
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decarboxylative sulfoximination of benzoic acids enabled by photoinduced ligand-to-copper charge transfer†
Peng
Xu
a,
Wanqi
Su
ab and
Tobias
Ritter
 *a
*a
aMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm Platz 1, D-45470 Mülheim an der Ruhr, Germany. E-mail: ritter@mpi-muelheim.mpg.de
bInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany
First published on 14th November 2022
Abstract
Sulfoximines are synthetically important scaffolds and serve important roles in drug discovery. Currently, there is no solution to decarboxylative sulfoximination of benzoic acids; although thoroughly investigated, limited substrate scope and harsh reaction conditions still hold back traditional thermal aromatic decarboxylative functionalization. Herein, we realize the first decarboxylative sulfoximination of benzoic acids via photo-induced ligand to copper charge transfer (copper-LMCT)-enabled decarboxylative carbometalation. The transformation proceeds under mild reaction conditions, has a broad substrate scope, and can be applied to late-stage functionalization of complex small molecules.
Introduction
Coordination of substrates including alcohols,1 halides,2 azides,3 and alkyl carboxylates4 to abundant 3d metal salts like Fe, Ni, or Cu can form photoactive metal complexes. Promoted to their excited states upon irradiation can result in intramolecular ligand-to-metal charge transfer (LMCT) within the excited state complexes to generate reactive open-shell radical intermediates with reactivity hardly reachable in the ground state.1–5 Conventional metal-catalysed or -mediated thermal decarboxylative cross-coupling reactions normally require high reaction temperature and ortho-substituents.6 Radical aromatic decarboxylation proceeds about three orders of magnitude slower than from aliphatic carboxyl radicals,7,8 which generally leads to undesirable side reactions such as hydrogen atom abstraction for benzoyl radicals.7 As a consequence, many aromatic decarboxylative bond forming reactions, including sulfoximination, are still out of reach for conventional reaction chemistry. Here we report the first decarboxylative sulfoximination of benzoic acids enabled by photo-induced ligand to copper charge transfer. Photoactive copper(II) carboxylates undergo a low-barrier radical CO2 extrusion upon irradiation, with the putative formed aryl radicals subsequently captured by copper complexes to generate CuAr(III) species for C–N reductive elimination. The synthetic utility of this method was exemplified by late-stage decarboxylative sulfoximination of several complex small-molecule benzoic acids, which are abundantly available from nature.Photonic excitation of copper(II) complexes have been known as an effective platform for generating reactive radicals for decades. Kochi first studied the addition and Csp3–H abstraction reactivity of chlorine radical generated by the photo-irradiation of CuCl2 in different organic solvents.2a Based on this initial finding, Wan and co-workers developed a vicinal dichlorination of alkenes catalyzed by CuCl2 under air,2b and the Rovis group realized a copper catalyzed olefination of unactivated Csp3–H bonds.2c In addition to chlorine radicals, copper-LMCT is also suitable for N- or C-centered radical generation. In 2018, Rehbein and Reiser found that copper-LMCT was effective for azide radical generation,3 and Wang and Xu suggested the formation of N-centered radical cations via intramolecular LMCT of quinolinyl-8-glycinate ester coordinated alkyl-Cu(III) adducts.9 For C-centered radicals, Gong and co-workers proposed the generation of alkyl radical intermediates via photolysis of Cu(II)-alkyl complexes.10 Although copper-LMCT in copper(II) carboxylate complexes was first described by DeGraff and co-workers during their research on the photolysis of copper(II)-malonate,4a,b it was not until 2021 that our group applied this copper-LMCT reactivity to synthetic applications.11 We realized the first aromatic decarboxylative fluorination11a and the decarboxylative hydroxylation11b of benzoic acids. Concurrently, the MacMillan group explored the copper-LMCT reactivity in aromatic decarboxylative borylation12a and halogenation,12b and achieved copper catalysis for transformations with single electron oxidants such as 1-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate (NFTPT). Stoichiometric copper is still required for nucleophiles such as fluoride.11a Nearly at the same time, the Yoon group developed a copper-mediated oxidative decarboxylative functionalization of aliphatic carboxylic acids.4e Most recently, Reiser and co-workers reported a copper-catalysed aliphatic decarboxylative oxygenation methodology, where oxygen was applied as the oxidant.4i While we and others have developed the concept of photo-induced copper-LMCT-enabled aromatic radical decarboxylation to achieve previously unknown reactivity, the coupling counterparts are generally limited to halides, carboxylates or boronate esters but strong coordinating NH-nucleophiles have not been shown to react (Fig. 1A).11,12 Given that copper-LMCT relies on the coordination of carboxylates to copper(II) species, strong coordination of NH-nucleophiles such as NH-sulfoximines will compete with carboxylates and form undesired copper species that can diminish the reaction efficiency. Though thermal aromatic decarboxylative C–N cross couplings under high reaction temperatures (normally more than 140 °C) have been explored by Jia, Gooβen, and Xie, electron-deficient ortho-substituted benzoic acids are required for efficient CO2 extrusion (Fig. 1A).13 Thus, a general aromatic decarboxylative C–N cross coupling still remains elusive.
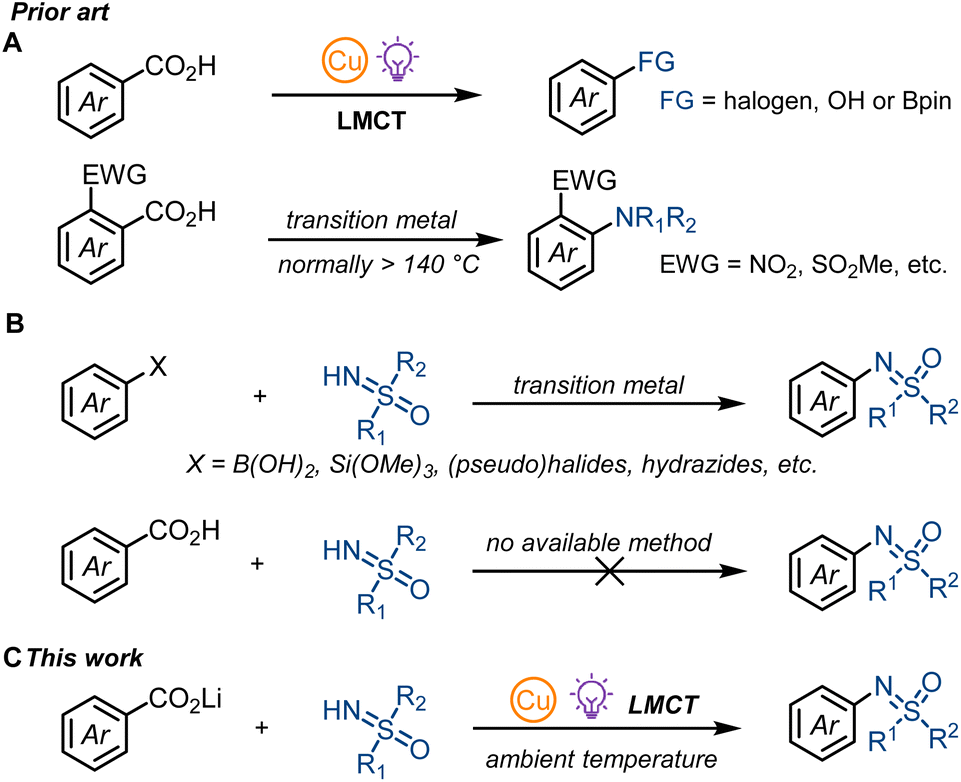 | ||
| Fig. 1 (A) Prior art of copper-LMCT enabled decarboxylation of benzoic acids and thermal aromatic decarboxylative C–N cross couplings. (B) Previous synthesis of N-arylated sulfoximines via N-arylation of NH-sulfoximines. (C) Aromatic decarboxylative sulfoximination via copper-LMCT. | ||
The study of sulfoximines dates back as far as 1949 when methionine sulfoximine was first synthesized by Bentley and Whitehead.14 Today, sulfoximines play a remarkable role in synthetic chemistry15 and drug discovery;16 they have been applied as chiral auxiliaries,17 chiral ligands,18 asymmetric organocatalysts19 and building blocks.15,20N-arylated sulfoximines are mainly prepared by transition-metal-catalyzed direct N-arylation of NH-sulfoximines with aryl (pseudo)halides,21 arylboronic acids,22 aryl siloxanes,23 acyl peroxides,24 arylsulfinates,25 and aryl hydrazides26 but cannot be accessed from benzoic acids (Fig. 1B). Herein, we present the first decarboxylative sulfoximination of benzoic acids by applying a photo-induced carboxylate-to-copper charge transfer strategy (Fig. 1C). We found that lithium carboxylates with 2,6-di-tert-butylpyridine (DTBP) and LiOMe as additives was able to overcome the challenging low reaction efficiency associated with copper-LMCT-enabled aromatic decarboxylative sulfoximination.
Results and discussion
Because of the enhanced N–H acidity, NH-sulfoximines can undergo facile deprotonation and readily coordinate with copper(II) species.16c In the copper-LMCT process, competing coordination of sulfoximines to copper(II) might hinder the formation of key copper(II) carboxylate intermediates and, in turn, decrease the reaction efficiency. We hypothesized that initial deprotonation of the benzoic acids to their carboxylate salts might facilitate the generation of photoactive copper(II) carboxylates, while the careful screening of additives can hinder the formation of undesired sulfoximine-ligated copper(II) species.As shown in Table 1, we verified our assumption via the decarboxylative sulfoximination of lithium 4-fluorobenzoate (1). A series of reaction condition optimization (see ESI for more details†) reveals that purple light irradiation of a mixture of 1, NH-sulfoximine 2, Cu(OTf)2, LiOMe and 2,6-di-tert-butylpyridine (DTBP) in MeCN can afford N-arylated sulfoximine 3 in 66% yield, together with side product ester 3a in 8% yield (Table 1, entry 1). Compared with 4-fluorobenzoic acid, 4-fluorobenzoate salts underwent more efficient decarboxylation, and the lithium salts gave the best yield (entries 1–3). The result is consistent with our hypothesis that the use of benzoate salts can promote the formation of copper carboxylates and increase the reaction efficiency. Notably, the combination of bulky DTBP and LiOMe is crucial to obtain a high yield. Replacing DTBP/LiOMe with other inorganic or organic bases generally led to lower yields, and a large amount of side oxydecarboxylation product 3a was detected (entries 4–6). Strong coordinating bi- or tridentate pyridine-based ligands or bidentate PyBOX ligand were also tested, yet no better yields were observed (see ESI for more details†). We assume that weak ligation of bulky DTBP to copper might favour C–N reductive elimination over C–O reductive elimination or help to form photoactive copper(II) carboxylates species. The role of LiOMe is not very clear; we propose that the addition of LiOMe might help decrease the concentration of free sulfoximines by forming poorly soluble sulfoximine lithium salts and in turn, accelerate the formation of copper(II) carboxylate species. Low conversion of starting substrates 1 and 2 was observed when copper sources including Cu(OAc)2 were used instead of Cu(OTf)2 (entry 7). Interestingly, only MeCN as solvent was productive, and DCM only afforded 12% protodecarboxylation side-product fluorobenzene (entry 8). Control experiments confirmed the essential use of 390 nm LEDs irradiation for CO2 extrusion, and no decarboxylation was observed under thermal reaction conditions.
|
|
||
|---|---|---|
| Entry | Variation | Yield of 3/3ab (%) |
| a Reaction conditions: 1 (0.05 mmol, 1.0 equiv.), 2 (2.5 equiv.), Cu(OTf)2 (2.5 equiv.), LiOMe (1.0 equiv.), 2,6-di-tert-butylpyridine (DTBP, 2.0 equiv.), MeCN (c = 25 mM), 18 h purple LEDs irradiation, 35 °C. b Yields were determined by 19F NMR using 2-fluorotoluene (2.0 equiv.) as an internal standard. c 12% yield of protodecarboxylation product fluorobenzene was observed. | ||
| 1 | None | 66/8 |
| 2 | H+ instead of Li+ | 44/8 |
| 3 | Na+ instead of Li+ | 60/12 |
| 4 | Li2CO3 instead of LiOMe/DTBP | 48/20 |
| 5 | KF instead of LiOMe/DTBP | 38/40 |
| 6 | 2,6-Difluoropyridine instead of LiOMe/DTBP | 22/36 |
| 7 | Cu(OAc)2 instead of Cu(OTf)2 | 0/0 |
| 8 | DCM instead of MeCN | 0/0c |
| 9 | No DTBP | 0/0 |
| 10 | No LiOMe | 58/8 |
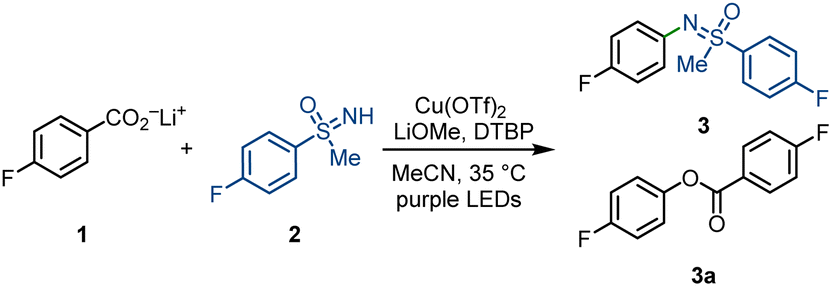
Subsequently, we next studied the substrate scope of the decarboxylative sulfoximination (Fig. 2). Electron-deficient (4, 5, 9, 17), electron-neutral (3, 7, 10) and electron-rich (6, 11, 16) benzoic acids underwent smooth decarboxylative sulfoximination to afford their corresponding N-arylated sulfoximines in moderate to good yields. Owing to the high oxidative potential, radical decarboxylation of electron-deficient benzoic acids is generally problematic,27 however, performed well under our present reaction condition. Ortho-fluoro-substituted benzoic acid (10) gave a moderate yield; yet, benzoic acids with large ortho-substituents failed to afford productive yields, possibly owing to the insufficient generation of copper(II) carboxylates. Heteroaromatic carboxylic acids such as CF3-substituted isonicotinic acid can also perform efficient decarboxylation to afford the corresponding N-arylated sulfoximine 12. Functional groups including aryl halides (7, 20), ketone (13), heterocycles (8, 12, 19), nitriles (9, 18) and sulfonamides (17, 19) were well tolerated. α-O or –N (11, 14, 17, 20), benzylic (15–17, 20), and tertiary (14) C–H bonds that are sensitive to HAT do not prevent efficient decarboxylative sulfoximination. In addition, strong coordinating or oxidizable functional groups like amines inhibit the transformation. The utility of this decarboxylative sulfoximination was further displayed by the late-stage decarboxylative sulfoximination of several complex small molecules (8, 14, 19, 20). NH-Sulfoximines with electron-rich (29, 30) and electron-neutral (21–24) arenes afforded good yields; however, electron-deficient NH-sulfoximines (25, 27) gave lower yields, possibly due to their weaker N-nucleophilicity. Dialkyl (31) and diaryl (32) NH-sulfoximines furnished their corresponding N-arylated sulfoximines in moderate yields. Benefiting from the mild reaction conditions, N-arylation of enantiopure NH-sulfoximines (21, 22) proceeded in good yields, and no racemization was observed. In most cases, low conversion of the starting benzoate salts accounts for the observed low yield.
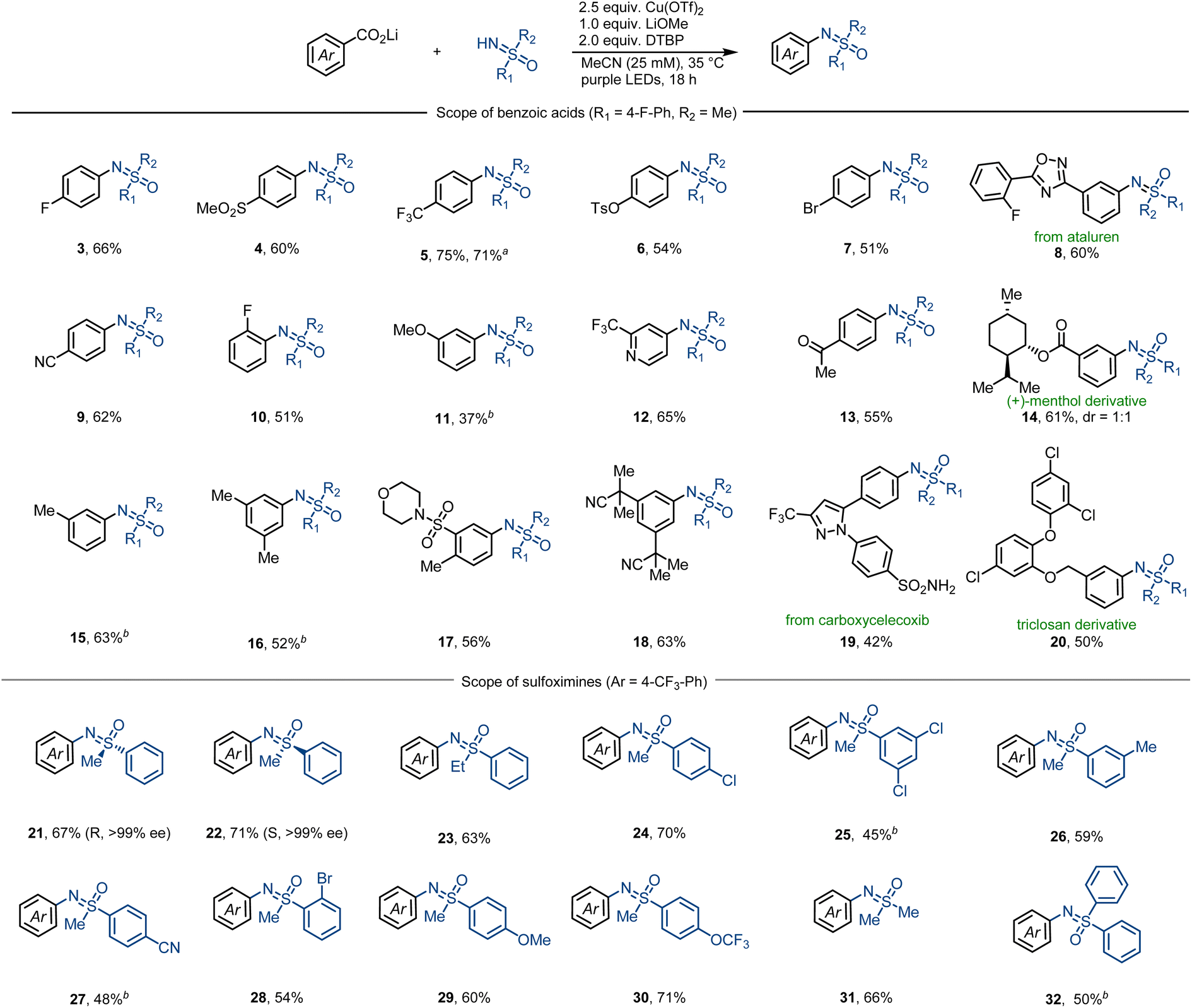 | ||
| Fig. 2 Substrate scope. Standard reaction condition: lithium benzoate (0.20 mmol, 1.0 equiv.), sulfoximine (2.5 equiv.), Cu(OTf)2 (2.5 equiv.), LiOMe (1.0 equiv.), DTBP (2.0 equiv.), MeCN (c = 25 mM), 18 h purple LEDs irradiation, 35 °C. aReaction was performed at 1.0 mmol scale. bReaction condition: lithium benzoate (0.20 mmol, 1.0 equiv.), sulfoximine (2.5 equiv.), Cu(MeCN)4BF4 (2.5 equiv.), 1-fluoro-2,4,6-trimethylpyridinium triflate (2.5 equiv.), DTBP (2.0 equiv.), MeCN (c = 25 mM), 18 h purple LEDs irradiation, 35 °C. DTBP = 2,6-di-tert-butylpyridine. | ||
Preliminary investigations to study the reaction mechanism are consistent with the mechanism shown in Fig. 3. In the UV-vis absorption spectrum of the mixture of lithium 4-fluorobenzoate (1) and Cu(OTf)2, a strong absorbance (370–470 nm) attributed to the LMCT band of copper(II) carboxylates was detected (Fig. 3A).28 The LMCT band overlaps with the purple LED emission spectrum, consistent with the excitation of copper(II) carboxylates under the reaction conditions. The coordination of sulfoximines to copper(II) and the coordination of 2,6-di-tert-butylpyridine (DTBP) to copper(II) are in agreement with the observation of an absorbance (370–470 nm) of a mixture of sulfoximine 2 and Cu(OTf)2, and a mixture of DTBP and Cu(OTf)2 (Fig. 3A). All copper(II)-containing mixtures display a broad d–d transitions absorbance at 550−900 nm, which decreased monotonously upon purple LED irradiation, consistent with the reduction of Cu(II) to Cu(I) (Fig. 3B). The formation of Cu(I) was confirmed by the observation of a characteristic purple [CuI(biq)2]+ complex when 2,2′-biquinoline (biq) was added to the irradiated reaction mixture (see ESI for more detail†).29 Additional experiments were then performed to explore the transformation of carboxylate ions upon irradiation. In radical trapping experiments, aryl carboxyl radical adduct phenyl 3-methoxybenzoate and aryl radical adduct 3-methoxy-1,1′-biphenyl were separated, which indicated the formation of aryl carboxyl and aryl radicals (see ESI for more details†). The presence of aryl carboxyl radical and aryl radical was further confirmed by 6-endo-trig intramolecular radical cyclisation (see ESI for more details†) and radical deuterodecarboxylation (Fig. 3C), respectively. N-Phenyl-sulfoximine, possibly formed by radical addition of sulfoximinyl radical to benzene, was also identified in the radical trapping experiments. We hypothesize that the sulfoximinyl radical is formed via a nitrogen to copper charge transfer in the copper(II) sulfoximine complex, with consumption of the copper(II) species. This result is consistent with competing coordination of sulfoximines to copper(II) and may also explain the low reaction reactivity caused by the competing coordination. Based on the above mechanistic investigation, we propose a mechanism as depicted in Fig. 3D. Photo-induced carboxylate to copper(II) charge transfer in copper(II) carboxylates I affords aryl carboxyl radical intermediates II, which then undergo low-barrier radical decarboxylation to afford aryl radicals III. Subsequent copper-assisted aryl radical capture generates arylcopper(III) intermediates IV that finally undergo C–N reductive elimination to afford N-arylated sulfoximines (Fig. 3D). Additionally, we found that other N-nucleophiles, such as ortho-sulphobenzamide, could also be coupled by the copper-LMCT approach (Fig. 3E).
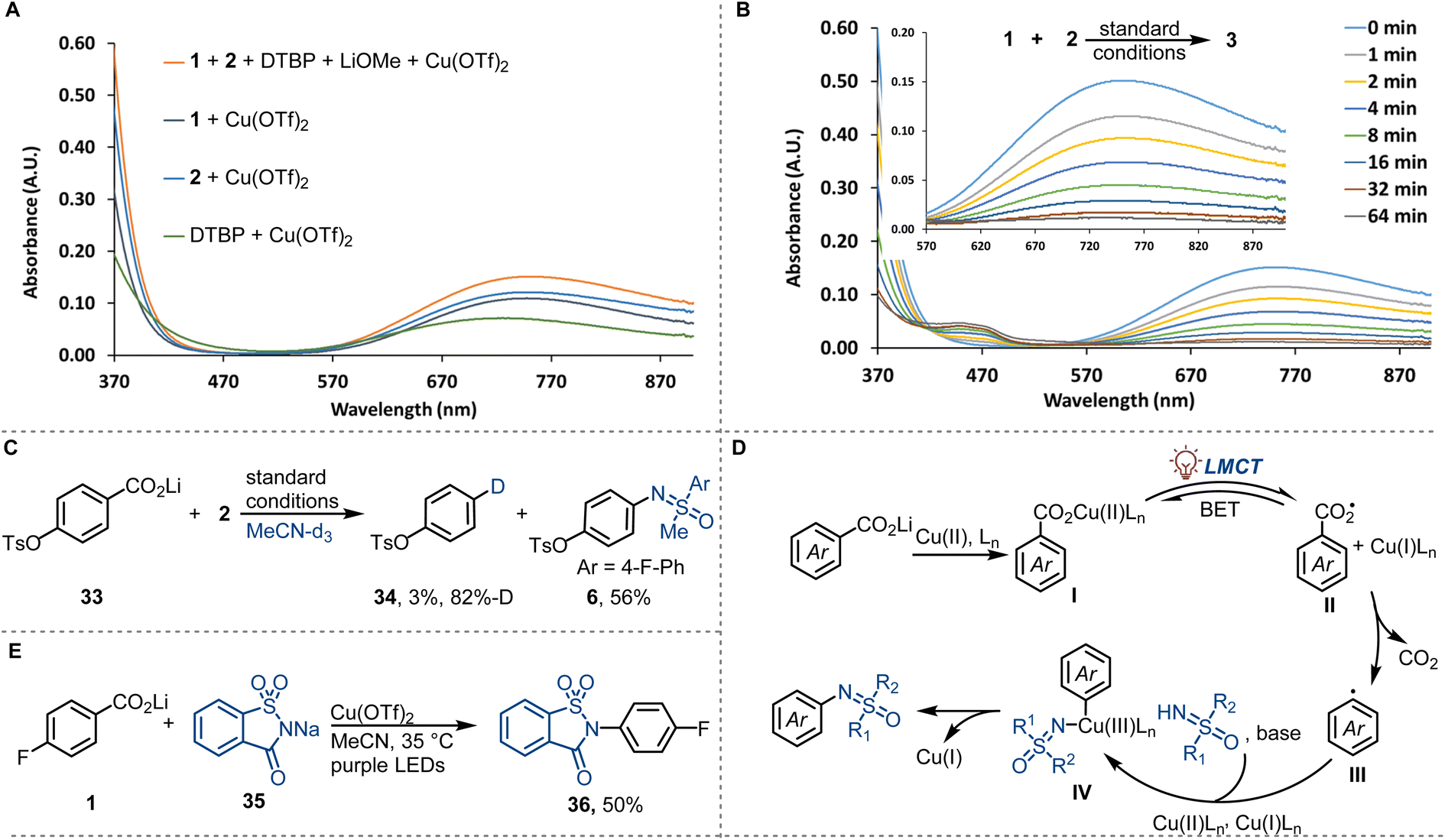 | ||
| Fig. 3 Mechanistic investigations and synthetic application. DTBP = 2,6-di-tert-butylpyridine. (A) UV-vis absorption spectra of reaction components. (B) UV-vis spectral changes observed upon photolysis of a mixture of 1 (1.0 mM), 2 (2.5 mM), Cu(OTf)2 (2.5 mM), LiOMe (1.0 mM), and DTBP (2.0 mM) in MeCN under purple LEDs irradiation (0–64 min). (C) Deuterodecarboxylation. (D) Proposed reaction mechanism. (E) Decarboxylative C–N cross-coupling with sodium saccharin as the N-nucleophile. | ||
Conclusions
Copper-LMCT based radical aromatic decarboxylative carbometalation enabled the first decarboxylative sulfoximination of benzoic acids. The broad substrate scope and good functional group tolerance demonstrate the generality of the copper-LMCT concept in aromatic decarboxylative sulfoximination. Conceptually, the success of this transformation demonstrates the expansion of the copper-LMCT concept for aromatic decarboxylative cross-couplings to reactions with strongly coordinating nucleophiles.Data availability
Procedures and compound characterization are provided in the ESI.†Author contributions
P. X. initiated the project and performed experiments and analyzed the data. W. S. performed and analyzed experiments regarding the mechanism. T. R. directed the project.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank Dr Sun Xiang (MPI KOFO) for helpful discussions.Notes and references
- (a) J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319–15322 CrossRef CAS PubMed; (b) A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed; (c) K. Zhang, L. Chang, Q. An, X. Wang and Z. Zuo, J. Am. Chem. Soc., 2019, 141, 10556–10564 CrossRef CAS PubMed; (d) W. Liu, Q. Wu, M. Wang, Y. Huang and P. Hu, Org. Lett., 2021, 23, 8413–8418 CrossRef CAS PubMed; (e) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed; (f) T. Xue, Z. Zhang and R. Zeng, Org. Lett., 2022, 24, 977–982 CrossRef CAS PubMed; (g) K. Wang and R. Zeng, Org. Chem. Front., 2022, 9, 3692–3696 RSC.
- (a) J. K. Kochi, J. Am. Chem. Soc., 1962, 84, 2121–2127 CrossRef CAS; (b) P. Lian, W. Long, J. Li, Y. Zheng and X. Wan, Angew. Chem., Int. Ed., 2020, 59, 23603–23608 CrossRef CAS PubMed; (c) S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729–2735 CrossRef CAS PubMed; (d) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed; (e) Y. C. Kang, S. M. Treacy and T. Rovis, ACS Catal., 2021, 11, 7442–7449 CrossRef CAS PubMed; (f) Q. Yang, Y.-H. Wang, Y. Qiao, M. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Science, 2021, 372, 847–852 CrossRef CAS PubMed; (g) Y. Jin, Q. Zhang, L. Wang, X. Wang, C. Meng and C. Duan, Green Chem., 2021, 23, 6984–6989 RSC; (h) G. Zhang, Z. Zhang and R. Zeng, Chin. J. Chem., 2021, 39, 3225–3230 CrossRef CAS; (i) C.-C. Wang, G.-X. Zhang, Z.-W. Zuo, R. Zeng, D.-D. Zhai, F. Liu and Z.-J. Shi, Sci. China: Chem., 2021, 64, 1487–1492 CrossRef CAS; (j) M. I. Gonzalez, D. Gygi, Y. Qin, Q. Zhu, E. J. Johnson, Y.-S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2022, 144, 1464–1472 CrossRef CAS PubMed; (k) S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed; (l) M. Yamane, Y. Kanzaki, H. Mitsunuma and M. Kanai, Org. Lett., 2022, 24, 1486–1490 CrossRef CAS PubMed; (m) N. Xiong, Y. Dong, B. Xu, Y. Li and R. Zeng, Org. Lett., 2022, 24, 4766–4771 CrossRef CAS PubMed; (n) Z.-Y. Dai, S.-Q. Zhang, X. Hong, P.-S. Wang and L.-Z. Gong, Chem. Catal., 2022, 2, 1211–1222 CrossRef; (o) L. Ding, K. Niu, Y. Liu and Q. Wang, ChemSusChem, 2022, 15, e202200367 CrossRef CAS PubMed.
- A. Hossain, A. Vidyasagar, C. Eichinger, C. Lankes, J. Phan, J. Rehbein and O. Reiser, Angew. Chem., Int. Ed., 2018, 57, 8288–8292 CrossRef CAS PubMed.
- (a) J. Y. Morimoto and B. A. DeGraff, J. Phys. Chem., 1972, 76, 1387–1388 CrossRef CAS; (b) P. Natarajan and G. Ferraudi, Inorg. Chem., 1981, 20, 3708–3712 CrossRef CAS; (c) Z. Li, X. Wang, S. Xia and J. Jin, Org. Lett., 2019, 21, 4259–4265 CrossRef CAS PubMed; (d) V. R. Yatham, P. Bellotti and B. König, Chem. Commun., 2019, 55, 3489–3492 RSC; (e) Q. Y. Li, S. N. Gockel, G. A. Lutovsky, K. S. DeGlopper, N. J. Baldwin, M. W. Bundesmann, J. W. Tucker, S. W. Bagley and T. P. Yoon, Nat. Chem., 2022, 14, 94–99 CrossRef CAS PubMed; (f) J.-L. Tu, H. Gao, M. Luo, L. Zhao, C. Yang, L. Guo and W. Xia, Green Chem., 2022, 24, 5553–5558 RSC; (g) Y. Zhang, J. Qian, M. Wang, Y. Huang and P. Hu, Org. Lett., 2022, 24, 5972–5976 CrossRef CAS PubMed; (h) S. Singh, N. Dagar and S. R. Roy, Chem. Commun., 2022, 58, 3831–3834 RSC; (i) A. Reichle, H. Sterzel, P. Kreitmeier, R. Fayad, F. N. Castellano, J. Rehbein and O. Reiser, Chem. Commun., 2022, 58, 4456–4459 RSC.
- (a) Y. Abderrazak, A. Bhattacharyya and O. Reiser, Angew. Chem., Int. Ed., 2021, 60, 21100–21115 CrossRef CAS PubMed; (b) F. Juliá, ChemCatChem, 2022, 14, e202200916 CrossRef.
- (a) R. Shang and L. Liu, Sci. China: Chem., 2011, 54, 1670–1687 CrossRef CAS; (b) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (c) J. Cornella and I. Larrosa, Synthesis, 2012, 44, 653–676 CrossRef CAS; (d) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed; (e) T. Patra and D. Maiti, Chem.–Eur. J., 2017, 23, 7382–7401 CrossRef CAS PubMed; (f) J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC; (g) A. Varenikov, E. Shapiro and M. Gandelman, Chem. Rev., 2021, 121, 412–484 CrossRef CAS PubMed.
- J. Chateauneuf, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 1988, 110, 2886–2893 CrossRef CAS.
- J. W. Hilborn and J. A. Pincock, J. Am. Chem. Soc., 1991, 113, 2683–2686 CrossRef CAS.
- R. Qi, C. Wang, Y. Huo, H. Chai, H. Wang, Z. Ma, L. Liu, R. Wang and Z. Xu, J. Am. Chem. Soc., 2021, 143, 12777–12783 CrossRef CAS PubMed.
- Y. Li, K. Zhou, Z. Wen, S. Cao, X. Shen, M. Lei and L. Gong, J. Am. Chem. Soc., 2018, 140, 15850–15858 CrossRef CAS PubMed.
- (a) P. Xu, P. López-Rojas and T. Ritter, J. Am. Chem. Soc., 2021, 143, 5349–5354 CrossRef CAS PubMed; (b) W. Su, P. Xu and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 24012–24017 CrossRef CAS PubMed.
- (a) N. W. Dow, P. S. Pedersen, T. Q. Chen, D. C. Blakemore, A.-M. Dechert-Schmitt, T. Knauber and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6163–6172 CrossRef CAS PubMed; (b) T. Q. Chen, P. S. Pedersen, N. W. Dow, R. Fayad, C. E. Hauke, M. C. Rosko, E. O. Danilov, D. C. Blakemore, A.-M. Dechert-Schmitt, T. Knauber, F. N. Castellano and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 8296–8305 CrossRef CAS PubMed.
- (a) W.-J. Sheng, Q. Ye, W.-B. Yu, R.-R. Liu, M. Xu, J.-R. Gao and Y.-X. Jia, Tetrahedron Lett., 2015, 56, 599–601 CrossRef CAS; (b) M. P. Drapeau, J. Bahri, D. Lichte and L. J. Gooßen, Angew. Chem., Int. Ed., 2019, 58, 892–896 CrossRef PubMed; (c) L. Shen, J. Zhang and J. Xie, Chin. J. Org. Chem., 2019, 39, 1153–1159 CrossRef CAS.
- H. R. Bentley, E. E. McDermott, J. Pace, J. K. Whitehead and T. Moran, Nature, 1949, 163, 675–676 CrossRef CAS PubMed.
- (a) M. Frings, I. Thomé and C. Bolm, Beilstein J. Org. Chem., 2012, 8, 1443–1451 CrossRef CAS PubMed; (b) S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS PubMed; (c) W. Zheng, X. Chen, F. Chen, Z. He and Q. Zeng, Chem. Rec., 2021, 21, 396–416 CrossRef CAS PubMed; (d) M. Andresini, A. Tota, L. Degennaro, J. A. Bull and R. Luisi, Chem.–Eur. J., 2021, 27, 17293–17321 CrossRef CAS PubMed; (e) D. Zeng, Y. Ma, W.-P. Deng, M. Wang and X. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202207100 CAS; (f) D. Zeng, Y. Ma, W.-P. Deng, M. Wang and X. Jiang, Nat. Synth., 2022, 1, 455–463 CrossRef.
- (a) M. Frings, C. Bolm, A. Blum and C. Gnamm, Eur. J. Med. Chem., 2017, 126, 225–245 CrossRef CAS PubMed; (b) P. Mäder and L. Kattner, J. Med. Chem., 2020, 63, 14243–14275 CrossRef PubMed; (c) Y. Han, K. Xing, J. Zhang, T. Tong, Y. Shi, H. Cao, H. Yu, Y. Zhang, D. Liu and L. Zhao, Eur. J. Med. Chem., 2021, 209, 112885–112912 CrossRef CAS PubMed.
- (a) H.-J. Gais, H. Mueller, J. Bund, M. Scommoda, J. Brandt and G. Raabe, J. Am. Chem. Soc., 1995, 117, 2453–2466 CrossRef CAS; (b) M. Nagatomo, S. Yoshioka and M. Inoue, Chem.–Eur. J., 2015, 10, 120–123 CAS; (c) G. Proietti, J. Kuzmin, A. Z. Temerdashev and P. Dinér, J. Org. Chem., 2021, 86, 17119–17128 CrossRef CAS PubMed.
- (a) C. R. Johnson and C. J. Stark Jr., Tetrahedron Lett., 1979, 20, 4713–4716 CrossRef; (b) C. Bolm and O. Simić, J. Am. Chem. Soc., 2001, 123, 3830–3831 CrossRef CAS PubMed; (c) M. Langner and C. Bolm, Angew. Chem., Int. Ed., 2004, 43, 5984–5987 CrossRef CAS PubMed.
- M. Frings, I. Thome and C. Bolm, Beilstein J. Org. Chem., 2012, 8, 1443–1451 CrossRef CAS PubMed.
- (a) T. Zhou, P.-F. Qian, J.-Y. Li, Y.-B. Zhou, H.-C. Li, H.-Y. Chen and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 6810–6816 CrossRef CAS PubMed; (b) Y.-F. Zhang, X.-Y. Dong, J.-T. Cheng, N.-Y. Yang, L.-L. Wang, F.-L. Wang, C. Luan, J. Liu, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, J. Am. Chem. Soc., 2021, 143, 15413–15419 CrossRef CAS PubMed.
- (a) C. Bolm and J. P. Hildebrand, Tetrahedron Lett., 1998, 39, 5731–5734 CrossRef CAS; (b) C. Bolm and J. P. Hildebrand, J. Org. Chem., 2000, 65, 169–175 CrossRef CAS PubMed; (c) G. Y. Cho, P. Rémy, J. Jansson, C. Moessner and C. Bolm, Org. Lett., 2004, 6, 3293–3296 CrossRef CAS PubMed; (d) J. Sedelmeier and C. Bolm, J. Org. Chem., 2005, 70, 6904–6906 CrossRef CAS PubMed; (e) A. Correa and C. Bolm, Adv. Synth. Catal., 2008, 350, 391–394 CrossRef CAS; (f) A. Wimmer and B. König, Org. Lett., 2019, 21, 2740–2744 CrossRef CAS PubMed; (g) D. Liu, Z.-R. Liu, C. Ma, K.-J. Jiao, B. Sun, L. Wei, J. Lefranc, S. Herbert and T.-S. Mei, Angew. Chem., Int. Ed., 2021, 60, 9444–9449 CrossRef CAS PubMed; (h) C. Zhu, A. P. Kale, H. Yue and M. Rueping, JACS Au, 2021, 1, 1057–1065 CrossRef CAS PubMed.
- (a) C. Moessner and C. Bolm, Org. Lett., 2005, 7, 2667–2669 CrossRef CAS PubMed; (b) C. Wang, H. Zhang, L. A. Wells, T. Liu, T. Meng, Q. Liu, P. J. Walsh, M. C. Kozlowski and T. Jia, Nat. Commun., 2021, 12, 932 CrossRef CAS PubMed.
- J. Kim, J. Ok, S. Kim, W. Choi and P. H. Lee, Org. Lett., 2014, 16, 4602–4605 CrossRef CAS PubMed.
- H. Zhu, F. Teng, C. Pan, J. Cheng and J.-T. Yu, Tetrahedron Lett., 2016, 57, 2372–2374 CrossRef CAS.
- Y. Jiang, Y. You, W. Dong, Z. Peng, Y. Zhang and D. An, J. Org. Chem., 2017, 82, 5810–5818 CrossRef CAS PubMed.
- W. Dong, C. Liu, X. Ma, Y. Zhang, Z. Peng, D. Xie and D. An, Tetrahedron, 2019, 75, 3886–3893 CrossRef CAS.
- (a) L. Candish, M. Freitag, T. Gensch and F. Glorius, Chem. Sci., 2017, 8, 3618–3622 RSC; (b) S. Kubosaki, H. Takeuchi, Y. Iwata, Y. Tanaka, K. Osaka, M. Yamawaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2020, 85, 5362–5369 CrossRef CAS PubMed.
- R. Cao, Q. Shi, D. Sun, M. Hong, W. Bi and Y. Zhao, Inorg. Chem., 2002, 41, 6161–6168 CrossRef CAS PubMed.
- (a) H. Kunkely and A. Vogler, Inorg. Chim. Acta, 2004, 357, 888–890 CrossRef CAS; (b) N. Ichiishi, A. J. Canty, B. F. Yates and M. S. Sanford, Organometallics, 2014, 33, 5525–5534 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05442f |
| This journal is © The Royal Society of Chemistry 2022 |