
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceXanthone-based solvatochromic fluorophores for quantifying micropolarity of protein aggregates†
Lushun
Wang‡
 a,
Chia-Heng
Hsiung‡
bc,
Xiaojing
Liu
b,
Shichao
Wang
a,
Axel
Loredo
a,
Xin
Zhang
*bc and
Han
Xiao
*ade
a,
Chia-Heng
Hsiung‡
bc,
Xiaojing
Liu
b,
Shichao
Wang
a,
Axel
Loredo
a,
Xin
Zhang
*bc and
Han
Xiao
*ade
aDepartment of Chemistry, Rice University, 6100 Main Street, Houston, Texas 77005, USA. E-mail: han.xiao@rice.edu
bDepartment of Chemistry, Pennsylvania State University, University Park, PA 16802, USA. E-mail: xuz31@psu.edu
cDepartment of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, PA 16802, USA
dDepartment of Biosciences, Rice University, 6100 Main Street, Houston, Texas 77005, USA
eDepartment of Bioengineering, Rice University, 6100 Main Street, Houston, Texas 77005, USA
First published on 12th October 2022
Abstract
Proper three-dimensional structures are essential for maintaining the functionality of proteins and for avoiding pathological consequences of improper folding. Misfolding and aggregation of proteins have been both associated with neurodegenerative disease. Therefore, a variety of fluorogenic tools that respond to both polarity and viscosity have been developed to detect protein aggregation. However, the rational design of highly sensitive fluorophores that respond solely to polarity has remained elusive. In this work, we demonstrate that electron-withdrawing heteroatoms with (d–p)–π* conjugation can stabilize lowest unoccupied molecular orbital (LUMO) energy levels and promote bathochromic shifts. Guided by computational analyses, we have devised a novel series of xanthone-based solvatochromic fluorophores that have rarely been systematically studied. The resulting probes exhibit superior sensitivity to polarity but are insensitive to viscosity. As proof of concept, we have synthesized protein targeting probes for live-cell confocal imaging intended to quantify the polarity of misfolded and aggregated proteins. Interestingly, our results reveal several layers of protein aggregates in a way that we had not anticipated. First, microenvironments with reduced polarity were validated in the misfolding and aggregation of folded globular proteins. Second, granular aggregates of AgHalo displayed a less polar environment than aggregates formed by folded globular protein represented by Htt–polyQ. Third, our studies reveal that granular protein aggregates formed in response to different types of stressors exhibit significant polarity differences. These results show that the solvatochromic fluorophores solely responsive to polarity represent a new class of indicators that can be widely used for detecting protein aggregation in live cells, thus paving the way for elucidating cellular mechanisms of protein aggregation as well as therapeutic approaches to managing intracellular aggregates.
Introduction
Folding into their correct three-dimensional structures is essential for the ability of proteins to acquire and express their native functions.1,2 Environmental perturbation, genetic mutations, chemical modifications, and aging may disrupt the balance required for normal proteostasis, leading to aberrant protein folding and aggregation. These aberrations result in signaling via the unfolded protein response pathway, as well as in the onset and progression of neurodegenerative diseases like Alzheimer's, Huntington's, and Parkinson's diseases.3–6 Generally, misfolding and aggregation of proteins are accompanied by changes in the polarity of protein complex.7–9 Aberrant polarity is also closely associated with neurodegenerative disease, cancer, and drug-induced tissue injury.10,11 To explore mechanistic details of protein misfolding and aggregation, Liu12–14 and our group15–17 developed a series of fluorogenic tools based on fluorescence activation. These probes respond to changes in both polarity and viscosity during protein aggregation (Fig. 1A, left panel),18–20 which mostly featured properties based on viscosity sensitivity. Increased viscosity in the local microenvironment of the protein aggregate restricts the internal rotation of fluorophores, resulting in fluorescence enhancement (Fig. 1A, left panel). Although useful, these probes lack the ability to detect changes in protein micro-polarity that are independent of viscosity, and thus cannot distinguish between different types of protein aggregates. For more complete understanding of the processes of protein misfolding and aggregation, it is therefore desirable to develop chemical strategies that enable live-cell fluorescence imaging for quantifying the micro-polarity of biomolecules (Fig. 1A, right panel).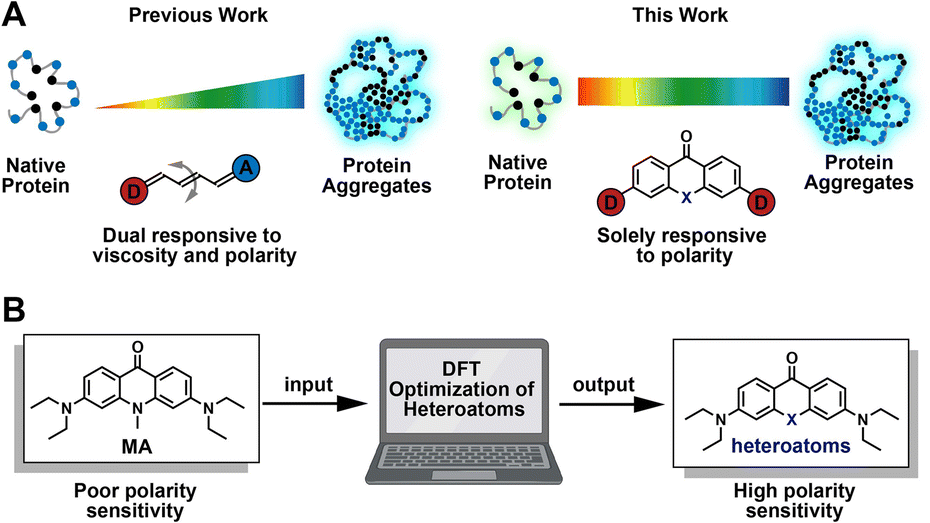 | ||
| Fig. 1 Rational design of solvatochromic fluorophores to detect protein aggregation in live cells. (A) Polymethine dyes had been previously developed to detect protein aggregation based on viscosity and polarity change. In this work, xanthone-based solvatochromic fluorophores with single-atom substitutions have been designed to quantify micropolarity of protein aggregates in live cells. (B) A single-atom replacement strategy based on TD-DFT optimization is employed for designing solvatochromic fluorophores with high sensitivity to protein polarity. | ||
The past several decades have witnessed the exploitation of a variety of “push–pull” based polarity probes,21–27 such as Fluoroprobe, 7-diethylamino-9,9-dimethyl-9H-fluorene-2-carbaldehyde (FR0), prodan, dansyl, 6-N,N-(dimethylamino)-2,3-naphthalimide (6DMN), nitrobenzoxadiazole (NBD), nile red, dapoxyl derivatives, etc. These probes have been widely used for a number of applications,28–30 including probing intrinsic biophysical properties of biomembranes,31,32 detecting protein conformational changes,33–36 and monitoring DNA interactions.37,38 Despite this progress, novel principles of chemical synthesis are still needed to guide the rational design of probes with well-defined sensitivity to macromolecular polarity. A typical solvatochromic fluorophore generally contains a donor-π-acceptor conjugation system, which undergoes an active excited state intramolecular charge transfer after excitation. As ambient polarity increases, the enhancement of the dipole–dipole interaction between fluorescent probes and solvent results in emission shifts to longer wavelengths. Even after extensive studies using the different probes described above, relatively few highly efficient strategies are available for the design of additional solvatochromic and fluorogenic dyes. At present, the most common design strategy involves modulating the “push–pull” effect by extension of the π-conjugation system, often requiring remodeling of the fluorophore scaffold.21,39–41 Recently, a novel study of the aggregation-enhanced emission and pH-sensitive emission features of the thioxanthone-based compound has been reported.42 Meanwhile, solvatofluorochromism was observed on this molecule. However, the mechanism of polarity change and potential biological applications of this molecule has not yet been explored.
In this work, we report a chemical strategy for modulating xanthone-based solvatochromic fluorophores using single-atom substitutions (Fig. 1B). The following factors were considered in the design of these fluorophores: (1) heteroatoms should contribute minimally to the highest occupied molecular orbitals (HOMOs) of xanthone derivatives, while the lowest unoccupied molecular orbitals (LUMOs) should be π*-type molecular orbitals that receive a significant contribution from the heteroatoms; (2) the LUMO energy level should be stabilized by electron-withdrawing heteroatoms more efficiently than by the electron-donating heteroatoms. As we expected, following the introduction of the electron-withdrawing heteroatom substituted sulfonyl group, the resulting novel probe exhibited the most significant bathochromic shift of 137 nm. To demonstrate the utility of the designed solvatochromic fluorophores, we constructed two different fluorescent probes for quantifying the polarity of the local microenvironment around misfolded and aggregated proteins based on the dielectric constants (ε). Using these fluorescent probes, we investigated the polarity of intracellular protein aggregates. We found unexpected hydration and heterogeneity among granular aggregates that arise as a result of different cellular stressors and genetic mutations. This observation provides insights that will guide future studies of intracellular aggregates, enabling the elucidation of cellular mechanisms underlying protein misfolding, and providing a therapeutic approach to managing misfolding-based diseases.
Results and discussion
For modulating the sensitivity to the polarity of a xanthone scaffold, we chose acridone due to its recent use in detecting polarity in biological contexts.43–46 These types of probes are generally characterized by relatively small emission shifts (ca. 40 nm), resulting in poor sensitivity to changes in polarity. In recent years, elegant strategies based on single-atom replacement have been applied to different fluorophore scaffolds to obtain fluorescent dyes with larger emission shifts.47–50 Learning from these studies, our rationale has been to develop a single-atom replacement strategy for designing minimally-modified solvatochromic probes capable of detecting changes in micro-polarity that occur during protein aggregation in live cells.Computational analysis of acridone reveals chemical mechanisms that promote its sensitivity to polarity
To determine whether a single atom substitution in acridone can lead to redshift emission and high sensitivity to polarity, we first synthesized 3,6-bis(diethylamino)-10-methylacridin-9(10H)-one (MA) (ESI Section 2†) and investigated its photophysical properties (Fig. 2A, Tables 1 and S1 and Fig. S1 and S2†). We initially measured the fluorescence emission spectra of MA in solvents with varying dielectric constants. Moving from hexane (nonpolar) to water (polar), the emission maximum of MA exhibited an obvious red-shift of 76 nm (Fig. 2B, Table S1, and Fig. S2†). We further evaluated the polarity sensitivity of MA by correlating the polarity dependence and relative emission maximum in standard solvents with varying ET30 parameters.51 The polarity sensitivity was quantified via the slope of the linear correlation between the emission maximum and the ET30 parameter. For MA, the slope values were determined as 2.49 in standard solvents (Fig. 2C, Table S1, and Fig. S1 and S2†). | ||
| Fig. 2 Rational design of xanthone derivative probes with sensitivity to polarity. (A) Structures of solvatochromic xanthone derivatives. (B) Emission spectra of MA in different solvents. (C) Polarity sensitivity calculated based on maximal emission over the ET30 in standard solvents. (D) HOMO and LUMO orbitals of xanthone derivatives. (E) Change in emission maximum of different solvatochromic xanthone derivatives going from hexane to water. (F) Quantitative polarity dependence of solvatochromic xanthone derivatives in protic and aprotic solvents. | ||
| Compounds a | λ abs (nm) | ε (M−1 cm−1) | λ em (nm) | Φ f |
|---|---|---|---|---|
| a Compounds were dissolved in MeOH. b λ abs peak wavelength of the absorption spectrum. c ε: molar extinction coefficients. d λ em: peak wavelength of the emission spectrum. e Fluorescence quantum yields were measured using quinine sulfate in 0.5 M H2SO4 as the reference. | ||||
| MA | 393 | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 662 662 |
437 | 0.47 |
| OX | 384 | 33747 |
441 | 0.80 |
| SX | 392 | 36049 |
468 | 0.42 |
| SeX | 394 | 36760 |
472 | 0.02 |
| CX | 396 | 37115 |
489 | 0.75 |
| SiX | 406 | 31036 |
496 | 0.39 |
| GeX | 402 | 21231 |
496 | 0.30 |
| POX | 347, 427 | 16760, 21246 |
526 | 0.19 |
| SO2X | 352, 430 | 8300, 13100 |
542 | 0.13 |
Chemical rules that regulate polarity sensitivity are rarely reported. To devise a rational strategy for endowing acridone with controllable sensitivity to polarity, we carried out a computational investigation of MA to determine the factors that control the polarity-sensitivity of xanthone derivatives. The time-dependent density functional theory (TD-DFT) was used to analyze frontier orbital distributions as a means of exploring how HOMO and LUMO energy levels might be controlled in xanthone derivatives containing a variety of replacement heteroatoms. We found that the HOMOs of most calculated xanthone derivatives were π-type molecular orbitals delocalized over the aromatic ring, wherein heteroatoms contributed minimally to the conjugate orbitals (Fig. 2D). By contrast, the LUMOs are π*-type molecular orbitals involving significant contributions from the heteroatoms (Fig. 2D). Thus, we varied heteroatoms in the computational study to understand the correlation between LUMO energy level and heteroatom identity. Based on the structure of MA, we introduced varying heteroatoms into the acridone scaffold (Fig. 2A), including oxygen group elements (O, S, Se, SO2), carbon group elements (C, Si, Ge), and nitrogen group elements (POPh). Interestingly, we found that electron-withdrawing heteroatoms stabilized the LUMO energy level more efficiently than electron-donating heteroatoms. This stabilization was achieved via two distinct conjugation modes: (1) the (p)–π* conjugation involving only the p orbitals of heteroatoms of xanthone derivatives (Fig. 2D); and (2) the (d–p)–π* conjugation involving the d orbitals of heteroatoms of xanthone derivatives (Fig. 2D). Of these two modes, the (d–p)–π* conjugation derivatives exhibited lower LUMO energy levels than the p–π* derivatives, because the in-phase p–d orbital interaction in the (d–p)–π* conjugation more effectively stabilizes the LUMO orbital. The results of our calculations, therefore, reveal that electron-withdrawing heteroatoms with (d–p)–π* conjugation is able to stabilize the LUMO energy level and promote the bathochromic shift. In addition, electron densities in the LUMOs of other xanthone derivatives exhibit significantly different distributions compared to those in the HOMOs (Fig. 2D), corresponding to the stronger intramolecular charge transfer (ICT) process. The excited states of these probes generally exhibit higher polarity than the ground states, which is attributable to ICT from the donor to the acceptor group. This ICT effect creates a large dipole moment in the excited state compared to the ground state, resulting in enhanced sensitivity to polarity.
SO2X acridone derivative exhibits improved polarity sensitivity via bathochromic shifts
Guided by the computational analysis, we synthesized the calculated acridone derivatives based on published methods,42,52–58 resulting in 3,6-bis(diethylamino)-9H-xanthen-9-one (OX), 3,6-bis(diethylamino)-9H-thioxanthen-9-one (SX), 3,6-bis(diethylamino)-9H-selenoxanthen-9-one (SeX), 3,6-bis(diethylamino)-9H-thioxanthen-9-one 10,10-dioxide (SO2X), 3,6-bis(diethylamino)-10,10-dimethylanthracen-9(10H)-one (CX), 3,7-bis(diethylamino)-5,5-dimethyldibenzo[b,e]silin-10(5H)-one (SiX), 3,7-bis(diethylamino)-5,5-dimethyldibenzo[b,e]germin-10(5H)-one (GeX), and 3,7-bis(diethylamino)-5-phenyl-10H-acridophosphin-10-one 5-oxide (POX). The chemical structures and purity of the synthesized acridone derivatives were verified by NMR and HRMS (ESI Section 2†). The photophysical properties of the acridone derivatives were characterized in methanol (Table 1), and their polarity responses were investigated in solvents of varying polarity (Tables S2–S9 and Fig. S3–S18†).With increasing polarity, all xanthone-based solvatochromic fluorophores exhibited red-shift in their absorption spectra from aprotic solvents to protic solvents as well as decreased extinction coefficient due to dipolar interactions with solvent molecules further relaxing the excited state (Tables S1–S9†). For carbon group derivatives, the extent of molar extinction coefficients and fluorescence quantum yields were CX < SiX < GeX, which may be due to the less efficient conjugation and larger delocalized structures with the increasing size of the atom. Oxygen family xanthone derivatives (OX, SX, SeX) showed similar molar extinction coefficients. The corresponding fluorescence quantum yields decreased as the atomic number increase, which could be attributed to the heavy-atom effect. Different with oxygen and nitrogen, sulfur and phosphorus could demonstrate +6 and +5 oxidation states, respectively. The corresponding oxides (SO2X and POX) with (d–p)–π* conjugations possess larger delocalized structures generating moderate molar absorption coefficients and fluorescence quantum yields.59 As polarity increased, all acridone derivatives exhibited a significant redshift in emission spectra, as measured by the shift in emission maximum going from nonpolar solvents (e.g. hexane) to polar solvents (e.g. water) (Fig. 2E). In all cases, Beer–Lambert plots were used to ensure that all acridone derivatives were soluble at the measurement conditions (Fig. S19†). Consistent with the computational analysis, the acridone derivative with the most electron-withdrawing heteroatom substitution SO2X exhibited the most significant bathochromic shift of 137 nm (Fig. 2E). Emission maxima of other acridone derivatives were also shifted to longer wavelengths by 96 nm, 108 nm, 112 nm, 116 nm, 105 nm, 117 nm, and 133 nm, for OX, SX, SeX, CX, SiX, GeX, and POX, respectively (Fig. 2E). To quantitatively describe the polarity dependence of the derivatives, we plotted the emission maximum over the ET30 in standard solvents (Fig. 2F and S20†), wherein the slope values indicate the extent of the bathochromic shift in response to polarity increase. We found that all acridone derivatives exhibited clear dependency with the ET30 parameters and SO2X demonstrated an extremely high slope value. All derivatives exhibited minimal dependence on solvent viscosity (Fig. S21†), making them ideal compounds to detect micropolarity without interference from the changes of microviscosity.
These results suggest that SO2X exhibits the most significant sensitivity towards polarity, as evidenced by its pronounced redshift in solvents with increasing ET30 parameters (Fig. 3A and B). We further carried out TD-DFT calculations to provide insights into the way that SO2X responds to polarity. In water, the LUMO orbital of SO2X in S1 is a (d–p)–π* conjugation with an in-phase p–d orbital, resulting in smaller emission energy than seen with SO2X in a vacuum representing a non-polar environment (Fig. 3C). Furthermore, the molecular electrostatic potential of SO2X in S1 also explains its polarity sensitivity (Fig. 3D) since the intramolecular charge transfer from the diethylamine moiety to the carbonyl oxygen and the SO2 moiety is enhanced more in water than in a vacuum. We also calculated the fluorescence emission energy of SO2X in solvents with varying dielectric constants (Fig. 3E). These calculations support the experimentally measured red-shift of the emission wavelength for SO2X in solvents with increasing polarity. Furthermore, the calculated dipole moments of SO2X in different solvents were proportional to the dielectric constants (Fig. 3E), providing additional evidence to account for the high polarity sensitivity of SO2X. Taken together, these results demonstrate the suitability of the single-atom replacement strategy for designing solvatochromic fluorophores. Due to its desirable sensitivity to polarity, SO2X was chosen for further biological applications aimed at quantifying micropolarity in protein aggregates.
 | ||
| Fig. 3 Sulfonyl substituted acridone derivative exhibits improved polarity sensitivity evidenced by bathochromic shifts. (A) The emission maximum of SO2X in different solvents. (B) Polarity sensitivity of SO2X calculated based on maximal emission over the ET30 in standard solvents. (C) TD-DFT calculated fluorescence emission energy of SO2X in water solvent environment. (D) Electrostatic potential maps based on the electron density of SO2X in water and vacuum. (E) Calculation showed that with solvent polarity increasement, the emission wavelength of SO2X underwent bathochromic shifts, accompanied by decreases in the energy band gap and increases in the dipole moment. | ||
Using SO2X to quantify polarity in stress-induced protein aggregates
The bathochromic shift of SO2X offers a unique opportunity to quantify changes in polarity when proteins undergo misfolding and aggregation in live cells. Polarity is an important physical property in the multi-step process of protein misfolding and aggregation. The surfaces of globular folded proteins are normally polar, as indicated by dielectric constants (ε) around 30. These protein surfaces largely display polar residues that are capable of making polarity-based interactions with water in the aqueous environment. Misfolding, however, is initiated by disruption of the hydrophobic core of folded proteins, thus exposing normally buried hydrophobic residues to the exterior surface. As a result, the surface of misfolded proteins becomes more non-polar, as indicated by decreased dielectric constant values. When misfolded proteins assemble into misfolded oligomers or insoluble aggregates, the resulting desolvation excludes water and leads to further reduction of polarity. Thus, the multi-step process of protein misfolding and aggregation should be accompanied by progressively decreased polarity, and the value of the dielectric constant should reflect the severity of protein aggregation.We chose the AgHalo proteome stress sensor to investigate polarity changes that occur in AgHalo when cells are subjected to various types of chemical stress. AgHalo is a two-component proteome stress sensor comprised of (a) a metastable HaloTag mutant K73T (ΔGfolding = −2.0 kcal mol−1, compared with ΔGfolding = −5.6 kcal mol−1 for the wild-type HaloTag) that only aggregates in cells treated with stressors and (b) the SO2X ligand that conjugates with HaloTag–K73T and undergoes changes in fluorescence emission when the K73T–SO2X conjugate misfolds or aggregates. To this end, we synthesized the Halo-SO2X compound (ESI Section 2, Fig. S23 and Table S10†) that reacts covalently with both wild-type HaloTag and the K73T mutant (Fig. 4A). An optimized sarcosine linker was added to the original linker of HaloTag ligands to extend the SO2X moiety away from HaloTag and position it in a hydrophilic environment. During changes in the folding of the K73T–SO2X conjugate, the fluorescence emission of the SO2X moiety reports on the polarity of the cytosol. In the presence of proteome stressors that cause K73T–SO2X misfolding and aggregation, the SO2X moiety exhibits quantifiable changes in fluorescence emission that reflect the extent of K73T–SO2X aggregation.
 | ||
| Fig. 4 Quantifying the polarity of stress-induced protein aggregates. (A) SO2X was used to label proteins for the detection of polarity changes in stress-induced protein aggregates. (B) Halo-SO2X probe was used to visualize stress-induced protein aggregation. K73T was expressed in HEK293T cells, in the presence of 1 μM Halo-SO2X, followed by treatment with PBS, sorbitol (0.5 M, 5 min), NaCl (1 M, 5 min), nilotinib (50 μM, 18 h). (C and D) Lambda scan imaging to reveal polarity changes in K73T–SO2X conjugates treated with different stressors as indicated in (B). Scale bar = 10 μm. Error bars in (D) = standard deviation of the maximum emission wavelength. | ||
Before we demonstrated the utility of biological imaging, we evaluated the photostability and cytotoxicity (Fig. S24 and S25†). Cytotoxicity was evaluated by Cell Counting Kit-8. Halo-SO2X didn't show significant cytotoxicity at concentrations up to 12 μM. K73T was expressed in HEK293T cells, in the presence of 1 μM of Halo-SO2X. After 24 h of expression, cells were washed free of excess dye. Subsequently, varying stressors were added to the culture media, and cells were incubated as indicated: 0.5 M sorbitol, 5 min; 1 M NaCl, 5 min; 50 μM nilotinib, 18 h. Fluorescence confocal imaging was carried out to visualize the aggregation status of K73T–SO2X in the absence and presence of stressors (Fig. 4B). In the absence of stressor, K73T–SO2X exhibited diffuse fluorescence in both cytosol and nucleus, consistent with previous reports.60,61 Sorbitol treatment resulted in a largely diffuse labeling pattern, but with the minor appearance of punctate structures (Fig. 4B). Increased numbers of punctate structures were found in cells treated with 1 M NaCl (Fig. 4B). In the presence of 50 μM Nilotinob, most of the K73T–SO2X labeling was granular, indicative of severe aggregation (Fig. 4B).
Next, we conducted lambda scan imaging to look more specifically at polarity changes when HEK293T cells expressing the K73T–SO2X conjugate were treated with different stressors. The lambda scan experiment provides a series of individual images within a range of fluorescence emission wavelength, with each imaging taken at a specific emission wavelength. The fluorescence intensity at each wavelength was analyzed, and images were plotted using a grey scale (Fig. 4C and S26†). Subsequently, the emission maximum was determined as a reflection of the polarity in the microenvironment of the K73T–SO2X conjugate. This value can be used to quantify the dielectric constant when combined with the linear correlation for the protic solvent (Fig. S22†) due to the protic, aqueous environment of the cytosol. In non-stressed cells, the emission maximum was determined to be 539 nm (Fig. 4C and D), corresponding to a dielectric constant of 29.1 that is consistent with the hydrophilic surface of a folded globular protein. When cells are treated with 0.5 M sorbitol, the dielectric constant was reduced to 22.1, as indicated by the emission maximum of 530 nm (Fig. 4C and D). This suggests that the stress-induced misfolding has exposed hydrophobic residues at the protein surface, decreasing polarity in the microenvironment of the SO2X moiety. Further reduction of polarity was observed when 1 M NaCl was used as a stressor, leading to a dielectric constant of 12.7 and an emission maximum of 512 nm (Fig. 4C and D). Treatment with nilotinib even more significantly decreased polarity, reflected by a dielectric constant of 3.3 and an emission maximum of 468 nm (Fig. 4C and D). The polarity of this microenvironment mimics that of non-polar solvents (e.g. dioxane), suggesting that the nilotinib-induced K73T aggregates possess a highly desolvated and non-polar structure.
Unexpected polarity of huntingtin polyglutamine aggregates as determined by SO2X
Encouraged by our results with chemically-induced protein aggregates, we extended our investigation of micro-polarity to protein aggregates caused by genetic mutations. For this study, we chose the well-known huntingtin (Htt) protein, whose N-terminus contains variable numbers of glutamine residues (polyQ). PolyQ variants with more than 36 N-terminal glutamine residues (36Q) form intracellular aggregates, a phenotype that is related to Huntington's disease. In particular, huntingtin with extended glutamine repeats, such as 110Q, form sizable punctate aggregates in the cytosol.62 Previous work has attempted to discover small molecule drugs that are able to prevent or disassemble the polyQ aggregates.16,63 In these studies, fluorescence morphology is used to determine whether the drugs are capable of preventing the formation of punctate aggregates. However, important information is missing in this traditional analysis. For example, punctate aggregates with similar appearances may nevertheless harbor dissimilar microenvironments in which the extent of desolvation or polarity may differ. In our work, the use of the SNAP-SO2X (ESI Section 2, Fig. S21 and Table S10†) reagent combined with lambda scan imaging provides a unique means of evaluating the effects of previously described drugs on the polarity of Htt–110Q aggregates.Following the established AggTag method, the C-terminus of Htt–110Q was genetically fused to SNAPf-tag and the resulting plasmid (110Q–SNAPf) was transiently expressed in HEK293T cells. The AggTag method is a generally approach to visualize aggregation of a variety of proteins in live cells.15,16 The small size (184 amino acid and 19.4 kDa) of SNAPf-tag minimizes its affect on the aggregation behavior of Htt–110Q. Treatment of these with 0.5 M SNAP-SO2X (0.5 μM, Fig. 5A) for 24 hours resulted in the formation of granular aggregates (Fig. 5B). Lambda scan imaging was used to evaluate the fluorescence intensity of these aggregates at each wavelength and thereby to determine the emission maximum (Fig. 5C and D). Via this analysis, the dielectric constant of the granular 110Q–SNAPf aggregates was determined as 22.1. The 110Q–SNAPf aggregates are thus much more polar than the nilotinib-induced AgHalo aggregates (ε = 3.3, Fig. 4C and D). This difference is likely due to the ability of the polyQ-rich protein to interact with polar water molecules. These polar interactions prevent the large degree of desolvation seen with AgHalo, in which hydrophobic residues from the non-polar core are exposed on the protein surface.
 | ||
| Fig. 5 Investigation of the micropolarity of protein aggregates resulted from genetic mutations. (A) Structure of SNAP-SO2X. (B) Granular 110Q–SNAPf aggregates (110Q–SNAPf) was transiently expressed in HEK293T cells with SNAP-SO2X (0.5 μM) and drug-induced aggregation elimination. (C and D) Lambda scan imaging to reveal whether drugs effectively ameliorated aggregation of 110Q-SNAPf. Scale bar = 10 μm. Error bars in (D) = standard deviation of the maximum emission wavelength. | ||
PD169316 was previously reported as a proteasome activator that increases the degradation of reporter peptides. YM-1 was reported to be an Hsp70 activator capable of disassembling Htt–polyQ aggregates. To test the effect of these drugs, on huntingtin, 110Q–SNAPf was expressed in HEK293T cells in the presence of PD169316 (1 μM), YM-1 (1 μM), or PD169316 combined with YM-1 (1 μM each) for 24 hours. Sizable aggregates as punctate fluorescent structures, albeit more diffusive fluorescence was observed compared to the control experiment (Fig. 5B). While it was unclear whether these drugs effectively ameliorated aggregation of 110Q–SNAPf, the lambda scan analysis nevertheless provided interesting insights (Fig. 5C and D). The polarity of 110Q–SNAPf aggregates was modestly affected by PD169316, with the dielectric constant increasing from 22.1 to 29.1. By contrast, YM-1 significantly increased the polarity of 110Q aggregates, resulting in a fluorescence emission maximum at 575 nm that is comparable to SO2X in water (Fig. 5C and D). These results suggest that the activation of Hsp70 greatly affected the microenvironment and physical properties of 110Q–SNAPf aggregates, even though granular aggregates still formed. It is possible that the activated Hsp70 interacts with 110Q to render aggregates more permeable and hydrated, and thus more polar. By contrast, the activation of proteasomes using PD169316 produced a minimal effect on 110Q aggregates, consistent with the knowledge that the ubiquitin-proteasome system acts more effectively on soluble and misfolded proteins than on insoluble aggregates. Consistent with this notion, 110Q aggregates treated with the combination of PD169316 and YM-1 exhibited a similar polarity to aggregates treated with YM-1 (Fig. 5C and D). These results suggest that Hsp70 activation might be a feasible approach to loosening and hydrating 110Q aggregates, providing promise for developing therapeutic strategies to ameliorate Huntington's disease.
Conclusion
Xanthone is a commonly used scaffold in constructing synthetic fluorophores. Using acridone as an example, we systematically studied the effect of heteroatoms on controlling the solvatochromism of xanthone-based fluorescent compounds. The electron-withdrawing heteroatoms with (d–p)–π* conjugation can stabilize the LUMO energy level and promote bathochromic shifts. Guided by this principle, we have synthesized a series of acridone derivatives with the introduction of heteroatom substitutions and found that SO2X exhibits an optimized solvatochromic response to polarity changes. Using the SO2X fluorophore, we investigated the polarity of intracellular protein aggregates. Our data reveal the following interesting concepts that have rarely been reported. First, misfolding and aggregation of folded globular proteins lead to a microenvironment with reduced polarity. AgHalo aggregates display different degrees of polarity when cells are subjected to distinct stressors. More severe stress is normally associated with greater desolvation and greater reduction of polarity. Second, granular aggregates of Htt–polyQ harbor a more polar environment than aggregates formed by folded globular protein represented by AgHalo. The ability of glutamine to interact with water may contribute to the reduced extent of desolvation in Htt–polyQ aggregates. Thus, the Htt–polyQ aggregates commonly regarded as being “insoluble” may not actually be insoluble, due to the extent of their hydration. Third, aggregates that are grossly regarded as granular may exhibit very different polarities, as exemplified by Htt–110Q granules in the presence of different drugs. The unexpected hydration and heterogeneity of granular aggregates encourage future studies aimed at elucidating the cellular mechanisms of intracellular aggregation and developing therapeutic approaches to managing these aggregates.Data availability
All the data including experimental procedures, synthetic methods, characterization spectra are recorded in the ESI.†Author contributions
L. W., C. H., X. Z., X. H. were responsible for experimental design and data analysis. L. W., C. H., X. L., S. W., A. L., performed research, L. W., C. H., X. Z., and X. H. contributed to writing and editing the manuscript. X. Z. and H. X. conceived and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Cancer Prevention Research Institute of Texas (CPRIT RR170014 to H. X.), NIH (R35-GM133706, R21-CA255894, and R01-AI165079 to H. X.), the Robert A. Welch Foundation (C-1970 to H. X.), US Department of Defense (W81XWH-21-1-0789 to H. X.), the John S. Dunn Foundation Collaborative Research Award (to H. X.), and the Hamill Innovation Award (to H. X.). We thank the support from the Burroughs Welcome Fund Career Award at the Scientific Interface 1013904 (X. Z.), Paul Berg Early Career Professorship (X. Z.), Lloyd and Dottie Huck Early Career Award (X. Z.), Sloan Research Fellowship FG-2018-10958 (X. Z.), PEW Biomedical Scholars Program 00033066 (X. Z.), and National Institute of Health R35-GM133484 (X. Z.). H. X. is a Cancer Prevention & Research Institute of Texas (CPRIT) scholar in cancer research.Notes and references
- C. B. Anfinsen, Science, 1973, 181, 223–230 CrossRef CAS PubMed.
- C. M. Dobson, Semin. Cell Dev. Biol., 2004, 15, 3–16 CrossRef CAS PubMed.
- W. E. Balch, R. I. Morimoto, A. Dillin and J. W. Kelly, Science, 2008, 319, 916–919 CrossRef CAS PubMed.
- C. A. R. M. A. Poirier, Nat. Rev. Mol. Cell Biol., 2005, 6, 891–898 CrossRef PubMed.
- R. M. Sebastian and M. D. Shoulders, Annu. Rev. Biochem., 2020, 89, 529–555 CrossRef CAS PubMed.
- L. W. Zhang, S. J. Peng, J. Y. Sun, J. Yao, J. Kang, Y. S. Hu and J. G. Fang, Chem. Sci., 2017, 8, 2966–2972 RSC.
- A. Aliyan, N. P. Cook and A. A. Martí, Chem. Rev., 2019, 119, 11819–11856 CrossRef CAS PubMed.
- M. A. Bhat, S. Izaddoost, Y. Lu, K.-O. Cho, K.-W. Choi and H. J. Bellen, Cell, 1999, 96, 833–845 CrossRef CAS.
- X. Michalet, S. Weiss and M. Jäger, Chem. Rev., 2006, 106, 1785–1813 CrossRef CAS PubMed.
- A. Wodarz and I. Nathke, Nat. Cell Biol., 2007, 9, 1016–1024 CrossRef CAS PubMed.
- R. G. Perez, H. Zheng, L. H. Van der Ploeg and E. H. Koo, J. Neurosci., 1997, 17, 9407–9414 CrossRef CAS.
- Y. Bai, W. Wan, Y. Huang, W. Jin, H. Lyu, Q. Xia, X. Dong, Z. Gao and Y. Liu, Chem. Sci., 2021, 12, 8468–8476 RSC.
- W. Wan, Y. Huang, Q. Xia, Y. Bai, Y. Chen, W. Jin, M. Wang, D. Shen, H. Lyu, Y. Tang, X. Dong, Z. Gao, Q. Zhao, L. Zhang and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 11335–11343 CrossRef CAS PubMed.
- D. Shen, W. Jin, Y. Bai, Y. Huang, H. Lyu, L. Zeng, M. Wang, Y. Tang, W. Wan, X. Dong, Z. Gao, H. L. Piao, X. Liu and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 16067–16076 CrossRef CAS PubMed.
- Y. Liu, C. H. Wolstenholme, G. C. Carter, H. Liu, H. Hu, L. S. Grainger, K. Miao, M. Fares, C. A. Hoelzel, H. P. Yennawar, G. Ning, M. Du, L. Bai, X. Li and X. Zhang, J. Am. Chem. Soc., 2018, 140, 7381–7384 CrossRef CAS PubMed.
- C. H. Wolstenholme, H. Hu, S. Ye, B. E. Funk, D. Jain, C. H. Hsiung, G. Ning, Y. Liu, X. Li and X. Zhang, J. Am. Chem. Soc., 2020, 142, 17515–17523 CrossRef CAS PubMed.
- S. Ye, H. Zhang, J. Fei, C. H. Wolstenholme and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 1339–1346 CrossRef CAS PubMed.
- S. Ye, C. H. Hsiung, Y. Tang and X. Zhang, Acc. Chem. Res., 2022, 55, 381–390 CrossRef CAS PubMed.
- Y. Y. Wang, X. F. Yang, Y. G. Zhong, X. Y. Gong, Z. Li and H. Li, Chem. Sci., 2016, 7, 518–524 RSC.
- L. Li, Z. Lv, Z. W. Man, Z. Z. Xu, Y. L. Wei, H. Geng and H. B. Fu, Chem. Sci., 2021, 12, 3308–3313 RSC.
- A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS PubMed.
- H. Xiao, P. Li and B. Tang, Coord. Chem. Rev., 2021, 427, 213582 CrossRef CAS.
- S. Wang, W. X. Ren, J.-T. Hou, M. Won, J. An, X. Chen, J. Shu and J. S. Kim, Chem. Soc. Rev., 2021, 50, 8887–8902 RSC.
- O. A. Kucherak, P. Didier, Y. Mély and A. S. Klymchenko, J. Phys. Chem. Lett., 2010, 1, 616–620 CrossRef CAS.
- H. B. Xiao, P. Li, W. Zhang and B. Tang, Chem. Sci., 2016, 7, 1588–1593 RSC.
- X. Y. Li, X. H. Li and H. M. Ma, Chem. Sci., 2020, 11, 1617–1622 RSC.
- S. J. Park, V. Juvekar, J. H. Jo and H. M. Kim, Chem. Sci., 2020, 11, 596–601 RSC.
- J. Zhou and H. M. Ma, Chem. Sci., 2016, 7, 6575 RSC.
- S. I. Reja, M. Minoshima, Y. Hori and K. Kikuchi, Chem. Sci., 2020, 12, 3437–3447 RSC.
- X. Tian, L. C. Murfin, L. L. Wu, S. E. Lewis and T. D. James, Chem. Sci., 2021, 12, 3406–3426 RSC.
- L. Wang, J. Hiblot, C. Popp, L. Xue and K. Johnsson, Angew. Chem., Int. Ed., 2020, 59, 21880–21884 CrossRef CAS.
- E. Prifti, L. Reymond, M. Umebayashi, R. Hovius, H. Riezman and K. Johnsson, ACS Chem. Biol., 2014, 9, 606–612 CrossRef CAS PubMed.
- B. E. Cohen, T. B. McAnaney, E. S. Park, Y. N. Jan, S. G. Boxer and L. Y. Jan, Science, 2002, 296, 1700–1703 CrossRef CAS PubMed.
- B. E. Cohen, A. Pralle, X. Yao, G. Swaminath, C. S. Gandhi, Y. N. Jan, B. K. Kobilka, E. Y. Isacoff and L. Y. Jan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 965–970 CrossRef CAS PubMed.
- G. Loving and B. Imperiali, J. Am. Chem. Soc., 2008, 130, 13630–13638 CrossRef CAS PubMed.
- T. C. Owyong, P. Subedi, J. Deng, E. Hinde, J. J. Paxman, J. M. White, W. Chen, B. Heras, W. W. H. Wong and Y. Hong, Angew. Chem., Int. Ed., 2020, 59, 10129–10135 CrossRef CAS PubMed.
- K. Tainaka, K. Tanaka, S. Ikeda, K.-I. Nishiza, T. Unzai, Y. Fujiwara, I. Saito and A. Okamoto, J. Am. Chem. Soc., 2007, 129, 4776–4784 CrossRef CAS PubMed.
- D. Dziuba, P. Pospisil, J. Matyasovsky, J. Brynda, D. Nachtigallova, L. Rulisek, R. Pohl, M. Hof and M. Hocek, Chem. Sci., 2016, 7, 5775–5785 RSC.
- X. P. Dong, W. Wan, L. G. Zeng, W. H. Jin, Y. A. Huang, D. Shen, Y. L. Bai, Q. Zhao, L. H. Zhang, Y. Liu and Z. M. Gao, Anal. Chem., 2021, 93, 16447–16455 CrossRef CAS.
- W. Wan, L. G. Zeng, W. H. Jin, X. X. Chen, D. Shen, Y. A. Huang, M. D. Wang, Y. L. Bai, H. C. Lyu, X. P. Dong, Z. M. Gao, L. Wang, X. J. Liu and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 25865–25871 CrossRef CAS.
- J. C. Er, M. K. Tang, C. G. Chia, H. Liew, M. Vendrell and Y. T. Chang, Chem. Sci., 2013, 4, 2168–2176 RSC.
- S. I. Hassan, A. Haque, Y. A. Jeilani, R. Ilmi, M. S. H. Faizi, I. Khan and M. Mushtaque, J. Mol. Struct., 2021, 1224, 129004 CrossRef CAS.
- C. M. Jones, Y. Venkatesh and E. J. Petersson, Methods Enzymol., 2020, 639, 37–69 CAS.
- Z. M. Hostetler, M. B. Cory, C. M. Jones, E. J. Petersson and R. M. Kohli, ACS Chem. Biol., 2020, 15, 1127–1133 CrossRef CAS PubMed.
- J. Tang, C. Yu, A. Loredo, Y. Chen and H. Xiao, Chembiochem, 2021, 22, 501–504 CrossRef CAS PubMed.
- M. C. Gonzalez-Garcia, T. Peña-Ruiz, P. Herrero-Foncubierta, D. Miguel, M. D. Giron, R. Salto, J. M. Cuerva, A. Navarro, E. Garcia-Fernandez and A. Orte, Sens. Actuators, B, 2020, 309, 127770 CrossRef CAS.
- J. B. Grimm, A. N. Tkachuk, L. Xie, H. Choi, B. Mohar, N. Falco, K. Schaefer, R. Patel, Q. Zheng, Z. Liu, J. Lippincott-Schwartz, T. A. Brown and L. D. Lavis, Nat. Methods, 2020, 17, 815–821 CrossRef CAS PubMed.
- J. Liu, Y. Q. Sun, H. Zhang, H. Shi, Y. Shi and W. Guo, ACS Appl. Mater. Interfaces, 2016, 8, 22953–22962 CrossRef CAS PubMed.
- L. Wang, S. Wang, J. Tang, V. B. Espinoza, A. Loredo, Z. Tian, R. B. Weisman and H. Xiao, Chem. Sci., 2021, 12, 15572–15580 RSC.
- J. Tang, M. A. Robichaux, K.-L. Wu, J. Pei, N. T. Nguyen, Y. Zhou, T. G. Wensel and H. Xiao, J. Am. Chem. Soc., 2019, 141, 14699–14706 CrossRef CAS PubMed.
- Y. Marcus, Chem. Soc. Rev., 1993, 22, 409–416 RSC.
- M. Yang, J. Fan, W. Sun, J. Du and X. Peng, Anal. Chem., 2019, 91, 12531–12537 CrossRef CAS PubMed.
- D. J. Del Valle, D. J. Donnelly, J. J. Holt and M. R. Detty, Organometallics, 2005, 24, 3807–3810 CrossRef CAS.
- X. Lv, C. Gao, T. Han, H. Shi and W. Guo, Chem. Commun., 2020, 56, 715–718 RSC.
- J. Tang, Z. Guo, Y. Zhang, B. Bai and W. H. Zhu, Chem. Commun., 2017, 53, 10520–10523 RSC.
- P. Horváth, P. Šebej, D. Kovář, J. Damborský, Z. Prokop and P. Klán, ACS Omega, 2019, 4, 5479–5485 CrossRef.
- M. Grzybowski, M. Taki, K. Senda, Y. Sato, T. Ariyoshi, Y. Okada, R. Kawakami, T. Imamura and S. Yamaguchi, Angew. Chem., Int. Ed., 2018, 57, 10137–10141 CrossRef CAS PubMed.
- G. Dejouy, M. Laly, I. E. Valverde and A. Romieu, Dyes Pigm., 2018, 159, 262–274 CrossRef CAS.
- F. Deng and Z. C. Xu, Chin. Chem. Lett., 2019, 30, 1667–1681 CrossRef CAS.
- Y. Liu, M. Fares, N. P. Dunham, Z. Gao, K. Miao, X. Y. Jiang, S. S. Bollinger, A. K. Boal and X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8672–8676 CrossRef CAS PubMed.
- Y. Liu, K. Miao, Y. H. Li, M. Fares, S. Y. Chen and X. Zhang, Biochemistry, 2018, 57, 4663–4674 CrossRef CAS PubMed.
- K. Miao and L. Wei, ACS Cent. Sci., 2020, 6, 478–486 CrossRef CAS PubMed.
- A. M. Wang, Y. Miyata, S. Klinedinst, H. M. Peng, J. P. Chua, T. Komiyama, X. Li, Y. Morishima, D. E. Merry, W. B. Pratt, Y. Osawa, C. A. Collins, J. E. Gestwicki and A. P. Lieberman, Nat. Chem. Biol., 2013, 9, 112–118 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05004h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |