
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A room-temperature antiferroelectric in hybrid perovskite enables highly efficient energy storage at low electric fields†
Yi
Liu
a,
Haojie
Xu
ab,
Xitao
Liu
 a,
Shiguo
Han
a,
Wuqian
Guo
ab,
Yu
Ma
ab,
Qingshun
Fan
ab,
Xinxin
Hu
ab,
Zhihua
Sun
*abc and
Junhua
Luo
*ab
a,
Shiguo
Han
a,
Wuqian
Guo
ab,
Yu
Ma
ab,
Qingshun
Fan
ab,
Xinxin
Hu
ab,
Zhihua
Sun
*abc and
Junhua
Luo
*ab
aState Key Laboratory of Structure Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, P. R. China. E-mail: sunzhihua@fjirsm.ac.cn; jhluo@fjirsm.ac.cn
bUniversity of Chinese Academy of Sciences Beijing, 100049, P. R. China
cFujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou, Fujian 350108, P. R. China
First published on 28th October 2022
Abstract
Molecular antiferroelectrics (AFEs) have taken a booming position in the miniaturization of energy storage devices due to their low critical electric fields. However, regarding intrinsic competitions between dipolar interaction and steric hindrance, it is a challenge to exploit room-temperature molecular AFEs with high energy storage efficiency. Here, we present a new 2D hybrid perovskite-type AFE, (i-BA)2(FA)Pb2Br7 (1), which shows ultrahigh energy storage efficiencies at room temperature. Most strikingly, the typical double P–E hysteresis loops afford an ultrahigh storage efficiency up to ∼91% at low critical electric fields (Ecr = 41 kV cm−1); this Ecr value is much lower than those of state-of-the-art AFE oxides, revealing the potential of 1 for miniaturized energy-storage devices. In terms of the energy storage mechanism, the dynamic ordering and antiparallel reorientation of organic cations trigger its AFE-type phase transition at 303 K, thus giving a large spontaneous electric polarization of ∼3.7 μC cm−2, while the increasement of steric hindrance of the organic branched-chain i-BA+ spacer cations stabilizes its antipolar sublattices. To the best of our knowledge, this exploration of achieving ultrahigh energy storage efficiency at such a low critical electric field is unprecedented in the AFE family, which paves a pathway for miniaturized energy storage applications.
Introduction
Solid-state energy storage materials have recently attracted increasing attention, due to the highly urgent demand for sustainable energy sources over the past decade.1–3 Conceptually, antiferroelectric (AFE) materials, characterized by the antiparallel arrangement of adjacent sublattice polarizations,4,5 are emerging as a remarkable candidate for efficient energy-storage applications.6 Benefiting from their typical polarization versus electric field (P–E) double hysteresis loops,7 the unique field-induced AFE-to-ferroelectric (FE) structural transition leads to prominent energy-storage performances,8 including superior energy density, fast charge–discharge rate, and excellent fatigue endurance. These advantages are far beyond linear dielectric and FE materials, as demonstrated by AFE oxides that possess high critical fields (Ecr), such as AgNbO3 (Ecr ∼150 kV cm−1), PbHfO3 (Ecr ∼270 kV cm−1), and PbZrO3 (Ecr ∼463 kV cm−1).9–11 By contrast, molecule-based soft AFEs with intrinsic merits of structural flexibility, facile processability and low operating voltage, are booming as alternatives for energy storage.12–16 It is emphasized that their low Ecr guarantees the possibility of high-level integration into miniaturized electronic devices. For most of the known molecular AFEs, however, an obvious drawback is their low energy-storage efficiency (η), such as TFMBI (Ecr ∼22 kV cm−1, η ∼ 44%) and TCMBI (Ecr ∼81 kV cm−1, η ∼ 62%).17,18 This is ascribed to the large hysteresis (ΔE) between forward and backward switching in the P–E loops. The low η value signifies high energy dissipation that transforms into thermal energy, thus drastically degrading the storage capability. Besides, the optimal η and lower Ecr values usually emerge near the Curie temperature (Tc) for the AFE-to-paraelectric phase transition, for molecular AFEs such as NH4H2PO4 (Tc = 148 K),19 2-difluoromethylbenzimidazole (Tc > 413 K)13 and cyclohexylmethylammonium bromide (Tc = 364 K),20 thus hindering their potential applications. In this context, it is urgently necessary to assemble new room-temperature AFE candidates that enable high energy-storage efficiency under low electric fields.Recently, the family of 2D Ruddlesden–Popper hybrid perovskites, showing rich and intriguing physicochemical properties,21–24 have provided new entries into diverse applications, such as light-emitting diodes,25,26 solar cells,27,28 lasers,29–31 and photodetectors.32–35 The most fascinating virtue of these 2D members is the breakthrough of the structural limitation of the tolerance factor, which permits an infinite possibility for incorporating bulky spacer cations between inorganic sheets.36 Dynamic motions or orientations of organic spacers are controllable by applying an external electric field; this will greatly facilitate the generation of molecular dipoles and electric order. Previous studies have concentrated on the mixed-cation alloying of flexible moieties to assemble new FE materials, such as (4,4-difluorohexahydroazepine)2PbI4,37 (isoamylammonium)2(EA)2Pb3Br10,38 and (4-aminomethyl-1-cyclohexanecarboxylate)2(EA)2Pb3Br10.39 Contrary to the robust advance of FEs, however, antiferroelectricity in this 2D hybrid perovskite system is still a virgin land with bright prospects. Regarding the antipolar structural and free energy criteria for AFEs, the incorporation of branched-chain organic cations allows a dramatic increasement of steric hindrance in a confined space. Hence, the repulsive interactions caused by the steric hindrance effect would stabilize the antipolar array of dipoles, i.e., the formation of the AFE structure. As verified by organic molecular AFEs, the steric hindrance of organic moieties reduces the distance between adjacent dipoles, thereby creating antipolar motifs.40–42 Despite continued efforts, it remains a challenge to explore room-temperature molecular AFEs in this 2D family, owing to the lack of knowledge regarding the competition balance between steric hindrance and dipolar interactions in homogeneous sublattices.
Here, we have successfully achieved a room-temperature molecular AFE with ultrahigh energy storage efficiency by alloying the branched-chain cation as an interlayered spacer of 2D multilayered hybrid perovskites, (i-BA)2(FA)Pb2Br7 (1, where i-BA+ is iso-butylammonium, and FA+ is formamidinium). Strikingly, the characteristic double P–E hysteresis loops enable ultrahigh energy-storage efficiency of ∼91% under low Ecr about 41 kV cm−1. Such merits reveal its great potential for miniaturized energy-storage devices. The antiparallel reorientations of i-BA+ and FA+ cations lead to its room-temperature antiferroelectricity with a Curie point (Tc) of 303 K, while the increasement of steric hindrance of i-BA+ spacer cations plays an important role in stabilizing its antipolar AFE motif. As far as we know, 1 is the first molecular antiferroelectric to achieve ultrahigh energy storage efficiency at such a low critical electric field, shedding light on exploration of new AFE candidates toward practical applications, e.g., high energy-storage capacitors.
Results and discussion
Variable-temperature structure analyses
Bulk crystals of 1 were prepared from its HBr solution by the slow temperature-cooling method (Fig. S1†), and the purity of the phase was verified by using powder X-ray diffraction (Fig. S2†). Variable-temperature crystal structures of 1 at 200 K (low-temperature phase, LTP) and 310 K (high-temperature phase, HTP) were determined by X-ray diffraction to understand the mechanism of phase transition and the origin of its antiferroelectricity. In the LTP, the structure of 1 is found to be in the orthorhombic space group Pnma (Table S1†). As depicted in Fig. 1a, the basic crystal architectures consist of two perovskite slabs of PbBr6 octahedra, separated by a thickness of two i-BA+ organic cations. A highly contorted perovskite framework verified by the unequal Br–Pb–Br bond angles (84.7–98.4°) allows for the alloying of the FA+ cation into perovskite cavities. For the organic components, the FA+ and i-BA+ cations are ordered and the NH3+ groups in them are antiparallelly aligned toward the a-axis utilizing the N–H⋯Br hydrogen bond interaction with the inorganic framework (Fig. S3†). Such an antiparallel configuration of neighboring dipoles results in an antipolar state (Ps = 0), which reveals the probable antiferroelectricity of 1.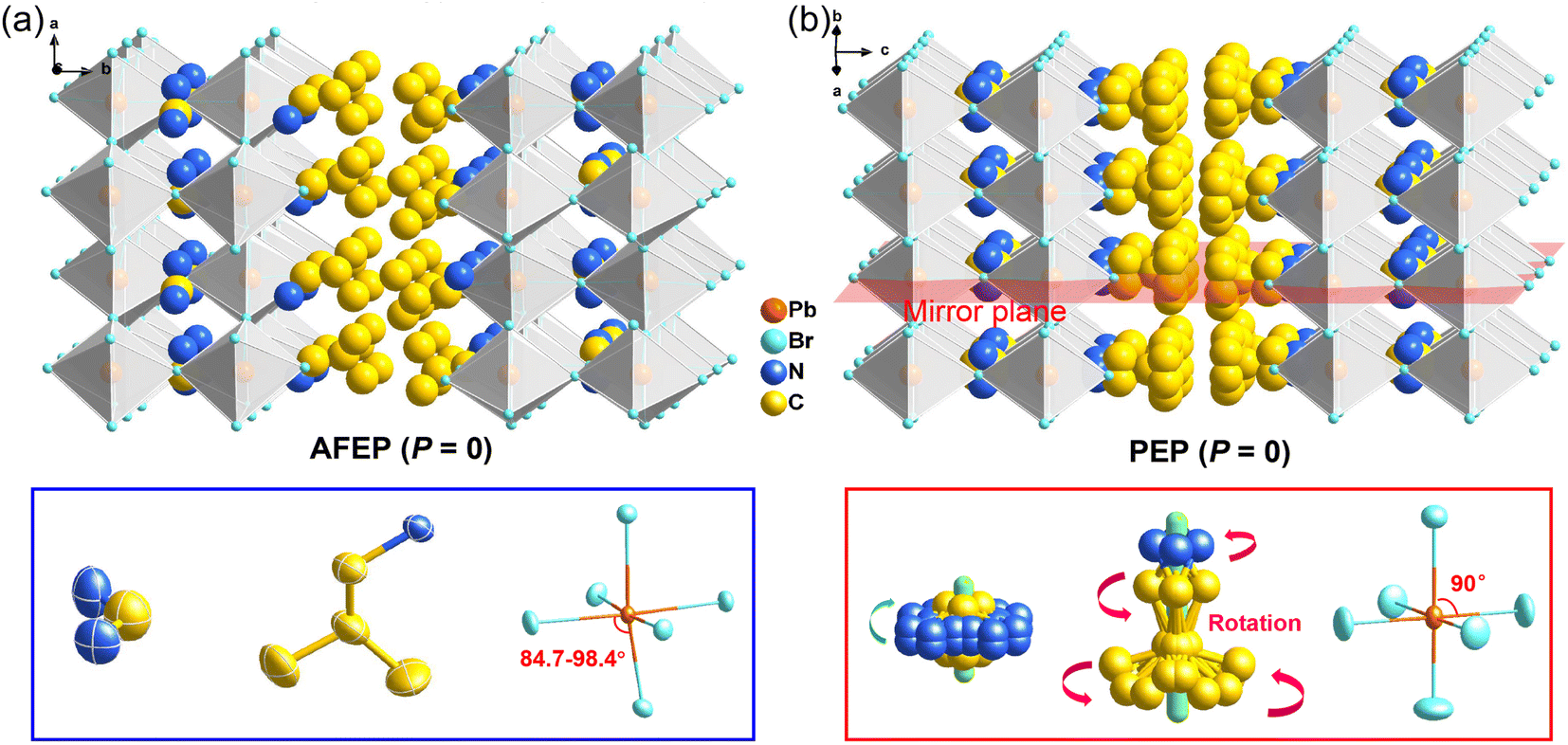 | ||
| Fig. 1 Crystal structure packing diagrams of 1 in the (a) LTP (left) at 200 K and (b) HTP (right) at 310 K. Upon heating, it is obvious that i-BA+ and FA+ cations become disordered with the dynamic reorientation, while the distorted PbBr6 octahedra return to be regular. | ||
At the HTP, 1 crystallizes in the tetragonal centrosymmetric space group I4/mmm. In terms of polarization, the characteristic of the HTP is that both i-BA+ and FA+ cations are located on a crystallographic mirror plane with the completely disordered patterns, thus cancelling the net polarization of the crystal (Fig. 1b). The rotational motion of these organic cations is easily activated at around room temperature, indicating the quite small potential energy barrier of 1, which can be conducive to the generation of molecular dipoles and electric order controlled by applying a low external electric field. Meanwhile, the distorted inorganic PbBr6 octahedra translate to the highly symmetric configurations. This evolution is driven by both their rotational motion around their respective centers with the rotation axis parallel to the b direction and anti-parallel tilting in adjacent inorganic layers (Fig. 2). Therefore, it is the thermally induced order–disorder transition of organic cations and dynamic distortion of inorganic PbBr6 octahedra that synergistically result in the room-temperature antiferroelectric-to-paraelectric phase transition behavior of 1 under low electric fields.
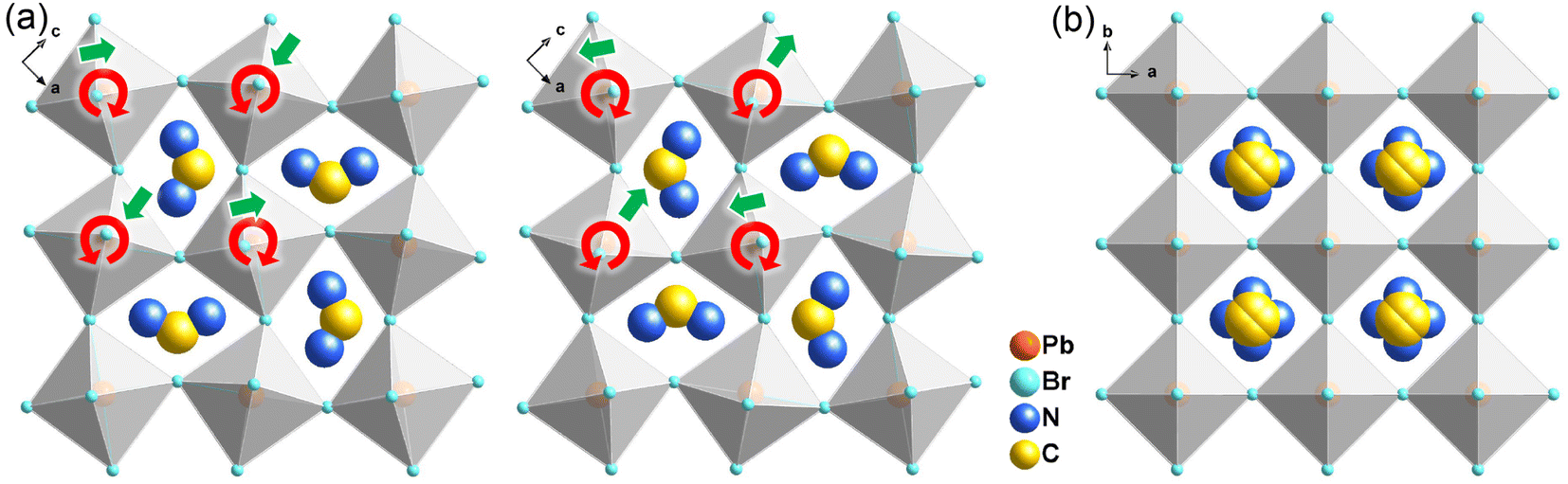 | ||
| Fig. 2 Schematic view of the rotational and tilted motions of PbBr6 octahedra between the (a) LTP and (b) HTP of 1. Red and green arrows represent the directions of rotation and tilting, respectively. | ||
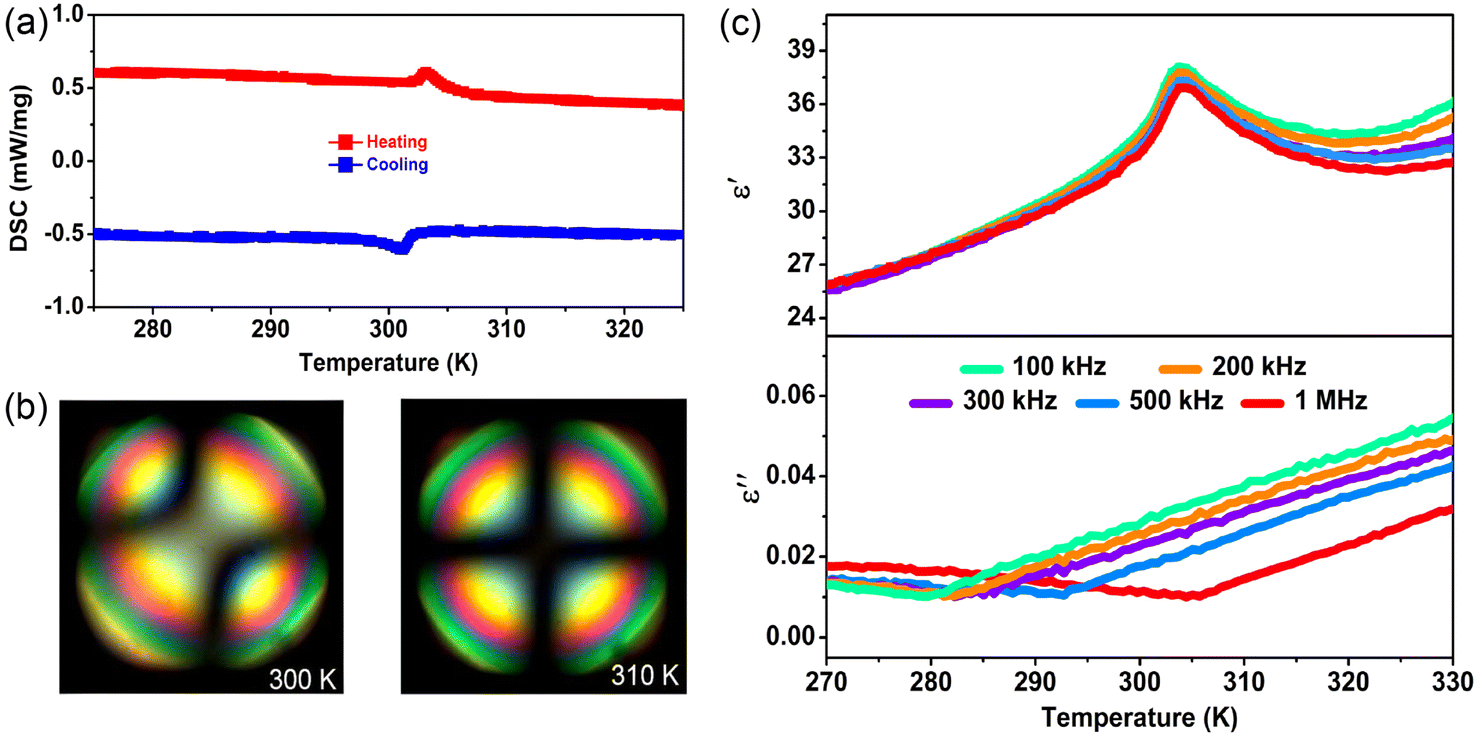 | ||
| Fig. 3 Phase transition properties of 1. (a) DSC curves of 1 measured in the heating and cooling runs. (b) Conoscopic images of the biaxial and uniaxial nematic phases at 300 and 310 K, respectively. (c) The real part (ε′) and imaginary part (ε′′) of the complex dielectric constant measured along the crystallographic c-axis direction at different frequencies upon heating. | ||
 | ||
| Fig. 4 The AFE properties of 1. (a) The P–E hysteresis loops and (b) corresponding J–E curves. (c) The fatigue endurance of 1 after ∼2 × 105 switching cycles under low field and room temperature conditions. | ||
 | ||
| Fig. 5 (a) EF, EA, and ΔE obtained at different temperatures. (b) Temperature dependence of energy density Wrec and storage efficiency η. (c) Energy storage efficiency (η) for various reported antiferroelectrics at room temperature. | ||
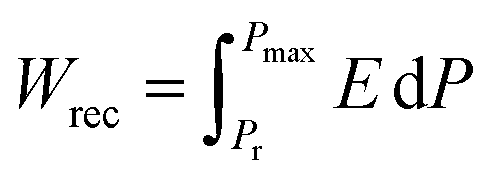 , where E is the applied electric field, Pr and Pmax are the remnant polarization and maximum polarization, respectively, and Wloss represents energy loss (green areas in Fig. S5†). At room temperature, the η and Wrec values are estimated to be ∼89% and 0.13 J cm−3 from the double P–E hysteresis loop. On increasing the temperature, a satisfactory thermal stability has been confirmed, with the variations of both Wrec and η being less than 5%. It is fascinating that the temperature dependence of η shows a gradual increase up to ∼91% at 302 K, due to the decrease of Wloss caused by the shrinking of P–E loops (Fig. 5b). From the perspective of practical application, the frequency dependence of energy storage performances is also of great significance. With the frequency decreasing from 100 to 40 Hz (at 302 K, 41 kV cm−1), the Pmax value slightly decreases, while the ΔE increases (Fig. S4†). This indicates the energy dissipation of 1 remains at a low level, thus accounting for the stability of Wrec and η (Fig. S6†). As far as we know, this energy storage efficiency is even higher than that of the majority of inorganic AFE ceramics, such as Hf0.3Zr0.7O2 (η ∼ 50%),50 PHS-0.075 (η ∼ 79%),51 and PLHT-0.05 (η ∼ 89%) (Fig. 5c and Table S5†).52 The high energy storage efficiency signifies the low energy dissipation, which will less easily transform into thermal energy, thus drastically promoting its energy storage capacity. Such an ultrahigh energy efficiency at room temperature, together with the low critical fields, demonstrates the great potential of 1 for the application of energy storage with high practicability. In terms of improving the Wrec, the chemical design of organic cations, such as H/D substitution, plays an important role in increasing the Tc and the Ps of molecular ferroelectrics, which provides a hint for the realization of high-performance molecular antiferroelectric candidates.53 Besides, ion doping, including halogen anions or skeleton cations, may be an effective approach to increase the Wrec due to the introduction of relaxor characteristics.45
, where E is the applied electric field, Pr and Pmax are the remnant polarization and maximum polarization, respectively, and Wloss represents energy loss (green areas in Fig. S5†). At room temperature, the η and Wrec values are estimated to be ∼89% and 0.13 J cm−3 from the double P–E hysteresis loop. On increasing the temperature, a satisfactory thermal stability has been confirmed, with the variations of both Wrec and η being less than 5%. It is fascinating that the temperature dependence of η shows a gradual increase up to ∼91% at 302 K, due to the decrease of Wloss caused by the shrinking of P–E loops (Fig. 5b). From the perspective of practical application, the frequency dependence of energy storage performances is also of great significance. With the frequency decreasing from 100 to 40 Hz (at 302 K, 41 kV cm−1), the Pmax value slightly decreases, while the ΔE increases (Fig. S4†). This indicates the energy dissipation of 1 remains at a low level, thus accounting for the stability of Wrec and η (Fig. S6†). As far as we know, this energy storage efficiency is even higher than that of the majority of inorganic AFE ceramics, such as Hf0.3Zr0.7O2 (η ∼ 50%),50 PHS-0.075 (η ∼ 79%),51 and PLHT-0.05 (η ∼ 89%) (Fig. 5c and Table S5†).52 The high energy storage efficiency signifies the low energy dissipation, which will less easily transform into thermal energy, thus drastically promoting its energy storage capacity. Such an ultrahigh energy efficiency at room temperature, together with the low critical fields, demonstrates the great potential of 1 for the application of energy storage with high practicability. In terms of improving the Wrec, the chemical design of organic cations, such as H/D substitution, plays an important role in increasing the Tc and the Ps of molecular ferroelectrics, which provides a hint for the realization of high-performance molecular antiferroelectric candidates.53 Besides, ion doping, including halogen anions or skeleton cations, may be an effective approach to increase the Wrec due to the introduction of relaxor characteristics.45
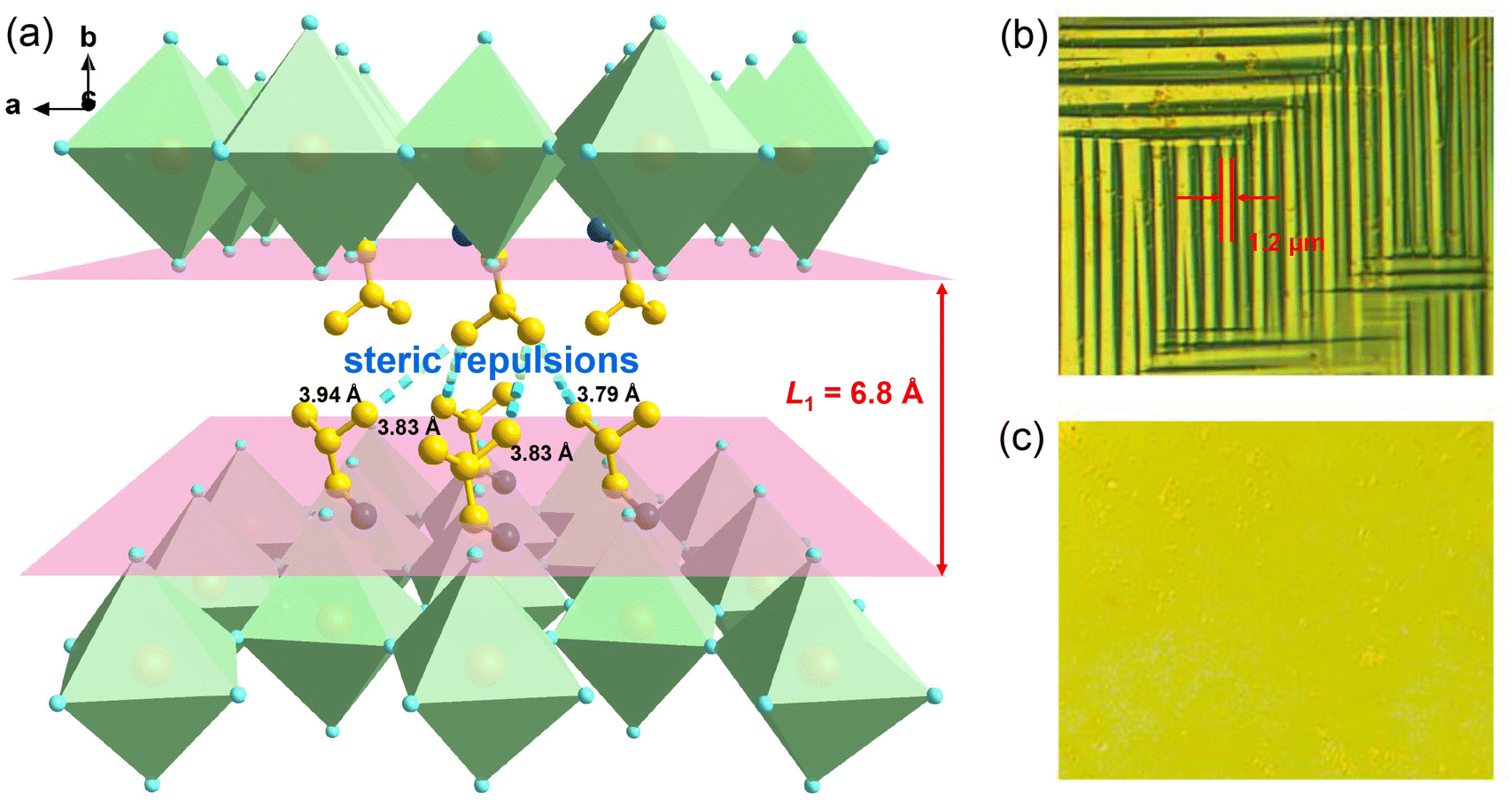 | ||
| Fig. 6 (a) The interlayered space and atomic distance between the adjacent layers of organic i-BA+ cations in 1. Time evolution of domain patterns observed at different temperatures: (b) 300 K and (c) 310 K. | ||
Further, we employed polarized light microscopy techniques to investigate the domain structure of 1, in order to better understand the microscopic origin of its excellent energy storage performance. Under perpendicular polarized light, the domains of different orientations possess different birefringence properties, thus exhibiting dark and bright patterns. As depicted in Fig. 6b and c, the crystalline sheets of 1 exhibit two types of periodic stripe domain walls with an average width ≈1.2 μm in the LTP, which are almost perpendicular to each other. In general, such domains of micrometer scale decrease the polarization hysteresis between the AFE–FE and FE–AFE phase transitions owing to the fast response of microscale domains to the applied electric field, induced by the weak interatomic interactions between the microdomain clusters.46,54–56 Consequently, a slim hysteresis loop with high energy storage efficiency can be acquired by regulating the domain size to the micrometer scale in AFEs. As the temperature increases to the HTP, the domain structures slowly disappear, corresponding to its paraelectric state. Subsequently, as the temperature cools down to the LTP, the crystalline sheet transforms back into the ferroelastic phase with the reconstructed domain structures. Such a temperature dependence of the domain patterns also coincides well with the AFE-to-paraelectric phase transition of 1. Furthermore, it is known that the formation of solid solutions with relaxor compounds is an efficiency-enhancement strategy for antiferroelectric ceramics with relaxor characteristics, as demonstrated in some inorganic antiferroelectric counterparts. The lattice distortion and reduction in the grain size are also observed for relaxor compound additions, which will lead to changes in the domain structure and size. It is proposed that nanosized domains characteristic of relaxor-antiferroelectrics can be achieved in 2D hybrid perovskite materials through constructing solid solutions using halogen doping methods.46,53
Conclusions
In summary, we have successfully designed a new 2D hybrid perovskite-type molecular AFE, (i-BA)2(FA)Pb2Br7, which demonstrates an ultrahigh energy storage efficiency under low electric fields at room temperature. Structurally, the dynamic ordering and antiparallel reorientation of organic dipoles induce its AFE-type phase transition at 303 K along with a large Ps of ∼3.7 μC cm−2. It is noteworthy that the antipolar sublattices of 1 have been stabilized through raising steric hindrance of organic i-BA+ spacer cations. Most strikingly, the characteristic double hysteresis loops of 1 allow a high energy storage efficiency ∼91% at a quite low Ecr ∼41 kV cm−1. This Ecr value is much lower than those of state-of-the-art AFE oxides, revealing its great potential for the miniaturization of energy-storage devices. This work sheds light on exploration of new AFE candidates toward practical applications.Author contributions
Y. L. prepared the samples, measured the antiferroelectric properties, and wrote the manuscript. H. J. X., X. T. L., and S. G. H. determined the structures. W. Q. G., Y. M., Q. S. F., and X. X. H. provided suggestions for research. Z. H. S. and J. H. L. designed and directed the studies. All authors contributed to writing and reviewing the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by NSFC (22125110, 21875251, 21833010, 22205233, 22193042, 22122507, 21921001, and U21A2069), the Key Research Program of Frontier Sciences of the Chinese Academy of Sciences (ZDBS-LY-SLH024), the Natural Science Foundation of Fujian Province (2021J01523 and 2020J01112), Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China (2021ZR126), the Youth Innovation Promotion of CAS (2019301, Y202069, and 2020307), the National Postdoctoral Program for Innovative Talents (BX2021315), the National Key Research and Development Program of China (2019YFA0210402), and China Postdoctoral Science Fund (2022TQ0337).Notes and references
- K. J. Griffith, K. M. Wiaderek, G. Cibin, L. E. Marbella and C. P. Grey, Nature, 2018, 559, 556–563 CrossRef CAS PubMed.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 CrossRef CAS PubMed.
- E. Pomerantseva and Y. Gogotsi, Nat. Energy, 2017, 2, 17089 CrossRef CAS.
- C. Kittel, Phys. Rev., 1951, 82, 729–732 CrossRef CAS.
- R. Blinc and B. Zeks, Soft Mode in Ferroelectrics and Antiferroelectrics, North-Holland, 1974 Search PubMed.
- Z. Liu, T. Lu, J. Ye, G. Wang, X. Dong, R. Withers and Y. Liu, Adv. Mater. Technol., 2018, 3, 1800111 CrossRef.
- P. Tolédano and M. Guennou, Phys. Rev. B, 2016, 94, 014107 CrossRef.
- X. Hao, J. Adv. Dielectr., 2013, 3, 1330001 CrossRef.
- J. Qi, N. Ma and Y. Yang, Adv. Mater. Interfaces, 2018, 5, 1701189 CrossRef.
- J. Wei, T. Yang and H. Wang, J. Eur. Ceram. Soc., 2019, 39, 624–630 CrossRef CAS.
- N. Ma and Y. Yang, Nano Energy, 2017, 40, 352–359 CrossRef CAS.
- S. Horiuchi and S. Ishibashi, J. Phys. Soc. Jpn., 2020, 89, 051009 CrossRef.
- S. Horiuchi, R. Kumai and S. Ishibashi, Chem. Sci., 2018, 9, 425–432 RSC.
- Z. Wu, X. Liu, C. Ji, L. Li, S. Wang, Y. Peng, K. Tao, Z. Sun, M. Hong and J. Luo, J. Am. Chem. Soc., 2019, 141, 3812–3816 CrossRef CAS PubMed.
- S. Han, X. Liu, Y. Liu, Z. Xu, Y. Li, M. Hong, J. Luo and Z. Sun, J. Am. Chem. Soc., 2019, 141, 12470–12474 CrossRef CAS PubMed.
- P.-F. Li, W.-Q. Liao, Y.-Y. Tang, H.-Y. Ye, Y. Zhang and R.-G. Xiong, J. Am. Chem. Soc., 2017, 139, 8752–8757 CrossRef CAS PubMed.
- S. Horiuchi and S. Ishibashi, Chem. Sci., 2021, 12, 14198–14206 RSC.
- S. Horiuchi, F. Kagawa, K. Hatahara, K. Kobayashi, R. Kumai, Y. Murakami and Y. Tokura, Nat. Commun., 2012, 3, 1308 CrossRef PubMed.
- T. Fukami, S. Akahoshi, K. Hukuda and T. Yagi, J. Phys. Soc. Jpn., 1989, 58, 3429–3430 CrossRef CAS.
- H. Xu, W. Guo, J. Wang, Y. Ma, S. Han, Y. Liu, L. Lu, X. Pan, J. Luo and Z. Sun, J. Am. Chem. Soc., 2021, 143, 14379–14385 CrossRef CAS PubMed.
- J. N. Wilson, J. M. Frost, S. K. Wallace and A. Walsh, APL Mater., 2019, 7, 010901 CrossRef.
- G. Grancini, A. R. S. Kandada, J. M. Frost, A. J. Barker, M. D. Bastiani, M. Gandini, S. Marras, G. Lanzani, A. Walsh and A. Petrozza, Nat. Photonics, 2015, 9, 695–701 CrossRef CAS PubMed.
- A. Walsh, D. O. Scanlon, S. Chen, X. G. Gong and S.-H. Wei, Angew. Chem., 2015, 127, 1811–1814 CrossRef.
- L. Dou, A. B. Wong, Y. Yu, M. Lai, N. Kornienko, S. W. Eaton, A. Fu, C. G. Bischak, J. Ma, T. Ding, N. S. Ginsberg, L.-W. Wang, A. P. Alivisatos and P. Yang, Science, 2015, 349, 1518–1521 CrossRef CAS PubMed.
- J. Byun, H. Cho, C. Wolf, M. Jang, A. Sadhanala, R. H. Friend, H. Yang and T.-W. Lee, Adv. Mater., 2016, 28, 7515–7520 CrossRef CAS PubMed.
- H. Tsai, C. Liu, E. Kinigstein, M. Li, S. Tretiak, M. Cotlet, X. Ma, X. Zhang and W. Nie, Adv. Sci., 2020, 7, 1903202 CrossRef CAS PubMed.
- G. Grancini, C. Roldán-Carmona, I. Zimmermann, E. Mosconi, X. Lee, D. Martineau, S. Narbey, F. Oswald, F. De Angelis, M. Graetzel and M. K. Nazeeruddin, Nat. Commun., 2017, 8, 15684 CrossRef CAS PubMed.
- T. Zhou, H. Lai, T. Liu, D. Lu, X. Wan, X. Zhang, Y. Liu and Y. Chen, Adv. Mater., 2019, 31, 1901242 CrossRef PubMed.
- C. Qin, A. S. D. Sandanayaka, C. Zhao, T. Matsushima, D. Zhang, T. Fujihara and C. Adachi, Nature, 2020, 585, 53–57 CrossRef CAS PubMed.
- L. Lei, D. Seyitliyev, S. Stuard, J. Mendes, Q. Dong, X. Fu, Y.-A. Chen, S. He, X. Yi, L. Zhu, C.-H. Chang, H. Ade, K. Gundogdu and F. So, Adv. Mater., 2020, 32, 1906571 CrossRef CAS PubMed.
- S. W. Eatona, M. Laia, N. A. Gibson, A. B. Wong, L. Dou, J. Ma, L.-W. Wang, S. R. Leone and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8 CrossRef PubMed.
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 1171–1190 CrossRef CAS PubMed.
- C. Katan, N. Mercier and J. Even, Chem. Rev., 2019, 119, 3140–3192 CrossRef CAS PubMed.
- Y. Liu, Z. Wu, X. Liu, S. Han, Y. Li, T. Yang, Y. Ma, M. Hong, J. Luo and Z. Sun, Adv. Opt. Mater., 2019, 7, 1901049 CrossRef CAS.
- Y. Liu, J. Wang, S. Han, X. Liu, M. Li, Z. Xu, W. Guo, M. Hong, J. Luo and Z. Sun, Chem.–Eur. J., 2020, 26, 3494–3498 CrossRef CAS PubMed.
- J. Hu, L. Yan and W. You, Adv. Mater., 2018, 30, 1802041 CrossRef PubMed.
- X.-G. Chen, X.-J. Song, Z.-X. Zhang, H.-Y. Zhang, Q. Pan, J. Yao, Y.-M. You and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 10212–10218 CrossRef CAS PubMed.
- Y. Ma, J. Wang, W. Guo, S. Han, J. Xu, Y. Liu, L. Lu, Z. Xie, J. Luo and Z. Sun, Adv. Funct. Mater., 2021, 31, 2103012 CrossRef CAS.
- Y. Liu, X. Pan, X. Liu, S. Han, J. Wang, L. Lu, H. Xu, Z. Sun and J. Luo, Small, 2022, 18, 2106888 CrossRef CAS PubMed.
- K. Takaea and H. Tanaka, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9917–9922 CrossRef PubMed.
- J. M. Luttinger and L. Tisza, Phys. Rev., 1946, 70, 954–964 CrossRef CAS.
- C. S. Zehe, J. A. Hill, N. P. Funnell, K. Kreger, K. P. van der Zwan, A. L. Goodwin, H.-W. Schmidt and J. Senker, Angew. Chem., Int. Ed., 2017, 56, 4432–4437 CrossRef CAS PubMed.
- V. L. Gurevich and A. K. Tagantsev, Adv. Phys., 1991, 40, 719–767 CrossRef CAS.
- Z. Yao, Z. Song, H. Hao, Z. Yu, M. Cao, S. Zhang, M. T. Lanagan and H. Liu, Adv. Mater., 2017, 29, 1601727 CrossRef PubMed.
- F. Li, S. Zhang, D. Damjanovic, L.-Q. Chen and T. R. Shrout, Adv. Funct. Mater., 2018, 28, 1801504 CrossRef.
- J. Li, F. Li, Z. Xu and S. Zhang, Adv. Mater., 2018, 30, 1802155 CrossRef PubMed.
- P. Mohapatra, Z. Fan, J. Cui and X. Tan, J. Eur. Ceram. Soc., 2019, 39, 4735–4742 CrossRef CAS.
- J. Gao, Y. Zhang, L. Zhao, K.-Y. Lee, Q. Liu, A. Studer, M. Hinterstein, S. Zhang and J.-F. Li, J. Mater. Chem. A, 2019, 7, 2225–2232 RSC.
- X. Hao, J. Zhai and X. Yao, J. Am. Ceram. Soc., 2009, 92, 1133–1135 CrossRef CAS.
- M. H. Park, H. J. Kim, Y. J. Kim, T. Moon, K. D. Kim and C. S. Hwang, Adv. Energy Mater., 2014, 4, 1400610 CrossRef.
- P.-Z. Ge, X.-G. Tang, K. Meng, X.-X. Huang, S.-F. Li, Q.-X. Liu and Y.-P. Jiang, Chem. Eng. J., 2022, 429, 132540 CrossRef CAS.
- P.-Z. Ge, X.-G. Tang, K. Meng, X.-X. Huang, Q.-X. Liu, Y.-P. Jiang, W.-P. Gong and T. Wang, Mater. Today Phys., 2022, 24, 100681 CrossRef CAS.
- H.-Y. Liu, H.-Y. Zhang, X.-G. Chen and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 15205–15218 CrossRef CAS PubMed.
- H. Qi, R. Zuo, A. Xie, A. Tian, J. Fu, Y. Zhang and S. Zhang, Adv. Funct. Mater., 2019, 29, 1903877 CrossRef.
- Q. Zhang, H. Tong, J. Chen, Y. Lu, T. Yang, X. Yao and Y. He, Appl. Phys. Lett., 2016, 109, 262901 CrossRef.
- G. Wang, J. Li, X. Zhang, Z. Fan, F. Yang, A. Feteira, D. Zhou, D. C. Sinclair, T. Ma, X. Tan, D. Wang and I. M. Reaney, Energy Environ. Sci., 2019, 12, 582–588 RSC.
- L. Li, X. Shang, S. Wang, N. Dong, C. Ji, X. Chen, S. Zhao, J. Wang, Z. Sun, M. Hong and J. Luo, J. Am. Chem. Soc., 2018, 140, 6806–6809 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2157694 and 2157696. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05285g |
| This journal is © The Royal Society of Chemistry 2022 |