
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA carbapenem antibiotic inhibiting a mammalian serine protease: structure of the acylaminoacyl peptidase–meropenem complex†
Anna J.
Kiss-Szemán
 a,
Luca
Takács
a,
Zoltán
Orgován
b,
Pál
Stráner
c,
Imre
Jákli
ac,
Gitta
Schlosser
d,
Simonas
Masiulis
e,
Veronika
Harmat
*ac,
Dóra K.
Menyhárd
*ac and
András
Perczel
*ac
a,
Luca
Takács
a,
Zoltán
Orgován
b,
Pál
Stráner
c,
Imre
Jákli
ac,
Gitta
Schlosser
d,
Simonas
Masiulis
e,
Veronika
Harmat
*ac,
Dóra K.
Menyhárd
*ac and
András
Perczel
*ac
aLaboratory of Structural Chemistry and Biology, Institute of Chemistry, Eötvös Loránd University, Pázmány Péter sétány 1/A, Budapest, Hungary
bMedicinal Chemistry Research Group, Research Centre for Natural Sciences, Budapest, Hungary
cELKH-ELTE Protein Modelling Research Group, Eötvös Loránd Research Network, Budapest, Hungary. E-mail: dora.k.menyhard@ttk.elte.hu; perczel.andras@ttk.elte.hu; veronika.harmat@ttk.elte.hu; Fax: +36-1-372-2500/1653; Tel: +36-1-372-2500/6547
dELKH-ELTE Lendület Ion Mobility Mass Spectrometry Research Group, Institute of Chemistry, Eötvös Loránd University, Budapest, Hungary
eMaterials and Structural Analysis Division, Thermo Fisher Scientific, Eindhoven, The Netherlands
First published on 8th November 2022
Abstract
The structure of porcine AAP (pAAP) in a covalently bound complex with meropenem was determined by cryo-EM to 2.1 Å resolution, showing the mammalian serine-protease inhibited by a carbapenem antibiotic. AAP is a modulator of the ubiquitin-proteasome degradation system and the site of a drug–drug interaction between the widely used antipsychotic, valproate and carbapenems. The active form of pAAP – a toroidal tetramer – binds four meropenem molecules covalently linked to the catalytic Ser587 of the serine-protease triad, in an acyl–enzyme state. AAP is hindered from fully processing the antibiotic by the displacement and protonation of His707 of the catalytic triad. We show that AAP is made susceptible to the association by its unusually sheltered active pockets and flexible catalytic triads, while the carbapenems possess sufficiently small substituents on their β-lactam rings to fit into the shallow substrate-specificity pocket of the enzyme.
Introduction
In 1997, it was reported that patients under treatment with the anticonvulsant agent sodium valproate (VPA) experienced serious relapse in condition when undergoing parallel treatment with carbapenem antibiotics for pulmonary infections, noting a drastic decrease of the serum concentration of valproate in the presence of the antibiotics.1 The adverse interaction of VPA, widely used for the treatment of neurological conditions such as epilepsy, migraine, bipolar disorder or neuropathic pain2, and a representative of one of the most prescribed antibiotic families has since been thoroughly documented and made its way into the general guidance provided for the use of either.3,4 In fact, the interaction is so fast and effective that the carbapenem antibiotic, meropenem (MEPM) could be successfully applied in cases of VPA overdose in emergency medicinal practice, inducing its fast clearance.5–7 The enzymatic site of this unexpected drug–drug interaction (DDI) has been identified over a decade later as the enzyme VPA-glucuronidase. VPA-glucuronidase is responsible for maintaining the serum concentration of VPA, by cleaving its major metabolic product, valproic acid-β-(D)-glucuronide (VPA-G),2 in a deglucuronidation/hydrolysis reaction, and releasing free VPA into the circulation, resulting in its reabsorption. It was this enzyme that was shown to be inhibited by carbapenems.8 Subsequently it was revealed that VPA-glucuronidase is in fact identical to a rather elusive enzyme that has also been referred to – indicating its unconnectedly discovered functions – as oxidized protein hydrolase (OPH), acylamino-acid-releasing enzyme, N-formylmethionyl-peptidase, acylpeptide-hydrolase (APEH) or as acylaminoacyl peptidase (AAP).9AAP, first and foremost, is an exopeptidase, catalyzing the removal of N-acetylated amino acids from the N-terminus of oligopeptides and proteins,10 contributing to the upkeep of healthy protein-homeostasis,11 and exerting an influence over the functioning of the proteasome,12,13 especially in regulating the degradation of oxidatively damaged proteins.14,15 As such, AAP has been indicated as a possible pharmacological target in certain types of ovarian,16 bone,17,18 skin19, colon,12 lung,20 renal21 and prostate22 cancers, and also as playing a role in the success of the radiotherapy of implicated patients.23
AAP can also function as an endopeptidase, taking part in the recognition and disassembly of damaged or misfolded proteins24,25 and has been proposed as being capable of cleaving amyloid aggregates of misfolded proteins26 through which it was suggested as a modulator of cognitive enhancement processes.27,28 It was shown to contribute to DNA damage repair mechanisms29 and to the membrane localization of oncogenic protein K-Ras possibly through its interaction with phosphatidylserine.30 Thus, AAP being inhibited by carbapenems at certain concentrations even irreversibly,31 is worthy of attention even beyond the aspects of the VPA–carbapenem interference.
The many functions and wide substrate range of AAP are served by a classical serine protease catalytic apparatus,32,33 which prompts two important questions: why – out of all the serine protease enzymes of the human physiology – has AAP proved to be the partner of carbapenems, and why do only carbapenems of the β-lactam family of antibiotics31 participate in this interaction?
Here, we present the structure of mammalian AAP in a covalently bound complex with meropenem, which is, to the best of our knowledge, the first structural information concerning the association between a β-lactam antibiotic and a mammalian enzyme. The structure allowed elucidation of the unique features that enable the binding of meropenem to AAP, as well as those that disable one of the most powerful hydrolytic machineries from fully processing the antibiotic and releasing the hydrolyzed products, thus leading to a permanently inhibited enzyme complex. In accordance with the clinical observations and biochemical characterization, our high-resolution results confirm meropenem to be a mechanism-based inhibitor of AAP. The structure can form the basis of a better understanding of AAP's function and its possible new roles in therapy, as well as provide a glimpse of the mechanisms by which antibiotics might produce side effects in human physiology.
Results and discussion
AAP was isolated from porcine liver (pAAP). Formation of an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with meropenem per monomer of pAAP was confirmed by mass spectrometry (MS) (Fig. S1†), while the inhibited state was demonstrated by the inability of pAAP to cleave the N-acetyl-L-alanine p-nitroanilide (AANA) substrate after incubation with meropenem. The structure of the tetrameric pAAP–MEPM covalent complex was determined using cryo-EM to 2.1 Å resolution (Fig. S2 and S3, and Table S1†). The tetrameric form of pAAP has an overall toroidal shape and we found that the tetramer binds four meropenem molecules (Fig. 1) covalently linked to to the Ser587 of the serine protease catalytic triad of each active site (Fig. S2†). The active site of each pAAP monomer is buried deep in the rather large cavity between their α/β-hydrolase and 7-bladed propeller domains and can be accessed through a double-gated shutter system33 created by tetramerization or through the narrow channel piercing the propeller domain. Both paths allow the entry of only small, and/or unstructured substrates (Fig. 1).
:1 complex with meropenem per monomer of pAAP was confirmed by mass spectrometry (MS) (Fig. S1†), while the inhibited state was demonstrated by the inability of pAAP to cleave the N-acetyl-L-alanine p-nitroanilide (AANA) substrate after incubation with meropenem. The structure of the tetrameric pAAP–MEPM covalent complex was determined using cryo-EM to 2.1 Å resolution (Fig. S2 and S3, and Table S1†). The tetrameric form of pAAP has an overall toroidal shape and we found that the tetramer binds four meropenem molecules (Fig. 1) covalently linked to to the Ser587 of the serine protease catalytic triad of each active site (Fig. S2†). The active site of each pAAP monomer is buried deep in the rather large cavity between their α/β-hydrolase and 7-bladed propeller domains and can be accessed through a double-gated shutter system33 created by tetramerization or through the narrow channel piercing the propeller domain. Both paths allow the entry of only small, and/or unstructured substrates (Fig. 1).
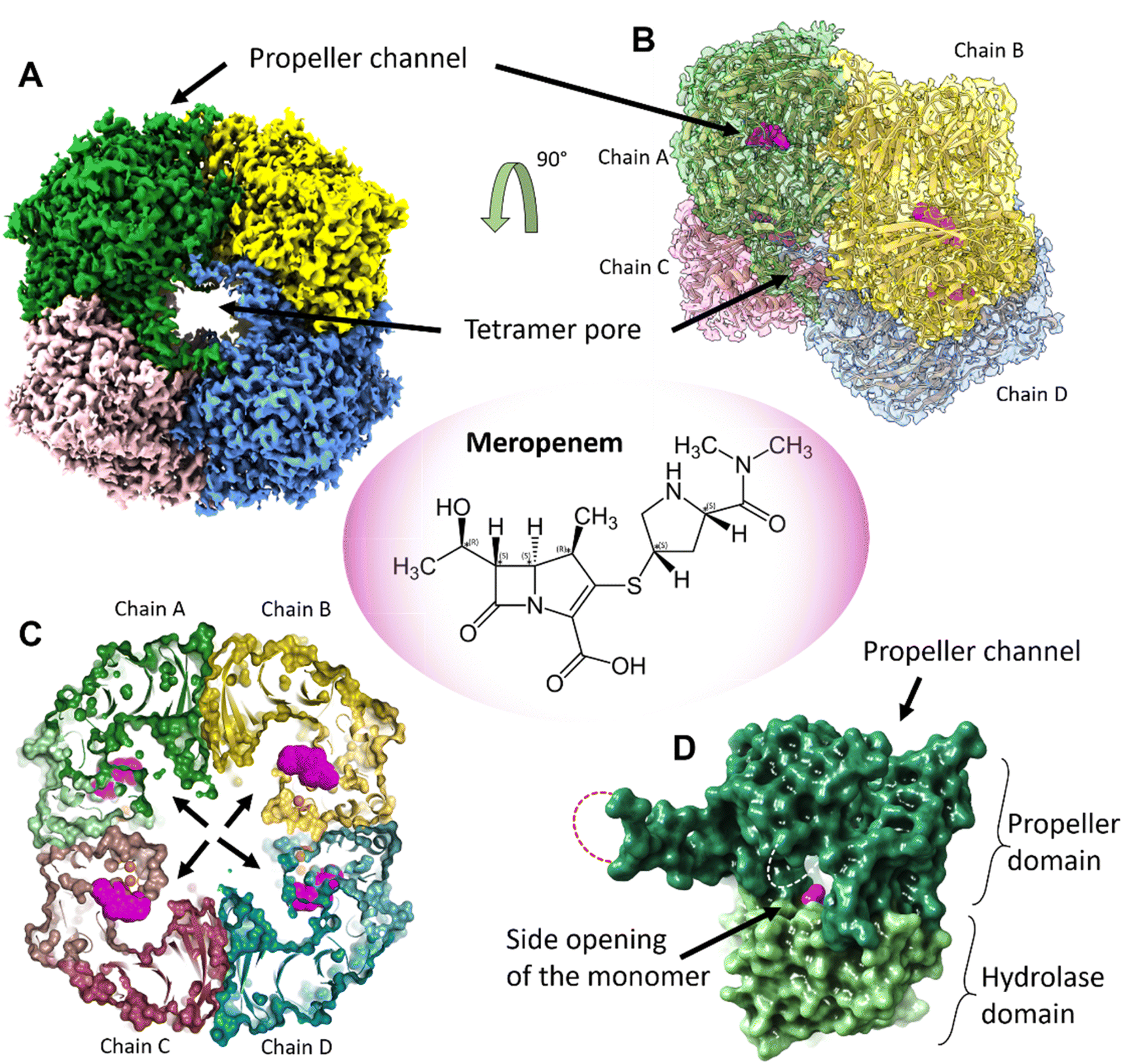 | ||
| Fig. 1 The tetrameric structure of AAP with four meropenem molecules (PDB id 7qun) bound to the deeply buried active sites. (A) The cryo-EM map (resolution = 2.1 Å, in C2 symmetry, and threshold level = 0.33) of the toroidal tetramer of pAAP showing the central pore (tetramer pore) and the location of the propeller channel – the possible gateways for substrate entry on the outer surface. (B) The cryo-EM map (side view and semi-transparent) with the fitted model of the AAP tetramer and the meropenem molecules bound to the active sites (magenta). (C) A cross section of the tetramer shows the large inner cavity: the four side openings of the monomers (black arrows) – the second frontier of the “double-gated channel and shutter” system created by the tetrameric assembly.33 The active site residues (Cα atoms of the catalytic triad are shown as orange spheres) are located between the propeller domain (dark colors) and the hydrolase domain (light colors) of the monomer units. Meropenem (magenta) is covalently bound to catalytic Ser587. (D) Both the tetramer pores on the outer surface and the side opening of the monomers that face the inner cavity are further shielded by flexible loops (dotted lines) that remained unresolved both in the pAAP-meropenem complex and in the structure of the uncomplexed enzyme (PBB id: 7px8). | ||
Complex formation – active site conformational changes
D,D-Transpeptidases and serine-β-lactamases of the known bacterial targets of meropenem (and β-lactam antibiotics, in general) share a similar fold and antibiotic-binding topology (Fig. S4†). Transpeptidases participate in the final steps of bacterial cell-wall synthesis and are effectively inhibited by β-lactam antibiotics, while lactamases are responsible for the rapid hydrolysis of β-lactams and through that, the rise of antibacterial resistance. These enzymes, just as AAP, employ a serine as the residue that performs the initial attack on the strained β-lactam ring. Their active site is defined by 3 short, conserved motifs that place the inhibitor/substrate between two facing Ser residues, one of which is the catalytic Ser. In their acyl–enzyme state, the acyl carbonyl-oxygen is coordinated by two backbone amide groups, that of the catalytic Ser (of motif I) and another residue in motif III. Motif III also provides a Ser/Thr hydroxyl and the amino group of a Lys to stabilize the carboxylate substituent of the dihydro-pyrrole ring, while its hydroxyethyl substituent and the ring N-atom are coordinated by members of motif II.34–37A different mode of association was found between bacterial L,D-transpeptidases (Fig. S4†) and β-lactams. Here, meropenem is covalently linked to the sulfur atom of the catalytic Cys, while the backbone amide of the same Cys forms a strong H-bond with the acyl carbonyl of the antibiotic, which is also linked – in a weaker association – to the backbone amide of the preceding Gly. In complexes formed with L,D-transpeptidases, meropenem displays a great conformational heterogeneity: several different binding modes were detected with the dihydro-pyrrole ring rotated along the CAM–CAL bond (atomic naming is compiled in Fig. S5 and Table S2†), coupled with the displacement of the substrate-stabilizing loop, suggesting that a number of different binding conformations of the antibiotic might exist within the neighboring pockets of these enzymes (Fig. S4†).38,39
In the pAAP–MEPM complex – also best described as an acyl–enzyme state, meropenem is covalently bound by catalytic Ser587 (Fig. 2A and B), while its acyl carbonyl (CAA![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OAC, Fig. S5†) is fixed and oriented by the oxyanion site conserved amongst AAPs: two backbone amide groups, those of Gly509 and His588 (the residue immediately following catalytic Ser587), in a binding mode similar to that seen in the case of D,D-transpeptidases and serine β-lactamases (Fig. S4†). The carboxylate substituent of the dihydro-pyrrole ring is coordinated by the hydroxyl group of Ser512.
OAC, Fig. S5†) is fixed and oriented by the oxyanion site conserved amongst AAPs: two backbone amide groups, those of Gly509 and His588 (the residue immediately following catalytic Ser587), in a binding mode similar to that seen in the case of D,D-transpeptidases and serine β-lactamases (Fig. S4†). The carboxylate substituent of the dihydro-pyrrole ring is coordinated by the hydroxyl group of Ser512.
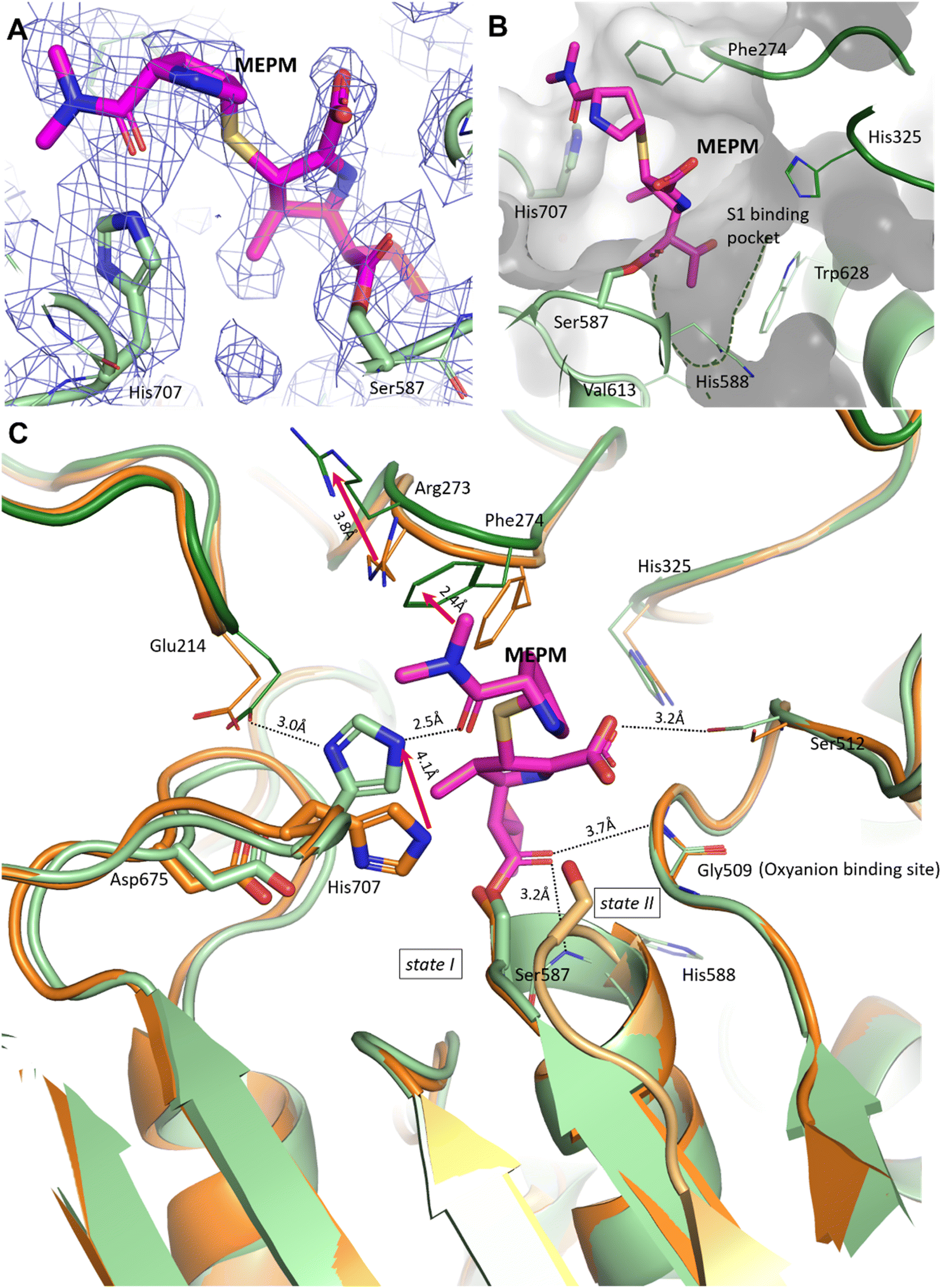 | ||
| Fig. 2 Binding site and binding conformation of meropenem in comparison with the uncomplexed pAAP structure. (A) Cryo-EM map (contoured in blue) around the covalently bound meropenem (magenta). (B) S1 binding pocket (green dotted line) is shallow, allowing the access of only smaller side chains. It is lined with residues (His325, His588, Val613 and Trp628) capable of accommodating both hydrophobic and polar groups. (C) Comparison of the meropenem complex of pAAP (hydrolase in light green, and propeller in dark green) and the uncomplexed pAAP structure (PDB id 7px8) showing the different conformations of catalytic Ser587 (state I: active, and dark orange and state II: inactive, and light orange).33 Binding of meropenem (MEPM, magenta) restores the active Ser-loop conformation, however, results in enzyme deactivation because of the displacement of catalytic His707. Loop 272–275 is also remodeled, with the side chains of Arg273 and Phe274 shifted. | ||
A further and rather striking feature of the pAAP–MEPM complex is that the oxo-group of the terminal carbamate forms a tight H-bond with the catalytic His707 residue, which is flipped (by approximately −100° along its χ1 (N–Cα–Cβ–Cγ) dihedral angle, accompanied by a 180° flip of the imidazole ring (χ2)) and is thus removed from the Ser-His-Asp catalytic triad – shifting 4.1 Å from its original position (measured at its Nε2 atom; Fig. 2C). EMRinger40 rotamer validation confirmed that the present orientation of the His707 side chain is a low-energy rotamer with no alternative conformation present in the obtained dataset (Fig. S6†), and the residual density in the position corresponding to another possible χ1 rotamer is most probably occupied by a water molecule.
In the complex, His707 is wedged between meropenem and Glu214, trapped in a doubly protonated state (with donor–acceptor distances of 2.5 Å and 3.0 Å, respectively) (Fig. 2C). Both its separation from the catalytic center and its protonation state prevent His707 from participating in the hydrolysis of the acyl–enzyme intermediate, where its role would be to act as a proton acceptor of the attacking water molecule. This might be the primary reason for the arrest of the hydrolysis at the acyl–enzyme state, affording the stable, covalently inhibited complex (Fig. 3A and B).
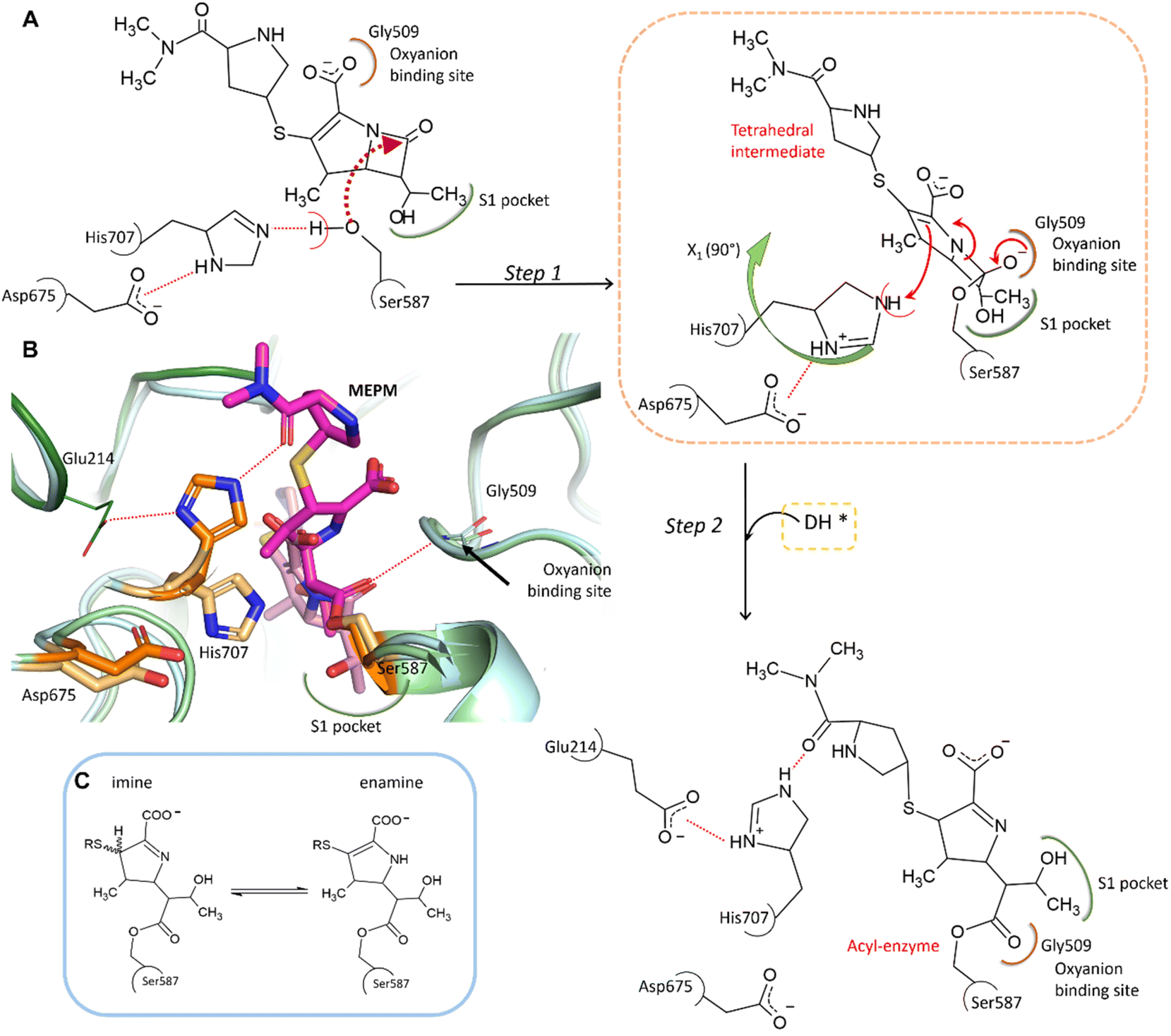 | ||
| Fig. 3 Proposed mechanism of meropenem binding to AAP. (A) Meropenem is coordinated to the catalytic triad of AAP, attacked by the activated side chain of Ser587 and stabilized in its bound form by the oxyanion binding site (according to our MCMM calculations the oxyanion binding site is initially occupied by the carboxyl-group of meropenem). During the docking and subsequent covalent linkage of the meropenem substrate, the sidechain of the catalytic His707 residue is dislocated to a position between Glu214 and the amide oxygen of meropenem, becoming doubly protonated. (B) Comparison of the non-covalently bound initial complex (derived by the MCMM protocol) (protein in light blue, catalytic triad in light orange, and the unhydrolyzed (intact) MEPM in light pink) and the cryo-EM determined structure of the covalent complex (hydrolase domain in light green, propeller domain in dark green, catalytic triad in dark orange, and hydrolyzed MEPM in magenta) (see also Fig. S5 and Table S2†). (C) The possible tautomerization states of the dihydro-pyrrole ring.41 | ||
To determine the protonation and tautomeric states of the two heterocycles of the pAAP–MEPM complex, QM/MM calculations were carried out (Fig. S7†). The experimentally determined binding pose was most closely reproduced by the calculations when the dihydro-pyrrole ring of meropenem is an imine and the pyrrolidine nitrogen atom is not protonated (thus the ring is neutral). The imine tautomer also happens to be the lower energy arrangement: the enamine form is +1.02 kcal mol−1 less favorable, suggesting an overwhelming presence (∼85%) of the former (Fig. 3C). This is a catalytically significant distinction. While the N–H group of the enamine tautomer may support the hydrolysis of the acyl–enzyme intermediate by H-bonding the acyl carbonyl moiety thus facilitating its attack by a water molecule, the imino form provides no such assistance (Fig. 3C).41 Therefore, the tautomeric form of meropenem stabilized by the protein matrix of pAAP may also contribute to trapping the complex in the acyl–enzyme state (Fig. 3A and B).
Besides the relocation of His707, a further significant difference can be seen between the meropenem-bound and uncomplexed forms of pAAP, namely the stabilization of the short loop holding the active serine (Ser-loop) in its active conformation. This segment was seen to alternate between active and latent conformers in the uncomplexed form of the enzyme. pAAP (and its human counterpart sharing a 92% of sequence identity) is unique among serine-proteases of the S9 oligopeptidase family in this respect – with a Pro (Pro506) inserted into the central β-sheet of its hydrolase domain, an unusual degree of conformational variability is granted to the Ser-loop. In the latent conformations, catalytic Ser587 is too far from His707 to establish an H-bond with it,33 thus the flip of His707 is made considerably easier in mammalian AAP than in other serine-proteases where the Ser-His-Asp triad is intact (Fig. 2C).
In the complex, both Arg273 and Phe274 shift (by 3.7 Å and 2.4 Å, respectively (both measured at Cξ atoms)), creating more space for the distal segment of the ligand (Fig. 2C). Phe274 also forms hydrophobic contacts with the sulfur atom of the thioether bridge, the pyrrolidine ring and the methyl groups of the terminal amide. The shallow and hydrophobic substrate specificity pocket of pAAP (S1 – serine protease nomenclature), known to favor Thr, Ala, Met, Ser, and Gly side chains as P1 residues,42 accommodates the hydroxyethyl substituent (CCAE(CCAF)OOA-group, Fig. S5†) of the dihydro-pyrrole ring of meropenem (Fig. 2B).
In this covalent acyl–enzyme complex pAAP binds meropenem in a nearly unstrained conformation with the antibiotic fitted into the substrate specificity pocket and spacious inter-domain cavity in a conformation that is also sampled as a low energy arrangement of the unbound, water-immersed, hydrolyzed meropenem (modeled using Monte Carlo Multiple Minimum (MCMM) calculations (Fig. S8†)).
Modelling the initial binding sites and conformations of valproic acid glucuronide and meropenem within the active pocket of pAAP
We carried out induced fit docking (IFD) calculations to identify initial binding sites of valproic acid glucuronide (VPA-G) and the unhydrolyzed (intact) MEPM within the catalytic cavity of pAAP (Fig. 4A and B). Using a large catalytic site area (of 45 Å box size) both VPA-G and MEPM were found to bind in four distinct locations within the active cavity (Fig. 4A and B): three different sites at the base of the propeller channel (clusters A–C) and one near the active site (cluster D). In a recent study using the homology model of the enzyme, topologically similar locations were identified for these two competing molecules.43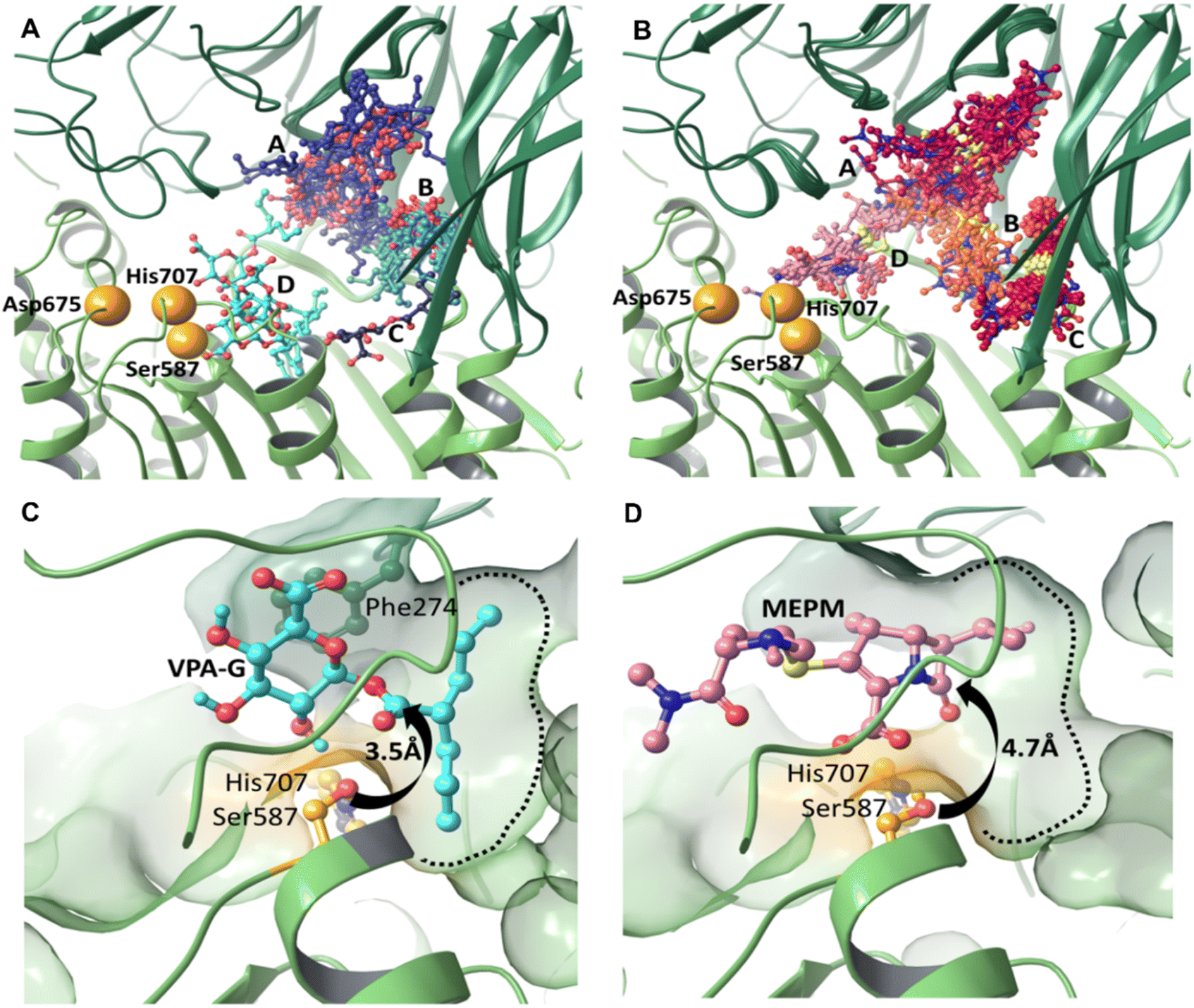 | ||
| Fig. 4 Non-covalent binding sites of valproic acid glucuronide (VPA-G) and meropenem (MEPM) in the catalytic cavity of pAAP explored by molecular modeling. Four clusters (A–D; (A) near residues 148–153, 358–367, 411–414 and 514; (B) at residues 36, 381–386, and 511–515; (C) at residues 21–25, 410, 424, 431, and 517–520, (D) in the vicinity of active Ser587) of unreacted (A) VPA-G (colored in different shades of blue) and (B) MEPM (colored in different shades of red) obtained using the induced-fit docking protocol. (C and D) Refined non-covalent binding poses near the catalytic apparatus. MCMM-derived lowest energy conformers in the case of VPA-G (C) and the unhydrolyzed meropenem (D) ligand. The wide and shallow S1 pocket (green dotted line) of pAAP accommodates both propyl-chains of VPA-G (cyan) and the threonine-like hydroxyethyl-group of MEPM (pink) (on all panels the hydrolase domain of pAAP is colored light green, the propeller dark green, while the residues of the catalytic triad orange). | ||
Subsequently, ligand positions in the proximity of the active-site (from cluster D) were chosen, and the non-covalent binding conformations of both VPA-G and intact MEPM were refined using restraint-free MCMM conformational searches.
We found that the lowest energy conformer of VPA-G is positioned in the catalytic pocket of pAAP so that Ser587 Oγ and the carbonyl carbon atom targeted by pAAP (of the ester connecting the valproate and glucose segments) are separated by 3.5 Å (Fig. 4C and S9†), while the carbonyl oxygen is H-bonded to the backbone amide of Gly509 – the oxyanion binding site of pAAP. The propyl chains of valproate dock into the S1 pocket, being quite similar in size to Ac-Thr or Ac-Met, the P1 residues preferred by the enzyme.42 Phe274 is positioned above the glucose ring at a 4.1 Å distance (measured at the centroids of the rings) in a near parallel orientation to the plane of the sugar (the angle between the two ring planes is 11.6°) forming a strong CH–π interaction44,45 with the axial C1–H1, C3–H3 and C5–H5 moieties of the sugar (Fig. 4D and S9†). Hydrogen bonds are formed by the hydroxyl group at the C2 atom of the pyranose ring with Oγ of Ser587 and by the carboxylate at C6 with Ser512 of pAAP. Thus VPA-G is coordinated via several strong interactions and oriented for hydrolytic attack within the undisturbed active site of pAAP (Fig. S10†).
In the pre-reaction (non-covalent) pAAP:MEPM complex, the carboxylate substituent (CAM–OAHOAT) of MEPM is coordinated by the backbone amides of His588 and Gly509 (Fig. 4D and S9†) of the oxyanion pocket, the hydroxyethyl group (-OAGH) is H-bonded by both Cys275 and Arg677, the N-atom of the pyrrolidine ring is coordinated by Ser512, and the oxygen of the terminal amide by Gly508. However, this initial docking of intact MEPM into the catalytic pocket of pAAP is not quite as favorable as that of VPA-G. While the antibiotic is accommodated in an orientation that is a prerequisite for forming the covalent association – with the hydroxyethyl substituent pointing toward the S1 pocket and the carbonyl moiety of the β-lactam ring in an orientation that allows its attack – the distance between the Oγ atom of Ser587 and the targeted CAA is 4.7 Å. This suggests that rearrangement of the active site must take place for the acylation step to proceed. This is in accordance with our finding that in the stable acyl–enzyme state (Fig. 3A) His707 is flipped away from the catalytic center and both Arg273 and Phe274 are relocated, widening the binding site.
Conclusion
MEPM is a broad spectrum carbapenem antibiotic, effective against Gram-positive, Gram-negative and anaerobic pathogens. It is indicated for the treatment of urinary tract infections, skin and soft tissue infections, respiratory tract infections – even multi-drug resistant tuberculosis – and infections of the central nervous system.37,46–48 MEPM contains (among other features) a fused β-lactam ring and a thioether linked pyrrolidine. On the other hand, VPA-G is a carbohydrate-functionalized short-chain fatty acid. In a very unlikely scenario, these two, quite different small molecules have selected AAP – a huge, toroidal shaped tetrameric enzyme with deeply buried active sites, equipped with a protein degradation machinery – as their common partner (Fig. S10†). Focusing on the antibiotic molecule, here we set out to understand what structural features of the enzyme contribute to this unusual drug–drug interaction and what factors restrict the association to the carbapenems from among the large family of β-lactam antibiotics.The monomeric units of pAAP contain two domains, the hydrolase that carries the serine protease apparatus and a propeller domain that shields it (Fig. 1). In the functional tetramer these monomers are arranged in such a way that the opening between the domains faces inward. The human enzyme carries a sequence that is 92% identical to that of pAAP (Fig. S11†) and was also shown to be tetrameric49 – thus it is expected to behave very similarly. The active sites of these mammalian AAPs are therefore doubly protected, their access is only possible via one of two well-protected pathways. One leads through the outer pore of the tetramer, on to the central antechamber and finally through the gate of the monomeric units, and the other through the narrow channel that passes through the propeller domains32,33 of the monomers (Fig. 1). We have found that within these secluded catalytic cavities several binding sites can accommodate MEPM, including the one in the immediate vicinity of the catalytic triad (Fig. 4). This allows for a far greater residence time for the antibiotic near the active site of pAAP than in the case of the surface-close, solvent-exposed active sites of most serine-proteases (Fig. S12†). Long residence is necessitated by the bulkiness of the fused and branched β-lactam core of the antibiotic (compared to a peptide bond of a protein chain, or the ester-carbonyl of VPA-G) which hinders the attack of the strained β-lactam ring by the catalytic Ser. In the case of pAAP, we have seen that the shift of Arg273 and Phe274 as well as the flip of catalytic His707 to a new position assist the emergence of the acyl–enzyme complex (Fig. 2C and 3).
The removal of His707 from the catalytic center results in the arrest of the catalytic reaction and the formation of a stable, covalently inhibited state. The next – deacylation – step of the hydrolysis reaction would require that the catalytic His coordinates and activates a water molecule that would attack the Ser-bound acyl–enzyme intermediate. However His707, in this case, is too far away and is also locked in a doubly protonated state by the proximity of both the carbonyl oxygen of the terminal amide of meropenem and the carboxylate moiety of Glu214. Since in this form the imidazole moiety of His707 is neither close to the active site, nor an H-acceptor, it is unable to assist the furthering of the catalytic process. In the case of both porcine and human AAPs, this relocation of His707 is made easier by the inherent flexibility of the Ser-loop, achieved by a unique Pro insertion nearby, which destabilizes the β-strand leading up to the active residue (Fig. 2C and 3). In fact, in the uncomplexed form of pAAP, multiple arrangements of the active triad were found to co-exist, with the Ser–His H-bond only partially present.33
Among the β-lactam antibiotics, carbapenems carry the smallest substituent on the β-lactam ring itself (Fig. S13†) – a hydroxyethyl group. The presence of such a small protuberance near the site of the serine protease attack seems to be a crucial requirement for binding to AAP, since the S1 pocket that hosts this segment of the enzyme-bound molecule is quite small – in agreement with AAP being specific for cleaving small Ac-Ala, Ac-Ser, Ac-Thr, and Ac-Gly residues or flexible Ac-Met from the N-terminus of oligopeptides and proteins.43 In the case of the MEPM–pAAP complex, this hydroxyethyl substituent – analogous to the side chain of Thr – is observed to be anchored within the S1 pocket. Since the hydroxyethyl substituent is a common structural feature of all carbapenems, its moderate size as compared to those of other β-lactams provides a plausible explanation for experiencing the DDI-effect during co-administration of VPA and not only meropenem (first clinical study reported in 1998),50 but other carbapenems such as panipenem,1 doripenem,51 ertapenem,52 biapenem53 and imipenem54 as well, but not in the case of any other β-lactam antibiotic.
Thus, AAP is made susceptible to this unlikely association by its unusually sheltered active pockets and flexible catalytic triad, while the carbapenems possess sufficiently small substituents on their β-lactam rings to fit into the shallow substrate-specificity pocket of the enzyme.
Our results indicate that the interaction between AAP and the carbapenems prevents VPA-G from binding to the catalytic site causing the DDI that led to the original discovery of AAP’s affinity toward this family of antibiotics. While VPA-G is hydrolyzed and released by the enzyme, carbapenems become permanently captured at the acyl–enzyme stage of the reaction – as it was seen in the case of bacterial transpeptidases that are also inhibited by these antibiotics. Our results gain curious actuality by VPA being currently repurposed for the treatment of COVID-19, proving effective in blocking infection and viral spreading.55–57
AAP has been singled out as a possible target for cancer-therapy in a number of widely different contexts, relying on its effect over the proteasome system,17,19 and the localization and function of oncogenic K-Ras variants30 or by being an effective esterase.22 Here, we showed that meropenem is a specific inhibitor of AAP, hindering its serine-protease function. Since no other mammalian target of the carbapenems has been identified as of yet, the AAP–meropenem interaction offers a unique possibility for designing safe in vivo studies of AAP inhibition (using a well-known antibiotic molecule) to clarify the exact role of this many-faceted enzyme, while the structure we presented here could be used as a starting point for the design of future therapeutics.
Materials and methods
Purification of porcine liver AAP and inhibition by meropenem (MEPM)
The preparation and purification of the mammalian AAP sample (from porcine liver) was based on a previous method58 with an additional size exclusion chromatographic step using an AKTA FPLC system, Superose 6 30/100 column (GE Healthcare, 20 mM TRIS, pH = 8.0, 0.15 M NaCl, 1 mM EDTA, and 1 mM DTT). Tetrameric composition was verified by size exclusion chromatography.33 To monitor that the catalytically competent form of the enzyme was preserved during the purification process, concentrated samples of pAAP were incubated with N-acetyl-alanine p-nitroanilide (AANA, eNovation Chemicals LLC) as a substrate49 (1.6 μM in 5% DMF/water) in buffer (50 mM phosphate, pH = 8.0, 0.3 M NaCl, 1 mM EDTA, and 5 mM mercaptoethanol) at 37 °C (reaction mixture: 10 μl of AANA solution, 985 μl buffer, and 5 μl protein sample). The formation of p-nitroaniline was measured spectrophotometrically by monitoring the increase in absorbance at 410 nm.Fractions from the final size exclusion chromatography step that showed specific activity were washed with buffer (100 ml, 10 mM TRIS, and pH = 7.5) to clear out dithiothreitol (DTT) and were concentrated to a final volume of 1 ml (Amicon Ultra-15 centrifugal filter (30 kDa MWCO)). The removal of DTT is necessitated since DTT, applied as a reducing agent during the purifying steps, reacts with MEPM leading to its degradation (Fig. S14†), possibly through the attack and opening of the beta-lactam ring59 – as noted during our previous crystallization experiments. The protein solution was further washed with 6 × 5 ml of 10 mM TRIS (pH = 7.5) buffer containing 0.1 mM MEPM, and 2 × 5 ml of 10 mM TRIS (pH = 7.5) and concentrated. AAP purified from porcine liver (1 mg ml−1, 0.01 mM solution) mixed with MEPM (added in 10-fold excess) was incubated at 37 °C (total reaction volume: 100 μl). After 1 h of incubation all the specific activity was lost. This sample was frozen in N2 (liq.) and stored at −80 °C.
Mass spectrometry
Cryo-EM sample preparation and measurement
Purified protein complexed with MEPM in 10 mM Tris (pH = 7.5) buffer was placed on a Quantifoil R1.2/1.3 grid (GIG, 1.0 μm hole size, 200 mesh) and was vitrified. After 6 s of blotting time, at 4 °C and 95% humidity, the grid was plunge-frozen in liquid ethane (VitrobotMkIV). Cryo-EM single particle data collection was performed using a Krios G4 microscope operated at 300 kV equipped with a Falcon 4 direct electron detector. Images were recorded at 130000-fold nominal magnification corresponding to 0.96 Å per pixel size using a 10 eV energy filter (Selectris X) with exposure time of 6 s and a total electron dose of ∼41 e Å−2. Defocus range was −0.5–1.5 μm. A total of 11407 micrographs were collected from a single grid (Table S1†).
Cryo-EM data processing
Movies were subjected to beam-induced motion correction using the CryoSPARC program package60 and contrast transfer function parameters were estimated using CTFFIND4.61 All of the following processes were performed using CryoSPARC. Particles were auto-picked and two rounds of reference-free two-dimensional (2D) classification were performed. In total, 1604467 particles from 11407 micrographs were auto-picked and subjected to 3D ab initio model building and 3D classification. 3D refinement was performed applying C1 and C2 symmetries. 654863 particles were used for the final reconstruction. Final 3D refinement and postprocessing yielded a map with an overall resolution of 2.17 Å (C1 symmetry) and 2.09 Å (C2 symmetry), estimated using the gold-standard FSC = 0.143 criterion (Fig. S2 and S3†).
Model building and refinement
Model building was carried out by docking the previously determined structure of the uncomplexed AAP (PDB id: 7px8), using Phenix Dock in Map.62 Manual finishing of the tetramer was carried out in Coot63 and the structure was refined with real space refinement.Validation and visualization
Refined structures were validated in Phenix64 (Table S1†) and EMRinger41 (Table S1 and Fig. S6†). Local resolution estimation of cryo-EM maps was performed with ResMap65 (Fig. S2 and S3†).Figures were generated using PyMOL (https://pymol.org/2), UCSF Chimera66 and UCSF ChimeraX.67 Sequence alignment was carried out using UniProt protein BLAST.68
Molecular docking simulation
Data availability
The cryo-EM map and atomic model have been deposited in the Electron Microscopy Data Bank (EMDB) and PDB, respectively, with the following accession codes: EMD-14149 and PDB: 7qun.Author contributions
A. J. K.-S., V. H., D. K. M. and A. P. conceived the project, A. J. K.-S., P. S., V. H. and D. K. M. designed the experiments. P. S. V. H., D. K. M. and A. P. provided funding and conceptual input. A. J. K.-S., V. H. and D. K. M. analyzed the data, prepared figures and tables and co-wrote the manuscript. A. J. K.-S. isolated and purified the protein with the help of P. S. S. M. conducted the cryo-EM experiment. I. J. helped with data transition and processing. Mass spectrometric experiments were conducted with the help of G. S. Docking was carried out by O. Z. and A. J. K.-S. Molecular modelling, MCMM and QM/MM calculations were carried out by D. K. M.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by project no. 2018-1.2.1-NKP-2018-00005 of the National Research Development and Innovation Fund of Hungary; No. VEKOP-2.3.2-16-2017-00014 and VEKOP-2.3.3-15-2017-00018 of the European Union and the State of Hungary, co-financed by the European Regional Development Fund; by MedInProt Grants from the Hungarian Academy of Sciences; and within the framework of the Thematic Excellence Program 2019 by the National Research, Development and Innovation Office under project “Synth+” as well as the Hungarian Scientific Research Fund (NKFIH-OTKA) grant K116305. The authors thank Zoltán Urbán (ComInnex Inc., Budapest, Hungary) for the LCMS measurements and analysis.References
- K. Nagai, T. Shimizu, A. Togo, M. Takeya, Y. Yokomizo, Y. Sakata, T. Matsuishi and H. Kato, Decrease in serum levels of valproic acid during treatment with a new carbapenem, panipenem/betamipron, J. Antimicrob. Chemother., 1997, 39, 295–296, DOI:10.1093/jac/39.2.295 , PMID: 9069562..
- M. K. Mishra, S. Kukal, P. R. Paul, S. Bora, A. Singh, S. Kukreti, L. Saso, K. Muthusamy, Y. Hasija and R. Kukreti, Insights into Structural Modifications of Valproic Acid and Their Pharmacological Profile, Molecules, 2022, 27(1), 104, DOI:10.3390/molecules27010104 , PMID: 35011339..
- H. Mori, K. Takahashi and T. Mizutani, Interaction between valproic acid and carbapenem antibiotics, Drug. Metab. Rev., 2007, 39(4), 647–657, DOI:10.1080/03602530701690341 , PMID: 18058328..
- S. Deshayes, A. Coquerel and R. Verdon, Neurological Adverse Effects Attributable to β-Lactam Antibiotics: A Literature Review, Drug Saf., 2017, 40(12), 1171–1198, DOI:10.1007/s40264-017-0578-2 , PMID: 28755095..
- D. Dreucean, K. Beres, A. McNierney-Moore and D. Gravino, Use of meropenem to treat valproic acid overdose, Am. J. Emerg. Med., 2019, 37(11), 2120.e5–2120.e7, DOI:10.1016/j.ajem.2019.158426 , PMID: 31500925..
- C. Thomas, J. Priano and T. L. Smith, Meropenem as an antidote for intentional valproic acid overdose, Am. J. Emerg. Med., 2020, 38(3), 690.e1–690.e2, DOI:10.1016/j.ajem.2019.09.011 , PMID: 31980292..
- D. Cunningham, K. Clark and K. Lord, Treatment of valproic acid overdose with meropenem in an epileptic patient, Am. J. Emerg. Med., 2022, 53, 284.e1–284.e3, DOI:10.1016/j.ajem.2021.09.033 , PMID: 34625331..
- Y. Nakamura, K. Nakahira and T. Mizutani, Decreased valproate level caused by VPA-glucuronidase inhibition by carbapenem antibiotics, Drug Metab. Lett., 2008, 2(4), 280–285, DOI:10.2174/187231208786734049 , PMID: 19356106..
- E. Suzuki, N. Yamamura, Y. Ogura, D. Nakai, K. Kubota, N. Kobayashi, S. Miura and O. Okazaki, Identification of valproic acid glucuronide hydrolase as a key enzyme for the interaction of valproic acid with carbapenem antibiotics, Drug Metab. Dispos., 2010, 38(9), 1538–1544, DOI:10.1124/dmd.110.032938 , PMID: 20551238..
- J. Perrier, A. Durand, T. Giardina and A. Puigserver, Catabolism of intracellular N-terminal acetylated proteins: involvement of acylpeptide hydrolase and acylase, Biochimie, 2005, 87(8), 673–685, DOI:10.1016/j.biochi.2005.04.002 , PMID: 15927344..
- A. Adibekian, B. R. Martin, C. Wang, K. L. Hsu, D. A. Bachovchin, S. Niessen, H. Hoover and B. F. Cravatt, Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors, Nat Chem Biol, 2011, 7(7), 469–478, DOI:10.1038/nchembio.579 , Erratum in: Nat. Chem. Biol., 2012, 8(3), 318, PMID: 21572424..
- G. Palmieri, P. Bergamo, A. Luini, M. Ruvo, M. Gogliettino, E. Langella, M. Saviano, R. N. Hegde, A. Sandomenico and M. Rossi, Acylpeptide hydrolase inhibition as targeted strategy to induce proteasomal down-regulation, PLoS One, 2011, 6(10), e25888, DOI:10.1371/journal.pone.0025888 , PMID: 22016782..
- C. S. Hwang, A. Shemorry and A. N. Varshavsky, N-terminal acetylation of cellular proteins creates specific degradation signals, Science, 2010, 327, 973–977, DOI:10.1126/science.1183147 , PMID: 20110468..
- K. Shimizu, T. Fujino, K. Ando, M. Hayakawa, H. Yasuda and K. Kikugawa, Overexpression of oxidized protein hydrolase protects COS-7 cells from oxidative stress-induced inhibition of cell growth and survival, Biochem. Biophys. Res. Commun., 2003, 304(4), 766–771, DOI:10.1016/s0006-291x(03)00657-0 , PMID: 12727222..
- K. Shimizu, Y. Kiuchi, K. Ando, M. Hayakawa and K. Kikugawa, Coordination of oxidized protein hydrolase and the proteasome in the clearance of cytotoxic denatured proteins, Biochem. Biophys. Res. Commun., 2004, 324(1), 140–146, DOI:10.1016/j.bbrc.2004.08.231 , PMID: 15464994..
- A. Tangri, K. Lighty, J. Loganathan, F. Mesmar, R. Podicheti, C. Zhang, M. Iwanicki, R. Drapkin, H. Nakshatri and S. Mitra, Deubiquitinase UCHL1 Maintains Protein Homeostasis through the PSMA7-APEH-Proteasome Axis in High-grade Serous Ovarian Carcinoma, Mol. Cancer Res., 2021, 19(7), 1168–1181, DOI:10.1158/1541-7786.MCR-20-0883 , PMID: 33753553..
- M. Gogliettino, E. Cocca, A. Sandomenico, L. Gratino, E. Iaccarino, L. Calvanese, M. Rossi and G. Palmieri, Selective inhibition of acylpeptide hydrolase in SAOS-2 osteosarcoma cells: is this enzyme a viable anticancer target?, Mol. Biol. Rep., 2021, 48(2), 1505–1519, DOI:10.1007/s11033-020-06129-4 , PMID: 33471263..
- R. Palumbo, M. Gogliettino, E. Cocca, R. Iannitti, A. Sandomenico, M. Ruvo, M. Balestrieri, M. Rossi and G. Palmieri, APEH Inhibition Affects Osteosarcoma Cell Viability via Downregulation of the Proteasome, Int. J. Mol. Sci., 2016, 17(10), 1614, DOI:10.3390/ijms17101614 , PMID: 27669226..
- P. Bergamo, E. Cocca, R. Palumbo, M. Gogliettino, M. Rossi and G. Palmieri, RedOx status, proteasome and APEH: insights into anticancer mechanisms of t10,c12-conjugated linoleic acid isomer on A375 melanoma cells, PLoS One, 2013, 8(11), e80900, DOI:10.1371/journal.pone.0080900 , PMID: 24260504..
- S. L. Naylor, A. Marshall, C. Hensel, P. F. Martinez, B. Holley and A. Y. Sakaguchi, The DNF15S2 locus at 3p21 is transcribed in normal lung and small cell lung cancer, Genomics, 1989, 4(3), 355–361, DOI:10.1016/0888-7543(89)90342-x , PMID: 2565880..
- R. Erlandsson, F. Boldog, B. Persson, E. R. Zabarovsky, R. L. Allikmets, J. Sümegi, G. Klein and H. Jörnvall, The gene from the short arm of chromosome 3, at D3F15S2, frequently deleted in renal cell carcinoma, encodes acylpeptide hydrolase, Oncogene, 1991, 6(7), 1293–1295 CAS , PMID: 1861871..
- C. A. McGoldrick, Y. L. Jiang, V. Paromov, M. Brannon, K. Krishnan and W. L. Stone, Identification of oxidized protein hydrolase as a potential prodrug target in prostate cancer, BMC Cancer, 2014, 14, 77, DOI:10.1186/1471-2407-14-77 , PMID: 24512522..
- A. Brzozowska, R. Mlak, I. Homa-Mlak, P. Gołębiowski, M. Mazurek, M. Ciesielka and T. Małecka-Massalska, Polymorphism of regulatory region of APEH gene (c.-521G>C, rs4855883) as a relevant predictive factor for radiotherapy induced oral mucositis and overall survival in head neck cancer patients, Oncotarget, 2018, 9(51), 29644–29653, DOI:10.18632/oncotarget.25662 , PMID: 30038710..
- T. Fujino, K. Watanabe, M. Beppu, K. Kikugawa and H. Yasuda, Identification of oxidized protein hydrolase of human erythrocytes as acylpeptide hydrolase, Biochim. Biophys. Acta, 2000, 1478(1), 102–112, DOI:10.1016/s0167-4838(00)00004-2 , PMID: 10719179..
- A. Sandomenico, M. Gogliettino, E. Iaccarino, C. Fusco, A. Caporale, M. Ruvo, G. Palmieri and E. Cocca, Oxidized Substrates of APEH as a Tool to Study the Endoprotease Activity of the Enzyme, Int. J. Mol. Sci., 2021, 23(1), 443, DOI:10.3390/ijms23010443 , PMID: 35008880..
- R. Yamin, C. Zhao, P. B. O'Connor, A. C. McKee and C. R. Abraham, Acyl peptide hydrolase degrades monomeric and oligomeric amyloid-beta peptide, Mol. Neurodegener., 2009, 4, 33, DOI:10.1186/1750-1326-4-33 , PMID: 19627603..
- P. G. Richards, M. K. Johnson and D. E. Ray, Identification of acylpeptide hydrolase as a sensitive site for reaction with organophosphorus compounds and a potential target for cognitive enhancing drugs, Mol. Pharmacol., 2000, 58(3), 577–583, DOI:10.1124/mol.58.3.577 , PMID: 10953051..
- G. García-Rojo, F. Gámiz, E. Ampuero, D. Rojas-Espina, R. Sandoval, C. Rozas, B. Morales, U. Wyneken and F. Pancetti, In Vivo Sub-chronic Treatment with Dichlorvos in Young Rats Promotes Synaptic Plasticity and Learning by a Mechanism that Involves Acylpeptide Hydrolase Instead of Acetylcholinesterase Inhibition. Correlation with Endogenous β-Amyloid Levels, Front. Pharmacol., 2017, 8, 483, DOI:10.3389/fphar.2017.00483 , PMID: 28790916..
- Z. Zeng, S. L. Rulten, C. Breslin, A. Zlatanou, V. Coulthard and K. W. Caldecott, Acylpeptide hydrolase is a component of the cellular response to DNA damage, DNA Repair, 2017, 58, 52–61, DOI:10.1016/j.dnarep.2017.08.008 , PMID: 28866241..
- L. Tan, K. J. Cho, W. E. Kattan, C. M. Garrido, Y. Zhou, P. Neupane, R. J. Capon and J. F. Hancock, Acylpeptide hydrolase is a novel regulator of KRAS plasma membrane localization and function, J. Cell Sci., 2019, 132(15), jcs232132, DOI:10.1242/jcs.232132 , PMID: 31266814..
- E. Suzuki, D. Nakai, N. Yamamura, N. Kobayashi, O. Okazaki and T. Izumi, Inhibition mechanism of carbapenem antibiotics on acylpeptide hydrolase, a key enzyme in the interaction with valproic acid, Xenobiotica, 2011, 41(11), 958–963, DOI:10.3109/00498254.2011.596582 , PMID: 21770850..
- A. J. Kiss-Szemán, V. Harmat and D. K. Menyhárd, Achieving Functionality Through Modular Build-up: Structure and Size Selection of Serine Oligopeptidases, Curr. Protein Pept. Sci., 2019, 20(11), 1089–1101, DOI:10.2174/1389203720666190925103339 , PMID: 31553292..
- A. J. Kiss-Szemán, P. Stráner, I. Jákli, N. Hosogi, V. Harmat, D. K. Menyhárd and A. Perczel, Cryo-EM structure of acylpeptide hydrolase reveals substrate selection by multimerization and a multi-state serine-protease triad, Chem. Sci., 2022, 13, 7132–7142, 10.1039/d2sc02276a , PMID: 35799812..
- S. Han, R. P. Zaniewski, E. S. Marr, B. M. Lacey, A. P. Tomaras, A. Evdokimov, J. R. Miller and V. Shanmugasundaram, Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(51), 22002–22007, DOI:10.1073/pnas.1013092107 , PMID: 21135211..
- Z. Lu, H. Wang, A. Zhang, X. Liu, W. Zhou, C. Yang, L. Guddat, H. Yang, C. J. Schofield and Z. Rao, Structures of Mycobacterium tuberculosis Penicillin-Binding Protein 3 in Complex with Five β-Lactam Antibiotics Reveal Mechanism of Inactivation, Mol. Pharmacol., 2020, 97(4), 287–294, DOI:10.1124/mol.119.118042 , PMID: 32086254..
- F. Wang, C. Cassidy and J. C. Sacchettini, Crystal structure and activity studies of the Mycobacterium tuberculosis beta-lactamase reveal its critical role in resistance to beta-lactam antibiotics, Antimicrob. Agents Chemother., 2006, 50(8), 2762–2771, DOI:10.1128/AAC.00320-06 , PMID: 16870770..
- J. E. Hugonnet, L. W. Tremblay, H. I. Boshoff, C. E. Barry 3rd and J. S. Blanchard, Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis, Science, 2009, 323(5918), 1215–1218, DOI:10.1126/science.1167498 , PMID: 19251630..
- W. J. Li, D. F. Li, Y. L. Hu, X. E. Zhang, L. J. Bi and D. C. Wang, Crystal structure of L,D-transpeptidase LdtMt2 in complex with meropenem reveals the mechanism of carbapenem against Mycobacterium tuberculosis, Cell Res., 2013, 23(5), 728–731, DOI:10.1038/cr.2013.53 , PMID: 23588382..
- H. S. Kim, J. Kim, H. N. Im, J. Y. Yoon, D. R. An, H. J. Yoon, J. Y. Kim, H. K. Min, S. J. Kim, J. Y. Lee, B. W. Han and S. W. Suh, Structural basis for the inhibition of Mycobacterium tuberculosis L,D-transpeptidase by meropenem, a drug effective against extensively drug-resistant strains, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69(3), 420–431, DOI:10.1107/S0907444912048998 , PMID: 23519417..
- B. A. Barad, N. Echols, R. Y. Wang, Y. Cheng, F. DiMaio, P. D. Adams and J. S. Fraser, EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy, Nat. Methods, 2015, 12(10), 943–946, DOI:10.1038/nmeth.3541 , PMID: 26280328..
- C. T. Lohans, E. I. Freeman, E. V. Groesen, C. L. Tooke, P. Hinchliffe, J. Spencer, J. Brem and C. J. Schofield, Mechanistic Insights into β-Lactamase-Catalysed Carbapenem Degradation Through Product Characterisation, Sci. Rep., 2019, 9, 13608, DOI:10.1038/s41598-019-49264-0 , PMID: 31541180..
- R. G. Krishna and F. Wold, Specificity determinants of acylaminoacyl-peptide hydrolase, Protein Sci., 1992, 1(5), 582–589, DOI:10.1002/pro.5560010504 , PMID: 1304357..
- T. Ishikawa, H. Otaki, S. Mizuta, M. Kuriyama, O. Onomura, N. Higuchi, M. N. Nakashima, M. Nakashima and K. Ohyama, Computational study of the competitive binding of valproic acid glucuronide and carbapenem antibiotics to acylpeptide hydrolase, Drug Metab. Pharmacokinet., 2017, 32(4), 201–207, DOI:10.1016/j.dmpk.2017.04.002 , PMID: 28734645..
- M. J. Plevin, D. L. Bryce and J. Boisbouvier, Direct detection of CH/pi interactions in proteins, Nat. Chem., 2010, 2(6), 466–471, DOI:10.1038/nchem.650 , PMID: 20489715..
- A. Kapros, A. Balázs, V. Harmat, A. Háló, L. Budai, I. Pintér, D. K. Menyhárd and A. Perczel, Configuration-Controlled Crystal and/or Gel Formation of Protected d-Glucosamines Supported by Promiscuous Interaction Surfaces and a Conformationally Heterogeneous Solution State, Chemistry, 2020, 26, 11643–11655, DOI:10.1002/chem.202000882 , PMID: 32333713..
- A. H. Diacon, L. van der Merwe, M. Barnard, F. von Groote-Bidlingmaier, C. Lange, A. L. García-Basteiro, E. Sevene, L. Ballell and D. Barros-Aguirre, β-Lactams against Tuberculosis--New Trick for an Old Dog?, N. Engl. J. Med., 2016, 375(4), 393–394, DOI:10.1056/NEJMc1513236 , PMID: 27433841..
- J. Rodríguez-Baño, B. Gutiérrez-Gutiérrez, I. Machuca and A. Pascual, Treatment of Infections Caused by Extended-Spectrum-Beta-Lactamase-, AmpC-, and Carbapenemase-Producing Enterobacteriaceae, Clin. Microbiol. Rev., 2018, 31(2), e000799, DOI:10.1128/CMR.00079-17 , PMID: 29444952..
- F. Schneider, A. Gessner and N. El-Najjar, Efficacy of Vancomycin and Meropenem in Central Nervous System Infections in Children and Adults: Current Update, Antibiotics, 2022, 11(2), 173, DOI:10.3390/antibiotics11020173 , PMID: 35203776..
- W. M. Jones, A. Scaloni and J. M. Manning, Acylaminoacyl-peptidase, Methods Enzymol., 1994, 244, 227–231, DOI:10.1016/0076-6879(94)44019-0 , PMID: 7845211..
- B. J. De Turck, M. W. Diltoer, P. J. Cornelis, V. Maes, H. D. Spapen, F. Camu and L. P. Huyghens, Lowering of plasma valproic acid concentrations during concomitant therapy with meropenem and amikacin, J. Antimicrob. Chemother., 1998, 42(4), 563–564, DOI:10.1093/jac/42.4.563 , PMID: 9818768..
- D. Clause, P. Y. Decleire, R. Vanbinst, A. Soyer and P. Hantson, Pharmacokinetic interaction between valproic acid and meropenem, Intensive Care Med., 2005, 31, 1293–1294, DOI:10.1007/s00134-005-2695-0 , PMID: 16170544..
- J. L. Lunde, R. E. Nelson and H. F. Storandt, Acute seizures in a patient receiving divalproex sodium after starting ertapenem therapy, Pharmacotherapy, 2007, 27(8), 1202–1205, DOI:10.1592/phco.27.8.1202 , PMID: 17655519..
- Y. Masuo, K. Ito, T. Yamamoto, A. Hisaka, M. Honma and H. Suzuki, Characterization of inhibitory effect of carbapenem antibiotics on the deconjugation of valproic acid glucuronide, Drug Metab. Dispos., 2010, 38(10), 1828–1835, DOI:10.1124/dmd.110.034231 , PMID: 20581094..
- S. G. Lee, J. H. Kim, J. Y. Joo and O. H. Kwon, [Seven cases of decreased serum valproic acid concentration during concomitant use of carbapenem antibiotics], Korean J. Lab. Med., 2007, 27(5), 338–343, DOI:10.3343/kjlm.2007.27.5.338 , PMID: 18094598..
- J. Collazos, P. Domingo, N. Fernández-Araujo, E. Asensi-Díaz, H. Vilchez-Rueda, A. Lalueza, E. Roy-Vallejo, R. Blanes, M. Raya-Cruz, J. Sanz-Cánovas, A. Artero, J. M. Ramos-Rincón, C. Dueñas-Gutiérrez, J. L. Lamas-Ferreiro and V. Asensi, Valproic Acid in COVID-19 Study Group. Exposure to valproic acid is associated with less pulmonary infiltrates and improvements in diverse clinical outcomes and laboratory parameters in patients hospitalized with COVID-19, PLoS One, 2022, 17(1), e0262777, DOI:10.1371/journal.pone.0262777 , PMID: 35085321..
- M. L. Saiz, M. L. DeDiego, D. López-García, V. Corte-Iglesias, A. Baragaño Raneros, I. Astola, V. Asensi, C. López-Larrea and B. Suarez-Alvarez, Epigenetic targeting of the ACE2 and NRP1 viral receptors limits SARS-CoV-2 infectivity, Clin. Epigenetics, 2021, 13(1), 209, DOI:10.1186/s13148-021-01168-5 , PMID: 34635175..
- B. Pitt, N. R. Sutton, Z. Wang, S. Clin. EpigeneticsN. Goonewardena and M. Holinstat, Potential repurposing of the HDAC inhibitor valproic acid for patients with COVID-19, Eur. J. Pharmacol., 2021, 898, 173988, DOI:10.1016/j.ejphar.2021.173988 , PMID: 33667455..
- A. L. Kiss, Z. Szeltner, V. Fülöp and L. Polgár, His507 of acylaminoacyl peptidase stabilizes the active site conformation, not the catalytic intermediate, FEBS Lett., 2004, 571, 17–20, DOI:10.1016/j.febslet.2004.06.054 , PMID: 15280010..
- A. S. Mendez, J. Dalomo, M. Steppe and E. E. Schapoval, Stability and degradation kinetics of meropenem in powder for injection and reconstituted sample, J. Pharm. Biomed. Anal., 2006, 41(4), 1363–1366, DOI:10.1016/j.jpba.2006.02.017 , PMID: 16533586..
- A. Punjani, J. L. Rubinstein, D. J. Fleet and M. A. Brubaker, cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination, Nat. Methods, 2017, 14, 290–296, DOI:10.1038/nmeth.4169 , PMID: 28165473..
- A. Rohou and N. Grigorieff, CTFFIND4: Fast and accurate defocus estimation from electron micrographs, Acta Crystallogr., Sect. D: Struct. Biol., 2015, 192(2), 216–221, DOI:10.1016/j.jsb.2015.08.008 , PMID: 26278980..
- D. Liebschner, et al., Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix, Acta Crystallogr., Sect. D: Struct. Biol., 2019, 75(10), 861–877, DOI:10.1107/S2059798319011471 , PMID: 31588918..
- P. Emsley and K. Cowtan, Coot: model-building tools for molecular graphics, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132, DOI:10.1107/S0907444904019158 , PMID: 15572765..
- P. V. Afonine, et al., Real-space refinement in PHENIX for cryo-EM and crystallography, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 531–544, DOI:10.1107/S2059798318006551 , PMID: 29872004..
- A. Kucukelbir, F. J. Sigworth and H. D. Tagare, Quantifying the local resolution of cryo-EM density maps, Nat. Methods, 2014, 11, 63–65, DOI:10.1038/nmeth.2727 , PMID: 24213166..
- E. F. Pettersen, et al., UCSF Chimera--a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25(13), 1605–1612, DOI:10.1002/jcc.20084 , PMID: 15264254..
- E. F. Pettersen, et al., UCSF ChimeraX: Structure visualization for researchers, educators, and developers, Protein Sci., 2021, 30(1), 70–82, DOI:10.1002/pro.3943 , PMID: 32881101..
- The UniProt Consortium, UniProt: the universal protein knowledgebase in 2021, Nucleic Acids Res., 2021, 49(D1), D480–D489, DOI:10.1093/nar/gkaa1100 , PMID: 33237286..
- Schrödinger Release 2021-3, Schrödinger, LLC, New York, NY, p. 2021 Search PubMed.
- R. Farid, T. Day, R. A. Friesner and R. A. Pearlstein, New Insights about HERG Blockade Obtained from Protein Modeling, Potential Energy Mapping, and Docking Studies, Bioorg. Med. Chem., 2006, 14(9), 3160–3173, DOI:10.1016/j.bmc.2005.12.032 , PMID: 16413785..
- W. Sherman, T. Day, M. P. Jacobson, R. A. Friesner and R. Farid, Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects, J. Med. Chem., 2005, 49(2), 534–553, DOI:10.1021/jm050540c , PMID: 16420040..
- W. Sherman, H. S. Beard and R. Farid, Use of an induced fit receptor structure in virtual screening, Chem. Biol. Drug Des., 2006, 67(1), 83–84, DOI:10.1111/j.1747-0285.2005.00327.x , PMID: 16492153..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05520a |
| This journal is © The Royal Society of Chemistry 2022 |