitrosamines are metabolites of nicotine formed during the growth, curing, aging and processing of tobaccos. Smoking of burned tobaccos and chewing of smokeless tobaccos viz. paan (tobaccos and other ingredients wrapped with betel quid) and Maras powder (extracted from Nicotiana rustica Linn tobacco) increase chronic inflammation and concentration of reactive oxygen species in human body. The free Sia concentration in the saliva of addicted paan consumers (@1.41 ± 0.65 pack per day for 4.23 ± 3.42 years) was found higher (23.21 ± 18.98 mg L−1; P < 0.001) compared to non-consumer controls (18.11 ± 16.5 mg L−1), but the total Sia concentration in saliva did not statistically differ upon consumption of paan.239 On the other hand, the saliva of young group of regular Maras powder consumers (@∼20 g per day) showed presence of higher (75.52 ± 6.86 mg L−1; P < 0.001) concentration of total Sia compared to that of young group of regular tobacco smokers (@∼20 cigarettes per day) (62.60 ± 3.91 mg L−1; P < 0.001) and unaddicted controls (51.60 ± 3.51 mg L−1).240
The activity of sialidase increases in the presence of ethanol and its metabolites such as acetaldehydes, reactive oxygen species and fatty-acid ethyl esters, whereas the activity of sialyltransferases in the Golgi body decreases. Hence, the concentration of total Sia in human serum is generally elevated for chronic alcoholics (∼1000 g ethanol per week) due to desialylation of apolipoprotein J, cholesteryl ester transfer protein and transferrin.241 The concentration of total Sia in serum of 51 chronic alcoholics was found specifically higher (1.449 ± 0.3019 mM) compared that of 20 non-alcoholics (1.154 ± 0.1702 mM). However, those with or without liver disease could not be statistically differentiated from the measured concentration of total Sia in serum.242 Apolipoprotein J is highly sialylated (∼26–28 Sia per molecule) glycoprotein found in human plasma and serum and long-term alcohol intake inhibits the hepatic glycolization process of apolipoprotein J. About 28 moles of Sia per mole of apolipoprotein J (known as Sia index) was found in the blood of non-alcoholic healthy human (n = 20), while the Sia index decreased to 12–14 moles of Sia per mole of apolipoprotein J in the plasma of alcoholics (n = 15) consuming alcohol @85–90 g per day (P < 0.001)243 and interestingly Sia index started partially recovering slowly after stopping alcohol consumption.244 Although, the concentration of total Sia was found to be distinctly elevated in heavy alcoholics, a noticeable difference was observed in the concentration of lipid bound Sia, which was not much influenced by the status of the liver cells.245 The Sia index or elevated total Sia concentration in the serum of both male and female alcoholics can be used for biomarking alcohol abuse as an alternative or a supplement to the existing laboratory biomarkers viz. carbohydrate-deficient transferrin, γ-glutamyltransferase, mean corpuscular volume, aspartate aminotransferase and alanine aminotransferase. Sia is highly specific (95.5%) and sensitive (57.7%) as an alcohol abuse biomarker for female alcoholics, where its sensitivity is slightly inferior to γ-glutamyltransferase, comparable to carbohydrate-deficient transferrin and superior to other alcohol biomarkers. However, Sia (81.3%) and γ-glutamyltransferase (87.5%) have low specificity to identify male alcoholics compared to carbohydrate-deficient transferrin (100%).246
2.3.2.2. Sia as biomarker of disease. The concentration of total Neu5Ac in the cerebrospinal fluid significantly increased to 38–1450 μM for pyogenic meningitis and 32–220 μM for tuberculous meningitis compared to 12–40 μM for healthy controls. The high concentration of free Neu5Ac in the range 80–620 μM is much distinctive for pyogenic meningitis patients compared to healthy control (6–32 μM), which entirely covers the range of free Neu5Ac concentrations found in tuberculous meningitis patients (13–26 μM).247 The dementia disease viz. Alzheimer is difficult to be diagnosed from the neurodegenerative disease viz. idiopathic normal pressure hydrocephalus. The elevated concentrations of free Neu5Ac and glycerate simultaneously with demoted concentration of serine and 2-hydroxybutyrate in the cerebrospinal fluid could easily discriminate Alzheimer disease from idiopathic normal pressure hydrocephalus disease. The concentrations of free Neu5Ac in the cerebrospinal fluid of Alzheimer and idiopathic normal pressure hydrocephalus patients were found to be 16.75 ± 3.89 and 13.35 ± 3.2 μM, respectively.248Sia has antioxidant properties and thus defends oxidative stress in brain cells. The concentration of total Sia in the plasma of Alzheimer patients were found to be higher (0.7133 ± 0.0201 ng L−1) compared to that of healthy control (0.5972 ± 0.01866 ng L−1). The total Sia concentrations in the plasma of 38 female and 22 male Alzheimer patients were found to be 0.6814 ± 0.14022 and 0.6755 ± 0.1985 ng L−1, respectively.249 A rare Sia syndrome known as cerebellar ataxia is difficult to be diagnosed with free Sia as its patients usually have similar free Sia concentration in urine, plasma and peripheral nerves compared to healthy controls. However, the free Sia concentration in the cerebrospinal fluid of cerebellar ataxia patients were found to be much higher (43.4 ± 11.0 μM) compared to other patients having well-defined neurological symptoms (8.2 ± 4.2 μM) and other diseases (9.9 ± 5.6 μM) including Salla disease (31.3 ± 4.9 μM).250 In-cell polymerization of Sia forms polySia glycans, which are associated with NCAM and control the molecular interactions during neural development, synaptic plasticity, etc. Thus, the concentration of Sia in the saliva of children having autism spectrum disorder is significantly less (0.100 ± 0.099 mM) compared to healthy children (0.160 ± 0.097 mM). The medication of risperidone @0.32 ± 0.013 mg per day to a group of children for 2–6 months slightly increased the Sia concentration in saliva to 0.102 ± 0.062 mM.251 Similar trends are also observed in the concentration of Sia in the plasma of children having autism spectrum disorder compared to the healthy control.252
Crook et al. reported that the total Sia concentration in the serum of 20 type-1 diabetic mellitus patients (having 10.6 ± 4.5 mM plasma glucose concentration and taking daily insulin dose of 54.6 ± 21.2 unit) was 2.00 ± 0.37 mM, which was not statistically significant (P < 0.02) compared to that of 20 non-diabetic healthy humans (1.98 ± 0.67 mM). The comorbidity of retinopathy to type-1 diabetes mellitus further increased the total Sia concentration in serum to 2.38 ± 0.33 mM (P < 0.01). The total serum Sia concentration of 16 age-gender matched type-1 diabetic mellitus patients, type-2 diabetic mellitus (T2DM) patients and non-diabetic patients were found to be 1.92 ± 0.35 mM (P < 0.001), 2.32 ± 0.41 mM and 1.85 ± 0.26 mM (P < 0.001), respectively.253 Nephropathy is a disease which interrupts the usual activity of kidney due to the damage of its cell membrane. The neuraminidase activity increases in the blood of nephropathic patients compared to the healthy individual and it further increases with the comorbidity of T2DM. Diabetic normoalbuminuria and diabetic microalbuminuria are two pre-stages of diabetic nephropathy and those are identified by the daily urinary albumin excretion rate ≤30 and 31–299 mg, respectively. Although total Sia concentration in both serum and urine of T2DM patients having either normoalbuminuria or microalbuminuria distinguishably increased compared to the non-diabetic healthy controls, while no correlation in the concentration of lipid bound Sia concentration in serum was observed.254 Furthermore, the higher neuraminidase activity in both serum and urine increased independently due to T2DM and nephropathy diseases compared to healthy controls and therefore, the comorbidity of these two diseases further increased the neuraminidase activity in serum and urine. Hence, the total concentration of Sia in serum and urine of T2DM-nephropathic patients, nephropathic patients, T2DM patients and healthy control follow the same decreasing order.255
The lipid membrane of RBC becomes damaged due to the oxidative stress in the blood of T2DM patients. Therefore, the lipid peroxidation of RBC increases the concentration of Sia in plasma but decreases that on RBC. This effect becomes more pronounced in T2DM patients having nephropathy. Therefore, simultaneous monitoring of lipid peroxidation of RBC and Sia level on RBC could be able to predict the appearance of nephropathy in T2DM patients.256 A large cohort study including 40 years of follow-up on 6718 T2DM patients (3445 men and 3273 women) in the age-group of 47.2 ± 13.0 years showed that the median of Sia concentration in blood is 680 mg L−1 for both men and women. After adjusting the covariates such as age, body mass index, systolic blood pressure, cholesterol, aspartate aminotransferase and alanine aminotransferase activities and socioeconomic status, it was found that the elevated Sia concentration in the blood of T2DM patients was statistically correlated to a higher chance of incidence responsible for diabetes-related hospitalizations and the risk-factor (P < 0.0001) was more to women compared to men.257 The gestational diabetes mellitus disease is associated with higher whole blood viscosity and reduced erythrocyte deformability in the 26th week of pregnancy. Gestational diabetic mellitus patients have blood glucose concentration more than 140 mg dL−1 in the ‘1 h–50 g glucose tolerance test’ and having the same exceeding 176 mg dL−1 in the next ‘3 h–100 g oral glucose tolerance test’. The amount of Sia in RBC protein increases during the development of the pregnancy, but due to the irregularities in the microcirculation of the blood in gestational diabetic mellitus patients, there exists an imbalance in the amount of Sia in the RBC.129,258 The blood vessel inflammation happened due to Behcet's syndrome and thus because of that the total Sia concentration in serum increased to 95.6 ± 15.3 mg dL−1 compared to that of patients having uveitis disease (75.8 ± 8.1 mg dL−1) and healthy humans (59.5 ± 7.5 mg dL−1). However, the resolution in the lipid bound Sia concentration for diagnosis of Behcet's syndrome and uveitis disease was very poor.259
Periodontal disease happens due to the attack of oral bacteria at the gum tissue around the teeth. The recoverable early stage of periodontal disease is known as gingivitis, whereas the destructive acute stage of this disease is known as periodontitis. Zipkin et al. reported that the free Sia concentration in the parotid and whole saliva of young patients having periodontal disease decreased to 0.0019% and 0.0040%, respectively, compared to those of healthy control (0.0024% and 0.0047%, respectively) owing to its utilization in the bacterial plaque formation. However, the opposite phenomenon was observed for patients having autoimmune rheumatoid arthritis (0.0035% and 0.0053%, respectively).260 The inflammatory periodontal disease increases the oxidative stress in human body and to defend that oxidative stress, the total Sia concentration in human saliva and serum increases owing to higher sialidase activity. Therefore, the total Sia concentration in both saliva and serum progressively increases from gingivitis to periodontitis diseases compared to healthy controls.261–263
The concentration of Sia in plasma increases during and immediately after the event of myocardial infraction. Hence, the total plasma Sia concentration, but not the lipid bound Sia, was statistically correlated to the event of the myocardial infraction compared to the healthy control. The total plasma Sia concentration increased continuously after 1, 2 and 5 day(s) to 0.88 ± 0.29 g L−1 (P < 0.05), 0.89 ± 0.30 g L−1 (P < 0.05) and 0.75 ± 0.19 g L−1 (P < 0.05) compared to that in the plasma of healthy humans (0.62 ± 0.13 g L−1) not encountered the event of myocardial infraction. However, no difference and correlation were observed in the concentration of total serum Sia between patients and survivors.264 Recently, it was observed that Neu5Ac concentration in serum has similar diagnostic efficacy compared to the common biomarkers viz. creatine kinase-MB and troponin I for acute coronary syndrome. Patients having myocardial infarction have higher concentration (median 297 μg L−1) of Neu5Ac in the serum compared to the that of patients having unstable angina pectoris (median 227 μg L−1) and healthy control (median 207 μg L−1), however it cannot statistically differentiate between the ST-elevation and non-ST-elevation myocardial infarction. The cut-off value of Neu5Ac in serum was reposted as 330.5 μg L−1 (optimal) and 351.5 μg L−1 (high-risk) acute coronary syndrome.265 A cohort study on 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 429 men and 19414 women of age 59.5 ± 6.5 years along with 40 years of follow-up revealed that the elevated blood Sia concentration (>median 700 mg L−1) and pulse pressure (>median 70 mm Hg) could both independently predict the risk of cardiovascular incident for patients of cardiovascular diseases (P < 0.0001).266 It has recently been reported that Neu5Ac fits well in the cavity of the protein RhoA and Cdc42 in human plasma and activates Rho/Rho-associated coiled-coil containing protein kinase (both 1 and 2), c-Jun N-terminal kinase and extracellular signal-regulated kinase signalling pathways in cardiomyocytes, but it does not fit with Rac1 protein and thus it does not follow p38 signalling pathway. Hence, the elevated concentration of Neu5Ac in the serum of patients having normal coronary artery, nonobstructive coronary atherosclerosis, stable angina found in the range 143.70–147.20 μg L−1 and it increased to 178.25 μg L−1 (P = 9.3 × 10−3) in patients having unstable angina and 182.34 μg L−1 (P = 1.7 × 10−4) in patients having acute myocardial infarction.267 The coronary artery disease also caused desialylation of low-density lipoprotein and increased the total Sia concentration in serum of the patients (mean 71.91 (68.33–75.60) mg dL−1; P < 0.001) compared to the healthy control (60.76 (57.04–65.92) mg dL−1).268 Furthermore, total Sia concentration in the serum and saliva of periodontitis patients increased in the comorbidity of cardiovascular diseases.269 The total Sia concentration in the serum can also statistically identify (P < 0.0001) the induction of carotid atherosclerosis in humans caused by plaque deposition in the brain arteries.270 Crook et al. studied the total and lipid bound serum Sia concentrations in the patients having lipoprotein patterns of Fredrickson phenotypes IIa (cholesterolemic) and IIb (hypertriglyceridaemic). The total Sia concentration of healthy controls was found to be higher compared to the cholesterolemic patients, but lower compared to the hypertriglyceridaemic patients. However, the lipid bound Sia concentration was highest in hypertriglyceridaemic patients (23.0 ± 4.3 mg dL−1) compared to cholesterolemic patients (14.9 ± 4.7 mg dL−1) and healthy controls (12.0 ± 3.2 mg dL−1).271
429 men and 19414 women of age 59.5 ± 6.5 years along with 40 years of follow-up revealed that the elevated blood Sia concentration (>median 700 mg L−1) and pulse pressure (>median 70 mm Hg) could both independently predict the risk of cardiovascular incident for patients of cardiovascular diseases (P < 0.0001).266 It has recently been reported that Neu5Ac fits well in the cavity of the protein RhoA and Cdc42 in human plasma and activates Rho/Rho-associated coiled-coil containing protein kinase (both 1 and 2), c-Jun N-terminal kinase and extracellular signal-regulated kinase signalling pathways in cardiomyocytes, but it does not fit with Rac1 protein and thus it does not follow p38 signalling pathway. Hence, the elevated concentration of Neu5Ac in the serum of patients having normal coronary artery, nonobstructive coronary atherosclerosis, stable angina found in the range 143.70–147.20 μg L−1 and it increased to 178.25 μg L−1 (P = 9.3 × 10−3) in patients having unstable angina and 182.34 μg L−1 (P = 1.7 × 10−4) in patients having acute myocardial infarction.267 The coronary artery disease also caused desialylation of low-density lipoprotein and increased the total Sia concentration in serum of the patients (mean 71.91 (68.33–75.60) mg dL−1; P < 0.001) compared to the healthy control (60.76 (57.04–65.92) mg dL−1).268 Furthermore, total Sia concentration in the serum and saliva of periodontitis patients increased in the comorbidity of cardiovascular diseases.269 The total Sia concentration in the serum can also statistically identify (P < 0.0001) the induction of carotid atherosclerosis in humans caused by plaque deposition in the brain arteries.270 Crook et al. studied the total and lipid bound serum Sia concentrations in the patients having lipoprotein patterns of Fredrickson phenotypes IIa (cholesterolemic) and IIb (hypertriglyceridaemic). The total Sia concentration of healthy controls was found to be higher compared to the cholesterolemic patients, but lower compared to the hypertriglyceridaemic patients. However, the lipid bound Sia concentration was highest in hypertriglyceridaemic patients (23.0 ± 4.3 mg dL−1) compared to cholesterolemic patients (14.9 ± 4.7 mg dL−1) and healthy controls (12.0 ± 3.2 mg dL−1).271
A perforating injury to the eyes after surgical or accidental trauma causes a bilateral granulomatous uveitis known as sympathetic ophthalmitis. The concentration of total Sia in the serum of sympathetic ophthalmia patients was significantly higher (120.2 ± 12.6 mg/100 mL; P < 0.001) compared to that of the healthy control (67.8 ± 7.9 mg/100 mL). However, the symptoms of traumatic uveitis (71.3 ± 8.4 mg/100 mL; P > 0.1) in humans could not be statistically diagnosed compared to healthy controls.272 The concentration of Sia in the serum of patients having overt hypothyroidism (58.2 ± 18.9 mg L−1) was statistically distinctively elevated compared to that observed in the patients having sub-clinical hypothyroidism (37.1 ± 15.5 mg L−1) and healthy individual (38.1 ± 12.0 mg L−1) might be due to the influence of atherogenesis.273 The inflammation due to the formation of ulcerations in the digestive tract happens in Crohn's disease. The total Sia concentration in the serum of patients having active ulcerations was significantly increased to 69.7 ± 9.4 mg dL−1 compared to that of the healthy humans (50.3 ± 4.4 mg dL−1). The successful surgery of the ulcerations could decrease it to 54.7 ± 5.5 mg dL−1, which was close to the normal levels.274
Sialylation of glycoproteins and glycolipids happens in the liver and thus chronic liver disease viz. hepatitis B and C alters the total Sia concentration in the serum. The median of Sia concentration in serum decreases to 1.32 mM in hepatitis B patients from 1.43 mM in healthy human due to reduced sialyltransferase activity by hepatitis B viral infection. In contrast, the median of Sia concentration in serum increases to 1.67 mM in hepatitis C patients due to elevated sialyltransferase activity by hepatitis C viral infection, but it cannot provide any indication about the progress of either hepatitis B or C.275 The total Sia concentration in serum of patients having jaundice with malignant tumours of ampulla of Vater, pancreas, liver, cholecystitis and bile ducts was found 7.95 mg L−1 (median) with the Sia to total protein ratio 1.20 compared to those of healthy individual (5.97 mg L−1 (median); 0.79; P < 0.001). The total Sia concentration (6.86 mg L−1) and Sia to total protein ratio (1.03) are statistically different (P = 0.041) for patience having non-malignant jaundice viz. choledocholithiasis, cholecystolithiasis, calculous or non-calculous chronic cholecystitis, exacerbation of chronic cholangitis, stricture of the common bile duct.276 The free Sia concentration in serum was reported as a potential biomarker for chronic liver diseases viz. alcoholic cirrhosis, although it could not statistically differentiate non-alcoholic patients having cirrhosis and other liver diseases viz. hepatitis C, etc.277
The concentration of pyruvate increases in chronic renal failure and chronic glomerulonephritis. Thus, the biosynthesis of Neu5Ac becomes predominant in renal disease. The total Sia concentrations in serum and urine of patients having chronic renal failure and glomerulonephritis are statistically differentiable compared to the healthy control, however the concentration of lipid bound Sia does not meet this trend.278 The kidney can filter free Sia, which cannot be reabsorbed there. The concentration of free Sia in the plasma of patients having GNE myopathy (glomerular filtration rate 85–155 mL min−1 per 1.73 m2) was not differentiable compared to healthy controls, but the concentration of free Sia in plasma of patients having very low glomerular filtration rate increased drastically to 310–1260 μg L−1 due to the glomerular dysfunction.279
Pregnant women sometimes suffer from hyperemesis gravidarum disease during the 22nd week of pregnancy and it can be predicted and diagnosed from the elevation of the concentration of total Sia in the serum of pregnant patients having hyperemesis gravidarum (28.370 ± 8.69 mg L−1) compared to that in the healthy control group (20.978 ± 9.96 mg L−1) with statistical significance P < 0.05.280 Pregnancy syndrome preeclampsia is another pregnancy related disease resulting to high blood pressure, damage of liver and kidney extending even to neonatal and maternal morbidity and mortality. Owing to high inflammation in preeclampsia patients, Sia was thought to be a potential biomarker for this disease. However, studies showed that plasma concentration of Sia could not be considered as suitable biomarker compared to neopterin for diagnosing preeclampsia during pregnancy.281
The bacterial infection brucellosis caused by the consumption of raw and unpasteurized dairy products increased the total serum Sia concentration to 2386 ± 82 μM compared to the healthy controls (1821 ± 44 μM).282 Oxidative stress in the cell produces reactive oxygen species including hydrogen peroxide, which can be arrested by Neu5Ac and polysia to form a decarboxylation product 4-(acetylamino)-2,4-dideoxy-D-glycero-D-galacto-octonic acid (ADOA) at neutral pH ∼ 7.5.283
It is also observed that long exposure (21.00 ± 11.75 years) of pesticides to non-alcoholic and non-smoker field workers increased the total Sia concentration in serum to 44.854 ± 36.48 mg L−1 compared to the healthy control (581.95 ± 134.36 mg L−1) due to induction of several diseases originated from the pesticides.284 Similarly, workers exposed to cadmium in nonferrous smelter for about 8 years had lower Sia in RBC membrane (22.6 ± 1.8 mg g−1 of protein; P < 0.01) compared to the healthy controls (25.8 ± 3.0 mg g−1 of protein), however they had elevated concentration of total Sia in plasma (761.8 ± 83.5 mg L−1; P < 0.01) and urine (276.7 ± 132.3 μg g−1 of creatinine; P < 0.05) compared to the control group (640.4 ± 70.7 mg L−1; 174.5 ± 70.9 μg g−1 of creatinine).285 Synovial fibroblasts are stromal cells present in synovium joint and they maintain the lubrication and cartilage integrity of the joints. Local inflammation stimulates tumour necrosis factor, which inhibits the activity of glycosyltransferase ST6Gal1 and diminished α2,6-sialylation on synovial fibroblasts leading to rheumatoid arthritis.286
The Neu5Gc is not synthesised in the human body, but it is eventually released in human cells mainly through diets of dairy products and animal meats. Interestingly, the human natural immune system does not identify it as fatal “xeno (foreign)-antigen”, but automatically produces anti-Neu5Gc antibody even during the first year of human life.287 The concentration of anti-Neu5Gc antibody in the blood of healthy human (n = 120; 20–80 years) is reported as 2.7 ± 0.3 ng μL−1.288 However, excessive consumption of Neu5Gc rich diets (lamb, pork and beef meats, goat and sheep milks)289–292 increases the interaction of anti-Neu5Gc antibody with xeno Neu5Gc antigen and thus eventually may promote inflammations leading to cancers.28,293,294
2.3.2.3. Sia as cancer biomarkers. The oral squamous cell carcinoma is an oral cancer that happens due to excessive smoking of tobacco and drinking alcohol. Sanjay et al. reported that the concentrations of both lipid bound and free Sia in the saliva of 30 oral squamous cell carcinoma patients were higher (3.02 ± 0.01 and 4.30 ± 0.01 mg dL−1, respectively) compared to 30 healthy humans (2.06 ± 0.01 and 3.40 ± 0.02 mg dL−1, respectively).295 Other oral cancers, pre-cancers and tumours can be diagnosed from the total serum Sia concentrations. Bose et al. did not find any statistically significant difference in total Sia serum concentration between healthy tobacco consumers (48.52 ± 6.49 mg dL−1) and non-consumers (48.77 ± 5.88 mg dL−1). However, the total serum Sia concentration increased to 60.30 ± 7.96 and 59.97 ± 6.03 mg dL−1 for pre-cancers leukoplakia and oral sub mucous fibrosis, respectively. It further increased to 94.69 ± 7.26 mg dL−1 for patients having oral cancers.296 The total serum Sia concentration also increased in patients having malignant fibrous histiocytoma (85.63 ± 5.45 mg dL−1), tongue and floor cancer (75.01 ± 12.00 mg dL−1), salivary gland tumours (70.58 ± 12.07 mg dL−1), mandibular cancer (69.72 mg dL−1), gingival, palatal and buccal cancer (69.46 ± 11.23 mg dL−1), sarcoma (64.06 ± 5.01 mg dL−1), lip and skin cancer (59.24 ± 12.10 mg dL−1) and benign tumours in oral and maxillofacial region (58.28 ± 8.34 mg dL−1) compared to healthy controls (55.02 ± 7.2 mg dL−1).298
The serum concentration of total Sia increased to 2.75 ± 0.67 mM (P < 0.001) in the case of cholangiocarcinoma patients compared to the healthy controls (1.89 ± 0.46 mM) and further it remained statistically different (2.33 ± 0.69 mM; P < 0.002) from the serum concentration of Sia for patients having benign hepatobiliary diseases viz. liver abscesses, liver cirrhosis, cholecystitis, cholangitis, bile duct stricture and stones, pancreatic stones, gallstones, intrahepatic stones, etc.298 The concentration of bound as well as total Sia in the serum of patients having colorectal cancer affecting the colon and rectum significantly increased compared to the healthy control. Therefore, the total Sia concentration in serum and Sia to total protein ratio can be used to diagnose the induction of colorectal cancer and Dukes' stage.299
The monoclonal premalignant endometrial glandular lesion, which is known as endometrial intraepithelial neoplasia, is a pre-stage of endometrial adenocarcinoma. The patients having intraepithelial neoplasia had higher (2.38 ± 0.72 mM and 27.91 ± 8.00 μmol g−1 of serum protein) total Sia concentration in serum compared to healthy women (1.52 ± 0.25 mM and 19.28 ± 2.24 μmol g−1 of protein). However, it could not statistically discriminate endometrial intraepithelial neoplasia from other neoplasia.300 Inoue et al. first reported by using HPLC-FLD that the ratio of free KDN to free Neu5Ac increased to 6.3% in the ovarian cancerous RBC compared to 1.5% in RBC of healthy human ovarian tissue and it further increased to 16% in the RBC of ascites cells. A positive correlation of the ratio of KDN/Neu5Ac (both in free form) was observed in different types and stages of ovarian cancers. This ratio was found as 18% in a young woman having ovarian mature cystic teratoma, while it was found as 5.3% in the ovarian cyst of a patient having mucinous adenocarcinoma. The ratio significantly changed in the ascites cell having mucinous adenocarcinoma from 0.55% at incidence to 33% in stage IIIa. Further, it was proposed that free KDN in the RBC of ovarian cancer patients might act as oncofetal antigen and thus it overexpressed (1.6 μg free KDN per mL RBC; 0.7 μg free Neu5Ac per mL RBC) in the foetal cord RBC of neonates compared to the maternal RBC of mothers having malignant ovarian cancer (0.9 μg free KDN per mL RBC; 0.4 μg free Neu5Ac per mL RBC).301 Wang et al., in 2015, by LC-MS/MS found that the overexpression of KDN in human cells was not limited to only ovarian cancer, rather it was also overexpressed in the human body for many types of cancers. They further identified the presence of Neu5Gc in cancer cells. They reported a higher amount of free KDN in throat tumours compared to lymph nodes and furthermore, the overexpression of free KDN in throat cancers even occurred in the absence of lymphatic metastasis. The amount of total KDN also varied upon the tumour sites in the order of hypopharynx (2.4 ± 0.8 μg g−1) > glottis (1.9 ± 0.3 μg g−1) > supraglottic region (1.7 ± 0.3 μg g−1) > sub-glottic region (0.8 ± 0.11 μg g−1).302 However, the mechanism of synthesis of KDN in the cancerous cells is still unconvinced to date.
The total serum Sia concentration of 3 patients having active level of lung cancer was found to be sufficiently high (4839 ± 443 μM) compared to that of healthy controls (1821 ± 44 μM). Thus, total serum Sia can be considered as a potential biomarker for lung cancer, but not for breast cancer as the total serum Sia concentration of 3 patients having breast cancer and 36 patients having secondary growth were found to be 2034 ± 135 and 1773 ± 58 μM, respectively.282 On the other hand, the total Sia concentrations in both serum and plasma of leukemia patients (2780 ± 110 and 2769 ± 142 μM, respectively) were found to be sufficiently higher compared to healthy humans (1821 ± 44 and 1715 ± 42 μM, respectively).282 The ratio of total Sia to creatinine concentration in the urine was a good marker for bladder tumours as this ratio significantly increased to 99.80 ± 35.80 μg g−1 (ref. 303) and 92.37 ± 42.55 μg g−1 (ref. 304) for the tumour patients compared to the healthy human (52.57 ± 15.60 μg g−1; P < 0.001) and interestingly with the progress of the treatment/therapy this ratio decreased and became closer to the control value indicating the disappearance of the tumour from the bladder.303,304 Further, a recent study on human glial cell line (1321N1) revealed that Sia assisted in the upregulation of interleukine-4,10 gene expressions resulting into metastasis of glioblastoma (an aggressive brain cancer), but in contrast lipopolysaccharide retarded the same.305
3. Commercial production of Sia
3.1 Extraction of Sia from natural resources
The EBN is one of the largest commercial resources of free Neu5Ac. Owing to high content i.e., ∼2–5% w/w of extractable Neu5Ac in boiling water, stew of EBN is very popular in eastern countries. In 1977, Martin et al. developed a facile method to extract 6.5 g of Neu5Ac from 300 g of EBN simply by refluxing the pulverized EBN in boiling water for 5 h followed by partial-freeze drying pre-concentration and subsequently ultra-filtration through Diaflo XM-50 and Diaflo UM-10 filters. The filtrate was again passed through Dowex 1 × 4 ion-exchange column for further purification and crude Neu5Ac was obtained by lyophilization of the purified filtrate. The crude Neu5Ac was then dissolved in water, passed through Dowex 1 × 4 and lyophilized to achieve pure Neu5Ac.306 In 2018, Wong et al. reported that 48 h of digestion of EBN in a simulated gastric fluid at pH 2 and 37 °C could completely release free Neu5Ac digestible by humans.307 Nowadays EBN has been used in foods, cosmetics and pharmaceuticals as one of the ingredients. It is being commercially produced and exported worldwide from Thailand, Indonesia, Malaysia and China generating a good revenue. However, commercially produced EBN in swiftlet farms generally contains nitrite, minerals, heavy metals, fungi, mites and bacteria contaminants originating from the farm, packing materials and adulterants such as egg white, swim bladder, porcine skin, white fungus. Thus direct consumption of commercial EBN has associated risk.308–311κ-Caseinoglycomacropeptide (cGMP) is a C-terminal sialylated phosphorylated glycopeptide having 64 amino acid residues and it is another recognised natural source of Sia.312 Chymosin enzyme releases cGMP during cheese making and then cGMP is isolated from the caseinate hydrolysate. The regular cGMP contains ∼81% (w/w) protein and 42–52 mg Sia per g of protein, however pre-treatment of bovines with exogenic Sia may enrich the Sia concentration in cGMP to 190–230 mg Sia per g of protein and reduce the protein content to 50–60% (w/w) in cGMP. Hence, mixing of normal or enriched cGMP in infant formula is a means to bring the bovine based infant formula closer to human breast milk.313
Sialex,314 which is a Sia concentrate production of Ecological Formulas,315 is commercial dietary supplement available in the market and it is derived from mucin extract. The commercially available Lacprodan® CGMP-10/20 consisting of ≥95% cGMP, >81% protein concentrate and a very low level of phenylalanine is a good source of protein for people suffering from phenylketonuria.316
3.2 Chemical, chemoenzymatic and whole cell biocatalytic synthesis of Neu5Ac
Danishefsky et al. reported the stereoselective method for chemical synthesis of Neu5Ac starting from lactic ester by using furan surrogate and boron trifluoride etherate catalyst. However, the yield of Neu5Ac was not encouraging for large scale production.317 The chemical synthesis of Sia glycoconjugates remains a challenging task despite impressive developments in this field which have allowed for very precise control of the stereochemical outcome in Sia's glycosylation reactions with α-anomers typically being the targets with biological relevance (Fig. 4).318 This often requires the multi-step synthesis of complex Neu5Ac and other Sia glycosyl donors.319,320 Some of these difficulties are related to the unusual structure of the 9-carbon backbone of Sia, the hindered tertiary anomeric centre, the presence of an electron-withdrawing carboxyl group directly linked to the anomeric carbon and the lack of neighbouring participating group in Sia.321 | ||
| Fig. 4 Schematic representation of a generic sialylation reaction. | ||
For these reasons, chemoenzymatic strategies have been found to be efficient approaches for the preparation of Sia and their derivatives.322–324 Neu5Ac can be produced in large scale in a two-step enzymatic process, where GlcNAc is initially converted to ManNAc by N-acetylglucosamine-2-epimerase (EC 5.1.3.8) under alkaline condition and then ManNAc produces Neu5Ac in the presence of excess pyruvate and sialic acid aldolase. Here, the aqueous alkaline solution (i.e., pH > 11) of GlcNAc mixed with a catalytic amount of epimerase (cloned and developed from porcine kidney325) is kept at 25–35 °C for 12–20 h to reach the equilibrium ratio 4:1 of GlcNAc:ManNAc followed by neutralisation with acetic acid. The neutralised mixture is diluted with 6 times volume of isopropanol at 20–25 °C for precipitating out GlcNAc and enriching supernatant with ManNAc. This step is usually repeated several times to reach the ratio 1:4 (for large scale) or 1:10 (for lab scale) of GlcNAc:ManNAc. The ManNAc enriched mixture is incubated at pH 7–8, 20–30 °C for 48 h with sodium pyruvate in the presence of aldolase cloned from TG1[pMexAld] and TG1[pPLAld] strains of E. coli. Here, the ratio of ManNAc:pyruvate should be maintained as 1:2.5 to get ∼95% conversion of ManNAc to Neu5Ac, which is separated from the equilibrium mixture by ion-exchange chromatography.326 The isolation and purification of Neu5Ac from the equilibrium mixture is indeed a difficult step and thus a membrane reactor containing two embedded enzymes could be an economic way to produce large quantities of Neu5Ac, but it often encounters low production yield.327 Furthermore, the use of rhRnBp, which is a recombinant human renin binding protein, in the reaction mixture enhances the activity of both enzymes.328 The epimerase, which is cloned and developed from porcine kidney, has limited solubility in the aqueous solution. This issue is recently overcome by the strain BT0453 cloned and isolated from Bacteroides thetaiotaomicron, which is a human gut symbiont.329 Bloemendal et al. prepared a continuous flow reactor by immobilising aldolase on immobead-150P to increase the process efficiency and conversion factor (up to 82%) of Neu5Ac.330
The company Glycom A/S,331 Denmark, which was acquired by the Royal DSM332 in 2020, developed (in 2013) a two-step chemical and chemicoenzymatic method to prepare Neu5Ac·2H2O by avoiding the epimerization reaction. In the first step, N-substituted-D-mannosamine was prepared from a chemical reaction of D-fructose with 3–4 equivalent excess of benzylamine via Heyns-rearrangement reaction followed by hydrogenation on palladium black catalyst. Then ManNAc was prepared by acetylation of N-substituted-D-mannosamine. The second step of this method was the usual chemicoenzymatic aldolase reaction between purified ManNAc and sodium pyruvate. The product Neu5Ac·2H2O was washed, purified and crystallized several times with acetic acid and acetone.333
Whole cell biocatalysis is another prospective means to prepare Neu5Ac on a large scale. This concept was developed with an objective to avoid the tedious extraction of aldolase and epimerase enzymes as well as excessive use of exogeneous pyruvate. Whole cell biocatalysis directly uses a culture medium having an engineered strain of microorganism, which is unable to decompose Neu5Ac compared to natural strain but capable of in situ production of both enzymes necessary for the synthesis of Neu5Ac from a precursor (such as GlcNAc) and a carbon source (such as glucose or fructose or glycerol) in the culture medium. The engineered strain E. coli NAN8-71/pLT4 (ref. 334) and E. coli SA-05/pDTrc-AB/pCDF-pck-ppsA335 could successfully produce 35 and 18.17 ± 0.27 g L−1 Neu5Ac, respectively, by whole cell biocatalysis.
4. Timeline of regulatory affairs relevant to Sia in food and nutritional products
Neu5Ac is the most abundant Sia in healthy humans and it is biosynthesised endogenously de novo in cytosol through an anabolic physiological process. It participates directly and indirectly in many physiological processes especially in the growth and development of neonatal and infants as discussed in Section 2. Either compromised biosynthesis or heavy loss of Neu5Ac at any stage of life can induce harmful effects to humans especially in their early stages of life. In this direction, a strong hypothesis exists to compensate the deficiency of Neu5Ac by an external dietary input, where the dietary Sia could be adsorbed in the human body by an action of neuraminidase in the small intestine and colon.336,337 The European Parliament and Council of European Union issued regulation EC/258/97 in January, 1997 on novel foods and food ingredients.338 Although, the presence of Neu5Ac in human breast milk and bovine milk derived infant formula was evident by several research groups, the Scientific Committee on Food of the Health and Consumer Protection Directorate-General, European Commission, in May 2003, did not find adequate scientific data to consider Neu5Ac as an essential component for infant and follow-on formula.339 Thus, Neu5Ac was not included in the global standard for infant formula composition recommended by the international expert group coordinated by the European Society for Paediatric Gastroenterology, Hepatology and Nutrition in 2005.340 In December 2006, the European Parliament and Council of European Union issued another regulation EC/1924/2006 on the nutrition and health claims on foods. Article 13 of this regulation deals with inclusion of compounds adding nutritional value to growth, development and other functions of the body, psychological benefits, slimming effects, etc.341 Following this, the European Commission requested the European Food Safety Authority (EFSA) panel on dietetic products, nutrition and allergies to provide an opinion addressing scientific substantiation of claims for including Sia in food products under Article 13 of the regulation EC/1924/2006 owing to its positive role in learning and memory developments (Question No. EFSA-Q-2008-2330 dated on 02/07/2009). The panel evaluated the contemporary relevant information provided by the Member States, but did not find any conclusive evidence of benefits by the dietary intake of Sia in relation to learning and memory.342 Hence, scientific opinion in 2014 on the essential composition of infant and follow-on formula was that although Sia was recognised as a source of energy and macronutrients originating from milk, it was not recommended for deliberate addition for uncertainty probability of sub-chronic dietary toxicity effects.141Later in 2014, the sub-chronic dietary toxicity effect of Neu5Ac·2H2O as prepared by Glycom A/S (briefly mentioned in Section 4.2) was tested on both male and female Sprague–Dawley rats for 13 weeks. No observed adverse effect level for the oral toxicity of Neu5Ac·2H2O was determined as 974 (for male rats) and 1246 (for female rats) mg kg(body weight)−1 day−1. It was also found safe to use up to 2% level (i.e., 1895 mg kg(body weight)−1 day−1) of maternal dietary intake. Furthermore, this compound was assessed as non-mutagenic and non-genotoxic through the bacterial-reverse-mutation and in vitro mammalian cell micronucleus tests with samples taken from a 23 year old adult male.343 It was experimentally proven in 2014 that the supplement of Sia in the form of either free Sia, gangliosides, 3′-sialyllactose, cGMP or their combination during at least one stage of preconception, pregnancy and lactation could improve the health and development of the foetus and/or child.344
The progress of research on the health benefits of Sia, results on the lack of mutagenic, genotoxic and sub-chronic toxicity effect of Sia up to 2% dietary level and industrially viable production of Sia could set the scene for its industrial application. Hence, on September 24, 2015, Glycom A/S applied for the pre-market “Generally Recognized as Safe Exemption” approval for Neu5Ac·2H2O (off-white crystalline powder; purity ≥97%, stable for 36 months without any special package), which was structurally identical to the human milk's free Neu5Ac, to the office of Food Additive Safety in the Food and Drug Administration.345 The Food Safety Authority of Ireland made an initial assessment on that application in March 2016 and they did not identify any safety concern in using of Neu5Ac·2H2O in food products.346 Hence, Dossier 187 was issued in April 2016 by the advisory Committee for Novel Foods and Processes on the initial assessment.347 The EFSA Panel on dietetic products, nutrition and allergies (NDA) in June 2017 published a scientific opinion on the safety of synthetic Neu5Ac·2H2O as a novel food pursuant to EC regulation 258/97. They found it a safe additive for foods and food supplements at proposed levels suitable for general population above 10 years of age.348 Following that opinion, the European Commission published a decision EU/2017/2375 on December 15, 2017 authorising use of Neu5Ac·2H2O in the market as a food ingredient at recommended levels (Table 2) under regulation EC/258/97.41
| Category of Food where | Recommended level of Neu5Ac·2H2O |
|---|---|
| Infant and follow-on formulae | 50 mg L−1 of reconstituted formula |
| Baby foods for infants; processed cereal-based foods; foods for young children | 50 mg kg−1 of solid food |
| Special medical purpose food for infants and young children | Should be considered case to case; but in any case, not exceeding the specified level for the category mentioned in this table |
| Dietary intakes for weight control | 200 mg L−1 (for liquid); 1.7 g kg−1 (for solid) |
| Foods suitable for people having intolerance to gluten | 1.25 g kg−1 |
| Unflavoured pasteurised and sterilised milk; flavoured drinks; fruit juices and nectars; vegetable juices and nectars | 50 mg L−1 |
| Table-top sweeteners | 8.3 g kg−1 |
| Coffee, tea, herbal and fruit infusions; chicory and its extracts | 200 mg kg−1 |
| Cereal bars | 500 mg kg−1 |
| Unflavoured and flavoured fermented milk-based products including heat treated products after fermentation | 50 mg L−1 (for liquid); 400 mg kg−1 (for solid) |
| Dairy analogues including beverage whiteners | 50 mg L−1 (for liquid); 250 mg kg−1 (for solid) |
| Food supplements in any form for infants (≤1 year old) | 55 mg per day |
| Food supplements in any form for young children (1–2 year(s) old) | 130 mg per day |
| Food supplements in any form for children (3–10 years old) | 250 mg per day |
| Food supplements in any form for general population (>10 years old) | 300 mg per day |
As mentioned in Section 2.2.1, Sia-binding adhesin of Helicobacter pylori bacteria often targets cell surface gangliosides causing gastric adenocarcinoma. Hence, food ingredients/supplements having anti-adhesive agents have good commercial importance in terms of prevention and therapy of H. pylori related diseases. Although, bovine milk and chicken egg white are promising dietary sources of anti-adhesive agents against H. pylori, commercial preventive and therapeutic supplements having lactoferrin, fat globule membrane fractions and 3′-sialyllactose sodium salt have high prospect in the food industry.349–351 In April 2019, Phipps et al. reported the toxicological safety studies of 3′-sialyllactose sodium salt352 and 6′-sialyllactose sodium salt,353 which were manufactured by Glycom A/S through microbial fermentation process with a genetically engineered strain E. coli K-12-DH1-MDO. They performed in vitro genotoxicity studies in addition to a 90 day-oral (gavage) toxicity study initiating test doses from 7th day from birth of neonatal rats, which can equivalently represent the development of the reproductive system and central nervous system of new-born human infants. New-born infants consume ∼600 mL human milk per day and thus they can consume up to <360 mg per day of 3′-sialyllactose and ∼72–324 mg per day of 6′-sialyllactose only from human milk. Therefore, it was proposed to be safer to include 0.2 g L−1 of 3′-sialyllactose352 and 0.4 g L−1 of 6′-sialyllactose in reconstituted products.353 On August 21, 2019, Glycom A/S again applied for the pre-market “Generally Recognized as Safe Exemption” approval for 3′-sialyllactose sodium salt (GRAS Notice No. 880)354 and 6′-sialyllactose sodium salt (GRAS Notice No. 881)355 to the office of Food Additive Safety in the Food and Drug Administration. The European Food Safety Authority has recently reviewed both the applications and concluded these materials as safe according to the novel food pursuant regulation EU/2015/2283 for using as food supplements, consumption of which should be avoided on the same day of consuming human and bovine milks, fermented milk-based products, curd cheese, etc. so as not to exceed daily intake limits of 3′-sialyllactose and 6′-sialyllactose from other natural sources.356,357 As per the latest update available on DSM's website, GlyCare™ 3SL (ref. 358) and GlyCare™ 6SL (ref. 359) will be commercially available soon in the market.
Herrmann et al. recently reported a COGNIS study (a neurocognitive and immunological study of a new formula for healthy infants) on 220 healthy infants by feeding breast milk (n = 50), standard infant formula (n = 75) and nutrient enriched infant formula (n = 75) for 18 initial months since their birth. The enriched infant formula had a different composition of arachidonic acid (10.2–15.8 mg dL−1), docosahexaenoic acid (10.2–11.2 mg dL−1); gangliosides (9 mg L−1); nucleotides (2.92–2.94 mg dL−1) and sialic acid (105 mg L−1) compared to the standard infant formula. This study revealed that the risk of respiratory tract and gastrointestinal infections was reduced by 30.2% and 32.5%, respectively, in the infants consumed enriched formula compared to the infants consumed standard formula. The enriched infant formula showed almost similar benefits compared to human breast milk.360 Ma et al. have recently published an article stating that oral administration of Neu5Ac @ 0.1–0.3 mg per day to mice for 3 weeks increased the liver weight, gut villus volume, number of goblet cell; decreased activity of aspartate aminotransferase and changed the microbial composition and activity in gastrointestinal tract of mice.361 However, the effect of oral administration of Sia in human gastrointestinal tract is yet unreported.
5. Analytical approaches to the assay of Sia in biological and commercial food samples
The sections above demonstrate the commercial and biological relevance of Sia in relation to the immune system, the central nervous system, pregnancy and lactation, their role in bacterial and viral infections in addition to disease biomarkers. This clearly conveys the medical and commercial importance of accurate determination of Sia in human plasma, serum, urine, brain-fluid; commercial dairy products, infant formula; plants tissues and animal organs. It can be seen from Fig. 5[A] that a majority of assays of Sia up to 2014 was based on colourimetry-spectrophotometry (15%), enzymatic analysis (15%), high-performance liquid chromatography (HPLC) coupled with fluorescence detection (FLD) (15%), UV-visible detection (UV-vis) (13%) and mass-spectrometry (MS) (12%). Assays with photoluminescence (8%) and electrophoresis (7%) were moderately studied and HPLC coupled with pulse amperometric detection (PAD), amperometry (4%), quartz crystal microbalance (QCM) (3%), potentiometry (1%), SPR (1%) and surface enhanced Raman spectroscopy (SERS) (1%) were introduced in late 90s and early 2000s. In the last 7 years (i.e., 2015–2021), a significant thrust is evident in the development of photoluminescence (21%) and electrochemical assay of Sia including voltammetry and electrochemical impedance spectroscopy (EIS) (15%), potentiometry (8%), photoelectrochemistry (PEC) and electrochemiluminescence (ECL) (8%) and organic electrochemical transistor (OECT) (3%) (Fig. 5[B]). The development of colourimetry–spectrophotometry (8%), HPLC-UV-vis (5%), HPLC-FLD (5%) is significantly decreased, whereas shares of HPLC-MS (10%), electrophoresis (5%) and QCM (3%) remain almost similar. A slight increase in interest of SPR (3%) and SERS (8%) assay methods has been observed in the last 7 years. Citing key observations of 124 landmark publications, this section will briefly discuss the evolution of different assays for Sia accounting for the limitations of preceding methods.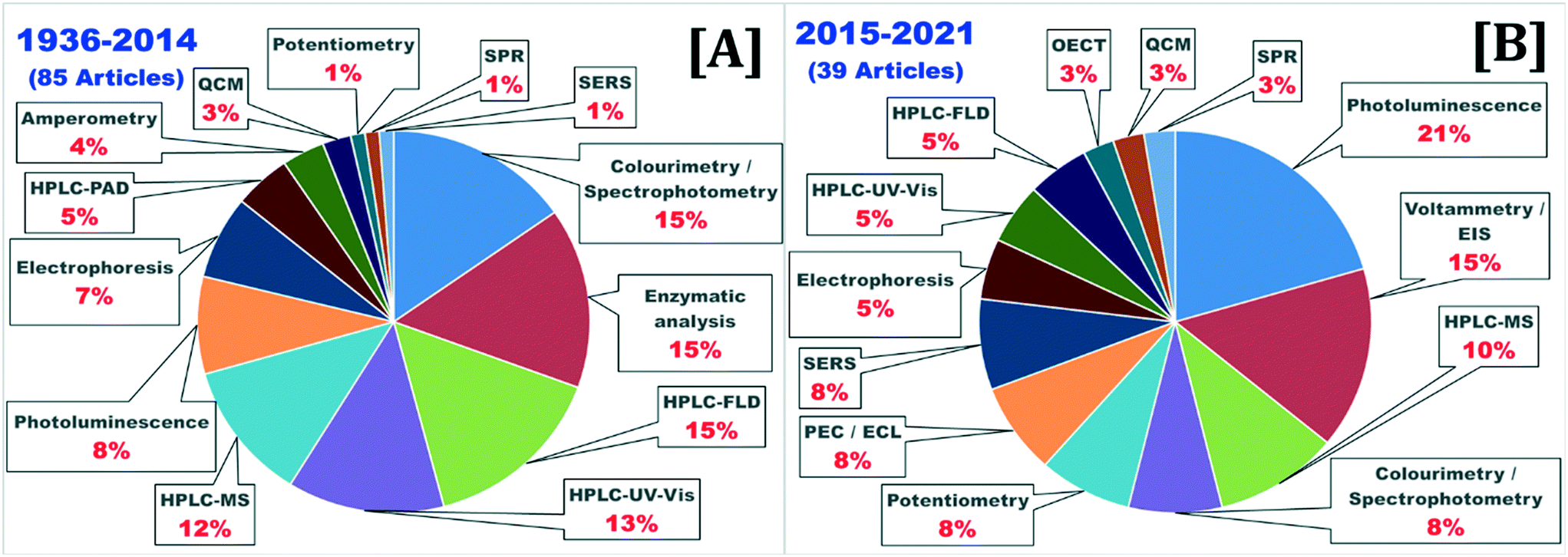 | ||
| Fig. 5 Statistical analysis of different analytical methods for assay of Sia in biological and commercial food samples reported during [A] 1936–2014 and [B] 2015–2021 (September). | ||
5.1 Exploitation of preferential interaction between Sia and boronic acid for Sia's assay
The boronic acid functional group, present in compounds known as “boronlectins”, chemically mimics the action of lectins for high affinity towards the diol of saccharides such as glucose at high pH.362 It is important to mention that many analytical methods exploited the preferential interactions of boronic acid functional group with Sia compared to other sugar molecules. Fig. 6[A] shows the molecules viz. 4-isoquinolineboronic acid, phenylboronic acid, 3-aminophenylboronic acid, 4-aminophenylboronic acid, 4-mercaptophenylboronic acid, p-vinylbenzeneboronic acid, 2-fluorophenylboronic acid, 1-decylboronic acid, 3-pyridylboronic acid, 3-(acrylamido)phenylboronic acid, etc. as mentioned in this review. The insights into specific interaction between Neu5Ac and phenylboronic acids (PBAs) are briefly discussed here. Otsuka et al. studied the complexes of free Neu5Ac and 3-(propionamido)phenylboronic acid in the pH range 4–10 by 1H, 11B, 13C and 15N nuclear magnetic resonance (NMR) spectroscopies. The binding constant (at physiological pH i.e., 7.4) of 3-(propionamido)phenylboronic acid complex with free Neu5Ac (37.6 ± 3.1 M−1) was found to be exceptionally high compared to that of galactose (15.0 ± 2.2 M−1), mannose (8.5 ± 0.9 M−1) and glucose (5.1 ± 1.2 M−1). They proposed that Neu5Ac possessed favourable intramolecular hydrogen bonding interaction between the glycerol moiety (involving –OH groups of C-7, C-8 and C-9) and amide group at C-5 (Fig. 6[A]). The –B(OH)2 functional group of the compound was proposed to interact with the glycerol side chain (C-7, C-8) of Neu5Ac.363 Later, Djanashvili et al., performed 11B, 13C and 17O NMR spectroscopy of phenylboronic acid (pKa 10.05 ± 0.03) complexes of glycolic acid, erythronic acid, threonic acid, Neu5Ac and 2α-methyl derivative of Neu5Ac in the pH range 2–12 and found that the stability constant of phenylboronic acid complex (at 25 °C and pH 7.4) of Neu5Ac (11.6 ± 1.9 M−1) is higher compared to that of glycolic acid (1.6 ± 0.2 M−1), erythronic acid (4.4 ± 0.5 M−1) and threonic acid (11.4 ± 1.5 M−1). They proposed, in contradiction to Otsuka et al.'s report,363 that phenylboronic acid interaction should be formed with the α-hydroxycarboxylate unit (i.e., C-1 and C-2) of Neu5Ac in the pH range 2–8 and with the glycerol tail (i.e., C-7/C-8 and C-9) of the side chain at pH > 8, but the involvement of C-7, C-8 glycerol tail was unfavourable due to the erythro configuration (Fig. 6[B]).364 Since then, the actual mechanism of boronic acid and Neu5Ac complexation was unresolved for a long period. Meanwhile it was also reported that this complexation might not occur for bound Neu5Ac in physiological conditions. Recently, Nishitani et al. performed 11B, 13C NMR spectroscopy and density functional theory simulations of phenylboronic acid complex with both α- and β-pyranoid structures of Neu5Ac and concluded that phenylboronic acid could bind with α-hydroxycarboxylate unit (i.e., C-1 and C-2) of β-pyranoid Neu5Ac, but could not interact with α-pyranoid Neu5Ac, which predominantly exists in membrane bound glycans. Furthermore, the C-2 position is also not available for the phenylboronic acid complexation due to blockage by glycoside bond. Therefore, phenylboronic acid or similar compounds although preferentially bind free Sia, they often fail to selectively recognize Sia at termini cell-surface glycans.365 The 4-isoquinolineboronic acid has recently showed highest binding affinity to Neu5Ac (formation constant 5390 ± 190 M−1) compared to common neutral saccharides (glucose, fructose, sorbitol, etc.), catechol, ribose-5-P, lactic acid and gluconic acid in aqueous phosphate buffer (pH 3) due to the formation of a ternary quaternary heterocyclic nitrogen containing 4-isoquinolineboronic acid-Neu5Ac-phosphate complex unlike vicinal diol complexes. Thus, at neutral pH, the affinity of 4-isoquinolineboronic acid to catechol, sorbitol, fructose became higher compared to Neu5Ac (formation constant 83 ± 23 M−1) and glucose (formation constant 8.9 ± 1.4 M−1).366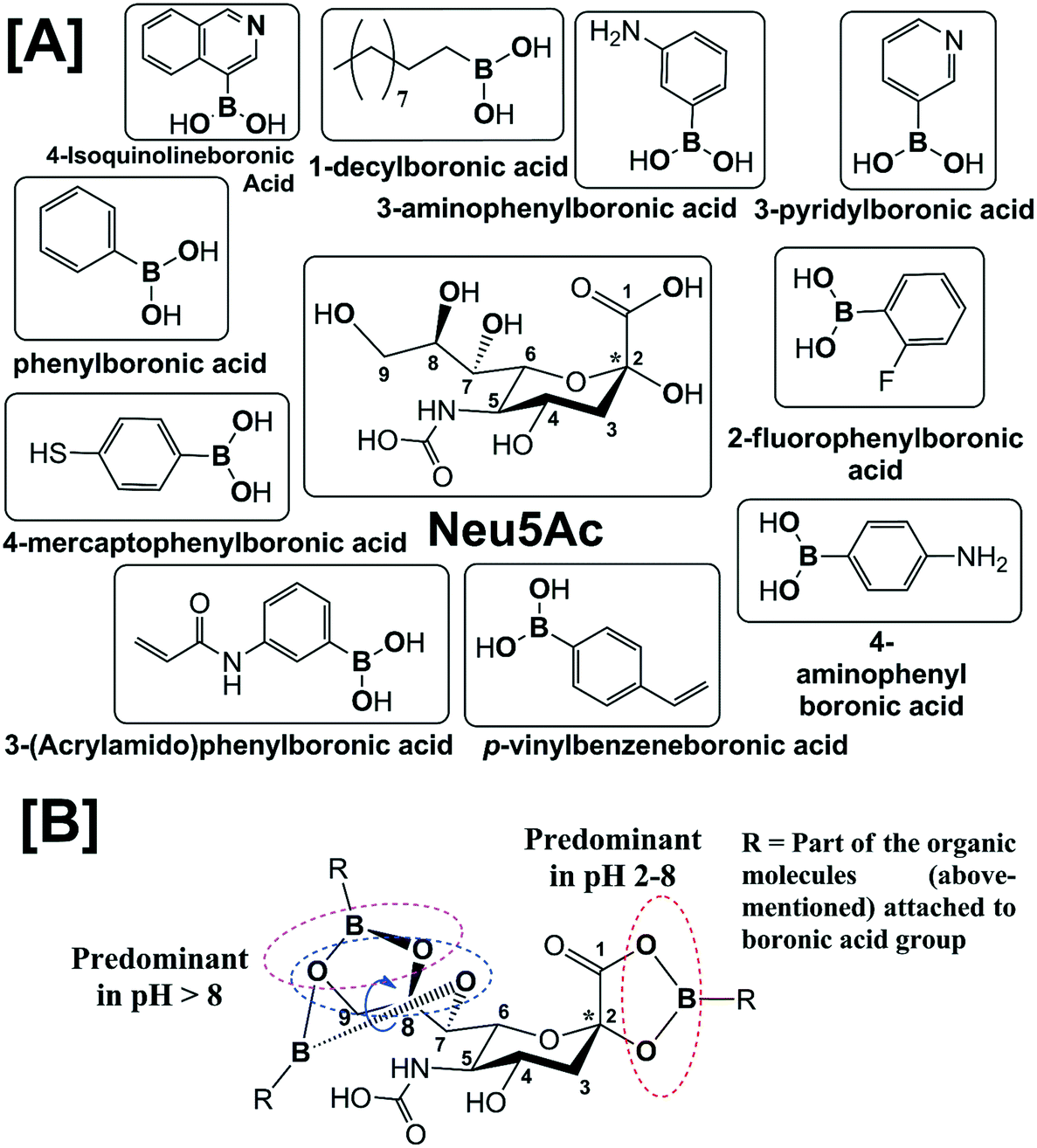 | ||
| Fig. 6 [A] Types of boronic acid molecules used in different assays of Neu5Ac as discussed in Section 5. [B] Change of binding sites of boronic acid functional group at Neu5Ac in different pH ranges. | ||
5.2 Colourimetry-spectrophotometry
Colourimetry and spectrophotometry were the dominant classical assays of Sia in early days. Blix, being one of the discoverers of Sia, used direct Ehrlich reagent (i.e., 5% w/v p-dimethylaminobenzaldehyde in 1:1 v/v ethanol (95% v/v) and concentrated HCl) to identify and quantify total Sia from a purple coloured product having absorbance maxima (λmax) at 565 nm.6 To avoid possible interference from pyrrole derivatives and alkali-treated mucopolysaccharides in direct Ehrlich assay, Klenk et al., the other pioneer of Sia, used modified Bial's method (i.e., orcinol, FeCl3, concentrated HCl, 100 °C) producing a red-violet product having λmax at 572 nm; but this colourimetric assay of Sia could not avoid the interference of pentose, fructose, sorbose, 2-furaldehyde, uronic acids, keto-hexoses.4 The modified Dische's assay with 0.5% diphenylamine solution in glacial acetic acid containing 5% H2SO4 was also used to quantify total Sia from a pink coloured supernatant having λmax at 530 nm, but it also suffered the interference from tryptophan and indole-3-acetic acid.367,368 Svennerholm proposed resorcinol in HCl with catalytic amount of CuSO4 for colourimetric assay of total Sia at λmax 580 nm, but encountered interferences from pentose, keto-hexoses and 2-deoxyhexose.369 Furthermore, the low molar absorption coefficients (ε) in the range 5300–6900 M−1 cm−1 (for Neu5Ac) of those chromophores resulted into poor sensitivity of these colorimetric assays. The HIO4-resorcinol assay followed by extraction of the chromophore (having λmax at 630 nm) in tertiary-butanol significantly improved the sensitivity of colourimetric assay owing to higher value of ε (27900 M−1 cm−1 for Nue5Ac).370 Furthermore, 10–60 μg of total Sia was determined by spectrophotometry at λmax 307 nm in 0.01% o-phenanthroline solution in 0.1 M NaCl (pH 6.0).371 To separately quantify free, bound and total Sia, Spyridaki et al. used different HIO4 treatments (i.e. at 0 °C for 10/30 min for free/total Sia; at 37 °C for 35 min for bound Sia) before the acidic resorcinol treatment followed by extraction of the chromophore (having λmax at 625 nm) with tertiary-butanol. The linear dynamic range (LDR) and a limit of detection (LOD) for Neu5Ac were also improved to 20–200 μM and 1 μM, respectively, due to an increase of ε to 35435 M−1 cm−1.372
Warren and Aminoff independently made a breakthrough in the spectrophotometric assay of Sia in 1959. Warren incubated Sia for 30 min at 37 °C with 0.2 M HIO4 in 9 M H3PO4 to produce aldehyde, which after removal of excess HIO4 by 10% NaAsO2 in 0.5 M Na2SO4 + 0.1 N H2SO4, reacted with 0.6% thiobarbituric acid in 0.5 M Na2SO4 while heating at 100 °C for 15 min. The chromophore extracted with cyclohexanone showed a red colour with λmax at 549 nm (ε = 61000 M−1 cm−1 for Nue5Ac; 50000 M−1 cm−1 for Neu5Gc).373 Aminoff slightly modified this method by incubating Sia for 30 min at 37 °C with 0.025 M HIO4 in 0.125 N H2SO4 at pH 1.2 to produce aldehyde, which after removal of excess HIO4 by 2% NaAsO2 in 0.5 N HCl, reacted with 0.1 M thiobarbituric acid in water (at pH 9.0) while heating at 100 °C for 15 min. The red colour chromophore extracted with 1-butanol containing 5% (v/v) of 12 N HCl had λmax at 549 nm and ε 70000 and 51000 M−1 cm−1 for Nue5Ac and Neu5Gc, respectively.374 These two methods were well adopted for several years by different research groups to determine Sia in biological samples owing to high sensitivity of assay by virtue of high ε values. However, both methods were limited to only free Sia and suffered interference from 2-keto-3-deoxygluconic acid and hexoses. Furthermore, these methods failed to produce reliable values in the concentration range 0.l–1.0 μg of Sia. The LDR of colourimetric assay of Sia was brought down to 225 ng–3.09 μg by modifying the thiobarbituric acid treatment followed by extraction steps of Aminoff. There, the aldehyde was allowed to react with 0.1 M thiobarbituric acid (at pH 9.0) in a boiling water bath for 10 min followed by cooling in an ice bath for 2 min. Then the reaction mixture was incubated in water bath at 37 °C for 2 min and the chromophore was extracted with 1-butanol containing 5.7% (v/v) of 10.5 N HCl.375
Massamiri et al. developed a 3-methyl-2-benzothiazolone hydrazone based method avoiding acid hydrolysis pre-treatment step. In this method, biological samples were shaken in 0.05–2.00 mM NaIO4 in phosphate buffer saline (PBS) at pH 7.4 in the dark for 15 min at 4 °C. Then formaldehyde in equivalent amount to bound Sia was released in the solution. The supernatant was successively treated with 10% (w/v) ZnSO4 in 1 N NaOH for 5 min and 0.01 M 3-methyl-2-benzothiazolone hydrazone in phosphate buffer for 20 min at room temperature. Then 5% (w/v) FeCl3 was added into the reaction mixture, vigorously shaken and left for 15 min before the absorbance was read at 625 nm. However, this assay was not specific for Sia as 2-deoxyribose and hexoses interfered with the measurement and there was a chance of over-estimation of Sia due to the presence of endogenous formaldehyde.376 Later, in another spectrophotometric assay, Sia was reacted with 2% (w/v) ninhydrin in a solution of DMSO-lithium acetate buffer (at pH 5.2) and glacial acetic acid on a boiling water bath for 10 min followed by cooling in ice-bath for 2 min. Its chromophore showed λmax at 470 nm (ε = 66400 M−1 cm−1 for Nue5Ac). In this method the interference from L-tryptophan (having λmax at 385 nm), L-cystine (having λmax at 485 nm), L-proline (having λmax at 510 nm) and L-cysteine (having λmax at 560 nm) could be eliminated by using a small column of cation-exchange resin before the spectrometric assay.377 This method, with a little modification, has recently been used for the spectrophotometry-based flow batch analysis of Sia in the whole, semi-skimmed and skimmed milks. The mixture of Sia in sample/standard, 0.1 M HCl and 0.16 M ninhydrin of equal volume was pumped into the batch flow chamber and mixed for 5 min at 75 °C, pH 1.6 and the absorbance of the product was recorded at 470 nm. The LDR and LOD of the method were reported as 1.0–10.0 mg L−1 and 0.239 mg L−1, respectively, for Nue5Ac with no significant interference from L-cystine, L-cystein, L-tryptophan and ammonia.378
No significant progress, except two recent works reported by Jayeoye et al., was observed in the development of spectrophotometric assays for the determination of Sia in present times. Aqueous suspension of gold nanoparticles (AuNPs) of average diameter 13 nm was prepared by citric acid reduction method and 3-aminophenylboronic acid was electrostatically assembled on the surface of AuNPs in 1 mM phosphate buffer solution at pH 5.6. The dispersion of boronic acid modified AuNPs was of red wine colour having λmax at 520 nm (ε = 2.7 × 108 M−1 cm−1), which after incubation with Sia for 5 min at room temperature changed to blue with appearance of a new absorbance peak at 700 nm due to aggregation of AuNPs linked through Sia (Fig. 7[A]). Hence, the ratio of absorbances at 700 nm to 520 nm was used for spectrophotometric assay of Sia in the LDR 0.15–1.0 mM and LOD 0.06 mM without significant interference from glucose, fructose, galactose, mannose and disaccharide sucrose. This assay was successfully tested with known concentration of spiked Sia in the simulated human saliva.379 Later, the LOD of this assay was further improved to 35 μM with two LDRs 80–250 and 300–700 μM by modifying the surface of AuNPs with dithiobis(succinimidylpropionate) through a ligand exchange mechanism at room temperature without changing the basic UV-vis absorbance profile of AuNPs having λmax at 520 nm. Again no significant interference was observed from glucose, mannose, lactose, fructose, sucrose, maltose and galactose in this assay, which was successfully tested with Sia spiked in the commercial human serum extracted from human male AB plasma.380
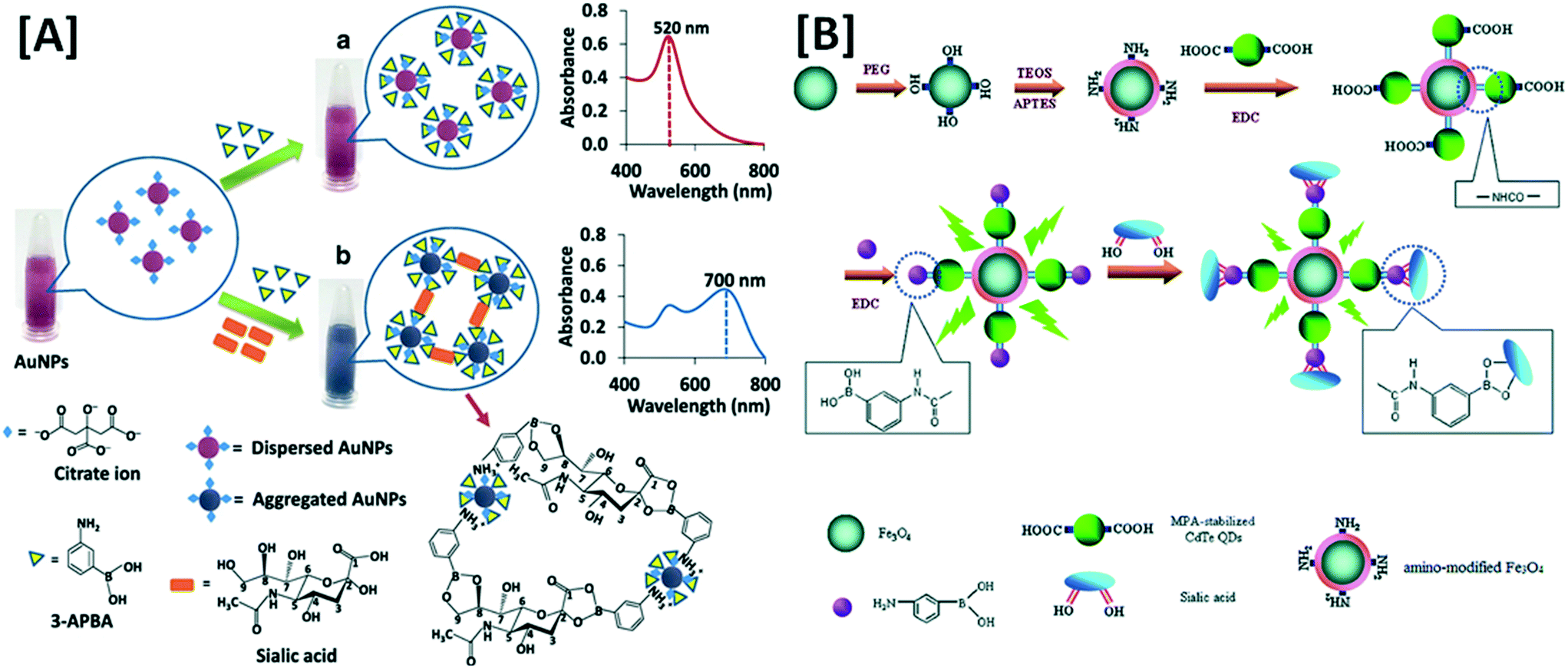 | ||
| Fig. 7 [A] Ratio-metric spectrophotometric assay of Neu5Ac by using relative absorbances at 520 and 700 nm of the aqueous dispersion of citrate capped-gold nanoparticles (AuNPs) modified with 3-aminophenylboronic acid (3-APBA) before and after incubation with Sia (figures reproduced with permission from ref. 379) Copyright 2018 Springer Nature. [B] Fluorometric assay of Neu5Ac by covalently linking both 3-APBA and Fe3O4 nanoparticles with CdTe fluorophores through 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (figures reproduced with permission from ref. 388) Copyright 2016 The Royal Society of Chemistry. | ||
Here, we summarize the perspective of colourimetric and spectrophotometric assay of Sia. For this purpose, we have compared the LDR and LOD mentioned in either M or g L−1 units in the above-mentioned publications considering 309.273 g mol−1 molar mass of Neu5Ac for interconversion of units, if required. The AuNPs-boronic acid based spectrophotometric method, although recently developed, could not produce lower LOD compared to classical dye-based colourimetric assay, but it minimized the interferences of other saccharides in the quantification of Neu5Ac. The lowest LOD reported for AuNPs-boronic acid based spectrophotometric assay was 35 μM having two separate LDRs of 80–250 and 300–700 μM.380 On the other hand, acidic ninhydrin dye based flow-batched spectrophotometric assay could provide lowest LOD of 0.77 μM (converted from 0.239 mg L−1) with narrow LDR of 3.23–32.3 μM (converted from 1.0–10.0 mg L−1) without interference from L-cystine, L-cystein, L-tryptophan and ammonia.378 Therefore, enough scope exists to develop new ultrasensitive spectrophotometric assay of Sia in real biological and food samples.
5.3 Photoluminescence
Photoluminescence assay showed higher sensitivity compared to spectrophotometric or colorimetric assay of Sia.381,382 In the reaction of both bound and free Sia with 5 mM 3,5-diaminobenzoic acid in 0.125 N HCl at 100 °C for 16 h, a green fluorescent product having excitation and photoluminescence maxima at 405 and 510 nm, respectively, was produced. While this fluorometric assay produced a good LDR of 0.15–2.0 μmol of Sia per 50 μL of 3,5-diaminobenzoic acid, it could not overcome strong interference of hexoses. Furthermore, dihydrochloride salt of 3,5-diaminobenzoic acid was commercially unavailable and thus had to be prepared from the commercially available precursors.381 Hammond followed the Aminoff's thiobarbituric acid process, but instead of measuring absorbance at 549 nm, the 1-butanol extract was excited at 550 nm. It emitted a fluorescence light having intensity peak at 565 nm suitable for fluorometric assay of Neu5Ac with LDR 10–250 ng (i.e., 32–800 pmol), but suffered interference of 2-deoxyribose.382 In contemporary times, another fluorometric assay was reported for the reaction of free Sia with pyridoxamine dihydrochloride, zinc acetate and pyridine in methanol at 70 °C for 45 min. The excitation of the fluorophore with 395 nm light resulted to a photoluminescence having intensity peak at 470 nm. This assay was suitable for the LDR 0.1–10.0 μg of free Nue5Ac, but α-ketoacids interfered in the measurement.383 On the other hand, mild oxidation of free Sia by 1 mM NaIO4 in PBS (pH 7.2) at 4 °C for 15 min in the dark could quantitatively produce formaldehyde, which after removal of excess periodate by 2% NaAsO2 in 0.5 N HCl, was allowed to incubate at 60 °C for 10 min with a mixture of acetylacetone, ammonium acetate, glacial acetic acid and distilled water. Free Nue5Ac was assayed from the fluorescence intensity of a light-yellow fluorophore (having excitation and luminescence maxima at 410 and 510 nm, respectively) with the LDR of 2–10 μg and a LOD of 0.1 μg (0.3 nmol). o-Acetylated (at either C-8 or C-9 position) Sia could not be oxidized by NaIO4, thus o-deacetylation by 0.1 M NaOH should be a pre-treatment step for this assay, which suffered interference from endogenous pyruvate.384 In 2008, Matsuno et al. reported a comparatively less hazardous fluorometric assay for both free and bound Sia by oxidising with 10 mM NaIO4 at 0 °C for 45 min to quantitatively produce formaldehyde followed by removal of excess periodate by reaction with 50 mM Na2S2O3 solution. The fluorophore, having excitation and luminescence maxima at 388 and 471 nm, respectively, was produced from the reaction of formaldehyde with 4 M ammonium acetate (at pH 7.5) and ethanolic solution of 0.1 M acetoacetanilide for 10 min at room temperature. The LDR and LOD of this fluorometric assay of Nue5Ac were reported as 0.3–27 nmol and 4 pmol, respectively. However, this photoluminescence assay suffered a strong interference from the glucose derived sorbitol, which could eventually produce two equivalents of formaldehydes.385 Alves et al. reported a solid-phase fluorometric assay of Nue5Ac with LDR and LOD 0.4–2000.0 and 0.4 mg L−1, respectively, by incubating Nue5Ac with Eu(III)-coordinated silica modified by sodium thenoyltrifluoroacetonate in ethanol (carbonate buffer pH 6) for 3 min and measuring the fluorescence intensity at 615 nm for a photoexcitation at 345 nm.386Several photoluminescence assays based on phenylboronic acid linked established fluorophores viz. CdTe, fluorescein isothiocyanate, etc. exploited the preferential affinity of phenylboronic acid to Nue5Ac. Wang et al. covalently conjugated CdTe (excitation and fluorescence maxima at 330 and 502 nm, respectively) with Sambucus nigra bark lectin, which specifically recognised Sia and increased the fluorescence intensity. The LDR and LOD of this assay were reported as 12–680 and 0.67 ng mL−1, respectively. This method was successfully implemented to quantify Sia in different egg products viz. whole egg, yolk and egg white powders, egg noodles, cake and steamed bun.387 Wang et al., on the one hand, prepared amine-functionalized magnetic Fe3O4 nanoparticles (NH2–Fe3O4) by modifying polyethylene glycol 400 coated Fe3O4 nanoparticles with 3-aminopropyl triethoxysilane and tetraethylorthosilicate. On the other hand, they prepared fluorescent 3-mercaptopropionic acid capped CdTe quantum dots from a solution of Cd(II), NaHTe and 3-mercaptopropionic acid. Then they covalently linked 3-mercaptopropionic acid capped CdTe quantum dots on modified Fe3O4 nanoparticles to prepare the fluorescence probe for assay of Sia (Fig. 7[B]). The probe was incubated with Nue5Ac in 5 mM PBS (pH 7.4) at 37 °C for 3 h and magnetically separated from the reaction mixture, washed and dispersed again in 5 mM PBS (pH 7.4) for measuring the fluorescence intensity at ∼550 nm. The fluorescence intensity of the probe was linearly quenched with the concentration of Nue5Ac resulting LDR and LOD 0.05–1.5 mg mL−1 and 16 μg mL−1 without any significant interference from Na+, K+, Mg2+, Ca2+, Zn2+, Cu2+, Hg2+, Pb2+, Mn2+, Fe3+, dopamine, glucose, galactose, mannose and fructose. Hence, this assay of Nue5Ac was validated with commercial infant formula.388 A comparatively simpler strategy was proposed by Hao et al., where aminophenylboronic acid was covalently linked to fluorescein isothiocyanate producing the fluorescence probe having excitation and luminescence intensity peak at 493 and 515 nm, respectively, in Tris-HCl (pH 7.40). The fluorescence intensity was completely quenched in the presence of 90 mg reduced graphene oxide dispersion due to the Förster resonance energy transfer from fluorescence probe stacks to reduced graphene oxide. The fluorescence intensity was regained, and simultaneously reduced graphene oxide was precipitated out upon incubation of this dispersion with Nue5Ac at room temperature for 2–3 h owing to the higher affinity of aminophenylboronic acid modified fluorescein isothiocyanate to Nue5Ac than reduced graphene oxide. The LDR and LOD of this assay for Neu5Ac were reported as 0.5–5.0 and 0.044 mM, respectively.389
Xu et al. hydrothermally synthesised boronic-acid-functionalized carbon dots of average diameter 3.5 nm having luminescence maxima at 390 nm by heating the aqueous solution of 3-pyridylboronic acid at 160 °C for 8 h and used it for fluorometric assay of Nue5Ac by incubating at room temperature for 4 min with Neu5Ac in 10 mM PBS (pH = 5.5) from the decrease of fluorescence intensity linearly to concentration of Nue5Ac in the LDR 0.08–4.0 mM. This method could detect Nue5Ac down to 54 μM without any interference from glucose, mannose and galactose. Hence, this fluorometric assay was validated to quantify Nue5Ac in real human serum samples.390 Wang et al. have recently reported a similar methodology by using dispersion of fluorescent B-, N-doped carbon dots of diameter 2–7 nm having excitation and luminescent maxima at 560 and 645 nm (quantum yield 8.56% with respect to rhodamin 6G), respectively, in 10 mM glycine buffer (pH 5.5). B-, N-doped carbon dots were prepared from a mixture of 10:1 (w/w) o-phenylenediamine:3-aminophenylboronic acid (w:w 10:1) by hydrothermal synthesis at 160 °C for 6 h. The fluorescence intensity of the dispersion at 645 nm increased upon incubation with Nue5Ac at 37 °C for 30 min because of the favourable complex formation between Sia and boronic acid groups present on the surface of B-, N-doped carbon dots. The LDR and LOD of Neu5Ac assay were reported as 0.02–1.0 mM and 9.24 μM, respectively, without any significant interference from urea, isoleucine, proline, galactose, maltose, Na+, K+, Ca2+, NH4+, SCN− and Cl−. Thus, the assay was validated for quantifying spiked known amount of Nue5Ac in simulated human saliva samples.391
Wang et al. have recently developed an indicator-displacement photoluminescence assay for Sia by preparing fluorescein sodium dye anchored covalent organic framework, TpPa-1, as fluorescent indicator having excitation and luminescence maxima at 437 and 514 nm, respectively. A hydrogel mould of the fluorescent indicator was prepared with sodium alginate and was immersed in Cr(NO3)3 solution for 2 h and then washed thoroughly. The luminescence of the fluorescent indicator was significantly quenched by Cr3+ by virtue of strong indicator-receptor interaction. The fluorescence intensity of the indicator hydrogel was partially regained in the presence of Sia in neutral water due to partial displacement of Cr3+ from the hydrogel matrix to the solution by the electrostatic attraction to negatively charged Sia present in the solution. The LDR and LOD of this fluorometric assay were reported as 10 nM–10 mM (in logarithmic scale) and 7.08 nM, respectively, with no significant interference from CaCl2, MgCl2, KCl, NaCl, NaHCO3, glucose, L-proline and urea. An on–off–on sequential action of this method was validated for quantifying Sia in real human serum samples.392 Yu et al. have recently reported the use of photoluminescent UiO–66-NH2@B(OH)2 (having excitation and luminescence maxima at 320 and ∼470 nm, respectively) to determine Neu5Ac in the LDR 0.05–2.5 mM with a LOD of 0.025 mM owing to the preferential interaction of the diol of Neu5Ac with the boronic acid functional group at pH 7.4.393 A molecular logic-gate has also been successfully designed by incorporating 4-carboxyphenylboronic acid in Eu(atpt)1.5(phen)(H2O)n metal–organic-framework to detect Sia from the fluorescence intensity ratio at λ470/λ614 nm.394 Wang et al. prepared a fluorescent polydiacetylene based liposome type structure by UV-light assisted polymerization with three monomers viz. 10,12-pentacosadiynoic acid-phenylboronic acid (as Sia recognising unit), 10,12-pentacosadiynoic acid-1,8-naphthalimide (as fluorescent unit with excitation and luminescence maxima at 420 and 535 nm, respectively) and 10,12-pentacosadiynoic acid-ethyl acrylate with molar ratio 4.5:1.0:4.5 in methylene chloride containing 10% methyl alcohol (v/v). The prepared liposome suffered from self-quenching fluorescence due to transfer of energy from 1,8-naphthalimide unit to polydiacetylene backbone. The pendent side chain configuration was disturbed in the presence of Sia-boronic acid complex and the original fluorescence of 1,8-naphthalimide was restored. This assay reported LDR up to 0.4 mM with a LOD of 14 μM with little spectral deformation in the presence of glucose, galactose and mannose.395 Yue et al. used 6-carboxyfluorescein (excitation and emission maxima at 494 and 520 nm, respectively) tagged aptamer (5′-6-carboxyfluorescein-TAGGGAATTCGTCGACGGAT-3) to quantify Neu5Ac in LDR 20–1000 nM with tolerable interference of Neu5Gc, KDN, glucose, sucrose and maltose. The aptamer tagged fluorophore lost photoluminescence after adsorbing on the dispersed graphene oxide in solution, but Neu5Ac brought the aptamers back into the solution due their preferential affinity resulting into the development of the photoluminescence of 6-carboxyfluorescein.396
Binding of different family members of Sia to lactose-specific lectin–peanut agglutinin slightly varies with respect to the core composition of binding domain Asp–Pro–Ala–Asp peptides. A panel consisting of 6 types of dipeptide combinations labelled with fluorescein (excitation and emission maxima at 470 and 514 nm, respectively) could discriminate Neu5Ac, Neu5Gc, KDN, N-acetylneuraminic acid methyl ester, N-acetyl-2,3-didehydro-2-deoxyneuraminic acid, 2,4,7,8,9-penta-OAc-N-acetylneuraminic acid methyl ester.397 Furthermore, the fluorescence intensity of pyrene-conjugated-histidine (excitation and emission maxima at 330 and 385/410 nm, respectively) decreased in the presence of Neu5Ac and 2,3′-sialyllactose due to hindrance in gelation, which usually occurred in the presence of other monosaccharides and sialic acid analogues. The fluorometric dynamics of gel-formation of pyrene-conjugated-histidine can be used to recognise Neu5Ac in complex matrix.398 Neu5Ac significantly enhanced the fluorescence intensity (at 658.8 nm) of phenyl-boronic acid functionalised squarylium cyanine dyes having shorter alkyl chain due to formation of J-aggregate (Nue5Ac:dye as 2:1) complex through multipoint interaction of dye with Neu5Ac compared to other monosaccharides. On the other hand, no such enhancement was observed by Neu5Ac for similar dyes having longer alkyl chain due to formation of H-aggregate (Nue5Ac:dye as 1:1) complex similar to other monosaccharides. This method is useful to discriminate Neu5Ac in biological samples.399
Here, we summarize the perspective of photoluminescence assay of Sia. For this purpose, we have compared the LDR and LOD mentioned in either M or g L−1 units in the above-mentioned publications considering 309.273 g mol−1 molar mass of Neu5Ac for interconversion of units, if required. Most of the photoluminescence assays of Sia reported LDR within ∼20–5000 μM with LOD ∼10–60 μM.388–391,393 The solid-phase fluorometric assay could provide a wide LDR 1.3–6500 μM with a LOD of 1.3 μM.386 The ultra-sensitive Cr(III)-displacement fluorometric assay with fluorescein sodium dye anchored TpPa-1 could provide a wide LDR of 10 nM–10 mM (although in logarithmic scale) with sufficiently low LOD (7.08 nM) without interference from L-proline and urea.392 However, the fluorescent CdTe quantum dots modified with Sambucus nigra bark lectin could provide lowest LOD (2.1 nM; converted from 0.67 ng mL−1) for fluorometric assay of Sia with a moderately wide LDR 3.88 nM–2.2 μM (converted from 12–680 ng mL−1) in linear scale without interference in commercial egg products.387 Therefore, the research towards the development of ultrasensitive photoluminescence assay of Sia in real biological and food samples is open to further investigations.
5.4 Enzymatic analysis
The enzymatic reactions of Sia can produce secondary assayable chemicals viz. H2O2, reduced nicotinamide adenine dinucleotide (NADH), etc. Exo-α-sialidase can release Sia from the terminal residue of sialylated glycoproteins, glycolipids and oligosaccharides having α2,3-, α2,6- and α-2,8-glycosidic bonds (reaction 1). However, 4O-acetylated Sia is irresponsive to this enzyme.400 Sialic acid aldolase can reversibly convert Neu5Ac to ManNAc and pyruvate during incubation at 37 °C and pH 7.1 for 15 min (reaction 2). At equilibrium, the molar ratio of Neu5Ac:ManNAc:pyruvate attained 1:9:9 with an equilibrium constant 0.064 ± 0.020 M at 37 °C.401,402 The optimum pH and Michaelis constant of the reaction were reported as 7.1–7.3 and 3.3 × 10−3 M, respectively. Neu5Ac almost completely cleaved at concentrations <1 mM, but a significant amount of un-reacted Neu5Ac remained in the solution at equilibrium at higher concentrations. The enzymatic cleavage reaction could be stopped by increasing the temperature of the incubated solution to 100 °C for 2 min.403 This enzymatic reaction is also equally effective for Neu5Gc producing N-glycolyl-D-mannosamine and pyruvate under similar condition but is ineffective for O-acetylated Sia. Therefore, Neu5Ac could be quantitatively assayed indirectly through either of four different options by measuring either ManNAc or pyruvate.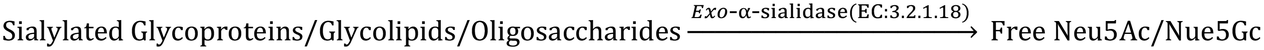 | (Reaction 1) |
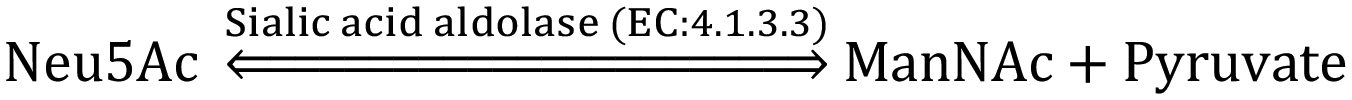 | (Reaction 2) |
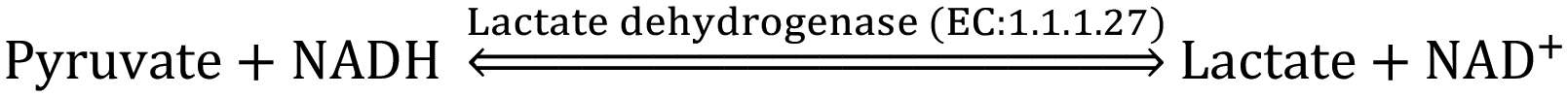 | (Reaction 3) |
 | (Reaction 4) |
 | (Reaction 5) |
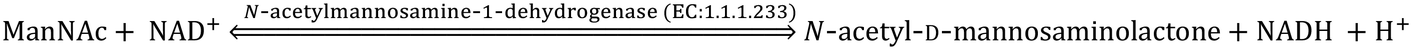 | (Reaction 6) |
 | (Reaction 7) |
 | (Reaction 8) |
The enzymatic assays of Sia are very specific and selective, but the high cost of purified enzymes and their stability in diverse sample conditions sometimes limit applications. Readers should be aware of the fact that except one publication,411 none of the above-mentioned enzymatic assay provided information about the LDR and LOD for Sia.
5.5 Chromatography
000 ng mL−1 for Neu5Ac.448 The ultra-HPLC coupled with positive-ion mode ESI source and selective reaction monitor tandem quadrupole MS/MS detector were also used to determine Neu5Ac in the milk-based commercial infant formulas with LDR 0.05–5.0 μg mg−1.449 However, the formation of Na+/K+/NH4+ adduct with the COO− functional group of Sia sometimes limits the direct HPLC-MS assay.
Fernando et al. protected the carboxyl group of Neu5Ac with fluorophore 1,2-diamine-4,5-dimethoxyl benzene dihydrochloride (excitation and luminescence maxima at 369 and 453 nm, respectively) and carried out the HPLC-MS assay coupled with scanning fluorescence detector, positive ion mode ESI source and quadrupole mass spectrometer with an internal standard α-keto glutaric acid. The LOD of this assay was calculated as 7 pg from the MS peak of fluorophore derivative of Neu5Ac at m/z 442.450 Xia et al. extracted glycoprotein from the root of a Tibetan plant Potentilla anserina L. and released Neu5Ac and Neu5Gc from that glycoprotein by acid hydrolysis. They used 3-methyl-1-(naphthalen-2-yl)-1H-pyrazol-5(4H)-one in ammonia solution to make pre-derivatives of the Sia and eluted the fluorophore derivatives of Sia through reversed phase HPLC coupled with UV detector (at 254 nm) for measurement and both positive- and negative-ion mode ESI sources, ion-trap mass spectrometer for identification. They could produce LDR 0.25–10.0 μM for both Neu5Ac and Neu5Gc with LOD 1.09 pmol (for Neu5Ac) and 1.06 pmol (for Neu5Gc).31 Hayama et al. pre-derivatised Neu5Ac and Neu5Gc with heptadecafluoroundecylamine in the presence of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride and used fluorous-phase fluophase reversed-phase HPLC column coupled with positive mode ESI source and Qtrap tandem mass spectrometer to assay Neu5Ac and Neu5Gc in the LDR 1–100 μM (for Neu5Ac) and 0.05–10 μM (for Neu5Gc).451 The pre-derivatization with HCl-n-butanol and reversed phase isotope dilution mass-spectrometry in positive-mode ESI source and tandem mass-spectrometer showed LDR and LOD 2–1000 and 0.42 μM, respectively, for Neu5Ac.452 Wang et al. followed a similar strategy i.e., isotope dilution of Neu5Ac with 1,2,3-13C3 Neu5Ac, inclusion of ketodeoxynonulosonic acid as an internal standard followed by pre-derivatisation with 3,4-diaminotoluene and eluting through ultra-HPLC coupled with positive ion mode ESI source and multiple reaction monitoring with Qtrap MS/MS detector. They used the m/z transition 396 → 253 at retention time 3.6 min for the assay of Neu5Ac with LDR 20.0–2.00 × 104 ng mL−1.453 Priego-Capote et al. used derivatives of Neu5Ac and Neu5Gc with DMB and pre-concentrated by a solid-phase extractor (hysphere polymeric polydi-vinylbenzene resin) online coupled with reversed phase chromatography having both positive- and negative-ion mode ESI source and multiple reaction monitoring with QqQ MS/MS detector. They reported LDR 0.2–500 ng mL−1 for both the target analytes in urine and serum samples with LOD 0.03 ng mL−1 for Neu5Ac and 0.04 ng mL−1 for Neu5Gc.454 Yesilyurt et al. used DMB derivative of Neu5Ac for HPLC-MS assay in the egg jelly coat of the sea urchin Paracentrotus lividus by using capillary liquid chromatography coupled with positive ion mode ESI source and ultra-ion trap tandem MS/MS detector.455
In the present day, the HPLC-FLD and HPLC-MS/MS (with or without fluorescence tagging) methods are the most reliable techniques for ultra-sensitive assays of Sia in the clinical and commercial food samples. The DMB based HPLC-FLD assay could produce a moderately wide LDR of 0.32–320 nM with a LOD of 0.32 nM of Neu5Ac.438 On the other hand, a DMB derivative of Neu5Ac preconcentrated by hysphere polymeric polydi-vinylbenzene resin online coupled with HPLC-ESI-MS/MS analyser resulted to wider LDR of 0.65 nm–1.6 μM of Neu5Ac with the lowest reported LOD of 97 pM.454
5.6 Capillary electrophoresis
Chen et al. used sialic acid aldolase for the enzymatic reaction of Neu5Ac to produce ManNAc, which was derivatised with the anionic fluorescent dye 8-aminopyrene-1,3,6-trisulfonic acid (having excitation and luminescence maxima at 425 and 503 nm, respectively) for the quantification of Neu5Ac by capillary electrophoretic separation followed by laser-induced fluorescence detection in the LDR 0.1–10 nmol.456 On the other hand, Neu5Ac was directly derivatised with 2-aminoacridone (having excitation and photoluminescence maxima at 429 and 529 nm, respectively), but monitored at 260 nm after capillary electrophoresis. The LDR and LOD of this assay were reported as 10–120 and 1 μM of Neu5Ac, respectively.457 Dong et al. used only high-performance capillary electrophoresis (HPCE) coupled with UV detection at 195 nm for assay of Neu5Ac without any pre- or post-derivatisation. The reported the LDR and LOD for Neu5Ac as 0.064–6.40 mM and 9.6 μM, respectively.458 Strousopoulou et al. derivatised Neu5Ac to per-O-benzoylated Neu5Ac by benzoic anhydride and quantified it with capillary zone electrophoresis coupled with diode-array detector fixed at 231 nm. They reported the LDR and LOD of Neu5Ac as 5–5000 μg mL−1 and 2 μM, respectively.459 Taga et al. developed a capillary electrophoresis assay featured with in-capillary sialidase digestion for the determination of Neu5Ac in sialoglycans and sialoglycoproteins. They introduced the samples into a running 50 mM acetate buffer (pH 5.0) containing exo-α-sialidase (of concentration 250 mU mL−1) from the cathodic end of the capillary and then applied 5 kV for 20 min followed by 20 kV for 5 min between two ends of the capillary. The authors achieved good linear response in the concentration range 0.025–10.0 mg mL−1 with a UV detector fixed at 200 nm.460Ortner et al. developed an assay method for the simultaneous determination of Neu5Ac and Neu5Gc by capillary zone electrophoresis coupled with negative-ion mode ESI source and ion-trap MS detector. They reported LDR and LOD 10–100 and 2 μg mL−1, respectively, for both Sia released from the glycoproteins after acid hydrolysis.461Zheng et al. oxidised Neu5Ac to β-formyl pyruvic acid by HIO4–H2SO4 treatment and then derivatised with electroactive 2-thiobarbituric acid. This derivatised Neu5Ac was assayed by sample-stacking capillary electrophoresis coupled with amperometric detection on a carbon disc electrode kept at 0.950 V vs. saturated calomel electrode. They reported the LDR and LOD as 2–200 and 0.5 μg mL−1, respectively, with distinguishable signal of excess 2-thiobarbituric acid, 2-deoxy-D-ribose, fucose, glucose, formaldehyde, acetaldehyde and propaldehyde.462 Wang et al. developed a capillary electrophoresis method in conjugation with capacitively coupled contactless conductivity detection based on synergistic electrophoretic stacking technique of field-amplified sample injection and moving chemical reaction boundary. This method was found to be suitable for the assay of both Neu5Ac and Neu5Gc in the commercial milk and milk powder samples with LDR 0.02–2.0 μg mL−1 (in milk) and 0.01–2.0 μg g−1 (in milk powder). The LOD of Neu5Ac and Neu5Gc in real samples was reported as 2.2 ng mL−1 (in milk); 1.7 ng mL−1 (in milk powder) and 2.0 ng mL−1 (in both milk and milk powder), respectively.463
In summarising electrophoretic analytical approaches to Sia quantitation, it could be conferred that although this method is not as ultra-sensitive as chromatographic assays, it is simpler to operate to quantify Sia in commercial food samples at moderately trace-levels. The pre-derivatization of Neu5Ac with fluorophores viz. 2-aminoacridone and benzoic anhydride followed by capillary electrophoresis could result in LDR of 10–120 and 16–16000 μM, respectively, with LOD of 1 and 2 μM, respectively.457,459 The lowest possible LDR (65–6500 nM; converted from 0.02–2.0 μg mL−1) and LOD (5.5 nM; converted from 1.7 ng mL−1) for capillary electrophoretic assay of Sia was achieved by coupling with capacitively coupled contactless conductivity detection technique.463 Hence, there is scope for scientific research and development for ultra-sensitive electrophoretic assays of Sia in clinical samples and food products.
5.7 Electrochemistry and electroluminescence
:1 sialic acid aldolase:pyruvate oxidase along with 5.0% glutaraldehyde in 0.1 M phosphate buffer (pH 7.0). Instead of probing H2O2, they probed the amperometric current of ferrocene redox reaction at 0.2 V vs. Ag/AgCl in 0.05 M phosphate buffer (pH 8). The pyruvate oxidase converted pyruvate to acetylphosphate, H2O2 and CO2 (see section 5.4) and itself was regenerated by ferrocinium/ferrocene redox reaction. The amperometric assay was suitable to determine Neu5Ac in the LDR 0.025–15.0 mM with a LOD of 18 μM without any significant interference of ascorbic acid, uric acid and pyruvic acid. This assay was successfully validated to quantify Sia in blood plasma samples.466
:2 w/w) was drop-casted on the glassy carbon electrode and dopamine was covalently linked on it by the carbodiimide route. Then 2-fluorophenylboronic acid was allowed to be adsorbed on the electrode surface owing to the interaction of the boronic acid functional group with the catecholamine dopamine. A DPV scan of the dopamine modified electrode in 0.1 M PBS (pH 7.0) showed an anodic peak at 0.25 V corresponding to oxidation of dopamine. After modification with 2-fluorophenylboronic acid, the anodic current at 0.25 V decreased and a new peak appeared at 0.6 V corresponding to the oxidation of an ester boronic acid–dopamine complex. In the presence of Neu5Ac in the electrolyte solution, the anodic peak at 0.6 V disappeared and the anodic current at 0.25 V increased due to the preferential bonding of 2-fluorophenylboronic acid with Neu5Ac in the solution compared to the diol unit of dopamine present on the electrode surface. The DPV peak current at 0.25 V proportionally increased with the concentration of Neu5Ac in the range 0.1–7.5 mM with a LOD of 28.5 μM. This voltammetric assay suffered significant interference from ascorbic acid, glucose and uric acid and thus samples were required to be pre-treated with an ion-exchange column.471 Lv et al. reported an aptamer based voltammetric sensor for the determination of Sia in hospital acquired human blood samples (Fig. 8[A]). Carbon cloth electrode was electrochemically modified with gold nanodendrites and incubated in the solution of aptamer sequence (i.e., TCCCT ACGGC GCTAA CCGAT AGGTG TAGCG TGGGG CACAT GTTCG CGCCA CCGTG CTACA AC) of Sia for 12 h at room temperature. The non-specific binding sites of this aptasensor base were blocked by 1-hexanethiol. On the other hand, an electroactive probe was prepared by 4-mercaptophenylboric acid and thionine modified and citrate-capped AuNPs. Then the aptasensor base was incubated in the aqueous solution of Neu5Ac in 0.1 M phosphate buffer (pH 4.5) for 3 h at room temperature followed by incubation with the electroactive probe in the same buffer for 5 h. Finally, Neu5Ac was assayed indirectly from the equivalent cathodic peak of thionine by DPV through potential scan from 0.4 V to −0.4 V in 0.1 M phosphate buffer (pH 4.5) in the LDR 0.1–440 μM with a LOD of 0.08 μM.472 Hai et al. synthesised pyridylboronic acid to specifically bind Sia in lower pH (5–6) and used it along with the poly-3,4-ethylenedioxythiophene to determine Neu5Ac in the LDR 0.1–3.0 mM with a LOD 0.1 mM by using ferri-/ferro-cyanide as a redox probe for the DPV assay.473 Ding et al. reported an electrochemical impedance spectroscopy (EIS) based stimuli-responsive polymer electrode hyphenated with in vivo microdialysis for electrochemical assay of Neu5Ac for Alzheimer disease in live rat brain microdialysates. They electrochemically deposited gold nanoflowers on a screen-printed carbon electrode and prepared a copolymer consisting of N-isopropylacrylamide, acryloyl-3-amidophenylboronic acid, acryloyl-3,5-bis(trifluoromethyl)-phenylthiourea and S-benzyl dithiobenzoate with the mole ratio of 170:15:15:2 through a reversible addition–fragmentation chain transfer polymerization mechanism. Here, S-benzyl dithiobenzoate anchored the copolymer on the gold nanoflowers, the amide group of poly-(N-isopropylacrylamide) made hydrogen bonds with the N–H functionality of acryloyl-3,5-bis(trifluoromethyl)-phenylthiourea and O–H functionality of acryloyl-3-amidophenylboronic acid provided a gated-channel polymeric network to the approach of the redox-probe (i.e., [Fe(CN)6]3−/4−) towards the working electrode surface. In the presence of Neu5Ac in the assay electrolyte, the hydrogen bond between poly-(N-isopropylacrylamide) and acryloyl-3-amidophenylboronic acid became weaker due to the higher affinity of Neu5Ac towards the boronic acid functional group and the polymeric network became more open towards the passage of the redox-probe to/from electrode surface. The authors used 0.1 M artificial cerebrospinal fluid buffer solution containing [Fe(CN)6]3−/4− redox probe and EIS assay was carried out at 0.14 V with a sine potential perturbation of 5 mV amplitude and frequency range 0.01–105 Hz. The charge-transfer-resistance linearly decreased with the concentration of Neu5Ac in the range 1 pM–10 μM with a LOD of 0.4 pM without any significant interference from glucose, mannose, galactose, fucose, xylose, fructose, K+, Na+, Ag+, Ca2+, Mg2+, Zn2+, Cu2+, Co2+, Fe2+, Mn2+, Ni2+, Cd2+, Fe3+, Al3+, amino acids (Glu, Gly, Phe, Met, Val, Cys, His, Iso, Lys, Leu, Tyr), ascorbic acid, uric acid, dopamine, lactate, 3,4-dihydroxyphenylacetic acid and 5-hydroxytryptamine.474 Broncová et al. electropolymerized 3,4-diaminobenzoic acid on a platinum electrode and covalently anchored 3-aminophenylboronic acid by a two-step carbodiimide modification. Authors performed EIS assay in the frequency range 0.1 Hz–100 kHz at 0 V with an amplitude of 10 mV in 0.04 M Britton–Robinson buffer (pH 7.0) and found that the difference between polarization and solution resistances linearly varied with the concentration (in logarithmic scale) of Neu5Ac in range 25 μM–0.998 mM.475
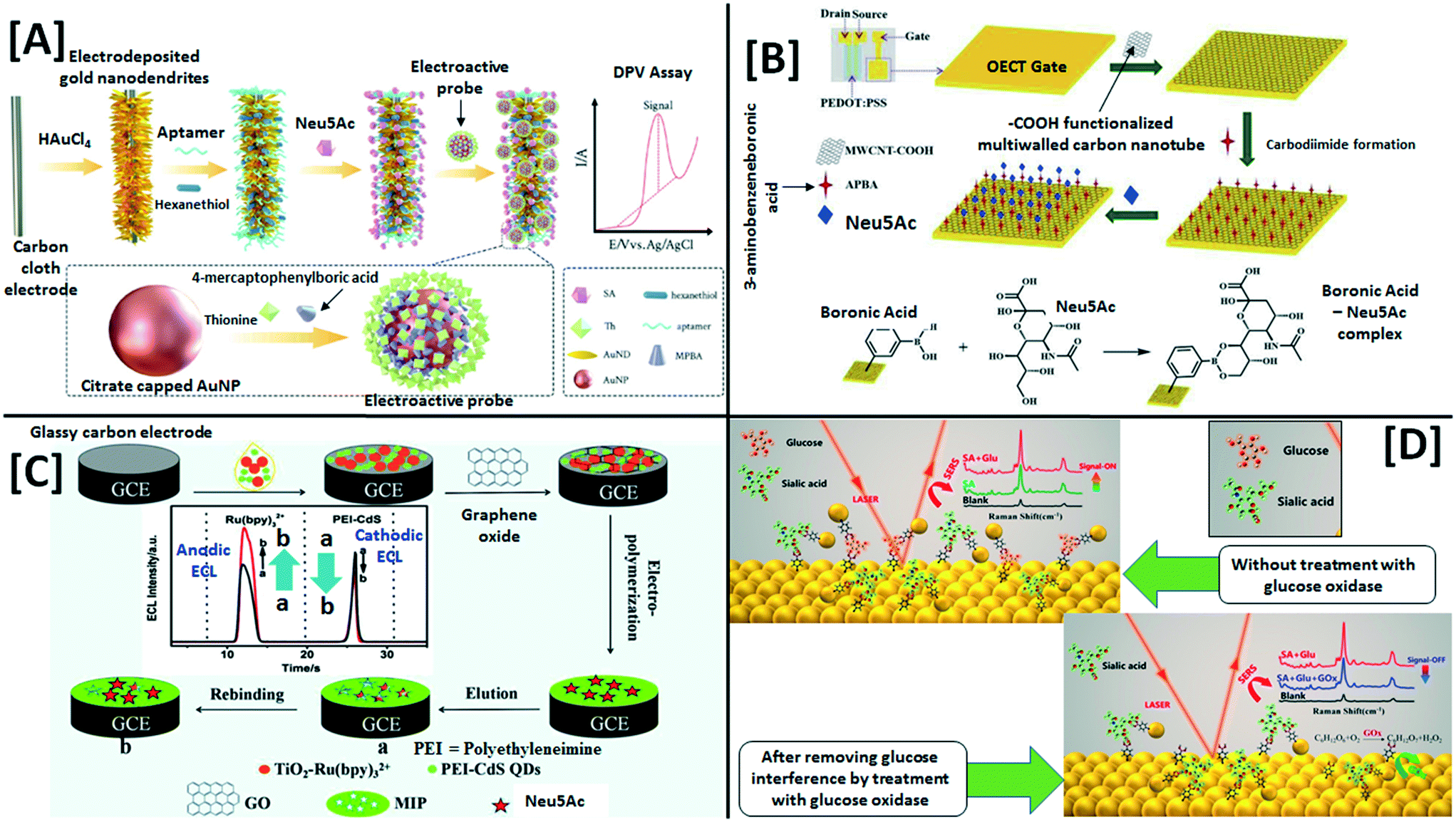 | ||
| Fig. 8 [A] DPV assay of Neu5Ac by using aptamer coated gold nanodendrites electrochemical platform and thionine-4-marcaptophenylboronic acid electroactive probe (figures reproduced with permission from ref. 472) Copyright 2020 John Wiley and Sons. [B] Interaction of Neu5Ac with 3-aminobenzeneboronic acid covalently linked to carboxylic acid functionalized multiwalled carbon nanotubes placed on the gate of OECT (figures reproduced with permission from ref. 476) Copyright 2020 Elsevier. [C] Ratiometric MIP based ECL assay of Neu5Ac on glassy carbon electrode modified with TiO2–Ru(bpy)32+ nanoparticles, polyethyleneimine (PEI) capped CdS quantum dots and graphene oxide (figures reproduced with permission from ref. 479) Copyright 2020 Elsevier. [D] SERS assay of Neu5Ac in the presence of glucose by using glucose oxidase pre-treatment (figures reproduced with permission from ref. 488) Copyright 2020 American Chemical Society. | ||
:4:1. Then the composite polymer was mixed with equal amount of rutile TiO2 and a monoclonal antibody was anchored to TiO2-composite polymer mixture. On the other hand, a glassy carbon electrode was modified with fullerene to increase the electrochemical current response and successively that was modified with AuNPs to chemically anchor 4-mercaptophenylboronic acid for the recognition of Sia. Then, Neu5Ac was incubated on the modified electrode surface for 40 min at room temperature in 0.1 M PBS (pH 7.4) and the Neu5Ac modified electrode was allowed to react with the monoclonal antibody anchored TiO2-composite polymer mixture by incubation for another 40 min at room temperature. The photocurrent (recorded in the potential range 0.5–0.6 V vs. Ag/AgCl reference electrode) and the temperature (recorded upon laser irradiation at 808 nm, 1.0 w cm−2) of the system linearly increased with concentration (in logarithmic scale) of Neu5Ac in the range 3.5 × 10−5–35.0 ng mL−1 with a LOD of 1.2 × 10−5 ng mL−1. The authors successfully employed this photoelectrochemical assay to measure Neu5Ac in blood samples collected from human volunteers.477
Fang et al. have recently developed a self-enhanced and renewable biosensor for ultra-sensitive detection of Sia in human serum though ECL assay. For this purpose, the ECL bio-probe was prepared by mixing Ru(bpy)32+ with TiO2 mesocrystals for 6 h followed by addition of 4-mercaptobenzoic acid and bismuth nano-belts. The monoclonal antibody was anchored to this surface via a carbodiimide route. On the other hand, fullerene followed by TiO2 mesocrystals were drop-casted on a glassy carbon electrode and 4-mercaptophenylboronic acid was also added on this surface. Furthermore, the nonspecific sites available on the modified electrode were blocked by adding 1% bovine serum albumin. For the ECL assay, the modified electrode was incubated with the solution of Neu5Ac in 0.1 M PBS (pH 8.0) for 40 min at room temperature and further incubated for another 40 min after adding the ECL bio-probe. The ECL response of Neu5Ac recorded in the potential range 0.8–1.5 V in 0.1 M PBS (pH 8.0) was found to be linear with concentration (in logarithmic scale) of Neu5Ac in the range 3.5×10−5–350 ng mL−1 with a LOD of 1.17 × 10−5 ng mL−1. Interestingly, the modified electrode could be regenerated by simply immersing into 0.1 M PBS (pH 6.0) when Neu5Ac conjugated with the ECL bio-probe detached from the modified electrode surface, which can be re-used for the next assay with fresh bio-probe.478
A ratiometric MIP based ECL sensor was developed by Cao et al. in 2020 for sensitive and selective detection of Neu5Ac in the commercially available synthetic human serum and EBN (Fig. 8[C]). Initially, TiO2–Ru(bpy)32+ nanoparticles and polyethyleneimine capped CdS quantum dots (w:w 1:4) were drop-casted on a glassy carbon electrode and covered with a few layers of graphene oxide to enhance the overall conductivity of the modified electrode suitable for electrochemical applications. The MIP was potentiodynamically electropolymerized on the modified electrode in a solution of 0.3 mM 3-aminophenylboronic acid and 0.2 mM Neu5Ac (acted as template) in 0.1 M PBS (pH 4.5). After removal of the template, the modified electrode was incubated in a solution of Neu5Ac (to be assayed) in 0.1 M PBS (pH 4.5) for 180 s followed by washing and drying. The ECL assay was carried out in 0.1 M PBS (pH 7.5) solution containing 50 mM H2O2 by cyclic voltammetry in the range −1.6 V to 1.2 V at a scan rate of 200 mV s−1. In this assay, Neu5Ac and H2O2 both acted as co-reactants for the anodic ECL of TiO2–Ru(bpy)32+; whereas only H2O2 acted as reactant for cathodic ECL of PEI-CdS quantum dots. Thus, in the presence of Neu5Ac, the intensity of the anodic ECL increased due to the availability of both co-reactants, but the intensity of the cathodic ECL of PEI-CdS quantum dots decreased due to the blockage of the available passage of H2O2 through imprinted cavities by Neu5Ac occupancy in the MIP.479
In the perspective of electrochemical assays as discussed in the above sub-sections, it should be mentioned that current electrochemical assays of Sia are competing well with expensive and time-consuming chromatographic and electrophoretic assays. The amperometric and potentiometric assays could provide a good LDR within about 20 μM–15 mM range for Neu5Ac.464,466–469 The aptamer based DPV assay could detect Sia at ultra-trace levels down to 80 nM in clinical samples472 and the EIS assay was able to bring the LOD further down to 0.4 pM with a LDR of 1 pM–10 μM.474 There are few reports published on the OECT, photoelectrochemical and ECL based electrochemical assays, although the ECL assay showed interesting results for quantifying Neu5Ac in the wide LDR 0.11 pM–1.13 μM (converted from 3.5 × 10−5–350 ng mL−1) with a very competitive LOD of ∼38 fM (converted from 1.17 × 10−5 ng mL−1) to HPLC-MS/SM assays.478 Therefore, this branch of electrochemical assay of Sia can inspire further research.
5.8 Quartz crystal microbalance (QCM)
In the year 2000, Kugimiya et al. coated two different types of MIPs on 9 MHz AT-cut platinum QCMs for assay of Sia in methanol (or water) in pyridine (99:1 v/v). The first type of MIP was prepared with p-vinylbenzeneboronic acid as a chemical receptor of Sia, whereas the second type of MIP was prepared with p-vinylbenzeneboronic acid and N,N,N-trimethylaminoethyl methacrylate, which could provide additional attractive electrostatic force to the carboxylate functional group of Sia. The p-vinylbenzeneboronic acid ester of Sia was used as a template molecule during formation of both MIPs. The resonance oscillation frequency of both the modified QCMs linearly varied with the concentration of Sia in the range 20–250 nmol.480 Twelve years later, Duan et al. used a gold QCM and modified it with a monolayer of 4-mercaptophenylboronic acid for a direct assay of Sia solely relying on the special affinity of boronic acid to Sia. The LDR and LOD of this QCM assay reported as 0.50–5.0 and 0.15 mM, respectively.481 In 2018, Qiu et al. modified gold QCM with allylmercaptane to introduce an allyl group on the QCM surface and then prepared a MIP on it by using Sia (as template molecule), 4-aminophenylboronic acid (as functional monomer), N,N-methylene-bis(acrylamide) (as cross-linker) and ammonium persulfate (as polymerisation initiator). This assay provided the best LDR and LOD of 0.025–0.50 μM and 1.0 nM, respectively, for Sia in 50 mM phosphate buffer (pH 7.7) among all QCM assays of Sia.482 Hence, the research on the QCM assay of Sia can be considered as rare, being less reported compared to other assays.
5.9 Surface plasmon resonance (SPR)
In 2001, Kugimiya et al. prepared p-vinylbenzeneboronic acid ester of Neu5Ac by azeotropic distillation in dry pyridine and used the ester as a template molecule for a thin MIP layer prepared on a gold-coated SPR chip. The MIP was prepared by co-polymerizing N,N,N-trimethylaminoethyl methacrylate, 2-hydroxyethyl methacrylate, ethyleneglycol dimethacrylate and 2,2′-azobis(dimethylvaleronitrile) in DMF. In the assay, the sample was passed along with 20 mM Tris-HCl buffer (pH 8.0) carrier solution over the SPR chip and the SPR resonance angle was monitored at 660 nm. The authors observed no change in the SPR resonance angle for free Neu5Ac compared to the control, but the SPR resonance angle linearly changed for sialylated gangliosides in the concentration range 0.1–1.0 mg mL−1.483 After a long gap until 2018, Li et al. developed a new SPR assay of Sia owing to the synchronous amplification of the opto-electric response of localised SPR during linear sweep of electrochemical potential. They sputter coated SiO2 layer of 100 nm thickness on the nanocone array of (poly)ethylene-terephthalate substrate fabricated by laser interference lithography followed by reaction ion etching. Then, AuNPs of diameter ∼40 nm were deposited on it by electron-beam evaporation technique. They also electrodeposited silver nanoparticles (AgNPs) of diameter ∼30–50 nm on it by using a potentiodynamic method to enhance the conductivity of the platform and hence sensitivity of the nanochip, while modifying the surface of AuNPs with mercaptophenyl boronic acid for selective adsorption of Neu5Ac. The localised SPR was recorded in the wavelength range 400–650 nm in 0.01 M PBS (pH 7.4) during the sweep of electrode potential in the range 0.0–0.3 V vs. Ag/AgCl at a scan rate of 0.05 V s−1. The applied electrochemical potential increased the sensitivity of localised SPR. The refractive index of the SPR-chip and water interface increased in the presence of Neu5Ac because of the preferential binding of Sia by the boronic acids and the SPR peak intensity decreased while shifting to higher wavelengths. At the same time, the adsorbed Sia on the SPR-chip hindered the movement of the redox-probe (the name was not specified in the manuscript) towards the electrode decreasing voltammetric peak current in the presence of Sia in the test solution. Hence, using localised SPR signal recorded during the potential sweep authors could quantify Neu5Ac in the LDR 0.05–5.00 mM with a LOD of 17 μM.484 However, it can be easily understood now that SPR based analysis of Sia is another neglected branch of science along with QCM assay.5.10 Surface-enhanced Raman spectroscopy (SERS)
In 2014, Vinogradova et al. first reported the possibility of SERS assay of Sia by using citrate capped AgNPs. Density functional calculation suggested that the negatively charged Neu5Ac could be chemisorbed on the surface of AgNPs via competitive adsorption of the carboxylate functional groups. The Raman peak of Neu5Ac at 1391 cm−1 enhanced significantly after the adsorption on citrate capped AgNPs upon excitation with 785 nm light.485 In order to understand the mechanism of adsorption of Neu5Ac on citrate capped AgNPs, Hernández-Arteaga et al. extended the experimental and DFT studies on Neu5Ac adsorbed on citrated capped AgNPs in comparison to propanethiol capped AgNPs. It was revealed that citrates could form aggregate structures on AgNPs leaving enough vacant sites available for the adsorption of Neu5Ac on silver by competitive surface adsorption of carboxylate functional groups, whereas propanethiol could uniformly coat AgNPs and hence did not allow adsorption of Nue5Ac on the silver surface.486 They further extended this study for the diagnosis of breast cancer through the SERS assay of Neu5Ac in human plasma. They used a special logarithmic equation with the average intensities of the SERS peaks at 1391 cm−1 (corresponding to stretching of –COOH), 1237 cm−1 (corresponding to C–N mode of amide III) and 1002 cm−1 (corresponding to breathing of pyranose ring) in the concentration range 1–200 mg dL−1 of Neu5Ac.487Recently, Teng et al. reported an enzyme assisted SERS assay of Neu5Ac in the human blood serum (Fig. 8[D]). They drop-casted citrate capped AuNPs on an indium-tin-oxide coated glass plate and submerged that into 0.1 mM 4-mercaptophenylboronic acid solution in dilute ethanol (pH 9.18) for 24 h at room temperature to make a sensing platform. Then, Neu5Ac and glucose were incubated on the sensing platform for 2 h followed by incubation with 0.1 mM 4-mercaptophenylboronic acid solution in dilute ethanol (pH 9.18) for another 24 h at room temperature. Then, that sensing platform was thoroughly washed and again immersed into the colloidal dispersion of citrate capped AuNPs for 18 h at room temperature to form a sandwich structure with AuNPs. To eliminate the interference of glucose, the same procedure was followed but after incubation of the test solution of Sia containing glucose with glucose oxidase at 37 °C for 2 h. The SERS signal intensities of Neu5Ac both with and without glucose interference were independently recorded using a confocal Raman instrument set with 785 nm laser line and subtracted to calculate the concentration of Neu5Ac. The LDR and LOD of this SERS assay were reported as 0.25–1.5 and 0.25 μM, respectively.488 The SERS detection of Sia, although promising for clinical applications, is understudied to date compared to other assays for detection and quantification of Sia.
Table 3 represents a summary of LDR, LOD and nature of tested samples of the analytical methods (as discussed in Section 5.2–5.10) validated to quality Sia in either biological or commercial samples.
| Assay method | LDR | LOD | Method validated by determination of Sia in | Ref. |
|---|---|---|---|---|
| Colourimetry and spectrophotometry | 1.0–10.0 mg L−1 | 0.239 mg L−1 | Whole, semi-skimmed and skimmed milks | 378 |
| 0.15–1.0 mM | 0.06 mM | Simulated human saliva | 379 | |
| 80–250, 300–700 μM | 35 μM | Commercial human serum extracted from human male AB plasma | 380 | |
| Photoluminescence | 12–680 ng mL−1 | 0.67 ng mL−1 | Whole egg, yolk and egg white powders, egg noodles, cake and steamed bun | 387 |
| 0.05–1.5 mg mL−1 | 16 μg mL−1 | Commercial infant formula | 388 | |
| 0.08–4.0 mM | 54 μM | Real human serum | 390 | |
| 0.02–1.0 mM | 9.24 μM | Simulated human saliva | 391 | |
| 10 nM–10 mM (in logarithmic scale) | 7.08 nM | Real human serum | 392 | |
| HPLC-UV–vis | 0.05–0.40 mM (Neu5Ac); 0.025–0.200 mM (Neu5Gc) | 5.5 μM (Neu5Ac); 0.4 μM (Neu5Gc) | Unprocessed bovine milk | 424 |
| HPLC-FLD | 30–1000 ng mL−1 | 2 ng mL−1 | Human serum | 426 |
| 3.5–28 pmol | — | Human pituitary gonadotropins viz. follitropin and lutropin | 432 | |
| 25–250 ng | 3.71 ng | Commercial infant formula | 433 | |
| 576 fmol–2.0 nmol (Neu5Ac); 556 fmol–2.0 nmol (ADOA) | 173 fmol (Neu5Ac); 167 fmol (ADOA) | Human saliva | 435 | |
| 50–500 μg mL−1 | 10 μg mL−1 | Fat globule membrane of yak milk | 436 | |
| — | 6.00 pg (Neu5Ac); 8.80 pg (Neu5Gc) | Foetal bovine serum and glycoprotein fetuin | 437 | |
| HPLC-MS | <7800 μmol L−1 | 1.7 (total); 0.3 μM (free) | Human urine | 445 |
| 0.1–3.0 ng μL−1 (Neu5Ac); 0.01–0.3 ng μL−1 (Neu5Gc) | 5 pg (Neu5Ac); 1 pg (Neu5Gc) | Glycoprotein and blood serum | 447 | |
| 25.0–10000 ng mL−1 |
— | Human plasma | 448 | |
| 0.05–5.0 μg mg−1 | — | Milk-based commercial infant formulas | 449 | |
| 0.25–10.0 μM (Neu5Ac; NeuGc) | 1.09 pmol (Neu5Ac); 1.06 pmol (Neu5Gc) | Extracted glycoprotein from the root of a Tibetan plant Potentilla anserina L. | 31 | |
| 20.0–20000 ng mL−1 |
20 ng mL−1 (LOQ) | Human plasma | 453 | |
| 0.2–500 ng mL−1 | 0.03 ng mL−1 (Neu5Ac); 0.04 ng mL−1 (Neu5Gc) | Urine and serum | 454 | |
| Capillary electrophoresis | 2–200 μg mL−1 | 0.5 μg mL−1 | Human saliva | 462 |
| 0.02–2.0 μg mL−1 (Neu5Ac, Neu5Gc in milk); 0.01–2.0 μg g−1 (Neu5Ac, Neu5Gc in milk powder) | 2.2 ng mL−1 (Neu5Ac in milk); 1.7 ng mL−1 (Neu5Ac in milk powder); 2.0 ng mL−1 (Neu5Gc in milk and milk powder) | Commercial milk and milk powder | 463 | |
| Amperometry | >200 μM | 10 μM | Epotin, blood, and serum samples of human | 464 |
| 0.025–15.0 mM | 18 μM | Blood plasma | 466 | |
| Potentiometry | 2 μM–1.38 mM | 0.8 μM | Human blood serum | 467 |
| 40–440 μM | 0.5 μM | Infant formulas | 468 | |
| 80 μM–8.2 mM | 60 μM | Human serum | 469 | |
| Voltammetry and EIS | 1–100 μM | — | Pea seedlings | 30 |
| 0.1–7.5 mM | 28.5 μM | Human blood and urine samples | 471 | |
| 0.1–440 μM | 0.08 μM | Human blood | 472 | |
| 1 pM–10 μM | 0.4 pM | Live rat brain microdialysates | 474 | |
| 25 μM–0.998 mM | Artificial human urine | 475 | ||
| OECT | 0.1–7.0 mM | — | Human serum | 476 |
| Photoelectrochemical and ECL | 3.5 × 10−5–35.0 ng mL−1 (logarithmic scale) | 1.2 × 10−5 ng mL−1 | Human blood | 477 |
| 3.5 × 10−5–350 ng mL−1 (logarithmic scale) | 1.17 × 10−5 ng mL−1 | Human serum | 478 | |
| 1.0 nM–0.1 mM (logarithmic scale) | 0.017 nM | Synthetic commercially available human serum and bird's nest | 479 | |
| QCM | 0.025–0.50 μM | 1.0 nM | Artificial human urine | 482 |
| SERS | 1–200 mg dL−1 | — | Human saliva | 487 |
| 0.25–1.5 μM | 0.25 μM | Human blood plasma | 488 |
5.11 Commercially available Sia assay kits
The commercially available assay kits of Sia are mostly limited to either colourimetric-spectrophotometric, fluorometric or HPLC-FLD methods. The colorimetric assay kit E-BC-K068489 and MBS2540448490 & MBS2563693491 are microplate-reader and spectrophotometer based reagents containing Sia standard (i.e., reagent 1) and a mixture of methyl resorcinol and an oxidant (i.e., reagent 2). The incubation of serum, plasma, saliva, urine, hydrothorax, tissue and cell samples containing Sia with reagent 2 produces a purplish red complex having λmax at 560 nm. The concentration of Sia in the sample can be determined within 60 min (total assay time) from the absorbance value of the sample with respect to standard in LDR 0.022–7.0 mM. The majority of colourimetric/fluorometric assay kits of Sia follow either of enzymatic or improved Warren method. The assay kits K566,492 EnzyChrom ESLA,493 LS-K187,494 PK-CA577-K566,495 ab83375,496 ABIN411705,497 CAK1183,498 MBS8309610,499 MBS841610,500 S1013-31Q,501 & A0535-01A,502 Kit-1099503 and Kit-2477504 produce equivalent amount of H2O2 from free Sia by the successive enzymatic reactions of sialic acid aldolase and pyruvate oxidase (reactions 2 and 4 of Section 5.4). Either absorbance (at 570 nm) or fluorescence (excitation and emission maxima at 535 and 587 nm, respectively) of the oxidised product of the reaction between H2O2 and amplex red (also known as amplisyn red, 10-acetyl-3,7-dihydroxyphenoxazine and Oxi-Red probe) in the presence of horseradish peroxidase enzyme (reaction 5 of Section 5.4) is used for quantification of free Sia in LDR 0.1–10 nmol (0.02–1.0 mM (colourimetric) and 0.002–0.100 mM (fluorometric)) in 40–60 min (assay time). On the other hand, the assay kits KA1655,505 DSLA,506 S1013-31P,507 MAK314,508 Kit-0783509 and BA0058510 oxidise Sia by periodate to formylpyruvic acid, which produces a pink-coloured product (having absorbance at 549 nm and excitation/emission maxima at 555/585 nm) from reaction with thiobarbituric acid. These kits can quantify free Sia in LDR 0.005–1.0 mM (colourimetric) and 0.0005–0.100 mM (fluorometric) in 40–60 min (assay time).The commercially available Sia assay kit SIALICQ can determine both bound and free Sia through successive enzymatic reactions of exo-α-sialidase, sialic acid aldolase and lactate dehydrogenase in the presence of NADH, whose absorbance is monitored at 340 nm (reactions 1–3 of Section 5.4).511
The Sia assay kits 4400512 and GKK-407513 use (ultra)HPLC-FLD methods with a pre-DMB derivatization step for the determination of free and bound (by additional hydrolysis step) Sia down to fmols. The assay kits LT-KDMB-A1514 and GS24-SAP515 follow similar procedure and it provided Sia reference panels containing Neu5Ac, Neu5Gc, Neu5,7Ac2, Neu5,9Ac2, Neu5,Gc9Ac, Neu5,7,(8),9Ac3Gc along with quantitative standards of Neu5Ac and Neu5Gc.
6. Indirect utilization of Sia in cytosensing, therapy and imaging of living cells
As discussed in the previous section, the SERS based assay is underutilised to date with respect to quantification of Sia, however Song et al. have recently used this technique to capture images of Sia on living cells. They prepared a SERS probe of gold nanoflower modified with 5,50-dithio-bis(2-nitrobenzoic acid) (as Raman reporter) and 3-mercaptophenylboronic acid (as Sia receptor). This modified nanoflower can bind to the cell surface Sia at physiological pH (i.e., 7.4) with the boronic acid group, which will also be available to make a sandwich detection system by anchoring another SERS probe made of polySia and 5,50-dithio-bis(2-nitrobenzoic acid) modified poly(amidoamine) encapsulated AuNPs. This sandwich-based SERS technique was used to image the cell surface over-expression of Sia on the human breast adenocarcinoma (MCF-7) cells in comparison to normal immortalized human keratinocytes (HaCaT) cells.516 Gong et al. synthesised 4-(dihydroxyborophenyl) acetylene for dual role. It results in an interference-free Raman signal as well as binding to Sia on cell surface. Upon loading the mixture of AuNPs and test cells incubated with 4-(dihydroxyborophenyl) acetylene at the side panel of the photonic crystal fibre and propagating laser light through central core, they could measure the average Sia concentration even on a single-cell by utilising the Raman peak at 2000 cm−1 corresponding to acetylene unit (Fig. 9[A]).517 Similarly, He et al. modified AgNPs of average diameter 60 nm, by both 4-mercaptophenylboronic acid (as Sia binding unit) and 4-mercaptobenzenitrile (as Raman reporter) to quantify the average cell surface Sia concentration from the interference-free SERS peak at 2232 cm−1 corresponding to cyanide unit.518 Liang et al. modified AgNPs of average diameter of 40 nm by 4-mercaptophenylboronic acid and used confocal SERS technique at 1074 and 1570 cm−1 for measuring and imaging the overexpression of Sia on HepG2, HeLa, BNL.CL2 cells to diagnose breast cancer.519 Shinde et al. prepared a SiO2 (as core of diameter ∼200 nm) and fluorescent-MIP (as shell of thickness ∼10 nm) based composite material for selective imaging of sialylated glycans on cell surfaces. They used nitrobenzoxadiazole (as fluorescence reporter having excitation and luminescence peaks at 411 and 509 nm, respectively), ester of polyvinylphenylboronic acid and Neu5Ac (as template) and 2-aminoethyl-methacrylate hydrochloride (as additional hydrogen bonding agent) and 2-(-3-(4-nitrobenzo[c][1,2,5]oxadiazo-7-yl)ureido)ethylmethacrylate (as additional carboxylate recognition site for Sia) in the MIP shell. This core–shell fluorescence probe was successfully used to image the sialylated glycans on surfaces of the prostate cancer cell lines DU145, PC3 and leukemic cell line Jurkat T cells.520 As discussed in section 5.3, the fluorescent polydiacetylene based liposome was prepared for the fluorescence-based assay of free Sia. 100 μM of this fluorescence probe was also used for imaging sialylated glycans expressed on MCF-7 cells through confocal microscopy (Fig. 9[B]).522 Peng et al. prepared tetraphenylethene tagged peptidylboronic acids having aggregation-induced emission at 460 nm (maxima). The change in luminescent intensity was selective for Sia compared to other monosaccharides and this phenomenon was used to label and image cancer cells owing to higher Sia expressions on their cell surfaces.521 Recently, Geng et al. modified AgNPs of average diameter ∼50 nm with 4-mercaptophenylboronic acid to recognise Sia and used it in a non-linear optical imaging technique known as four-wave-mixing to visualize Sia on A549, HeLa, HepG2 and MCF-7 cells.522 Almeida-Marrero et al. recently synthesised a photosensitizer by covalently linking dendritic zinc(II) phthalocyanine with 3 units of Neu5Ac for the photodynamic therapy through in situ generation of photoinduced singlet oxygen and other reactive species in the lysosome. The amphiphilic Neu5Ac modified photosensitizer formed non-photoresponsive self-assembled nano-aggregates and enters into the cellular lysosome, where it could regain its photoresponse.523 Yang et al. modified a gold coated QCM with mercaptobutanedioic acid followed by concanavalin A lectin to specifically attach mannose of the cell surface of RBC. Then AuNPs modified with 4-aminobenzeneboronic acid were added on the modified QCM with RBC to measure the average number of Sia 2.1 ± 0.2 × 108 on RBC of healthy person compared to 8.2 ± 0.7 × 107 on RBC of a diabetic patient.524 | ||
| Fig. 9 [A] Illustration of highly sensitive SERS method for detection and quantification method of Sia on single cell using photonic-crystal fiber with gold nanoparticles: (a) chemical structure of Sia on cell surface; (b) chemical structure of 4-(dihydroxyborophenyl) acetylene (DBA); (c) DBA-bonded Sia on the membrane of a cell at the C-8,9 diol of Sia; (d) Raman spectrum detected from DBA tagged cell in a solid core photonic crystal fiber, the 2000 cm−1 peak corresponded to alkyne group from DBA; (e) schematic of solid core photonic crystal fiber as a SERS platform for detection of Sia on a single cell (figures reproduced with permission from ref. 517) Copyright 2015 Elsevier. [B] UV-light assisted self-assembly of 10,12-pentacosadiynoic acid–phenylboronic acid (PCDA–pBA as Sia recognising unit), 10,12-pentacosadiynoic acid–1,8-naphthalimide (PCDA–Nap as fluorescent unit with excitation and luminescence maxima at 420 and 535 nm, respectively) and 10,12-pentacosadiynoic acid–ethyl acrylate (PCDA–EA) to form a composite polydiacetylene (PDA) based liposome and the schematic illustration of the as-prepared PDA liposomes for specific cell-glycan imaging (figures reproduced with permission from ref. 395) Copyright 2018 Royal Society of Chemistry. [C] Preparation of a phenylboronic acid (PBA) end functionalised poly-(ethylene glycol) (PEG) onto a cantilever of atomic force microscope (AFM) for dynamic Sia specific imaging on human breast cancer cell surfaces with sub-micrometer scale resolution: (a) synthetic scheme for PBA end-functionalized polymer; (b) installation of the polymer to an AFM cantilever by click chemistry. (c) Scanning electron microscopy image of the prepared PBA-modified AFM cantilever and schematic representation of its interaction with Sia on cell surface (figures reproduced with permission from ref. 530) Copyright 2020 American Chemical Society. [D] (a) The appearance of liposome (L) composed of hydrogenated soy phosphatidylcholine, cholesterol and Sia-octadecylamine in solution of epirubicin (S-E-L), zoledronate (S-Z-L), mixture of epirubicin and zoledronate (S-ZE-L) in comparison to solution of epirubicin (E-S), zoledronate (Z-S), mixture of epirubicin and zoledronate (ZE-S); (b) stable complex of epirubicin and zoledronate with water molecules, (c) transmission electron micrographs of the liposomes S-Z-L, S-E-L and S-ZE-L (scale bar: 100 nm, the red arrows mark the low-solubility aggregates); (d) in vitro drug release from S-E-L, S-Z-L and S-ZE-L liposomes in comparison to E-S and Z-S (n = 3) (figures reproduced with permission from ref. 531) Copyright 2021 Elsevier. | ||
Qian et al. demonstrated a multivalent recognition and signal amplification strategy for cancer cell cytosensing and dynamic evaluation of cell surface Sia using graphene oxide functionalised with 3-aminophenylboronic acid for cell capture and Sambucus nigra agglutinin and thionine conjugated to gold nanoparticles. The DPV based electrochemical method was used to monitor the changes in expression of Sia in response to addition of sialidase enzyme.525 A similar approach combined 3-aminophenylboronic acid modified carbon nanospheres for Sia recognition with horseradish peroxidase decorated gold nanoparticles for signal amplification. The electrochemical cytosensor realised an identification system using Sambucus nigra agglutinin and 3-aminophenylboronic acid modified carbon nanospheres for cancer cell capture with measurement of the expression level of Sia on MCF-7 cells vs. control cell lines (LOD of 25 cells per mL).526 Zhang et al. electrochemically prepared poly-pyrrole on a gold electrode allowing physical inclusion of bovine serum albumin incorporated silver submicron particles into the polymer matrix. Then the electrode was modified with 3-aminophenylboronic acid through the cross-linking agent p-phenylene-diisothiocyanate. This modified electrode was used to diagnose renal cell carcinoma from the change of charge-transfer resistance of [Fe(CN)6]3−/4− electrochemical reaction with LDR and LOD 17–1.7 × 106 and 6 Cell786-O per mL (S/N = 3), respectively.527 Matsumoto et al. used 3-aminophenylboronic acid end-functionalised poly(ethylene glycol) modified gold electrode for potentiometric determination of a blood-circulating glycoprotein fetuin at pH 7.4.528
Landa et al. modified AuNPs of average diameter 20 nm with Neu5Ac to generate a negatively charged nanoprobe of zeta potential −13.21 ± 1.87 mV and absorption maxima at 520 nm in neutral pH. This nanoprobe was agglomerated in the presence of gram-positive bacteria viz. Staphylococcus aureus and methicillin resistant Staphylococcus aureus extending the absorption to longer wavelengths, but did not show any changes in the presence of Gram-negative bacteria viz. Pseudomonas aeruginosa. This method was effective to discriminate Gram-positive and Gram-negative bacteria at high concentration ∼105 CFU mL−1 in tryptic soy broth medium and serum sample.529
Finally, a phenylboronic acid end functionalised poly-(ethylene glycol) was decorated onto a cantilever of atomic force microscope for dynamic Sia specific imaging on human breast cancer cell surfaces with sub-micrometer scale resolution (Fig. 9[C]). A greater extent of probe-surface interaction was observed for the high Sia expressed MCF-7 as compared to health control cell lines, taking advantage of the reversible and pH dependant boronic acid-Sia binding chemistry.530Sia receptors are usually over-expressed on the surface of tumour-associated macrophages. Sui et al. reported that sequential administration of Sia (0.2 mg mL−1) modified epirubicin (0.056 mg mg−1 of lipid) liposome followed by (after 24 h) Sia modified zoledronate (0.08 mg mg−1 of lipid) liposome (Fig. 9[D]) could kill the tumour-associated macrophages and restrict tumour growth in mice.531
The commercially available EZClick™ sialic acid (ManAz) modified glycoprotein assay kit K441 uses direct feeding of modified ManAc precursor into the cell for biosynthesis of Sia followed by its participation in N-glycosylation of cell surface protein, which can be mapped and quantified by using fluorescence-activated cell sorting method following the click reaction with an alkyne-containing dye. This assay is very useful to identify the altered sialylation of tumour-cell surfaces.532 Similarly, the GlyS Kit is used to screen the terminal Sia content of glycans in protein of interest even in the presence of host cell glycoproteins in crude samples by high throughput Octet system.533
7. Future direction
Sia is widely distributed in free, polymeric and conjugated forms in living creatures. However, the discovery and identification of new members of the Sia family is yet an open science. Furthermore, with the advancement of analytical methodologies to achieve ultra-trace LODs, exploration of Sia in different parts of plants has a great opportunity to either discover green resources of Sia or confirm the absence of Sia in plants, fruits and vegetables.The involvement of Sia in important physiology including brain development and formation of central nervous system during foetal and neonatal stages has been studied mostly on animals viz. laboratory rats and pigs and correlated to human physiology. The participation of Sia in the human immune system has been studied, and has keen ongoing interest, especially given the inconclusive evidence of Sia's involvement in several bacterial and viral infections including SARS-CoV-2 causing COVID-19 disease. The over-/under-expression of Sia in human cells/tissues/fluids during physiological disorders and onset of cancers are now in high surveillance to promote Sia as one of the potential biomarkers for a number of human diseases.
The correlated case studies of Sia in human lactation and infant cognition produced sufficient evidence to receive regulatory approval for use of Neu5Ac.2H2O, 3′-sialyllactose sodium salt and 6′-sialyllactose sodium salt in infant formula and commercial food products as nutrition additives. This has definitely boosted the demand of Sia in commercial domain, although the number of economic and large-scale production of Sia suitable for safe consumption by humans is very limited at present.
The emerging biological relevance of Sia in human health and clinical sciences along with its commercial role in human health and nutrition has increased the importance of development of robust sensors and diagnostic methods for Sia in complex clinical and food matrices. There is a vast scope of research and development in this direction as presented in this review. In summary, it should be mentioned that knowledge of the biological role and participation of Sia in human physiology is expanding day-by-day. Hence, the extension of the analytical horizons of Sia beyond the current state of the art will underpin further explorations in the expansive Sia galaxy.
Symbols and abbreviations
Symbols
| ε | Molar absorption coefficient |
| λ Max | Absorbance maxima |
| C | Carbon |
| n | Number of samples/persons/patients |
Abbreviations
| ACE2 | Angiotensin-converting enzyme 2 |
| ADOA | 4-(Acetylamino)-2,4-dideoxy-D-glycero-D-galacto-octonic acid |
| AgNPs | Silver nanoparticles |
| AuNPs | Gold nanoparticles |
| cGMP | Caseinoglycomacropeptide |
| CMP | Cytidine-5′-monophosphate |
| CoV | Corona virus |
| CTD | C-Terminal domain |
| DMB | 1,2-Diamino-4,5-methyleneoxybenzene |
| DPV | Differential pulse voltammetry |
| EBN | Edible bird's nest |
| ECL | Electrochemiluminescence |
| EIS | Electrochemical impedance spectroscopy |
| ESI | Electron spray ionization |
| HMOs | Human milk oligosaccharides |
| HPLC-FLD | High performance liquid chromatography coupled with fluorescence detection |
| HPLC-MS | High performance liquid chromatography coupled with mass-spectrometry detection |
| HPLC-PAD | High performance liquid chromatography coupled with pulsed-amperometry detection |
| HPLC-UV–vis | High performance liquid chromatography coupled with UV–Vis detection |
| KDN | 2-Keto-3-deoxy-D-glycero-D-galacto-nononic acid |
| LDR | Linear dynamic range |
| LOD | Limit of detection |
| ManNAc | N-Acetyl-D-mannosamine |
| MERS-CoV | Middle east respiratory syndrome coronavirus |
| MIP | Molecularly imprinted polymer |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NCAM | Neural cell adhesion molecule |
| ND | Not detected |
| Neu5Ac | N-Acetyl-D-neuraminic acid |
| Neu5Gc | N-Glycolyl-D-neuraminic acid |
| NMR | Nuclear magnetic resonance |
| NR | Not reported |
| NTD | N-terminal domain |
| OECT | Organic electrochemical transistors |
| PBS | Phosphate buffer saline |
| PEC | Photoelectrochemistry |
| polySia | Poly-sialic acid |
| QCM | Quartz crystal microbalance |
| RBC | Red blood cells |
| SAMPs | Self-associated molecular patterns |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 (novel corona virus) |
| SERS | Surface-enhanced Raman spectroscopy |
| Sia | Sialic acid |
| Siglec | Sialic acid binding immunoglobulin like lectin |
| SPR | Surface plasmon resonance |
| ST8SiaII | ST8-α-N-Acetyl-neuraminide-α-2,8-sialyltransferase-2 |
| ST8SiaIV | ST8-α-N-Acetyl-neuraminide-α-2,8-sialyltransferase-4 |
| T2DM | Type 2 diabetic mellitus |
| UDP-GlcNAc | Uridine diphosphate-N-acetylglucosamine |
8. Funding source
This project has received funding from the Enterprise Ireland and the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie Career FIT PLUS grant agreement no. 847402 (Project No. MF20200140).Author contributions
Dr. Saurav K. Guin has completed the literature survey and written the original draft. Dr. Eithne Dempsey and Dr. Trinidad Velasco-Torrijos reviewed and edited the manuscript. Dr. Dempsey and Dr. Guin are jointly involved in conceptualisation, funding acquisition and administration of this project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr. Guin wishes to acknowledge the Open-Access Publication Policy of Enterprise Ireland and the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie Career FIT PLUS fellowship for dissemination of work to wider community. The authors acknowledge Dr. Fergal Lawless and Glanbia Ingredients Ireland. Dr. Guin wishes to thank Dr. P.K. Pujari, Director of Radiochemistry and Isotope Group and Dr. S. Kannan, Head of Fuel Chemistry Division in Bhabha Atomic Research Centre for approving his visit (on leave) in Ireland.References
- R. Schauer and J. P. Kamerling, in Glycoproteins II, ed. J. Montreuil, J. F. G. Vliegenthart and H. Schachter, Elsevier Science Publishers B.V, Amsterdam, 1997, pp. 243–372 Search PubMed.
- T. Janas and T. Janas, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2923–2932 CrossRef CAS PubMed.
- E. Klenk, Biol. Chem., 1935, 235, 24–36 CrossRef CAS.
- E. Klenk, H. Langerbeins and E. Schumann, Biol. Chem., 1941, 270, 185–193 CrossRef CAS.
- E. Klenk, Biol. Chem., 1941, 268, 50–58 CrossRef CAS.
- F. G. Blix, Biol. Chem., 1936, 240, 43–54 CrossRef.
- G. Blix, L. Svennerholm and I. Werner, Acta Chem. Scand., 1952, 6, 358–362 CrossRef CAS.
- T. Yamakawa and S. Suzuki, J. Biochem., 1951, 38, 199–212 CrossRef CAS.
- A. Gottschalk, Nature, 1951, 176, 845–847 CrossRef PubMed.
- J. R. E. Hoover, G. A. Braun and P. György, Arch. Biochem. Biophys., 1953, 47, 216–217 CrossRef CAS PubMed.
- F. Zilliken, G. A. Braun and P. György, Arch. Biochem. Biophys., 1955, 54, 564–566 CrossRef CAS PubMed.
- K. Richard, R. Brossmer and M. von W. Schulz, Chem. Ber., 1954, 87, 123–127 CrossRef.
- A. Gottschalk, Nature, 1955, 176, 881–882 CrossRef CAS.
- F. G. Blix, A. Gottschalk and E. Klenk, Nature, 1957, 179, 1088 CrossRef CAS PubMed.
- L. Warren and H. Felsenfeld, J. Biol. Chem., 1962, 237, 1421–1431 CrossRef CAS.
- M. E. Tanner, Bioorg. Chem., 2005, 33, 216–228 CrossRef CAS PubMed.
- L. Shaw and R. Schauer, Biol. Chem. Hoppe-Seyler, 1988, 369, 477–486 CrossRef CAS PubMed.
- T. Hayakawa, Y. Satta, P. Gagneux, A. Varki and N. Takahata, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11399–11404 CrossRef CAS PubMed.
- P. S. K. Ng, R. Böhm, L. E. Hartley-Tassell, J. A. Steen, H. Wang, S. W. Lukowski, P. L. Hawthorne, A. E. O. Trezise, P. J. Coloe, S. M. Grimmond, T. Haselhorst, M. Von Itzstein, A. W. Paton, J. C. Paton and M. P. Jennings, Nat. Commun., 2014, 5, 5750 CrossRef CAS PubMed.
- S. A. Springer, S. L. Diaz and P. Gagneux, Immunogenetics, 2014, 66, 671–674 CrossRef CAS PubMed.
- M. Jahan, P. C. Thomson, P. C. Wynn and B. Wang, Food Chem., 2021, 343, 128439 CrossRef CAS PubMed.
- S. Inoue and K. Kitajima, Glycoconjugate J., 2006, 23, 277–290 CrossRef CAS PubMed.
- S. Ghosh, Sialic acid and biology of life: An introduction, Elsevier B.V., 2020/01/17, 2020 Search PubMed.
- M. Yang, S. H. Cheung, S. C. Li and H. Y. Cheung, Food Chem., 2014, 151, 271–278 CrossRef CAS PubMed.
- O. Mahaq, M. A. Mohd, M. Jaoi Edward, N. Mohd Hanafi, S. Abdul Aziz, H. Abu Hassim, M. H. Mohd Noor and H. Ahmad, Brain Behav., 2020, 10, e01817 Search PubMed.
- G. K. Chan, K. Y. Zheng, K. Y. Zhu, T. T. Dong and K. W. Tsim, The Journal of Ethnobiology and Traditional Medicine, 2013, 120, 620–628 Search PubMed.
- R. Zeleny, D. Kolarich, R. Strasser and F. Altmann, Planta, 2006, 224, 222–227 CrossRef CAS PubMed.
- A. N. Samraj, O. M. T. Pearce, H. Läubli, A. N. Crittenden, A. K. Bergfeld, K. Band, C. J. Gregg, A. E. Bingman, P. Secrest, S. L. Diaz, N. M. Varki, A. Varki and S. A. Kornfeld, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 542–547 CrossRef CAS PubMed.
- L. Ye, L. Mu, G. Li and Y. Bao, J. Food Compos. Anal., 2020, 87, 103393 CrossRef CAS.
- L. J. Sun, Y. Xie, Y. F. Yan, H. Yang, H. Y. Gu and N. Bao, Sens. Actuators, B, 2017, 247, 336–342 CrossRef CAS.
- L. Xia, L. Liu, F. Qu, R. Kong, G. Li and J. You, Chromatographia, 2017, 80, 861–872 CrossRef CAS.
- R. Schauer and J. P. Kamerling, Exploration of the Sialic Acid World, Elsevier Inc., 1st edn, 2018, vol. 75 Search PubMed.
- H. Yang, L. Lu and X. Chen, Biotechnol. Adv., 2021, 46, 107678 CrossRef CAS PubMed.
- M. Huizing, M. E. Hackbarth, D. R. Adams, M. Wasserstein, M. C. Patterson, S. U. Walkley, W. A. Gahl, K. Dobrenis, J. Foglio, B. Gasnier, M. Hackbarth, M. Lek, M. C. V. Malicdan, L. E. Paavola, R. Reimer, R. Y. Wang and R. Zoncu, Neurosci. Lett., 2021, 755, 135896 CrossRef CAS PubMed.
- R. Heida, Y. C. Bhide, M. Gasbarri, Ö. Kocabiyik, F. Stellacci, A. L. W. Huckriede, W. L. J. Hinrichs and H. W. Frijlink, Drug Discovery Today, 2021, 26, 122–137 CrossRef CAS PubMed.
- P. Burzy, K. Mikołajczyk, M. Jodłowska and E. Ja, Biomolecules, 2021, 11, 831 CrossRef PubMed.
- E. Gianchecchi, A. Arena and A. Fierabracci, Int. J. Mol. Sci., 2021, 22, 5774 CrossRef CAS PubMed.
- X. Zhou, G. Yang and F. Guan, Cell, 2020, 9, 273 CrossRef CAS PubMed.
- R. Lacomba, J. Salcedo, A. Alegría, M. Jesús Lagarda, R. Barberá and E. Matencio, J. Pharm. Biomed. Anal., 2010, 51, 346–357 CrossRef CAS PubMed.
- J. Cheeseman, G. Kuhnle, D. I. R. Spencer and H. M. I. Osborn, Bioorg. Med. Chem., 2021, 30, 115882 CrossRef CAS PubMed.
- V. Andriukaitis, Official Journal of the European Union, 2017, L 337, 63–67 Search PubMed.
- A. B. Engin, E. D. Engin and A. Engin, Environ. Toxicol. Pharmacol., 2020, 79, 103436 CrossRef CAS PubMed.
- P. Wielgat, K. Rogowski, K. Godlewska and H. Car, Cell, 2020, 9, 1963 CrossRef CAS PubMed.
- X.-L. Sun, Glycobiology, 2021, cwab032 Search PubMed.
- W. Zhao, T. L. L. Chen, B. M. Vertel and K. J. Colley, J. Biol. Chem., 2006, 281, 31106–31118 CrossRef CAS PubMed.
- F. Tokumasu, G. R. Ostera, C. Amaratunga and R. M. Fairhurst, Exp. Parasitol., 2012, 131, 245–251 CrossRef CAS PubMed.
- S. E. Quaggin, J. Clin. Invest., 2007, 117, 1480–1483 CrossRef CAS PubMed.
- A. Varki, Glycobiology, 2011, 21, 1121–1124 CrossRef CAS PubMed.
- X. Wang, N. Mitra, I. Secundino, K. Banda, P. Cruz, V. Padler-Karavani, A. Verhagen, C. Reid, M. Lari, E. Rizzi, C. Balsamo, G. Corti, G. De Bellis, L. Longo, W. Beggs, D. Caramelli, S. A. Tishkoff, T. Hayakawa, E. D. Green, J. C. Mullikin, V. Nizet, J. Bui and A. Varki, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9935–9940 CrossRef CAS PubMed.
- P. R. Crocker, J. C. Paulson and A. Varki, Nat. Rev. Immunol., 2007, 7, 255–266 CrossRef CAS PubMed.
- H. Läubli and A. Varki, Cell. Mol. Life Sci., 2020, 77, 593–605 CrossRef PubMed.
- S. Pillai, I. A. Netravali, A. Cariappa and H. Mattoo, Annu. Rev. Immunol., 2012, 30, 357–392 CrossRef CAS PubMed.
- B. S. Blaum, J. P. Hannan, A. P. Herbert, D. Kavanagh, D. Uhrín and T. Stehle, Nat. Chem. Biol., 2015, 11, 77–82 CrossRef CAS PubMed.
- C. Q. Schmidt, A. L. Hipgrave Ederveen, M. J. Harder, M. Wuhrer, T. Stehle and B. S. Blaum, Glycobiology, 2018, 28, 765–773 CrossRef PubMed.
- B. A. H. Smith and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2021, 20, 217–243 CrossRef CAS PubMed.
- K. Sołkiewicz, H. Krotkiewski, M. Jędryka and E. M. Kratz, Sci. Rep., 2021, 11, 5586 CrossRef PubMed.
- J. Finne, J. Biol. Chem., 1982, 257, 11966–11970 CrossRef CAS.
- C. Weisgerber, M. Husmann, M. Frosch, C. Rheinheimer, T. W. Peuckert and I. Gorgen, J. Neurochem., 1990, 55, 2063–2071 CrossRef CAS PubMed.
- J. Ahrens, N. Foadi, A. Eberhardt, G. Haeseler, R. Dengler, A. Leffler, M. Mühlenhoff, R. Gerardy-Schahn and M. Leuwer, Pharmacology, 2011, 87, 311–317 CrossRef CAS PubMed.
- J. Nakayama, M. N. Fukuda, B. Fredette, B. Ranscht and M. Fukuda, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7031–7035 CrossRef CAS PubMed.
- M. S. Skog, J. Nystedt, M. Korhonen, H. Anderson, T. A. Lehti and M. I. Pajunen, Stem Cell Res. Ther., 2016, 7, 113 CrossRef PubMed.
- P. M. Drake, J. K. Nathan, C. M. Stock, P. V. Chang, M. O. Muench, D. Nakata, J. R. Reader, P. Gip, K. P. K. Golden, B. Weinhold, R. Gerardy-Schahn, F. A. Troy and C. R. Bertozzi, J. Immunol., 2008, 181, 6850–6858 CrossRef CAS PubMed.
- T. M. Villanueva-Cabello, L. D. Gutiérrez-Valenzuela, D. V. López-Guerrero, M. E. Cruz-Muñoz, H. M. Mora-Montes and I. Martínez-Duncker, Glycobiology, 2019, 29, 557–564 CrossRef CAS PubMed.
- S. Werneburg, F. F. R. Buettner, M. Mühlenhoff and H. Hildebrandt, Stem Cell Res., 2015, 14, 339–346 CrossRef CAS PubMed.
- U. Yabe, C. Sato, T. Matsuda and K. Kitajima, J. Biol. Chem., 2003, 278, 13875–13880 CrossRef CAS PubMed.
- S. Curreli, Z. Arany, R. Gerardy-schahn, D. Mann and N. M. Stamatos, J. Biol. Chem., 2007, 282, 30346–30356 CrossRef CAS PubMed.
- R. W. Nelson, P. A. Bates and U. Rutishauser, J. Biol. Chem., 1995, 270, 17171–17179 CrossRef CAS PubMed.
- S. S. Mendiratta, N. Sekulic, A. Lavie and K. J. Colley, J. Biol. Chem., 2005, 280, 32340–32348 CrossRef CAS PubMed.
- S. S. Mendiratta, N. Sekulic, F. G. Hernandez-Guzman, B. E. Close, A. Lavie and K. J. Colley, J. Biol. Chem., 2006, 281, 36052–36059 CrossRef CAS PubMed.
- J. L. Brusés and U. Rutishauser, J. Cell Biol., 1998, 140, 1177–1186 CrossRef PubMed.
- P. C. Hallenbeck, E. R. Vimr, F. Yu, B. Basslero and A. Troyn, J. Biol. Chem., 1987, 262, 3553–3561 CrossRef CAS.
- C. D. Hunter and C. Cairo, ChemRxiv, 2021 DOI:10.26434/chemrxiv.14065025.v1.
- A. Mori, M. Hane, Y. Niimi, K. Kitajima and C. Sato, Glycobiology, 2017, 27, 834–846 CrossRef CAS PubMed.
- F. Miragall, G. Kadmon, M. Husmann and M. Schachner, Dev. Biol., 1988, 129, 516–531 CrossRef CAS PubMed.
- L. Bonfanti, Prog. Neurobiol., 2006, 80, 129–164 CrossRef CAS PubMed.
- U. Rutishauser, Nat. Rev. Neurosci., 2008, 9, 26–35 CrossRef CAS PubMed.
- C. Sato and K. Kitajima, Sialic Acids in Neurology, Elsevier Inc., 1st edn, 2019, vol. 76 Search PubMed.
- P. Simon, S. Bäumner, O. Busch, R. Röhrich, M. Kaese, P. Richterich, A. Wehrend, K. Müller, R. Gerardy-schahn, M. Mühlenhoff, H. Geyer, R. Geyer, R. Middendorff and S. P. Galuska, J. Biol. Chem., 2013, 288, 18825–18833 CrossRef CAS PubMed.
- R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2010, 9, 308–324 CrossRef CAS PubMed.
- Y. Wang and H. Neumann, J. Neurosci., 2010, 30, 3482–3488 CrossRef CAS PubMed.
- J. Roth, C. Zuber, P. Komminoth, T. Sata, W. P. Li and P. U. Heitz, Histochem. Cell Biol., 1996, 106, 131–148 CrossRef CAS PubMed.
- C. Sato and K. Kitajima, Mol. Aspects Med., 2020, 100892 Search PubMed , In press.
- K. F. Boligan, J. Oechtering, C. W. Keller, B. Peschke, R. Rieben, N. Bovin, L. Kappos, R. D. Cummings, J. Kuhle, S. von Gunten and J. D. Lünemann, Neurol. Neuroimmunol. Neuroinflamm., 2020, 7, e676 CrossRef PubMed.
- R. Sandhoff and K. Sandhoff, FEBS Lett., 2018, 592, 3835–3864 CrossRef CAS PubMed.
- L. Svennerholm, in Cholera and Related Diarrheas 43rd Nobel Symposium, Stockholm, 1978, pp. 80–87 Search PubMed.
- L. Svennerhom, Gangliosides and Synaptic Transmission, Springer, Boston, MA, 1980 Search PubMed.
- L. Svennerholm, Prog. Brain Res., 1994, 101, xi–xiv CAS.
- R. K. Yu, T. Ariga, M. Yanagisawa and G. Zeng, in Glycoscience, ed. B. O. Fraser-Reid, K. Tatsuta and J. Thiem, 2008, pp. 1671–1695 Search PubMed.
- R. K. Yu, Y. Nakatani and M. Yanagisawa, J. Lipid Res., 2009, 50, S440–S445 CrossRef PubMed.
- S. Ando, N. C. Chang and R. K. Yu, Anal. Biochem., 1978, 89, 437–450 CrossRef CAS PubMed.
- M. Sarbu, A. C. Robu, R. M. Ghiulai, Ž. Vukelić, D. E. Clemmer and A. D. Zamfir, Anal. Chem., 2016, 88, 5166–5178 CrossRef CAS PubMed.
- M. Sarbu, Ž. Vukelić, D. E. Clemmer and A. D. Zamfir, Analyst, 2018, 143, 5234–5246 RSC.
- M. Sarbu, L. Dehelean, C. V. A. Munteanu, Ž. Vukelić and A. D. Zamfir, Anal. Biochem., 2017, 521, 40–54 CrossRef CAS PubMed.
- M. Sarbu, S. Raab, L. Henderson, D. Fabris, Z. Vukelic, D. E. Clemmer and A. D. Zam, Biochimie, 2020, 170, 36–48 CrossRef CAS PubMed.
- N. Sasaki, M. Toyoda and T. Ishiwata, Int. J. Mol. Sci., 2021, 22, 5076 CrossRef CAS PubMed.
- M. Sarbu, L. Petrica, D. E. Clemmer, Ž. Vukelić and A. D. Zamfir, J. Am. Soc. Mass Spectrom., 2021, 32, 1249–1257 CrossRef CAS PubMed.
- M. Sarbu, D. E. Clemmer and A. D. Zamfir, Biochimie, 2020, 177, 226–237 CrossRef CAS PubMed.
- E. Chiricozzi, E. Di Biase, M. Maggioni, G. Lunghi, M. Fazzari, D. Y. Pomè, R. Casellato, N. Loberto, L. Mauri and S. Sonnino, J. Neurochem., 2019, 149, 231–241 CrossRef CAS PubMed.
- N. Sasaki, Y. Itakura and M. Toyoda, Int. J. Mol. Sci., 2019, 20, 1–12 Search PubMed.
- S. Sipione, J. Monyror, D. Galleguillos, N. Steinberg and V. Kadam, Front. Neurosci., 2020, 14, 572965 CrossRef PubMed.
- T. Mitsuda, K. Furukawa, S. Fukumoto, H. Miyazaki, T. Urano and K. Furukawa, J. Biol. Chem., 2002, 277, 11239–11246 CrossRef CAS PubMed.
- Y. Ohkawa, H. Momota, A. Kato, N. Hashimoto, Y. Tsuda, N. Kotani, K. Honke, A. Suzumura, K. Furukawa, Y. Ohmi, A. Natsume, T. Wakabayashi and K. Furukawa, J. Biol. Chem., 2015, 290, 16043–16058 CrossRef CAS PubMed.
- R. París, A. Morales, O. Coll, A. Sánchez-Reyes, C. García-Ruiz and J. C. Fernández-Checa, J. Biol. Chem., 2002, 277, 49870–49876 CrossRef PubMed.
- R. De Maria, L. Lenti, F. Malisan, B. Tomassini, A. Zeuner, M. R. Rippo and R. Testi, Science, 1997, 277, 1652–1655 CrossRef CAS PubMed.
- Y. Makino, K. Hamamura, Y. Takei, R. Hasan, Y. Ohkawa and G. Lauc, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 1753–1763 CrossRef CAS PubMed.
- K. Furukawa, M. Kambe, M. Miyata, Y. Ohkawa, O. Tajima and K. Furukawa, Cancer Sci., 2014, 105, 52–63 CrossRef CAS PubMed.
- K. Hamamura, M. Tsuji, H. Hotta, Y. Ohkawa, M. Takahashi, H. Shibuya, H. Nakashima, Y. Yamauchi, N. Hashimoto, H. Hattori, M. Ueda, K. Furukawa and K. Furukawa, J. Biol. Chem., 2011, 286, 18526–18537 CrossRef CAS PubMed.
- Y. Liang, C. Wang, I. Wang, Y. Chen, L. Li, M. Ho, T. Chou, Y. Wang, S. Chiou, Y. Lin and J. Yu, Oncotarget, 2017, 8, 47454–47473 CrossRef PubMed.
- B. L. Mirkin, S. H. Clark and C. Zhang, Cell Proliferation, 2002, 35, 105–115 CrossRef CAS PubMed.
- N. Kawashima, Y. Nishimiya, S. Takahata and K. I. Nakayama, J. Biol. Chem., 2016, 291, 21424–21433 CrossRef CAS PubMed.
- S. Kawamura, I. Sato, T. Wada, K. Yamaguchi, Y. Li, D. Li, X. Zhao, S. Ueno, H. Aoki, T. Tochigi, M. Kuwahara, T. Kitamura, K. Takahashi, S. Moriya and T. Miyagi, Cell Death Differ., 2012, 19, 170–179 CrossRef CAS PubMed.
- H. Wang, T. Isaji, M. Satoh, D. Li, Y. Arai and J. Gu, Urology, 2013, 81, 210.E11–210.E15 CrossRef PubMed.
- K. Shiga, K. Takahashi, I. Sato, K. Kato, S. Saijo, S. Moriya, M. Hosono and T. Miyagi, Cancer Sci., 2015, 106, 1544–1553 CrossRef CAS PubMed.
- H. J. Choi, T. W. Chung, S. K. Kang, Y. C. Lee, J. H. Ko, J. G. Kim and C. H. Kim, Glycobiology, 2006, 16, 573–583 CrossRef CAS PubMed.
- K. Takahashi, M. Hosono, I. Sato, K. Hata, T. Wada, K. Yamaguchi, K. Nitta, H. Shima and T. Miyagi, Int. J. Cancer, 2015, 137, 1560–1573 CrossRef CAS PubMed.
- T. W. Chung, H. J. Choi, S. J. Kim, C. H. Kwak, K. H. Song, U. H. Jin, Y. C. Chang, H. W. Chang, Y. C. Lee, K. T. Ha and C. H. Kim, PLoS One, 2014, 9, e92786 CrossRef PubMed.
- N. Sasaki, K. Hirabayashi, M. Michishita, K. Takahashi, F. Hasegawa, F. Gomi, Y. Itakura, N. Nakamura, M. Toyoda and T. Ishiwata, Sci. Rep., 2019, 9, 19369 CrossRef CAS PubMed.
- K. Nguyen, Y. Yan, B. Yuan, A. Dasgupta, J. Sun, H. Mu, K. A. Do, N. T. Ueno, M. Andreeff and V. Lokesh Battula, Mol. Cancer Ther., 2018, 17, 2689–2701 CrossRef CAS PubMed.
- A. Cazet, M. Bobowski, Y. Rombouts, J. Lefebvre, A. Steenackers, I. Popa, Y. Guérardel, X. Le Bourhis, D. Tulasne and P. Delannoy, Glycobiology, 2012, 22, 806–816 CrossRef CAS PubMed.
- T. Iwasawa, P. Zhang, Y. Ohkawa, H. Momota, T. Wakabayashi, Y. Ohmi, R. H. Bhuiyan, K. Furukawa and K. Furukawa, Int. J. Oncol., 2018, 52, 1255–1266 CAS.
- S. Yoshida, S. Fukumoto, H. Kawaguchi, S. Sato, R. Ueda and K. Furukawa, Cancer Res., 2001, 61, 4244–4252 CAS.
- W. Aixinjueluo, K. Furukawa, Q. Zhang, K. Hamamura, N. Tokuda, S. Yoshida, R. Ueda and K. Furukawa, J. Biol. Chem., 2005, 280, 29828–29836 CrossRef CAS PubMed.
- S. Hyuga, N. Kawasaki, M. Hyuga, M. Ohta, R. Shibayama, T. Kawanishi, S. Yamagata, T. Yamagata and T. Hayakawa, Int. J. Cancer, 2001, 94, 328–334 CrossRef CAS PubMed.
- S. H. Ha, J. M. Lee, K. M. Kwon, C. H. Kwak, F. Abekura, J. Y. Park, S. H. Cho, K. Lee, Y. C. Chang, Y. C. Lee, H. J. Choi, T. W. Chung, K. T. Ha, H. W. Chang and C. H. Kim, Int. J. Mol. Sci., 2016, 17, 652 CrossRef PubMed.
- J. H. Hwang, J. S. Sung, J. M. Kim, Y. H. Chung, J. S. Park, S. H. Lee and I. S. Jang, Am. J. Cancer Res., 2014, 4, 801–810 Search PubMed.
- R. Rajan, A. R. Sheth and S. S. Rao, Eur. J. Obstet. Gynecol. Reprod. Biol., 1983, 16, 37–46 CrossRef CAS.
- M. H. Alvi, N. A. Amer and I. Sumerin, Obstet. Gynecol., 1988, 72, 171–174 CAS.
- E. Salvolini, R. Di Giorgio, A. Curatola, L. Mazzanti and G. Fratto, Br. J. Obstet. Gynaecol., 1998, 105, 656–660 CrossRef CAS PubMed.
- N. Moretti, R. A. Rabini, L. Nanetti, G. Grechi, M. C. Curzi, N. Cester, L. A. Tranquilli and L. Mazzanti, Metabolism, 2002, 51, 605–608 CrossRef CAS PubMed.
- M. Orczyk-Pawiłowicz, J. Floriański, J. Zalewski and I. Kątnik-Prastowska, Glycoconjugate J., 2005, 22, 433–442 CrossRef PubMed.
- Y. Zhu and M. Li, Int. J. Clin. Exp. Med., 2017, 10, 12322–12328 Search PubMed.
- S. Martín-Sosa, M. J. Martín, L. A. García-Pardo and P. Hueso, J. Pediatr. Gastroenterol. Nutr., 2004, 39, 499–503 CrossRef PubMed.
- B. Wang, J. Brand-Miller, P. McVeagh and P. Petocz, Am. J. Clin. Nutr., 2001, 74, 510–515 CrossRef CAS PubMed.
- E. B. Isaacs, B. R. Fischl, B. T. Quinn, W. K. Chong, D. G. Gadian and A. Lucas, Pediatr. Res., 2010, 67, 357–362 CrossRef PubMed.
- M. K. Kim and J.-W. Choi, Int. Breastfeed J., 2020, 15, 83 CrossRef PubMed.
- S. Mills, R. P. Ross, C. Hill, G. F. Fitzgerald and C. Stanton, Int. Dairy J., 2011, 21, 377–401 CrossRef CAS.
- J. Lis-Kuberka and M. Orczyk-Pawiłowicz, Nutrients, 2019, 11, 306 CrossRef CAS PubMed.
- T. Urashima, J. Hirabayashi, S. Sato and A. Kobata, Trends Glycosci. Glycotechnol., 2018, 30, SE51–SE65 CrossRef.
- M. K. McGuire, C. L. Meehan, M. A. McGuire, J. E. Williams, J. Foster, D. W. Sellen, E. W. Kamau-Mbuthia, E. W. Kamundia, S. Mbugua, S. E. Moore, A. M. Prentice, L. J. Kvist, G. E. Otoo, S. L. Brooker, W. J. Price, B. Shafii, C. Placek, K. A. Lackey, B. Robertson, S. Manzano, L. Ruíz, J. M. Rodríguez, R. G. Pareja and L. Bode, Am. J. Clin. Nutr., 2017, 105, 1086–1100 CrossRef CAS PubMed.
- D. S. Newburg, Am. J. Clin. Nutr., 2017, 105, 1027–1028 CrossRef PubMed.
- EFSA NDA Panel (EFSA Panel on Dietetic Products Nutrition and Allergies), EFSA J., 2014, 12, 3760 CrossRef.
- T. A. Lehti, M. I. Pajunen, M. S. Skog and J. Finne, Nat. Commun., 2017, 8, 1915 CrossRef PubMed.
- S. Sakarya, G. T. Ertem, S. Oncu, I. Kocak, N. Erol and S. Oncu, FEMS Immunol. Med. Microbiol., 2003, 39, 45–50 CrossRef CAS PubMed.
- F. Schwarz, C. S. Landig, S. Siddiqui, I. Secundino, J. Olson, N. Varki, V. Nizet and A. Varki, EMBO J., 2017, 36, 751–760 CrossRef CAS PubMed.
- Z. Han, P. S. Thuy-Boun, W. Pfeiffer, V. F. Vartabedian, A. Torkamani, J. R. Teijaro and D. W. Wolan, Sci. Rep., 2021, 11, 4763 CrossRef CAS PubMed.
- T. D. Mubaiwa, E. A. Semchenko, L. E. Hartley-tassell, C. J. Day, M. P. Jennings and K. L. Seib, Pathog. Dis., 2017, 75, ftx063 Search PubMed.
- G. A. Jarvis and N. A. Vedros, Infect. Immun., 1987, 55, 174–180 CrossRef CAS PubMed.
- C. Jones, M. Virji and P. R. Crocker, Mol. Microbiol., 2003, 49, 1213–1225 CrossRef CAS PubMed.
- S. L. Xiang, M. Zhong, F. C. Cai, B. Deng and X. P. Zhang, J. Neuroimmunol., 2006, 174, 126–132 CrossRef CAS PubMed.
- R. Huizinga, W. Van Rijs, J. J. Bajramovic, M. L. Kuijf, J. D. Laman, J. N. Samsom and C. Jacobs, J. Immunol., 2013, 191, 5636–5645 CrossRef CAS PubMed.
- M. Bax, M. L. Kuijf, A. P. Heikema, W. van Rijs, S. C. M. Bruijns, J. J. García-Vallejo, P. R. Crocker, B. C. Jacobs, S. J. van Vliet and Y. van Kooyk, Infect. Immun., 2011, 79, 2681–2689 CrossRef CAS PubMed.
- S. Uchiyama, J. Sun, K. Fukahori, N. Ando, M. Wu, F. Schwarz and S. S. Siddiqui, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7465–7470 CrossRef CAS PubMed.
- S. Weiman, S. Uchiyama, F.-Y. C. Lin, D. Chaffin, A. Varki, V. Nizet and A. L. Lewis, Biochem. J., 2013, 428, 163–168 CrossRef PubMed.
- P. S. K. Ng, C. J. Day, J. M. Atack, L. E. Hartley-Tassell, L. E. Winter, T. Marshanski, V. Padler-Karavani, A. Varki, S. J. Barenkamp, M. A. Apicella and M. P. Jennings, MBio, 2019, 10, e00422-19 CrossRef PubMed.
- K. Hentrich, J. Löfling, A. Pathak, V. Nizet, A. Varki and B. Henriques-Normark, Cell Host Microbe, 2016, 20, 307–317 CrossRef CAS PubMed.
- B. Khatua, A. Ghoshal, K. Bhattacharya, C. Mandal, B. Saha, P. R. Crocker and C. Mandal, FEBS Lett., 2010, 584, 555–561 CrossRef CAS PubMed.
- B. Khatua, K. Bhattacharya and C. Mandal, J. Leukocyte Biol., 2012, 91, 641–655 CrossRef CAS PubMed.
- S. Ram, J. Shaughnessy, R. B. De Oliveira, L. A. Lewis, S. Gulati and P. A. Rice, Pathog. Dis., 2017, 75, ftx049 Search PubMed.
- S. M. Spinola, W. Li, K. R. Fortney, D. M. Janowicz, B. Zwickl, B. P. Katz and R. S. Munson, Infect. Immun., 2012, 80, 679–687 CrossRef CAS PubMed.
- F. M. Tatum, L. B. Tabatabai and R. E. Briggs, Microb. Pathog., 2009, 46, 337–344 CrossRef CAS PubMed.
- A. Varki and P. Gagneux, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14739–14740 CrossRef CAS PubMed.
- I. Moustafa, H. Connaris, M. Taylor, V. Zaitsev, J. C. Wilson, M. J. Kiefel, M. Von Itzstein and G. Taylor, J. Biol. Chem., 2004, 279, 40819–40826 CrossRef CAS PubMed.
- S. Almagro-Moreno and E. F. Boyd, Infect. Immun., 2009, 77, 3807–3816 CrossRef CAS PubMed.
- M. Unemo, M. Aspholm-Hurtig, D. Ilver, J. Bergström, T. Borén, D. Danielsson and S. Teneberg, J. Biol. Chem., 2005, 280, 15390–15397 CrossRef CAS PubMed.
- P. Zhou, J. Liu, X. Li, Y. Takahashi and F. Qi, PLoS One, 2015, 10, e0143898 CrossRef PubMed.
- H. Geyer, C. Holschbach, G. Hunsmann and J. Schneider, J. Biol. Chem., 1988, 263, 11760–11767 CrossRef CAS.
- Z. Zou, A. Chastain, S. Moir, J. Ford, K. Trandem, E. Martinelli, C. Cicala, P. Crocker, J. Arthos and P. D. Sun, PLoS One, 2011, 6, e24559 CrossRef CAS PubMed.
- S. Varchetta, P. Lusso, K. Hudspeth, J. Mikulak, D. Mele, S. Paolucci, R. Cimbro, M. Malnati, A. Riva, R. Maserati, M. U. Mondelli and D. Mavilio, Retrovirology, 2013, 10, 154 CrossRef PubMed.
- J. Wang, Q. Fan, T. Satoh, J. Arii, L. L. Lanier, P. G. Spear, Y. Kawaguchi and H. Arase, J. Virol., 2009, 83, 13042–13045 CrossRef CAS PubMed.
- Q. Lu, G. Lu, J. Qi, H. Wang, Y. Xuan, Q. Wang, Y. Lia, Y. Zhang, C. Zheng, Z. Fana, J. Yan and G. F. Gao, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8221–8226 CrossRef CAS PubMed.
- A. S. Kondratowicz, N. J. Lennemann, P. L. Sinn, R. A. Davey, C. L. Hunt, S. Moller-Tank, D. K. Meyerholz, P. Rennert, R. F. Mullins, M. Brindley, L. M. Sandersfeld, K. Quinn, M. Weller, P. B. McCray, J. Chiorini and W. Maury, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8426–8431 CrossRef CAS PubMed.
- S. Yuan, L. Cao, H. Ling, M. Dang, Y. Sun, X. Zhang, Y. Chen, L. Zhang, D. Su, X. Wang and Z. Rao, Protein Cell, 2015, 6, 814–824 CrossRef CAS PubMed.
- D. Perez-Zsolt, I. Erkizia, M. Pino, M. García-Gallo, M. T. Martin, S. Benet, J. Chojnacki, M. T. Fernández-Figueras, D. Guerrero, V. Urrea, X. Muñiz-Trabudua, L. Kremer, J. Martinez-Picado and N. Izquierdo-Useros, Nat. Microbiol., 2019, 4, 1558–1570 CrossRef CAS PubMed.
- C. W. Tan, C. H. Huan Hor, S. Sen Kwek, H. K. Tee, I. C. Sam, E. L. K. Goh, E. E. Ooi, Y. F. Chan and L. F. Wang, Emerging Microbes Infect., 2019, 8, 426–437 CrossRef CAS PubMed.
- J. Cime-Castillo, P. Delannoy, G. Mendoza-Hernández, V. Monroy-Martínez, A. Harduin-Lepers, H. Lanz-Mendoza, F. D. L. C. Hernández-Hernández, E. Zenteno, C. Cabello-Gutiérrez and B. H. Ruiz-Ordaz, BioMed Res. Int., 2015, 504187 Search PubMed.
- G. Zocher, N. Mistry, M. Frank, I. Hähnlein-Schick, J. O. Ekström, N. Arnberg and T. Stehle, PLoS Pathog., 2014, 10, e1004401 CrossRef PubMed.
- T. Imamura, M. Okamoto, S.-I. Nakakita, A. Suzuki, M. Saito, R. Tamaki, S. Lupisan, C. N. Roy, H. Hiramatsu, K.-E. Sugawara, K. Mizuta, Y. Matsuzaki, Y. Suzuki, H. Oshitani and S. Perlman, J. Virol., 2014, 88, 2374–2384 CrossRef PubMed.
- L. T. Jae, M. Raaben, A. S. Herbert, A. I. Kuehne, A. S. Wirchnianski, T. K. Soh, S. H. Stubbs, H. Janssen, M. Damme, P. Saftig, S. P. Whelan, J. M. Dye and T. R. Brummelkamp, Science, 2014, 344, 1506–1510 CrossRef CAS PubMed.
- F. Superti, B. Hauttecoeur, M. J. Morelec, P. Goldoni, B. Bizzini and H. Tsiang, J. Gen. Virol., 1986, 67, 47–56 CrossRef CAS PubMed.
- G. Ayora-Talavera, J. Recept., Ligand Channel Res., 2018, 10, 1–11 CrossRef.
- K. Shinya, M. Ebina, S. Yamada, M. Ono, N. Kasai and Y. Kawaoka, Nature, 2006, 440, 435–436 CrossRef CAS PubMed.
- N. Limsuwat, O. Suptawiwat, C. Boonarkart, P. Puthavathana, W. Wiriyarat and P. Auewarakul, Arch. Virol., 2016, 161, 649–656 CrossRef CAS PubMed.
- Z. Chen, W. Chen, Q. Wang, Y. Qin, X. Wang, T. Ma, P. Zhang, X. Li, X. Wang, L. Ding and Z. Li, J. Mol. Struct., 2021, 1230, 129859 CrossRef CAS.
- L. Ding, X. Fu, W. Guo, Y. Cheng, X. Chen, K. Zhang, G. Zhu, F. Yang, H. Yu, Z. Chen, X. Wang, X. Wang, X. Wang and Z. Li, Int. J. Biol. Macromol., 2021, 184, 339–348 CrossRef CAS PubMed.
- T. Ginex and F. J. Luque, Expert Opin. Ther. Pat., 2021, 31, 53–66 CrossRef CAS PubMed.
- R. E. Forgione, C. Di Carluccio, M. Kubota, Y. Manabe, K. Fukase, A. Molinaro, T. Hashiguchi, R. Marchetti and A. Silipo, Sci. Rep., 2020, 10, 1589 CrossRef CAS PubMed.
- M. Amonsen, D. F. Smith, R. D. Cummings and G. M. Air, J. Virol., 2007, 81, 8341–8345 CrossRef CAS PubMed.
- I. V. Alymova, A. Portner, V. P. Mishin, J. A. McCullers, P. Freiden and G. L. Taylor, Glycobiology, 2012, 22, 174–180 CrossRef CAS PubMed.
- L.-Y. Huang, A. Patel, R. Ng, E. B. Miller, S. Halder, R. McKenna, A. Asokan and M. Agbandje-McKenna, J. Virol., 2016, 90, 5219–5230 CrossRef CAS PubMed.
- R. Burger-Calderon and J. Webster-Cyriaque, Cancers, 2015, 7, 1244–1270 CrossRef CAS PubMed.
- N. J. Bayer, D. Januliene, G. Zocher, T. Stehle, A. Moeller and B. S. Blaum, J. Virol., 2020, 94, e01664-19 CrossRef PubMed.
- C. K. Liu, G. Wei and W. J. Atwood, J. Virol., 1998, 72, 4643–4649 CrossRef CAS PubMed.
- D. M. Reiter, J. M. Frierson, E. E. Halvorson, T. Kobayashi, T. S. Dermody and T. Stehle, PLoS Pathog., 2011, 7, e1002166 CrossRef CAS PubMed.
- K. Reiss, J. E. Stencel, Y. Liu, B. S. Blaum, D. M. Reiter, T. Feizi, T. S. Dermody and T. Stehle, PLoS Pathog., 2012, 8, e1003078 CrossRef CAS PubMed.
- T. Haselhorst, F. E. Fleming, J. C. Dyason, R. D. Hartnell, X. Yu, G. Holloway, K. Santegoets, M. J. Kiefel, H. Blanchard, B. S. Coulson and M. Von Itzstein, Nat. Chem. Biol., 2009, 5, 91–93 CrossRef CAS PubMed.
- S. M. Cashman, D. J. Morris and R. Kumar-Singh, Virology, 2004, 324, 129–139 CrossRef CAS PubMed.
- E. C. Nilsson, R. J. Storm, J. Bauer, S. M. C. Johansson, A. Lookene, J. Ångström, M. Hedenström, T. L. Eriksson, L. FräCurrency Signngsmyr, S. Rinaldi, H. J. Willison, F. P. Domellöf, T. Stehle and N. Arnberg, Nat. Med., 2011, 17, 105–109 CrossRef CAS PubMed.
- A. Lenman, A. Manuel Liaci, Y. Liu, L. Frängsmyr, M. Frank, B. S. Blaum, W. Chai, I. I. Podgorski, B. Harrach, M. Benko, T. Feizi, T. Stehle and N. Arnberg, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4264–E42736 CrossRef CAS PubMed.
- D. X. Liu, J. Q. Liang and T. S. Fung, Encyclopedia of Virology, 2021, vol. 2, pp. 428–440 Search PubMed.
- L. Guruprasad, Prog. Biophys. Mol. Biol., 2021, 161, 39–53 CrossRef CAS PubMed.
- A. H. M. Wong, A. C. A. Tomlinson, D. Zhou, M. Satkunarajah, K. Chen, C. Sharon, M. Desforges, P. J. Talbot and J. M. Rini, Nat. Commun., 2017, 8, 1735 CrossRef PubMed.
- X. Zhao, F. Guo, M. A. Comunale, A. Mehta, M. Sehgal, P. Jain, A. Cuconati, H. Lin, T. M. Block, J. Chang and J. T. Guo, Antimicrob. Agents Chemother., 2015, 59, 206–216 CrossRef PubMed.
- R. J. G. Hulswit, Y. Lang, M. J. G. Bakkers, W. Li, Z. Li, A. Schouten, B. Ophorst, F. J. M. Van Kuppeveld, G. J. Boons, B. J. Bosch, E. G. Huizinga and R. J. De Groot, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2681–2690 CrossRef CAS PubMed.
- W. Li, R. J. G. Hulswit, I. Widjaja, V. S. Raj, R. McBride, W. Peng, W. Widagdo, M. A. Tortorici, B. Van Dieren, Y. Lang, J. W. M. Van Lent, J. C. Paulson, C. A. M. De Haan, R. J. De Groot, F. J. M. Van Kuppeveld, B. L. Haagmans and B. J. Bosch, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8508–E8517 CrossRef CAS PubMed.
- M. Örd, I. Faustova and M. Loog, Sci. Rep., 2020, 10, 16944 CrossRef PubMed.
- M. Seyran, K. Takayama, V. N. Uversky, K. Lundstrom, G. Palù, S. P. Sherchan, D. Attrish, N. Rezaei, A. A. A. Aljabali, S. Ghosh, D. Pizzol, G. Chauhan, P. Adadi, T. Mohamed Abd El-Aziz, A. G. Soares, R. Kandimalla, M. Tambuwala, S. S. Hassan, G. K. Azad, P. Pal Choudhury, W. Baetas-da-Cruz, Á. Serrano-Aroca, A. M. Brufsky and B. D. Uhal, FEBS J., 2021, 288, 5010–5020 CrossRef CAS PubMed.
- M. Awasthi, S. Gulati, D. P. Sarkar, S. Tiwari, S. Kateriya, P. Ranjan and S. K. Verma, Viruses, 2020, 12, 909 CrossRef CAS PubMed.
- L. Pruimboom, Med. Hypotheses, 2021, 146, 110368 CrossRef CAS PubMed.
- E. Milanetti, M. Miotto, L. Di Rienzo, M. Nagaraj, M. Monti, T. W. Golbek, G. Gosti, S. J. Roeters, T. Weidner, D. E. Otzen and G. Ruocco, Front. Mol. Biosci., 2021, 8, 690655 CrossRef CAS PubMed.
- B. Li, L. Wang, H. Ge, X. Zhang, P. Ren, Y. Guo, W. Chen, J. Li, W. Zhu, W. Chen, L. Zhu and F. Bai, Front. Chem., 2021, 9, 659764 CrossRef PubMed.
- V. C. Divya and B. Saravanakarthikeyan, J. Oral Biosci., 2021 DOI:10.1016/j.job.2021.08.004 , In Press.
- N. Z. Cuervo and N. Grandvaux, eLife, 2020, 9, e61390 CrossRef CAS PubMed.
- W. Hao, B. Ma, Z. Li, X. Wang, X. Gao, Y. Li, B. Qin, S. Shang, S. Cui and Z. Tan, Sci. Bull., 2021, 66, 1205–1214 CrossRef CAS PubMed.
- A. Shajahan, S. Archer-Hartmann, N. T. Supekar, A. S. Gleinich, C. Heiss and P. Azadi, Glycobiology, 2021, 31, 410–424 CrossRef CAS PubMed.
- Q. Yang, T. A. Hughes, A. Kelkar, X. Yu, K. Cheng, S. J. Park, W. C. Huang, J. F. Lovell and S. Neelamegham, eLife, 2020, 9, e61552 CrossRef CAS PubMed.
- H. Chu, B. Hu, X. Huang, Y. Chai, D. Zhou, Y. Wang, H. Shuai, D. Yang, Y. Hou, X. Zhang, T. T. T. Yuen, J. P. Cai, A. J. Zhang, J. Zhou, S. Yuan, K. K. W. To, I. H. Y. Chan, K. Y. Sit, D. C. C. Foo, I. Y. H. Wong, A. T. L. Ng, T. T. Cheung, S. Y. K. Law, W. K. Au, M. A. Brindley, Z. Chen, K. H. Kok, J. F. W. Chan and K. Y. Yuen, Nat. Commun., 2021, 12, 134 CrossRef CAS PubMed.
- C. Dhar, A. Sasmal, S. Diaz, A. Verhagen, H. Yu, W. Li, X. Chen and A. Varki, Glycobiology, 2021, 31, 1068–1071 CrossRef PubMed.
- L. Nguyen, K. A. McCord, D. T. Bui, K. M. Bouwman, E. N. Kitova, M. Elaish, D. Kumawat, G. C. Daskhan, I. Tomris, L. Han, P. Chopra, T.-J. Yang, S. D. Willows, A. L. Mason, L. K. Mahal, T. L. Lowary, L. J. West, S.-T. D. Hsu, T. Hobman, S. M. Tompkins, G.-J. Boons, R. P. de Vries, M. S. Macauley and J. S. Klassen, Nat. Chem. Biol., 2021 DOI:10.1038/s41589-021-00924-1.
- E. Song, C. Zhang, B. Israelow, A. Lu-Culligan, A. Vieites Prado, S. Skriabine, P. Lu, O.-E. Weizman, F. Liu, Y. Dai, K. Szigeti-Buck, Y. Yasumoto, G. Wang, C. Castaldi, J. Heltke, E. Ng, J. Wheeler, M. M. Alfajaro, E. Levavasseur, B. Fontes, N. G. Ravindra, D. V. Dijk, S. Mane, M. Gunel, A. Ring, S. A. J. Kazmi, K. Zhang, C. B. Wilen, T. L. Horvath, I. Plu, S. Haik, J.-L. Thomas, A. Louvi, S. F. Farhadian, A. Huttner, D. Seilhean, N. Renier, K. Bilguvar and A. Iwasaki, J. Exp. Med., 2021, 218, e20202135 CrossRef CAS PubMed.
- C. M. Salvatore, J.-Y. Han, K. P. Acker, P. Tiwari, J. Jin, M. Brandler, C. Cangemi, L. Gordon, A. Parow, J. DiPace and P. DeLaMora, Lancet Child Adolesc. Health, 2020, 4, 721–727 CrossRef PubMed.
- J. C. Silva-Filho, C. G. F. de Melo and J. L. de Oliveira, Med. Hypotheses, 2020, 144, 110155 CrossRef CAS PubMed.
- R. Uraki and Y. Kawaoka, Nat. Chem. Biol., 2021 DOI:10.1038/s41589-021-00923-2.
- T. A. Altalhi, K. Alswat, W. F. Alsanie, M. M. Ibrahim, A. Aldalbahi and H. S. El-Sheshtawy, J. Mol. Struct., 2021, 1228, 129459 CrossRef CAS PubMed.
- P. Colson, J. M. Rolain, J. C. Lagier, P. Brouqui and D. Raoult, Int. J. Antimicrob. Agents, 2020, 55, 105932 CrossRef CAS PubMed.
- C. Prodromos and T. Rumschlag, New Microbes New Infect., 2020, 38, 100776 CrossRef CAS PubMed.
- N. Sinha and G. Balayla, Postgrad. Med. J., 2020, 96, 550–555 CrossRef CAS PubMed.
- J. K. Sun, Y. T. Chen, X. De Fan, X. Y. Wang, Q. Y. Han and Z. W. Liu, Postgrad. Med., 2020, 132, 604–613 CrossRef PubMed.
- M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi, Z. Hu, W. Zhong and G. Xiao, Cell Res., 2020, 30, 269–271 CrossRef CAS PubMed.
- J. A. Al-Tawfiq, A. H. Al-Homoud and Z. A. Memish, Travel Med. Infect. Dis., 2020, 34, 101615 CrossRef PubMed.
- A. N. Baker, S. J. Richards, C. S. Guy, T. R. Congdon, M. Hasan, A. J. Zwetsloot, A. Gallo, J. R. Lewandowski, P. J. Stansfeld, A. Straube, M. Walker, S. Chessa, G. Pergolizzi, S. Dedola, R. A. Field and M. I. Gibson, ACS Cent. Sci., 2020, 6, 2046–2052 CrossRef CAS PubMed.
- M. Renlund, M. A. Chester, A. Lundblad, P. Aula, K. O. Raivio, S. Autio and S.-L. Koskela, Eur. J. Biochem., 1979, 101, 245–250 CrossRef CAS PubMed.
- L. W. Hancock, M. M. Thaler, A. L. Horwitz and G. Dawson, J. Neurochem., 1982, 38, 803–808 CrossRef CAS PubMed.
- L. W. Hancock, A. L. Horwitz and G. Dawson, Biochim. Biophys. Acta, 1983, 760, 42–52 CrossRef CAS.
- G. H. Thomas, J. Scocca, C. S. Miller and L. Reynolds, Clin. Genet., 1989, 36, 242–249 CrossRef CAS PubMed.
- G. Strecker, M.-C. Peers, J.-C. Michalski, T. Hondi-Assah, B. Fournet, G. Spik, J. Montreuil, J.-P. Farriaux, P. Maroteaux and P. Durand, Eur. J. Biochem., 1977, 75, 391–403 CrossRef CAS PubMed.
- M. F. Goldberg, E. Cotlier, L. G. Fichenscher, K. Kenyon, R. Enat and S. A. Borowsky, Arch. Intern. Med., 1971, 128, 387–398 CrossRef CAS PubMed.
- T. Yardeni, T. Choekyi, K. Jacobs, C. Ciccone, K. Patzel, Y. Anikster, W. A. Gahl, N. Kurochkina and M. Huizing, Biochemistry, 2011, 50, 8914–8925 CrossRef CAS PubMed.
- C. Tran, L. Turolla, D. Ballhausen, S. C. Buros, T. Teav, H. Gallart-Ayala, J. Ivanisevic, M. Faouzi, D. J. Lefeber, I. Ivanovski, S. Giangiobbe, S. G. Caraffi, L. Garavelli and A. Superti-Furga, Mol. Genet. Metab. Rep., 2021, 28, 100777 CrossRef CAS PubMed.
- L. F. Mollashahi, M. Honarmand, A. Nakhaee and G. Mollashahi, Int. J. High Risk Behav. Addict., 2016, 5, e27969 Search PubMed.
- N. Kurtul and E. Gkpnar, Mediators Inflammation, 2012, 2012, 619293 Search PubMed.
- N. Waszkiewicz, S. D. Szajda, A. Kȩpka, A. Szulc and K. Zwierz, Biochem. Soc. Trans., 2011, 39, 365–369 CrossRef CAS PubMed.
- J. Romppanen, K. Punnonen, P. Anttila, T. Jakobsson, J. Blake and O. Niemelä, Alcohol.: Clin. Exp. Res., 2002, 26, 1234–1238 CrossRef CAS.
- P. Ghosh, E. A. Hale and M. R. Lakshman, Alcohol, 2001, 25, 173–179 CrossRef CAS PubMed.
- F. M. Wurst, N. Thon, W. Weinmann, S. Tippetts, P. Marques, J. A. Hahn, C. Alling, S. Aradottir, S. Hartmann and R. Lakshman, Alcohol.: Clin. Exp. Res., 2012, 36, 251–257 CrossRef CAS PubMed.
- B. Cylwik, L. Chrostek, A. Krawiec, Z. Supronowicz, A. Koput and M. Szmitkowski, Alcohol, 2010, 44, 457–462 CrossRef CAS PubMed.
- P. Sillanaukee, M. Pönniö and K. Seppä, Alcohol.: Clin. Exp. Res., 1999, 23, 1039–1043 CAS.
- T. Alam, R. Cherian, P. T. Raman and A. S. Balasubramanian, J. Neurol., Neurosurg. Psychiatry, 1976, 39, 1201–1203 CrossRef CAS PubMed.
- Y. Nagata, A. Hirayama, S. Ikeda, A. Shirahata, F. Shoji, M. Maruyama, M. Kayano, M. Bundo, K. Hattori, S. Yoshida, Y.-I. Goto, K. Urakami, T. Soga, K. Ozaki and S. Niida, Biomark. Res., 2018, 6, 5 CrossRef PubMed.
- J. Yadav, A. K. Verma, R. K. Garg, K. Ahmad, Shiuli, A. A. Mahdi and S. Srivastava, Exp. Gerontol., 2020, 141, 111092 CrossRef CAS PubMed.
- F. Mochel, F. Sedel, A. Vanderver, U. F. H. Engelke, J. Barritault, B. Z. Yang, B. Kulkarni, D. R. Adams, F. Clot, J. H. Ding, C. R. Kaneski, F. W. Verheijen, B. W. Smits, F. Seguin, A. Brice, M. T. Vanier, M. Huizing, R. Schiffmann, A. Durr and R. A. Wevers, Brain, 2009, 132, 801–809 CrossRef CAS PubMed.
- E. Demirci, Y. Guler, S. Ozmen, M. Canpolat and S. Kumandas, Clin. Psychopharmacol. Neurosci., 2019, 17, 415–422 CrossRef CAS PubMed.
- X. Yang, S. Liang, L. Wang, P. Han, X. Jiang, J. Wang, Y. Hao and L. Wu, Brain Res., 2018, 1678, 273–277 CrossRef CAS PubMed.
- M. A. Crook, P. Tutt, H. Simpson and J. C. Pickup, Clin. Chim. Acta, 1993, 219, 131–138 CrossRef CAS.
- T. Ozben, S. Nacitarhan and N. Tuncer, Ann. Clin. Biochem., 1995, 32, 303–306 CrossRef PubMed.
- J. Roozbeh, A. Merat, F. Bodagkhan, R. Afshariani and H. Yarmohammadi, Int. Urol. Nephrol., 2011, 43, 1143–1148 CrossRef CAS PubMed.
- S. Shahvali, A. Shahesmaeili, M. Sanjari and S. Karami-Mohajeri, Diabetol. Int., 2020, 11, 19–26 CrossRef PubMed.
- P. Khalili, J. Sundström, J. Jendle, F. Lundin, I. Jungner and P. M. Nilsson, Prim. Care Diabetes, 2014, 8, 352–357 CrossRef PubMed.
- R. B. Findik, F. M. Yilmaz, G. Yilmaz, H. Yilmaz and J. Karakaya, Arch. Gynecol. Obstet., 2012, 286, 913–916 CrossRef CAS PubMed.
- H. Dogan and H. Pagaoglu, Acta Ophthalmol., 1992, 70, 790–794 CrossRef CAS PubMed.
- I. Zipkin, S. E. Mergenhagen and R. Kass, Biochem. Biophys. Res. Commun., 1961, 4, 76–78 CrossRef CAS PubMed.
- S. R. Rathod, F. Khan, A. P. Kolte and M. Gupta, J. Clin. Diagn. Res., 2014, 8, 19–21 Search PubMed.
- A. Hernández-Cedillo, M. G. García-Valdivieso, A. C. Hernández-Arteaga, N. Patiño-Marín, Á. A. Vértiz-Hernández, M. José-Yacamán and H. R. Navarro-Contreras, Oral Dis., 2019, 25, 1627–1633 CrossRef PubMed.
- S. Oktay, Ö. Ö. Bal, L. Kuru, A. Yarat and Ü. Noyan, Niger. J. Clin. Pract., 2020, 23, 603–609 CAS.
- M. Crook, M. Haq, S. Haq and P. Tutt, Angiology, 1994, 45, 709–715 CrossRef CAS PubMed.
- M. N. Li, S. H. Qian, Z. Y. Yao, S. P. Ming, X. J. Shi, P. F. Kang, N. R. Zhang, X. J. Wang, D. S. Gao, Q. Gao, H. Zhang and H. J. Wang, BMC Cardiovasc. Disord., 2020, 20, 404 CrossRef CAS PubMed.
- P. Khalili, J. Sundström, S. S. Franklin, J. Jendle, F. Lundin, I. Jungner and P. M. Nilsson, J. Hypertens., 2012, 30, 1718–1724 CrossRef CAS PubMed.
- L. Zhang, T. T. Wei, Y. Li, J. Li, Y. Fan, F. Q. Huang, Y. Y. Cai, G. Ma, J. F. Liu, Q. Q. Chen, S. L. Wang, H. Li, R. N. Alolga, B. Liu, D. S. Zhao, J. H. Shen, X. M. Wang, W. Zhu, P. Li and L. W. Qi, Circulation, 2018, 137, 1374–1390 CrossRef CAS PubMed.
- M. T. Goodarzi, M. Rezaei, E. Moadel, S. Homayounfar and M. R. Safari, Eur. J. Lipid Sci. Technol., 2008, 110, 302–306 CrossRef CAS.
- S. Oktay, I. Basar, E. Emekli-Alturfan, E. Malali, E. Elemek, F. Ayan, L. Koldas, U. Noyan and N. Emekli, Pathophysiol. Haemostasis Thromb., 2009, 37, 67–71 CrossRef CAS PubMed.
- L. Råstam, G. Lindberg, A. R. Folsom, G. L. Burke, P. Nilsson-Ehle and A. Lundblad, Int. J. Epidemiol., 1996, 25, 953–958 CrossRef PubMed.
- M. Crook and W. U-test, Clin. Sci., 1992, 83, 593–595 CrossRef CAS PubMed.
- P. A. Larnba, P. K. Pandey, G. S. Sarin and M. D. Mathur, Acta Ophthalmol., 1993, 71, 833–835 Search PubMed.
- M. Altay, M. A. Karakoç, N. Çakır, C. Yılmaz Demirtaş, E. T. Cerit, M. Aktürk, İ. Ateş, N. Bukan and M. Arslan, J. Clin. Lab. Anal., 2017, 31, e22034 CrossRef PubMed.
- R. Baba, K. Yashiro, K. Nagasako and H. Obata, Gastroenterol. Jpn., 1992, 27, 604–610 CrossRef CAS PubMed.
- E. Gruszewska, B. Cylwik, M. Gudowska, A. Panasiuk, R. Flisiak and L. Chrostek, Ann. Clin. Biochem., 2019, 56, 118–122 CrossRef CAS PubMed.
- B. Cylwik, L. Chrostek, B. Zalewski, A. Dabrowski and M. Szmitkowski, Dig. Dis. Sci., 2007, 52, 2317–2322 CrossRef PubMed.
- B. Cylwik, L. Chrostek, A. Panasiuk and M. Szmitkowski, Clin. Chem. Lab. Med., 2010, 48, 137–139 CAS.
- T. Özben, Ann. Clin. Biochem., 1991, 28, 44–48 CrossRef PubMed.
- F. Fuentes, N. Carrillo, K. J. Wilkins, J. Blake, P. Leoyklang, W. A. Gahl, J. B. Kopp and M. Huizing, Kidney360, 2020, 1, 957–961 CrossRef PubMed.
- S. S. Demir, H. Ç. Özcan, Ö. Balat, E. Öztürk, M. G. Uğur, R. Gündüz and S. Taysi, J. Obstet. Gynaecol., 2018, 38, 532–535 CrossRef CAS PubMed.
- F. M. Von Versen-Hoeynck, C. A. Hubel, M. J. Gallaher, H. S. Gammill and R. W. Powers, Am. J. Hypertens., 2009, 22, 687–692 CrossRef CAS PubMed.
- R. O'Kennedy, G. Berns, E. Moran, H. Smyth, K. Carroll, R. D. Thornes, A. O'Brien, J. Fennelly and M. Butler, Cancer Lett., 1991, 58, 91–100 CrossRef.
- R. Iijima, H. Takahashi, S. Ikegami and M. Yamazaki, Biol. Pharm. Bull., 2007, 30, 580–582 CrossRef CAS PubMed.
- N. Kurtul and B. C. Arikan, Acta Med., 2006, 49, 97–100 CAS.
- A. Cardenas, A. M. Bernard and R. R. Lauwerys, Toxicol. Appl. Pharmacol., 1991, 108, 547–558 CrossRef CAS PubMed.
- Y. Wang, A. Khan, A. Antonopoulos, L. Bouché, C. D. Buckley, A. Filer, K. Raza, K. P. Li, B. Tolusso, E. Gremese, M. Kurowska-Stolarska, S. Alivernini, A. Dell, S. M. Haslam and M. A. Pineda, Nat. Commun., 2021, 12, 2343 CrossRef CAS PubMed.
- R. E. Taylor, C. J. Gregg, V. Padler-Karavani, D. Ghaderi, H. Yu, S. Huang, R. U. Sorensen, X. Chen, J. Inostroza, V. Nizet and A. Varki, J. Exp. Med., 2010, 207, 1637–1646 CrossRef CAS PubMed.
- L. Le Berre, J. Rousse, P. A. Gourraud, B. M. Imbert-Marcille, A. Salama, G. Evanno, G. Semana, A. Nicot, E. Dugast, P. Guérif, C. Adjaoud, T. Freour, S. Brouard, F. Agbalika, R. Marignier, D. Brassat, D. A. Laplaud, E. Drouet, V. Van Pesch and J. P. Soulillou, Clin. Immunol., 2017, 180, 128–135 CrossRef CAS PubMed.
- Y. Chen, L. Pan, N. Liu, F. A. Troy and B. Wang, J. Geophys. Res. Oceans, 2014, 111, 332–341 CAS.
- L. Tolenaars, D. Romanazzi, E. Carpenter, S. Gallier and C. G. Prosser, Int. Dairy J., 2021, 117, 105012 CrossRef CAS.
- H. Li and X. Fan, Open J. Prev. Med., 2014, 04, 57–63 CrossRef.
- H. L. Yao, L. P. Conway, M. M. Wang, K. Huang, L. Liu and J. Voglmeir, Glycoconjugate J., 2016, 33, 219–226 CrossRef CAS PubMed.
- M. Hedlund, V. Padler-Karavani, N. M. Varki and A. Varki, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18936–18941 CrossRef CAS PubMed.
- A. N. Samraj, H. Läubli, N. Varki and A. Varki, Front. Oncol., 2014, 4, 33 Search PubMed.
- P. R. Sanjay, K. Hallikeri and A. R. Shivashankara, Indian J. Dent. Res., 2008, 19, 288–291 CrossRef CAS PubMed.
- K. S. C. Bose, P. V. Gokhale, S. Dwivedi and M. Singh, J. Nat. Sci., Biol. Med., 2013, 4, 122–125 CrossRef CAS PubMed.
- R. D. Xing, Z. S. Wang, C. Q. Li, Q. Y. Tang, C. B. Jiang and Y. Z. Zhang, Int. J. Biol. Markers, 1994, 9, 239–242 CrossRef CAS PubMed.
- S. Wongkham, C. Boonla, S. Kongkham, C. Wongkham, V. Bhudhisawasdi and B. Sripa, Clin. Biochem., 2001, 34, 537–541 CrossRef CAS PubMed.
- C. Feijoo-Carnero, F. J. Rodríguez-Berrocal, M. Páez de la Cadena, D. Ayude, A. de Carlos and V. S. Martínez-Zorzano, Int. J. Biol. Markers, 2004, 19, 38–45 CrossRef CAS PubMed.
- A. Semczuk, M. Cybulski, A. Paszkowska and H. Berbec, Eur. J. Obstet. Gynecol. Reprod. Biol., 1998, 76, 211–215 CrossRef.
- S. Inoue, S. L. Lin, T. Chang, S. H. Wu, C. W. Yao, T. Y. Chu, F. A. Troy and Y. Inoue, J. Biol. Chem., 1998, 273, 27199–27204 CrossRef CAS PubMed.
- F. Wang, B. Xie, B. Wang and F. A. Troy, Glycobiology, 2015, 25, 1362–1374 CrossRef CAS PubMed.
- D. Konukoglu, T. Akqay and A. Ergzenci, Cancer Lett., 1995, 94, 97–100 CrossRef CAS PubMed.
- T. Akqay, D. Konukoglua, S. Atausb, A. Dirican and C. Uygur, Cancer Lett., 1994, 78, 7–9 CrossRef.
- M. Payazdan, S. Khatami, H. Galehdari, N. Delfan, M. Shafiei and S. Heydaran, Gene Rep., 2021, 24, 101218 CrossRef.
- J. E. Martin, S. W. Tanenbaum and M. Flashner, Carbohydr. Res., 1977, 56, 423–425 CrossRef CAS PubMed.
- Z. C. F. Wong, G. K. L. Chan, K. Q. Y. Wu, K. K. M. Poon, Y. Chen, T. T. X. Dong and K. W. K. Tsim, Food Funct., 2018, 9, 5139–5149 RSC.
- N. H. Jamalluddin, N. A. Tukiran, N. Ahmad Fadzillah and S. Fathi, Food Control, 2019, 104, 247–255 CrossRef CAS.
- X. Ma, J. Zhang, J. Liang, X. Ma, R. Xing, J. Han, L. Guo and Y. Chen, Food Res. Int., 2019, 125, 108639 CrossRef CAS PubMed.
- N. Daud, S. Mohamad Yusop, A. S. Babji, S. J. Lim, S. R. Sarbini and T. Hui Yan, Food Rev. Int., 2021, 37, 177–196 CrossRef.
- Y. Dai, J. Cao, Y. Wang, Y. Chen and L. Jiang, Food Res. Int., 2021, 140, 109875 CrossRef CAS PubMed.
- T. Nakano and M. Betti, J. Dairy Res., 2020, 87, 364–367 CrossRef CAS PubMed.
- R. J. McMahon, M. F. Locniskar, S. C. Rumsey, J. C. Anthony and R. Wantanagorn, US7951410B2, 2011.
- https://www.thegreenpharmacy.com/sialex-90-caps-p6399.html (Accessed November 2021).
- https://www.ecologicalformulas.net/ (Accessed November 2021).
- https://www.ulprospector.com/en/na/Food/Detail/5331/190810/Lacprodan-CGMP---10 (Accessed November 2021).
- S. J. Danishefsky, M. P. DeNinno and S. Hui Chen, J. Am. Chem. Soc., 1988, 110, 3929–3940 CrossRef CAS.
- C. De Meo and B. T. Jones, in Advances in Carbohydrate Chemistry and Biochemistry, ed. D. C. Baker, Elsevier, 75th edn, 2018, pp. 215–316 Search PubMed.
- N. Komura, K. Kato, T. Udagawa, S. Asano, H. N. Tanaka, A. Imamura, H. Ishida, M. Kiso and H. Ando, Science, 2019, 364, 677–680 CrossRef CAS PubMed.
- A. M. Vibhute, N. Komura, H. N. Tanaka, A. Imamura and H. Ando, Chem. Rec., 2021, 21, 1–31 CrossRef.
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 163–176 CrossRef CAS PubMed.
- C. C. Yu and S. G. Withers, Adv. Synth. Catal., 2015, 357, 1633–1654 CrossRef CAS.
- W. Li, J. B. McArthur and X. Chen, Carbohydr. Res., 2019, 472, 86–97 CrossRef CAS PubMed.
- A. S. Kooner, H. Yu and X. Chen, Front. Immunol., 2019, 10, 2004 CrossRef CAS PubMed.
- I. Maru, J. Ohnishi, Y. Ohta and Y. Tsukada, J. Biosci. Bioeng., 2002, 93, 258–265 CrossRef CAS PubMed.
- M. J. Dawson, D. Noble and M. Mahmoudian, USOO6156544A, 2000.
- U. Kragl, D. Gygax, O. Ghisalba and C. Wandrey, Angew. Chem., Int. Ed. Engl., 1991, 30, 827–828 CrossRef.
- J. Lee, J. Yi, S. Lee, S. Takahashi and B. Kim, Enzyme Microb. Technol., 2004, 35, 121–125 CrossRef CAS.
- X. Gao, F. Zhang, M. Wu, Z. Wu and G. Shang, J. Agric. Food Chem., 2019, 67, 6285–6291 CrossRef CAS PubMed.
- V. R. L. J. Bloemendal, S. J. Moons, J. J. A. Heming, M. Chayoua, O. Niesink, J. C. M. Van Hest, T. J. Boltje and F. P. J. T. Rutjes, Adv. Synth. Catal., 2019, 361, 2443–2447 CrossRef CAS PubMed.
- https://www.glycom.com/ (Accessed November 2021).
- https://www.dsm.com/corporate/home.html (Accessed November 2021).
- A. Schroven, G. Dekany and I. Vrasidas, WO2013088267Al, 2013.
- M. Shiba and S. Koizumi, EP1484406B1, 2004.
- D. Zhu, W. Xiao, L. Zhu and Y. Jiang, J. Ind. Microbiol. Biotechnol., 2019, 46, 125–132 CrossRef CAS PubMed.
- B. Wang, Annu. Rev. Nutr., 2009, 29, 177–222 CrossRef CAS PubMed.
- B. Wang, Adv. Nutr., 2012, 3, 465–472 CrossRef PubMed.
- J. M. Gil-Robles and G. Zalm, Official Journal of the European Communities, 1997, L43, 1–6 Search PubMed.
- Report of the Scientific Committee on Food on the Revision of Essential Requirements of Infant Formulae and Follow-on Formulae, 2003.
- B. Koletzko, S. Baker, G. Cleghorn, U. F. Neto, S. Gopalan, O. Hernell, Q. S. Hock, P. Jirapinyo, B. Lonnerdal, P. Pencharz, H. Pzyrembel, J. Ramirez-Mayans, R. Shamir, D. Turck, Y. Yamashiro and D. Zong-Yi, J. Pediatr. Gastroenterol. Nutr., 2005, 41, 584–599 CrossRef PubMed.
- J. B. Fontelles and J. Korkeaoja, Official Journal of the European Union, 2007, L12, 3–18 Search PubMed.
- J.-L. Bresson, A. Flynn, M. Heinonen, K. Hulshof, H. Korhonen, P. Lagiou, M. Løvik, R. Marchelli, A. Martin, B. Moseley, H. Przyrembel, S. Salminen, S. (J. J.) Strain, S. Strobel, I. Tetens, H. van den Berg, H. van Loveren and H. Verhagen, EFSA J., 2009, 7, 1277 CrossRef.
- S. S. H. Choi, N. Baldwin, V. O. Wagner, S. Roy, J. Rose, B. A. Thorsrud, P. Phothirath and C. H. Röhrig, Regul. Toxicol. Pharmacol., 2014, 70, 482–491 CrossRef CAS PubMed.
- J. P. Zimmer and C. M. Butt, US8703716B2, 2014.
- Glycom A/S, GRAS Exemption Claim for N-Acetyl-D-neuraminic acid (NANA), http://www.fda.gov/Food/IngredientsPackagingLabeling/GRAS/NoticeInventory/default.htm.
- Food Safety Authority of Ireland, Safety Assessment of N-acetyl-D-neuraminic acid dihydrate (NANA), 2016 Search PubMed.
- ADVISORY COMMITTEE FOR NOVEL FOODS AND PROCESSES, N-Acetyl-D-Neuraminic Acid (NANA, Sialic Acid) DOSSIER 187, https://acnfp.food.gov.uk/sites/default/files/nana.pdf.
- EFSA Panel on Dietetic Products, EFSA J., 2017, 15, 4918 Search PubMed.
- S. Peled and Y. D. Livney, Food Hydrocolloids, 2021, 120, 106911 CrossRef CAS.
- X. Sun, S. Zhang, J. Ren and C. C. Udenigwe, Crit. Rev. Food Sci. Nutr., 2020, 1846157 Search PubMed.
- X. Sun and J. Wu, Trends Food Sci. Technol., 2017, 69, 148–156 CrossRef CAS.
- K. R. Phipps, Š. Alica, N. J. Baldwin, B. Gilby, B. Lynch, D. R. Stannard, M. H. Mik and C. H. Röhrig, J. Appl. Toxicol., 2019, 39, 1378–1393 CrossRef CAS PubMed.
- K. R. Phipps, Š. Alica, N. J. Baldwin, B. Gilby, B. Lynch, D. R. Stannard, M. H. Mik and C. H. Röhrig, J. Appl. Toxicol., 2019, 39, 1444–1461 CrossRef CAS PubMed.
- Glycom A/S, GRAS Notice for 3 ’ -Sialyllactose, https://www.fda.gov/food/generally-recognized-safe-gras/gras-notice-inventory.
- Glycom A/S, GRAS Notice for 6 ’-Sialyllactose, https://www.fda.gov/food/generally-recognized-safe-gras/gras-notice-inventory.
- EFSA Panel on Nutrition, EFSA J., 2020, 18, 6098 Search PubMed.
- EFSA Panel on Nutrition, EFSA J., 2020, 18, 6097 Search PubMed.
- https://www.dsm.com/human-nutrition/en/early-life-nutrition/human-milk-oligosaccharides/GlyCare-3SL.html (Accessed November 2021).
- https://www.dsm.com/human-nutrition/en_AP/early-life-nutrition/human-milk-oligosaccharides/GlyCare-6SL.html (Accessed November 2021).
- F. Herrmann, A. Nieto-Ruiz, N. Sepúlveda-Valbuena, M. T. Miranda, E. Diéguez, J. Jiménez, R. De-Castellar, M. García-Ricobaraza, J. A. García-Santos, M. G. Bermúdez and C. Campoy, J. Funct. Foods, 2021, 83, 104529 CrossRef CAS.
- J. Ma, S. Gong, Y. He, W. Gao, W. Hao and X. Lan, Lett. Appl. Microbiol., 2021, 73, 20–25 CrossRef CAS PubMed.
- X. Wu, Z. Li, X. X. Chen, J. S. Fossey, T. D. James and Y. B. Jiang, Chem. Soc. Rev., 2013, 42, 8032–8048 RSC.
- H. Otsuka, E. Uchimura, H. Koshino, T. Okano and K. Kataoka, J. Am. Chem. Soc., 2003, 125, 3493–3502 CrossRef CAS PubMed.
- K. Djanashvili, L. Frullano and J. A. Peters, Chem. – Eur. J., 2005, 11, 4010–4018 CrossRef CAS PubMed.
- S. Nishitani, Y. Maekawa and T. Sakata, ChemistryOpen, 2018, 7, 513–519 CrossRef CAS PubMed.
- N. Wellington, S. Macklai and P. Britz-McKibbin, Chem. – Eur. J., 2019, 25, 15277–15280 CrossRef CAS PubMed.
- E. L. Hess, J. W. Hahn and W. Ayala, Exp. Biol. Med., 1956, 91, 528–531 CrossRef CAS PubMed.
- E. L. Hess, A. F. Coburn, R. C. Bates and P. Murphy, J. Clin. Invest., 1957, 36, 449–455 CrossRef CAS PubMed.
- L. Svennerholm, Biochim. Biophys. Acta, 1957, 24, 604–611 CrossRef CAS.
- G. W. Jourdian, L. Dean and S. Roseman, J. Biol. Chem., 1971, 246, 430–435 CrossRef CAS.
- G. D. Dimitrov, Hoppe-Seyler's Z. Physiol. Chem., 1973, 354, 121–124 CrossRef CAS PubMed.
- M. H. E. Spyridaki and P. A. Siskos, Anal. Chim. Acta, 1996, 327, 277–285 CrossRef CAS.
- L. Warren, J. Biol. Chem., 1959, 234, 1971–1975 CrossRef CAS.
- D. Aminoff, Virology, 1959, 7, 355–357 CrossRef CAS PubMed.
- I. E. Horgan, Clin. Chim. Acta, 1981, 116, 409–415 CrossRef CAS.
- Y. Massamiri, G. Durand, A. Richard, J. Féger and J. Agneray, Anal. Biochem., 1979, 97, 346–351 CrossRef CAS PubMed.
- K. Yao and T. Ubuka, Acta Med. Okayama, 1987, 41, 237–241 CAS.
- J. B. Joicy, N. T. G. de Paula, P. A. B. da Silva, G. C. S. de Souza, A. P. S. Paim and A. F. Lavorante, Microchem. J., 2019, 147, 782–788 CrossRef.
- T. J. Jayeoye, W. Cheewasedtham, C. Putson and T. Rujiralai, Microchim. Acta, 2018, 185, 409 CrossRef PubMed.
- T. J. Jayeoye, W. Cheewasedtham, C. Putson and T. Rujiralai, J. Mol. Liq., 2019, 281, 407–414 CrossRef CAS.
- H. Hess and E. Rolde, J. Biol. Chem., 1964, 239, 3215–3220 CrossRef CAS.
- K. S. Hammond and D. S. Papermaster, Anal. Biochem., 1976, 74, 292–297 CrossRef CAS PubMed.
- J. I. Murayama, M. Tomita, A. Tsuji and A. Hamada, Anal. Biochem., 1976, 73, 535–538 CrossRef CAS PubMed.
- A. K. Shukla and R. Schauer, Hoppe-Seyler's Z. Physiol. Chem., 1982, 363, 255–262 CrossRef CAS PubMed.
- K. Matsuno and S. Suzuki, Anal. Biochem., 2008, 375, 53–59 CrossRef CAS PubMed.
- A. D. A. Alves, M. F. Belian and A. F. Lavorante, Luminescence, 2014, 29, 779–783 CrossRef PubMed.
- Q. Wang, B. Wang, M. Ma and Z. Cai, J. Food Sci., 2014, 79, C2434–C2440 CrossRef CAS PubMed.
- J. Wang, G. Xu, F. Wei, J. Yang, P. Zhou and Q. Hu, RSC Adv., 2016, 6, 481–488 RSC.
- X. Hao, Q. Zhang, L. Li and L. Zhou, Mater. Lett., 2018, 227, 165–168 CrossRef CAS.
- S. Xu, S. Che, P. Ma, F. Zhang, L. Xu, X. Liu, X. Wang, D. Song and Y. Sun, Talanta, 2019, 197, 548–552 CrossRef CAS PubMed.
- N. Wang, M. Wang, Y. Yu, G. Yang and X. Su, New J. Chem., 2020, 44, 2350–2356 RSC.
- J. Wang, L. Zhao and B. Yan, ACS Appl. Mater. Interfaces, 2020, 12, 12990–12997 CrossRef CAS PubMed.
- H. Yu, Y. Li and A. Huang, Talanta, 2021, 232, 122434 CrossRef CAS PubMed.
- Y. Zhang, H. Lu and B. Yan, Sens. Actuators, B, 2021, 349, 130736 CrossRef CAS.
- D. E. Wang, J. Yan, J. Jiang, X. Liu, C. Tian, J. Xu, M. Sen Yuan, X. Han and J. Wang, Nanoscale, 2018, 10, 4570–4578 RSC.
- H. Yue, J. Chen, X. Chen, X. Wang, Y. Zhang and N. Zhou, Sens. Actuators, B, 2021, 344, 130270 CrossRef CAS.
- Q. Lu, M. Zhan, L. Deng, G. Qing and T. Sun, Analyst, 2017, 142, 3564–3568 RSC.
- X. Wang, S. Qian, D. Wang, C. Wang, H. Qin, L. Peng, W. Lu, Y. Zhang and G. Qing, J. Mater. Chem. B, 2021, 9, 4690–4699 RSC.
- K. Ouchi, C. L. Colyer, M. Sebaiy, J. Zhou, T. Maeda, H. Nakazumi, M. Shibukawa and S. Saito, Anal. Chem., 2015, 87, 1933–1940 CrossRef CAS PubMed.
- J. A. Cabezas, Biochem. J., 1991, 278, 311–312 CrossRef CAS PubMed.
- S. Roseman and D. G. Comb, J. Am. Chem. Soc., 1958, 80, 3166–3167 CrossRef CAS.
- D. G. Comb and S. Roseman, J. Am. Chem. Soc., 1958, 80, 497–499 CrossRef CAS.
- D. G. Comb and S. Roseman, J. Biol. Chem., 1960, 235, 2529–2537 CrossRef CAS.
- A. D. Winer and G. W. Schwert, Science, 1958, 128, 660–661 CrossRef CAS PubMed.
- O. H. Lowry, N. R. Roberts and J. I. Kapphahn, J. Biol. Chem., 1957, 224, 1047–1064 CrossRef CAS.
- P. Brunetti, A. Swanson and S. Roseman, Methods Enzymol., 1963, 6, 465–473 CAS.
- D. G. Comb and S. Roseman, Methods Enzymol., 1962, 5, 391–394 CAS.
- K. Sugahara, K. Sugimoto, O. Nomura and T. Usui, Clin. Chim. Acta, 1980, 108, 493–498 CrossRef CAS.
- S. Teshima, K. Tamai, Y. Hayashi and S. Emi, Clin. Chem., 1988, 34, 2291–2294 CrossRef CAS.
- H. Simpson, G. D. Chusney, M. A. Crook and J. C. Pickup, Br. J. Biomed. Sci., 1993, 50, 164–167 CAS.
- T. Horiuchi and T. Kurokawa, Clin. Chim. Acta, 1989, 182, 117–121 CrossRef CAS.
- J. P. Johnston, A. G. Ogston and J. E. Stanier, Analyst, 1951, 76, 88–89 RSC.
- L. Svennerholm, Acta Chem. Scand., 1958, 12, 547–554 CrossRef CAS.
- M. J. Krantz and Y. C. Lees, Anal. Biochem., 1975, 63, 464–469 CrossRef CAS PubMed.
- D. A. Craven and C. W. Gehrke, J. Chromatogr., 1968, 37, 414–421 CrossRef CAS PubMed.
- J. Casals-Stenzel, H.-P. Buscher and R. Schauer, Anal. Biochem., 1975, 65, 507–524 CrossRef CAS PubMed.
- J. P. Kamerling, J. F. G. Vliegenthart, C. Versluis and R. Schauer, Carbohydr. Res., 1975, 41, 7–17 CrossRef CAS PubMed.
- H. K. B. Silver, K. A. Karim, M. J. Gray and F. A. Salinas, J. Chromatogr., 1981, 224, 381–388 CrossRef CAS.
- A. K. Shukla, N. Scholz, E. H. Reimerdes and R. Schauer, Anal. Biochem., 1982, 123, 78–82 CrossRef CAS PubMed.
- A. K. Shukla and R. Schauer, J. Chromatogr., 1982, 244, 81–89 CrossRef CAS.
- N. K. Karamanos, B. Wikström, C. A. Antonopoulos and A. Hjerpe, J. Chromatogr., 1990, 503, 421–429 CrossRef CAS.
- F. Unland and J. Muthing, Biomed. Chromatogr., 1992, 6, 155–159 CrossRef CAS PubMed.
- E. L. Romero, M. F. Pardo, S. Porro and S. Alonso, J. Biochem. Biophys. Methods, 1997, 35, 129–134 CrossRef CAS PubMed.
- S. M. Levonis, J. Pittet, B. C. M. Pointon and S. S. Schweiker, J. Chromatogr., B, 2019, 1104, 130–133 CrossRef CAS PubMed.
- K. Kobayashi, Y. Akiyama, K. Kawaguchi, T. Shinzo and T. Imanari, Anal. Sci., 1985, 1, 81–84 CrossRef CAS.
- K. Li, J. Chromatogr., 1992, 579, 209–213 CrossRef CAS PubMed.
- S. Honda, S. Iwase, S. Suzuki and K. Kakehi, Anal. Biochem., 1987, 160, 455–461 CrossRef CAS PubMed.
- K. R. Anumula, Anal. Biochem., 1995, 230, 24–30 CrossRef CAS PubMed.
- S. Hara, M. Yamaguchi, Y. Takemori and M. Nakamljra, J. Chromatogr., 1986, 377, 111–119 CrossRef CAS PubMed.
- S. Hara, Y. Takemori, M. Yamaguchi, M. Nakamura and Y. Ohkura, Anal. Biochem., 1987, 164, 138–145 CrossRef CAS PubMed.
- S. Hara, M. Yamaguchi, Y. Takemori, K. Furuhata, H. Ogura and M. Nakamura, Anal. Biochem., 1989, 179, 162–166 CrossRef CAS PubMed.
- P. G. Stanton, Z. Shen, E. A. Kecorius, P. G. Burgon, D. M. Robertson and M. T. W. Hearn, J. Biochem. Biophys. Methods, 1995, 30, 37–48 CrossRef CAS PubMed.
- M. J. Martín, E. Vázquez and R. Rueda, Anal. Bioanal. Chem., 2007, 387, 2943–2949 CrossRef PubMed.
- A. Kawasaki, M. Yasuda, K.-I. Mawatari, T. Fukuuchi, N. Yamaoka, K. Kaneko, R. Iijima, S. Yui, M. Satoh and K. Nakagomi, Anal. Sci., 2018, 34, 841–844 CrossRef CAS PubMed.
- T. Ota, M. Yasuda, R. Iijima, S. Yui, T. Fukuuchi, N. Yamaoka, K. Mawatari, K. Kaneko and K. Nakagomi, J. Chromatogr., B, 2013, 932, 152–157 CrossRef CAS PubMed.
- D. Li, Food Sci. Biotechnol., 2012, 21, 1317–1320 CrossRef CAS.
- L. Wang, D. Wang, X. Zhou, L. Wu and X. Sun, RSC Adv., 2014, 4, 45797–45803 RSC.
- M. I. Orozco-Solano, F. Priego-Capote and M. D. Luque de Castro, Anal. Chim. Acta, 2013, 766, 69–76 CrossRef CAS PubMed.
- F. Qu, L. Xia, C. Wu, L. Liu, G. Li and J. You, RSC Adv., 2016, 6, 64895–64901 RSC.
- A. E. Manzi, S. Diaz and A. Varki, Anal. Biochem., 1990, 188, 20–32 CrossRef CAS PubMed.
- M. R. Hardy, Methods Enzymol., 1989, 179, 76–82 CAS.
- J. S. Rohrer, J. Thayer, M. Weitzhandler and N. Avdalovic, Glycobiology, 1998, 8, 35–43 CrossRef CAS PubMed.
- R. D. Rocklin, A. P. Clarke and M. Weitzhandler, Anal. Chem., 1998, 70, 1496–1501 CrossRef CAS.
- C. J. Shaw, H. Chao and B. Xiao, J. Chromatogr. A, 2001, 913, 365–370 CrossRef CAS PubMed.
- M. van der Ham, B. H. C. M. T. Prinsen, J. G. M. Huijmans, N. G. G. M. Abeling, B. Dorland, R. Berger, T. J. de Koning and M. G. M. de Sain-van der Velden, J. Chromatogr., B, 2007, 848, 251–257 CrossRef CAS PubMed.
- P. Allevi, E. A. Femia, M. L. Costa, R. Cazzola and M. Anastasia, J. Chromatogr. A, 2008, 1212, 98–105 CrossRef CAS PubMed.
- L. A. Hammad, D. Z. Derryberry, Y. R. Jmeian and Y. Mechref, Rapid Commun. Mass Spectrom., 2010, 24, 1565–1574 CrossRef CAS PubMed.
- Y. Shi, X. Xu, M. Fang, M. Zhang, Y. Li, B. Gillespie, S. Yorke, N. Yang, J. C. McKew, W. A. Gahl, M. Huizing, N. Carrillo-Carrasco and A. Q. Wang, J. Chromatogr., B, 2015, 1000, 105–111 CrossRef CAS PubMed.
- C. Li, L. Liu, H. Xie and N. Liu, Int. J. Dairy Technol., 2015, 68, 166–173 CrossRef CAS.
- S. F. Fernando and B. W. Woonton, J. Food Compos. Anal., 2010, 23, 359–366 CrossRef CAS.
- T. Hayama, Y. Sakaguchi, H. Yoshida, M. Itoyama, K. Todoroki, M. Yamaguchi and H. Nohta, Rapid Commun. Mass Spectrom., 2010, 24, 2868–2874 CrossRef CAS PubMed.
- A. Tebani, D. Schlemmer, A. Imbard, O. Rigal, D. Porquet and J. F. Benoist, J. Chromatogr., B, 2011, 879, 3694–3699 CrossRef CAS PubMed.
- D. Wang, X. Zhou, L. Wang, S. Wang and X. L. Sun, J. Chromatogr., B, 2014, 944, 75–81 CrossRef CAS PubMed.
- F. Priego-Capote, M. I. Orozco-Solano, M. Calderón-Santiago and M. D. Luque de Castro, J. Chromatogr. A, 2014, 1346, 88–96 CrossRef CAS PubMed.
- B. Yesilyurt, U. Sahar and R. Deveci, Mol. Reprod. Dev., 2015, 82, 115–122 CrossRef CAS PubMed.
- F. T. A. Chen, T. S. Dobashi and R. A. Evangelista, Glycobiology, 1998, 8, 1045–1052 CrossRef CAS PubMed.
- F. Y. Che, X. X. Shao, K. Y. Wang and Q. C. Xia, Electrophoresis, 1999, 20, 2930–2937 CrossRef CAS PubMed.
- X. Dong, X. Xu, F. Han, X. Ping, X. Yuang and B. Lin, Electrophoresis, 2001, 22, 2231–2235 CrossRef CAS PubMed.
- K. Strousopoulou, M. Militsopoulou, K. Stagiannis, F. N. Lamari and N. K. Karamanos, Biomed. Chromatogr., 2002, 16, 146–150 CrossRef CAS PubMed.
- A. Taga, M. Sugimura, S. Suzuki and S. Honda, J. Chromatogr. A, 2002, 954, 259–266 CrossRef CAS PubMed.
- K. Ortner and W. Buchberger, Electrophoresis, 2008, 29, 2233–2237 CrossRef CAS PubMed.
- Y. Zheng, T. Wang, J. Kong, Y. Ma, Y. Heng, Y. Ren, J. Ye and Q. Chu, Chin. J. Chem., 2016, 34, 925–930 CrossRef CAS.
- Y. Wang, J. Kong, Z. Chen, D. Luo, J. Ye and Q. Chu, Food Anal. Methods, 2018, 11, 1105–1112 CrossRef.
- S. A. M. Marzouk, S. S. Ashraf and K. A. Al Tayyari, Anal. Chem., 2007, 79, 1668–1674 CrossRef CAS PubMed.
- S. A. M. Marzouk, J. D. Haddow and A. Amin, Sens. Actuators, B, 2011, 157, 647–653 CrossRef CAS.
- A. Fatoni, A. Numnuam, P. Kanatharana, W. Limbut and P. Thavarungkul, Electrochim. Acta, 2014, 130, 296–304 CrossRef CAS.
- Y. Zhou, H. Dong, L. Liu, J. Liu and M. Xu, Biosens. Bioelectron., 2014, 60, 231–236 CrossRef CAS PubMed.
- Y. Zhou, H. Huangfu, J. Yang, H. Dong, L. Liu and M. Xu, Analyst, 2019, 144, 6432–6437 RSC.
- F. Huang, B. Zhu, H. Zhang, Y. Gao, C. Ding, H. Tan and J. Li, Microchim. Acta, 2019, 186, 1–11 CrossRef CAS PubMed.
- T. V. Shishkanova, G. Broncová, Z. Němečková, V. Vrkoslav, V. Král and P. Matějka, J. Electroanal. Chem., 2019, 832, 321–328 CrossRef CAS.
- T. Liu, B. Fu, J. Chen, Z. Yan and K. Li, Electrochim. Acta, 2018, 269, 136–143 CrossRef CAS.
- Y. Lv, Y. Zhou, H. Dong, L. Liu, G. Mao, Y. Zhang and M. Xu, ChemElectroChem, 2020, 7, 922–927 CrossRef CAS.
- W. Hai, S. Pu, X. Wang, L. Bao, N. Han, L. Duan, J. Liu, T. Goda and W. Wu, Langmuir, 2020, 36, 546–553 CrossRef CAS PubMed.
- S. Ding, S. Cao, Y. Liu, Y. Lian, A. Zhu and G. Shi, ACS Sens., 2017, 2, 394–400 CrossRef CAS PubMed.
- G. Broncová, P. Matějka, Z. Němečková, V. Vrkoslav and T. V. Shishkanova, Electroanalysis, 2018, 30, 672–680 CrossRef.
- L. Chen, N. Wang, J. Wu, F. Yan and H. Ju, Anal. Chim. Acta, 2020, 1128, 231–237 CrossRef CAS PubMed.
- J. Wang, S. Zhang, H. Dai, H. Zheng, Z. Hong and Y. Lin, Biosens. Bioelectron., 2019, 142, 111567 CrossRef CAS PubMed.
- D. Fang, S. Zhang, H. Dai, Z. Hong and Y. Lin, Electrochim. Acta, 2019, 326, 134956 CrossRef CAS.
- N. Cao, F. Zhao and B. Zeng, Sens. Actuators, B, 2020, 313, 128042 CrossRef CAS.
- A. Kugimiya, H. Yoneyama and T. Takeuchi, Electroanalysis, 2000, 12, 1322–1326 CrossRef CAS.
- S.-J. Duan, X.-W. He, L.-X. Chen and Y.-K. Zhang, Gaodeng Xuexiao Huaxue Xuebao, 2012, 33, 464–469 CAS.
- X. Qiu, X. Y. Xu, X. Chen, Y. Wu and H. Guo, Anal. Bioanal. Chem., 2018, 410, 4387–4395 CrossRef CAS PubMed.
- A. Kugimiya and T. Takeuchi, Biosens. Bioelectron., 2001, 16, 1059–1062 CrossRef CAS PubMed.
- S. Li, J. Liu, Y. Lu, L. Zhu, C. Li, L. Hu, J. Li, J. Jiang, S. Low and Q. Liu, Biosens. Bioelectron., 2018, 117, 32–39 CrossRef CAS PubMed.
- E. Vinogradova, A. Tlahuice-Flores, J. J. Velazquez-Salazar, E. Larios-Rodriguez and M. Jose-Yacaman, J. Raman Spectrosc., 2014, 45, 730–735 CrossRef CAS.
- A. C. Hernández-Arteaga, F. C. Delgado-Nieblas, H. J. Ojeda-Galván, J. J. Velázquez-Salazar, E. Vinogradova, M. José-Yacamán, R. A. Guirado-López and H. R. Navarro-Contreras, J. Phys. Chem. C, 2017, 121, 21045–21056 CrossRef.
- A. Hernández-Arteaga, J. de Jesús Zermeño Nava, E. S. Kolosovas-Machuca, J. J. Velázquez-Salazar, E. Vinogradova, M. José-Yacamán and H. R. Navarro-Contreras, Nano Res., 2017, 10, 3662–3670 CrossRef.
- X. Teng, F. Chen, Y. Gao, R. Meng, Y. Wu, F. Wang, Y. Ying, X. Liu, X. Guo, Y. Sun, P. Lin, Y. Wen and H. Yang, Anal. Chem., 2020, 92, 3332–3339 CrossRef CAS PubMed.
- https://www.elabscience.com/p-sialic_acid_sa_colorimetric_assay_kit-40365.html (Accessed November 2021).
- https://www.mybiosource.com/assay-kits/sialic-acid/2540448 (Accessed November 2021).
- https://www.mybiosource.com/general-assay-kits/sialic-acid-sa/2563693 (Accessed November 2021).
- https://www.biovision.com/sialic-acid-nana-colorimetric-fluorometric-assay-kit.html (Accessed November 2021).
- https://www.bioassaysys.com/Sialic-acid-Assay-Kit-(ESLA-100).html (Accessed November 2021).
- https://www.lsbio.com/assaykits/sialic-acid-assay-kit-colorimetric-fluorometric-ls-k187/187 (Accessed November 2021).
- https://www.promocell.com/product/sialic-acid-nana-assay-kit/ (Accessed November 2021).
- https://www.abcam.com/sialic-acid-nana-assay-kit-ab83375.html (Accessed November 2021).
- https://www.antibodies-online.com/kit/411705/Sialic+Acid+NANA+Colorimetric/Fluorometric+Assay+Kit/ (Accessed November 2021).
- http://www.cohesionbio.com/goods.php?id=127272 (Accessed November 2021).
- https://www.mybiosource.com/general-assay-kits/sialic-acid/8309610 (Accessed November 2021).
- https://www.mybiosource.com/assay-kits/sialic-acid-nana/841610 (Accessed November 2021).
- https://www.usbio.net/kits/S1013-31Q/sialic-acid-assay-kit-bioassaytrade-colorimetricfluorometric-high-sensitivity (Accessed November 2021).
- https://www.usbio.net/kits/A0535-01A/n-acetylneuraminic-acid-nana-sialic-acid-bioassaytrade-kit (Accessed November 2021).
- https://www.creativebiomart.net/sialic-acid-nana-colorimetricfluorometric-assay-kit-463657.htm (Accessed November 2021).
- https://www.creativebiomart.net/sialic-acid-assay-kit-502969.htm (Accessed November 2021).
- http://www.abnova.com/products/products_detail.asp?catalog_id=KA1655 (Accessed November 2021).
- https://www.bioassaysys.com/Sialic-acid-Assay-Kit-(DSLA-100).html (Accessed November 2021).
- https://www.usbio.net/kits/S1013-31P/sialic-acid-assay-kit-bioassaytrade-colorimetricfluorometric (Accessed November 2021).
- https://www.sigmaaldrich.com/IE/en/product/sigma/mak314?context=product (Accessed November 2021).
- https://www.creativebiomart.net/sialic-acid-assay-kit-463740.htm (Accessed November 2021).
- https://www.assaygenie.com/sialic-acid-assay-kit-colorimetric-or-fluorometric-ba0058/ (Accessed November 2021).
- https://www.sigmaaldrich.com/IE/en/product/sigma/sialicq?context=product (Accessed November 2021).
- https://www.takarabio.com/products/protein-research/glycobiology/sialic-acid-kit?catalog=4400 (Accessed November 2021).
- https://www.agilent.com/store/productDetail.jsp?catalogId=GKK-407 (Accessed November 2021).
- https://www.ludger.com/dmb-sialic-acid-analysis/index.php (Accessed November 2021).
- https://www.agilent.com/store/productDetail.jsp?catalogId=GS24-SAP (Accessed November 2021).
- W. Song, L. Ding, Y. Chen and H. Ju, Chem. Commun., 2016, 52, 10640–10643 RSC.
- T. Gong, Y. Cui, D. Goh, K. K. Voon, P. P. Shum, G. Humbert, J. L. Auguste, X. Q. Dinh, K. T. Yong and M. Olivo, Biosens. Bioelectron., 2015, 64, 227–233 CrossRef CAS PubMed.
- X. N. He, Y. N. Wang, Y. Wang and Z. R. Xu, Talanta, 2020, 209, 120579 CrossRef CAS PubMed.
- L. Liang, Y. Shen, J. Zhang, S. Xu, W. Xu, C. Liang and B. Han, Anal. Chim. Acta, 2018, 1033, 148–155 CrossRef CAS PubMed.
- S. Shinde, Z. El-Schich, A. Malakpour, W. Wan, N. Dizeyi, R. Mohammadi, K. Rurack, A. Gjörloff Wingren and B. Sellergren, J. Am. Chem. Soc., 2015, 137, 13908–13912 CrossRef CAS PubMed.
- N. Peng, R. Xu, M. Si, A. Victorious, E. Ha, C. Y. Chang and X. D. Xu, RSC Adv., 2017, 7, 11282–11285 RSC.
- Y. Geng, L. Cong, Y. Tian, Z. Huo, Y. Wang, L. Xing, S. Xu, C. Liang and W. Xu, Sens. Actuators, B, 2019, 301, 127074 CrossRef CAS.
- V. Almeida-Marrero, M. Mascaraque, M. Jesús Vicente-Arana, Á. Juarranz, T. Torres and A. de la Escosura, Chem. – Eur. J., 2021, 27, 9634–9642 CrossRef CAS PubMed.
- X. Yang, L. Zhou, Y. Hao, B. Zhou and P. Yang, Analyst, 2017, 142, 2169–2176 RSC.
- D. Qian, F. Han, W. Li, N. Bao, C. Yu and H. Gu, Microchim. Acta, 2017, 184, 3841–3850 CrossRef CAS.
- K. Liu, Q. Sun, L. Zhu, C. Feng, J. Zhang, Y. Wang, F. Zhang, Z. Xu and W. Zhang, Anal. Methods, 2015, 7, 3819–3826 RSC.
- L. Zhang, C. Yu, R. Gao, Y. Niu, Y. Li, J. Chen and J. He, Biosens. Bioelectron., 2017, 92, 434–441 CrossRef CAS PubMed.
- A. Matsumoto, S. Osawa, T. Arai, Y. Maejima, H. Otsuka and Y. Miyahara, Bioconjugate Chem., 2021, 32, 239–244 CrossRef CAS PubMed.
- G. Landa, L. G. Miranda-Calderón, V. Sebastian, S. Irusta, G. Mendoza and M. Arruebo, Talanta, 2021, 234, 122644 CrossRef CAS PubMed.
- S. Osawa, A. Matsumoto, Y. Maejima, T. Suzuki, Y. Miyahara and H. Otsuka, Anal. Chem., 2020, 92, 11714–11720 CrossRef CAS PubMed.
- D. Sui, X. Tang, J. Ding, Y. Wang, Y. Qin, N. Zhang, X. Liu, Y. Deng and Y. Song, Int. J. Pharm., 2021, 602, 120552 CrossRef CAS PubMed.
- https://www.biovision.com/ezclicktm-sialic-acid-manaz-modified-glycoprotein-assay-kit-facs-microscopy-green-fluorescence.html (Accessed November 2021).
- https://www.sartorius.com/en/products/protein-analysis/biosensors-chips-kits/sialic-acid-glys-kit (Accessed November 2021).
| This journal is © The Royal Society of Chemistry 2022 |