
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium-catalyzed enantioselective alkynylation and kinetic resolution of alkyl allylic alcohols†‡
Jia
Guo
a,
Hao-Ran
Ma
ab,
Wen-Bin
Xiong
a,
Luoyi
Fan
a,
You-Yun
Zhou
*ad,
Henry N. C.
Wong
 *abc and
Jian-Fang
Cui
*a
*abc and
Jian-Fang
Cui
*a
aDepartment of Chemistry, Southern University of Science and Technology, 1088 Xueyuan Blvd., Shenzhen 518055, China. E-mail: cuijf@sustech.edu.cn; hncwong@cuhk.edu.hk; zhouyy@sustech.edu.cn
bSchool of Science and Engineering, The Chinese University of Hong Kong (Shenzhen), 2001 Longxiang Blvd., Shenzhen 518172, China
cDepartment of Chemistry, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong SAR, China
dGuangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, 1088 Xueyuan Blvd., Shenzhen 518055, China
First published on 1st November 2022
Abstract
Herein, we report an efficient kinetic resolution of alkyl allylic alcohols enabled by an iridium-catalyzed enantioselective alkynylation of alkyl allylic alcohols with potassium alkynyltrifluoroborates. A wide range of chiral 1,4-enynes bearing various functional groups and unreacted enantioenriched allylic alcohols were obtained with excellent enantioselectivities and high kinetic resolution performance (s-factor up to 922). Additionally, this method is particularly effective for preparing some useful optically pure alkyl allylic alcohols, such as the key components towards the synthesis of prostaglandins and naturally occurring matsutakeols, which are difficult to access via other asymmetric reactions. Mechanistic studies revealed that the efficient kinetic resolution might be due to the significant distinction of the η2-coordination between the (R)- and (S)-allylic alcohols with the iridium/(phosphoramidite, olefin) complex.
Introduction
1,4-Enynes are important and versatile synthons in chemical synthesis due to the easy elaboration of the alkenyl and alkynyl groups towards the construction of fine chemicals, pharmaceuticals, and natural products.1 Accordingly, various synthetic approaches to 1,4-enynes have been well developed so far.2 In particular, the synthesis of enantioenriched 1,4-enynes via transition metal (Cu, Rh and Ir)–catalyzed asymmetric allylic alkynylation of allylic substrates has drawn considerable attention in the past decade (Scheme 1A).3–8 In 2011, Hoveyda pioneered the asymmetric allylic alkynylation of allylic phosphates employing Cu-catalysis, but moisture-sensitive pre-activated alkynylaluminum reagents were required as nucleophiles.3 Subsequently, significant progress of Cu-catalyzed asymmetric allylic alkynylation for (Z)-allylic phosphates by directly using terminal alkynes as nucleophiles was achieved by Sawamura and Ohmiya in 2013,4 leading to moderate product enantioselectivities. In 2018, to expand the chemistry of chiral guanidine, Tan and Lee developed a Cu/guanidine complex-catalyzed highly enantioselective allylic alkynylation of cyclic allylic bromides with terminal alkynes.5 Recently, Li reported an elegant synergistic Rh/Cu-catalyzed regio- and enantioselective allylic alkynylation by direct addition of terminal alkynes to alkyl allylic carbonates under neutral conditions.6 On the other hand, chiral phosphoramidite ligands have aptly demonstrated their excellent performance in iridium-catalyzed asymmetric allylic substitutions over the past 20 years.7 Consequently, in 2013, Carreira utilized [Ir(cod)Cl]2 and the Carreira-type chiral (phosphoramidite, olefin) ligand as a catalyst, and successfully achieved the synthesis of enantioenriched 1,4-enynes by means of asymmetric alkynylation of non-activated racemic secondary allylic alcohols with potassium alkynyltrifluoroborates, albeit with a limited substrate scope involving only aryl-substituted allylic alcohols (Scheme 1B).8 Despite extensive efforts that have been devoted to accessing chiral 1,4-enynes by asymmetric allylic alkynylation, to date, successful examples are still quite rare and in most cases pre-activated allylic substrates are required. Therefore, it is still a challenging and desirable task to perform asymmetric allylic alkynylation reactions by directly employing readily available non-activated allylic alcohols.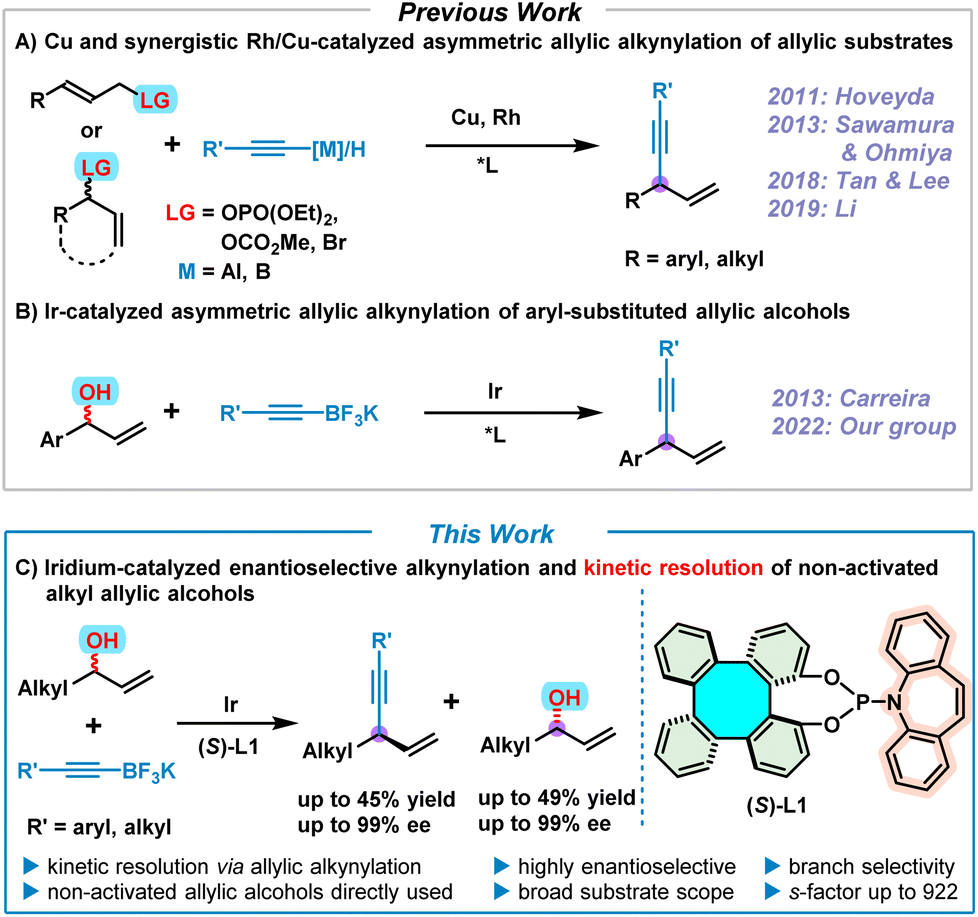 | ||
| Scheme 1 (A) Cu and synergistic Rh/Cu-catalyzed asymmetric allylic alkynylation of allylic substrates; (B) Ir-catalyzed asymmetric allylic alkynylation of aryl-substituted allylic alcohols; (C) iridium-catalyzed enantioselective alkynylation and kinetic resolution of non-activated alkyl allylic alcohols. | ||
Over the years, we have been developing tetraphenylene-derived chiral ligands/catalysts and investigating their applications in asymmetric catalysis.9 Amongst these tetraphenylene-based asymmetric catalysts, enantioenriched 1,16-dihydroxytetraphenylene (DHTP) stands out.9f–h,10 More recently, our group developed a series of phosphoramidite ligands based on the (S)-DHTP framework, of which the (S)-DHTP derived (phosphoramidite, olefin) ligand (S)-L1 exhibited excellent efficiency in the iridium-catalyzed asymmetric allylic alkynylation of allylic alcohols.9h This Ir/(S)-L1-catalyzed asymmetric allylic alkynylation led to the formation of a variety of 1,4-enynes with high regio- and enantioselectivities. However, both our work and that of Carreira are limited to aryl-substituted allylic alcohols. Iridium-catalyzed asymmetric allylic substitution of alkyl-substituted non-activated allylic alcohols still remains a challenge due to the fact that these reactions usually proceed with moderate regio- and enantio-selectivities,7c although significant developments have been achieved by Krische via π-allyliridium-C,O-benzoate-catalyzed allylic substitution of alkyl-substituted allylic acetates.11 It is therefore our aim to use alkyl allylic alcohols to expand the scope of this allylic alkynylation, making use of our Ir/(S)-L1 catalyst system. Herein, we report an efficient iridium-catalyzed enantioselective alkynylation and kinetic resolution7p,12 of non-activated alkyl allylic alcohols, providing both 1,4-enynes and unreacted enantioenriched alkyl allylic alcohols with high enantioselectivities (Scheme 1C).
Results and discussion
We commenced our investigation by allowing the reaction to proceed between a racemic alkyl allylic alcohol (±)-1a and potassium (4-methoxyphenyl)ethynyl trifluoroborate 2a under the conditions previously developed for aryl allylic alcohols (Table 1).9h Thus, in the presence of the Ir/(S)-L1 catalyst, KHF2 (1.5 equiv.), CF3CO2H (1.5 equiv.) and nBu4NBr (10 mol%), the treatment of (±)-1a (0.4 mmol) with 2a (0.6 mmol, 1.5 equiv.) in 1,4-dioxane (0.4 mL) at room temperature (25 °C) for 8 hours afforded (R)-3a in 28% yield with 78% ee, while the unreacted (S)-1a was recovered in 58% yield with 80% ee (s = 20, Table 1, entry 1). Preliminary results indicated that kinetic resolution occurred during the reaction.7p,12 We then attempted to improve the kinetic resolution performance by reducing the quantity of trifluoroborate 2a (Table 1, entries 2–5). Gratifyingly, significant improvement was achieved when 1.2 equivalents of 2a were used, leading to the formation of the alkynylation product (R)-3a in 34% yield with 94% ee, and (S)-1a in 45% yield with 96% ee (s = 127, Table 1, entry 3). When the reaction was carried out at 0 °C under the above optimal conditions, to our delight, product (R)-3a and recovered (S)-1a were obtained in excellent yields and enantioselectivities with the best selectivity factor (s = 180, Table 1, entry 6). A variety of (S)-DHTP-derived phosphoramidite ligands were further examined (Table 1, entries 7–10). In contrast to (S)-L1, ligand (S)-L2 without an olefin moiety was not effective in this reaction. Product (R)-3a in 14% yield with −56% ee and racemic 1a in 63% yield were provided, indicating that no kinetic resolution occurred (Table 1, entry 7). Ligands (S)-L3, (S)-L4, and (S,S,S)-L5 failed to promote similar reactions (Table 1, entries 8–10). Moreover, when the Carreira-type (phosphoramidite, olefin) ligand (S)-L6 was used, a slightly lower selectivity factor (s = 124) was achieved, as compared with the result of (S)-L1 (Table 1, entry 11). These results indicate that the use of a (phosphoramidite, olefin) ligand is essential for this kinetic resolution/allylic alkynylation. Deduction of the loading of [Ir(cod)Cl]2 and (S)-L1 to 2 mol% and 8 mol%, respectively, led to a lower selectivity factor (s = 109, Table 1, entry 12). Finally, we found that both the increase and decrease of reaction time were detrimental to the kinetic resolution performance (s = 12–65, Table 1, entries 13–15). The absolute configuration of 1,4-enyne 3a was assigned to be R,2k,6 while the configuration of unreacted 1a was assigned as S,13,14 by comparison with literature data.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 2a × [eq.] | Ligand | Temp./time [°C]/[h] | (R)-3a | (S)-1a | C [%] | s |
| Yield (ee) [%] | Yield (ee) [%] | ||||||
| a Reaction conditions: (±)-1a (0.4 mmol, 1.0 equiv.), [Ir(cod)Cl]2 (4 mol%), ligand (16 mol%), KHF2 (equivalent to 2a), CF3CO2H (1.5 equiv.), nBu4NBr (10 mol%), 1,4-dioxane (0.4 mL). Notes: nBu4NBr acted as a phase transfer catalyst, CF3CO2H acted as an activator of the –OH group and KHF2 acted as a stabilizer of potassium alkynyltrifluoroborates. Yield of the isolated product. ee% was determined by chiral HPLC analysis. b Yield was determined by 1H NMR with 1,3,5-trimethylbenzene as the internal standard. c [Ir(cod)Cl]2 (2 mol%), ligand (8 mol%). d Selectivity factor (s) = In[(1 − C)(1 − ees)]/ln[(1 − C)(1 + ees)], conversion (C) = ees/(ees + eep).12 “—”: without testing. | |||||||
| 1 | 1.5 | (S)-L1 | rt/8 | 28(78) | 58(80) | 50.6 | 20 |
| 2 | 1.3 | (S)-L1 | rt/8 | 31(86) | 51(86) | 50.0 | 37 |
| 3 | 1.2 | (S)-L1 | rt/8 | 34(94) | 45(96) | 50.5 | 127 |
| 4 | 1.0 | (S)-L1 | rt/8 | 34(90) | 44(88) | 49.4 | 55 |
| 5 | 0.8 | (S)-L1 | rt/8 | 37(52) | 43(79) | 60.3 | 7 |
| 6 | 1.2 | (S)-L1 | 0/8 | 39(95) | 43(98) | 50.8 | 180 |
| 7b | 1.2 | (S)-L2 | 0/8 | 14(−56) | 63(0) | — | — |
| 8b | 1.2 | (S)-L3 | 0/8 | 0(—) | 78(0) | — | — |
| 9b | 1.2 | (S)-L4 | 0/8 | 0(—) | 99(0) | — | — |
| 10b | 1.2 | (S,S,S)-L5 | 0/8 | 0(—) | 96(0) | — | — |
| 11b | 1.2 | (S)-L6 | 0/8 | 36(95) | 45(91) | 48.9 | 124 |
| 12b,c | 1.2 | (S)-L1 | 0/8 | 33(95) | 51(86) | 47.5 | 109 |
| 13 | 1.2 | (S)-L1 | 0/9 | 41(91) | 43(90) | 49.7 | 65 |
| 14 | 1.2 | (S)-L1 | 0/10 | 33(70) | 50(71) | 50.4 | 12 |
| 15 | 1.2 | (S)-L1 | 0/6 | 30(69) | 58(79) | 53.4 | 13 |

With the optimal conditions established, we next investigated the substrate scope of potassium alkynyltrifluoroborates (Table 2). Thus, a range of aromatic and aliphatic alkynyltrifluoroborates 2 were allowed to react with (±)-1a to give the corresponding 1,4-enynes (R)-3a–p and unreacted (S)-1a in good to excellent kinetic resolution performance (s-factor up to 922). Aromatic alkynyltrifluoroborates 2a–j bearing either electron-donating groups (methoxyl and tert-butyl) or electron-withdrawing groups (F, Cl, Br and CF3) on the phenyl ring were applicable reaction partners, affording the corresponding products (R)-3a–j in good to excellent selectivities (s = 58–922), accompanied by unreacted (S)-1a in high yields (43–49%) and excellent enantioselectivities (90–99% ee). 2-Naphthyl substituted alkynyltrifluoroborate 2k could also be employed to give (R)-3k and unreacted (S)-1a, albeit with a slightly lower selectivity (s = 74). A heteroaromatic substrate, such as (3-thienyl)alkynyltrifluoroborate 2l, was also tolerated, furnishing 1,4-enyne (R)-3l (25% yield and 96% ee) and unreacted (S)-1a (49% yield and 98% ee) with an outstanding selectivity factor (s = 226). Alternatively, aliphatic alkynyltrifluoroborates with different substituents were also allowed to react under the standard conditions, leading to the corresponding 1,4-enynes (R)-3m–o and unreacted (S)-1a with excellent selectivities (s = 303–790). It is noteworthy that an aliphatic alkynyltrifluoroborate, bearing a conjugated C–C double bond, also efficiently gave (R)-3p and (S)-1a in good yields, as well as excellent enantioselectivities and selectivity (s = 328).
| a Reaction conditions: (±)-1a (0.4 mmol, 1.0 equiv.), 2 (0.48 mmol, 1.2 equiv.), [Ir(cod)Cl]2 (4 mol%), (S)-L1 (16 mol%), KHF2 (1.2 equiv.), CF3CO2H (1.5 equiv.), nBu4NBr (10 mol%), 1,4-dioxane (0.4 mL). Yield of the isolated product. ee% was determined by chiral HPLC analysis. Selectivity factor (s) = ln[(1 − C)(1 − ees)]/ln[(1 − C)(1 + ees)], conversion (C) = ees/(ees + eep). |
|---|

|
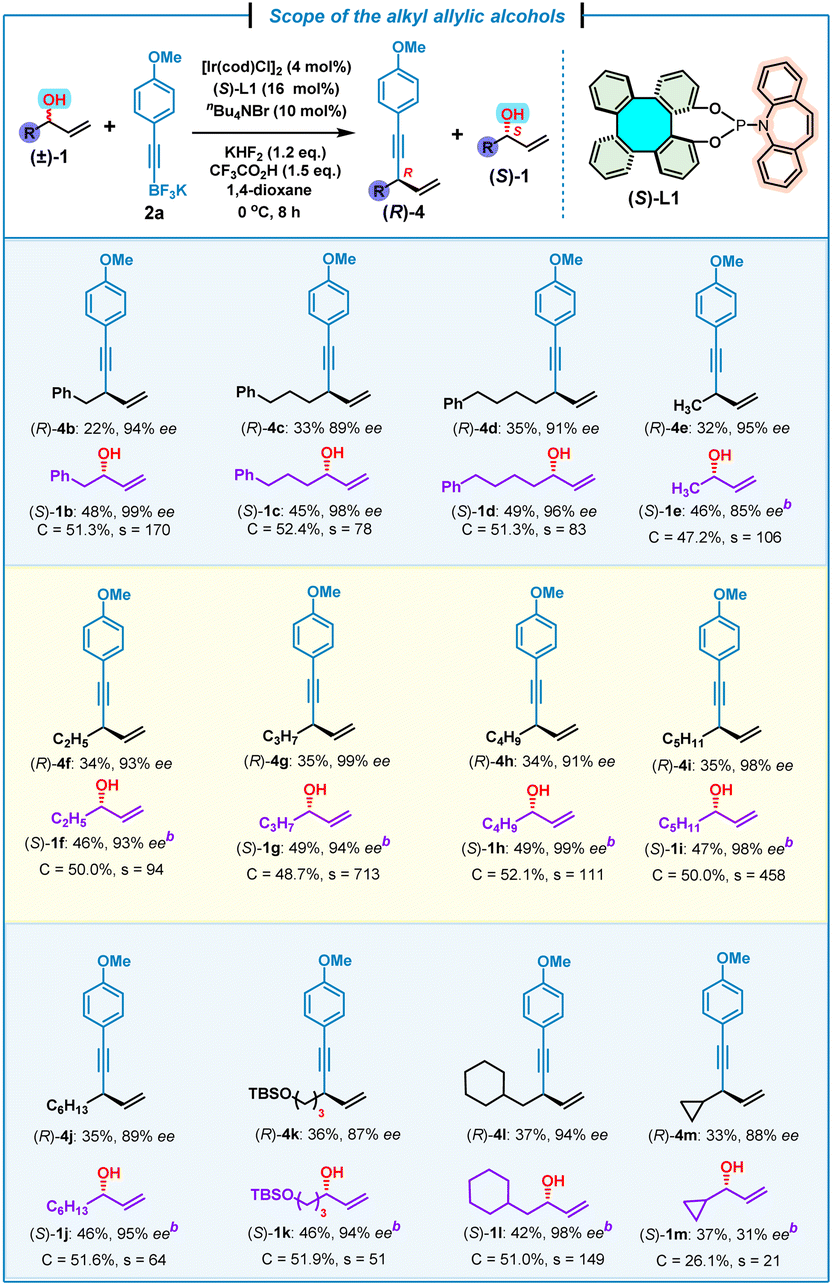
Next, the scope of alkyl allylic alcohols (±)-1 was examined by using potassium (4-methoxyphenyl)ethynyl trifluoroborate 2a as the model substrate (Table 3). Various alkyl allylic alcohols bearing (phenyl)methyl, (phenyl)propyl and (phenyl)butyl groups were all suitable for this kinetic resolution process, affording the corresponding 1,4-enynes (R)-4b–d and (S)-1b–d in good yields and high enantioselectivities (s = 83–170). Methyl-, ethyl-, propyl-, butyl-, pentyl- and hexyl-substituted chiral 1,4-enynes (R)-4e–j were also obtained successfully together with (S)-1e–j with good to excellent kinetic resolution performance factors (s = 64–713). An allylic alcohol bearing a tert-butyldimethylsilyl (TBS)–protected terminal hydroxyl group at the alkyl was found to be compatible with this kinetic resolution/allylic alkynylation, affording 1,4-enyne (R)-4k and (S)-1k in comparable yields and enantioselectivities, but with a moderate selectivity of 51. In addition, a β-cyclohexyl-substituted allylic alcohol was also proved to be suitable with this reaction, leading to the expected (R)-4l and (S)-1l in a high kinetic resolution selectivity (s = 149). It was uncovered that a more sterically hindered cyclopropyl-substituted allylic alcohol led to the corresponding (R)-4m and (S)-1m with a relatively low kinetic resolution performance (s = 21), which pointed to the significant steric hindrance effect for this kinetic resolution/allylic alkynylation.
| a Reaction conditions: (±)-1 (0.4 mmol, 1.0 equiv.), 2a (0.48 mmol, 1.2 equiv.), [Ir(cod)Cl]2 (4 mol%), (S)-L1 (16 mol%), KHF2 (1.2 equiv.), CF3CO2H (1.5 equiv.), nBu4NBr (10 mol%), 1,4-dioxane (0.4 mL). Yield of the isolated product. ee% was determined by chiral HPLC analysis. b ee% of (S)-1e–m was determined by chiral HPLC analysis after derivatization (see the ESI). Selectivity factor (s) = ln[(1 − C)(1 − ees)]/ln[(1 − C)(1 + ees)], conversion (C) = ees/(ees + eep). TBS = tert-butyldimethylsilyl. |
|---|
|
It is well known that chiral alkyl allylic alcohol fragments are important structural motifs prevalent in many bioactive natural products and pharmaceutical molecules, such as prostaglandins and their analogs14 as well as matsutakeols15 (Scheme 2A). To demonstrate the synthetic utilities of this kinetic resolution process, several scale-up reactions for preparing some useful optically pure alkyl allylic alcohols were conducted. For example, (S)-1a is the key synthon for the asymmetric synthesis of bimatoprost and latanoprost.14 Thus, a scale-up reaction of (±)-1a (4.0 mmol) and 2a (4.8 mmol) was performed under the standard conditions, affording (S)-1a in 43% yield (0.28 g) with 96% ee. Subsequently, on treatment of the obtained (S)-1a with 3,5-dinitrobenzoyl chloride in the presence of Et3N,15a (S)-1a′ as a white solid was obtained, and the absolute configuration (S) was further confirmed by an X-ray crystallographic analysis (Scheme 2B).16 Alcohol (S)-1i, also named (S)-matsutakeol,15b,c is the key synthon of prostaglandins PGE2 and PGF2α.14 Likewise, a scale-up preparation by treatment of (±)-1i (4.0 mmol) with 2a (4.8 mmol) under the standard conditions afforded (S)-1i in 48% yield (0.246 g) with an excellent enantioselectivity (97% ee) (Scheme 2C). In addition, (R)-matsutakeol is the major flavor substance that occurs naturally in matsutake and has a stronger mushroom smell than (S)-matsutakeol.15a,b Although remarkable progress has been made in the synthesis of (R)-matsutakeol,15d–f the development of a concise and scalable synthetic route to (R)-matsutakeol is still required. We were delighted to find that the use of (R)-L1 instead of (S)-L1 under the standard conditions led to the reaction between (±)-1i (4.0 mmol) and 2a (4.8 mmol), affording successfully (R)-matsutakeol [(R)-1i] in 47% yield with 99% ee. These results indicate that our methodology indeed offers an efficient route to valuable optically pure alkyl allylic alcohols, which are otherwise difficult to access via other asymmetric methods.
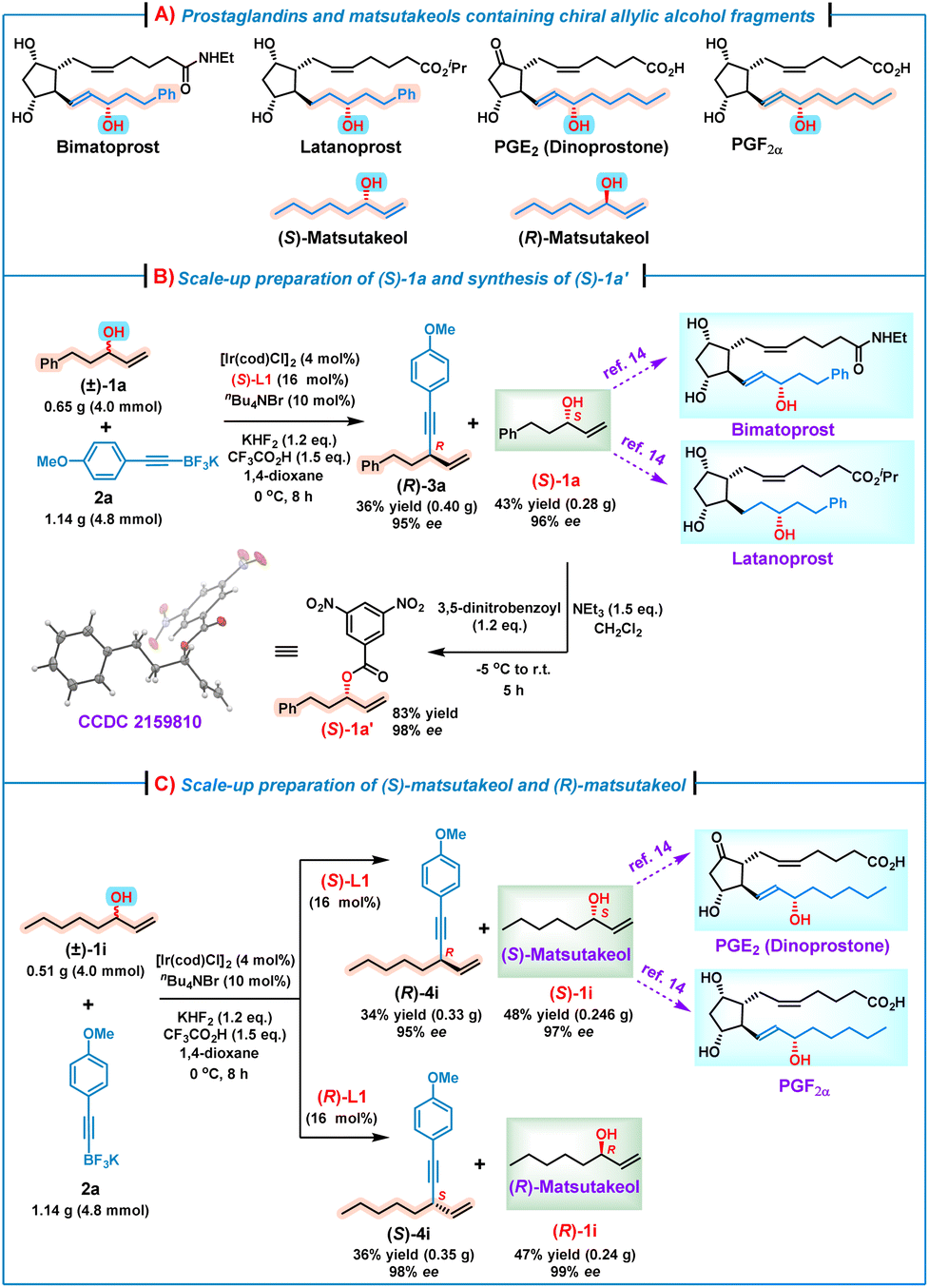 | ||
| Scheme 2 The scale-up reactions and synthetic utilities of this kinetic resolution/allylic alkynylation. | ||
To further demonstrate the synthetic utilities of the accessible enantiopure allylic alcohols, synthetic transformations of enantiopure (S)-1a (98% ee) were investigated (Scheme 3). In the presence of PPh3 and diethyl azodicarboxylate, (S)-1a could undergo a Mitsunobu reaction with N-hydroxyphthalimide, giving (R)-5 in 84% yield with 99% ee.17 In addition, treatment of (S)-1a with trichloroacetyl isocyanate followed by hydrolysis furnished carbamate (S)-6 in 86% yield with >99% ee.18 Moreover, the double bond of (S)-1a could be converted to a cyclopropyl group via the Simmons–Smith reaction, affording (S)-7 in 85% yield with 99% ee.17b,19 Finally, hydrogenation of the alkene moiety of (S)-1a on Pd/C by using H2 (1.0 atm balloon) gave an enantiopure dialkyl alcohol (R)-8 in 98% yield with 99% ee.20
 | ||
| Scheme 3 Synthetic transformations of (S)-1a. See the ESI‡ for detailed conditions. DEAD = diethyl azodicarboxylate. DCM = dichloromethane. DME = dimethoxyethane. | ||
To obtain an insight into the mechanism, several control experiments and 31P NMR experiments were conducted (Scheme 4). First, in the presence of (S)-L1, treatment of substrate (S)-1a (96% ee) with 2a under the standard conditions gave (S)-1a in 78% yield with 98% ee, while only a trace of alkynylation product 3a was observed. For comparison, the use of (R)-L1 as the chiral ligand triggered the reaction between (S)-1a (96% ee) and 2a under the standard conditions to generate (S)-3a in 74% yield with 99% ee by consuming all (S)-1a (Scheme 4A). These results suggest that (S)-1a mismatched with the Ir/(S)-L1 catalyst system, leading to no alkynylation product 3a and (S)-1a was recovered. In contrast, (S)-1a matched with the Ir/(R)-L1 catalyst system to allow for the reaction of all (S)-1a to afford 1,4-enyne (S)-3a in a high yield and an excellent enantioselectivity. In addition, mixing of (R)-1a (0.096 mmol) with [Ir(cod)Cl]2 (0.016 mmol) and (S)-L1 (0.064 mmol) in CDCl3 produced an (η2-allylic alcohol)iridium(I) complex (S,S,R)-A, which was evidenced by 31P NMR spectroscopy (Scheme 4B). As shown in Scheme 3B, two doublets at δ = 108.6 (2JP–P = 30.2 Hz) and 103.1 ppm (2JP–P = 29.9 Hz) indicate two non-equivalent phosphorus donors at the iridium center. For comparison, the 31P NMR chemical shift of (S)-L1 is δ = 134.8 ppm and the 31P NMR spectrum of the complex Ir[(S)-L1]2Cl shows only a singlet at δ = 135.6 ppm (see ESI, Fig. S9‡). These features strongly suggest that the complex (S,S,R)-A consists of two (S)-L1 coordinated to one iridium center, of which one (S)-L1 coordinated in a P, olefin-bidentate fashion and the other (S)-L1 acted as a P-monodentate ligand, while the allylic alcohol coordinated with iridium in an η2-fashion via the olefin motif.7h Nonetheless, treatment of (S)-1a (0.096 mmol) with [Ir(cod)Cl]2 (0.016 mmol) and (S)-L1 (0.064 mmol) under the same conditions did not lead to the formation of the expected (η2-allylic alcohol)iridium(I) complex (S,S,S)-A (Scheme 4C), while the 31P NMR spectrum (only one singlet at δ = 135.7 ppm) and HRMS (ESI) ([M–Cl]+m/z = 1307.2744) analyses indicate that the complex Ir[(S)-L1]2Cl was formed.
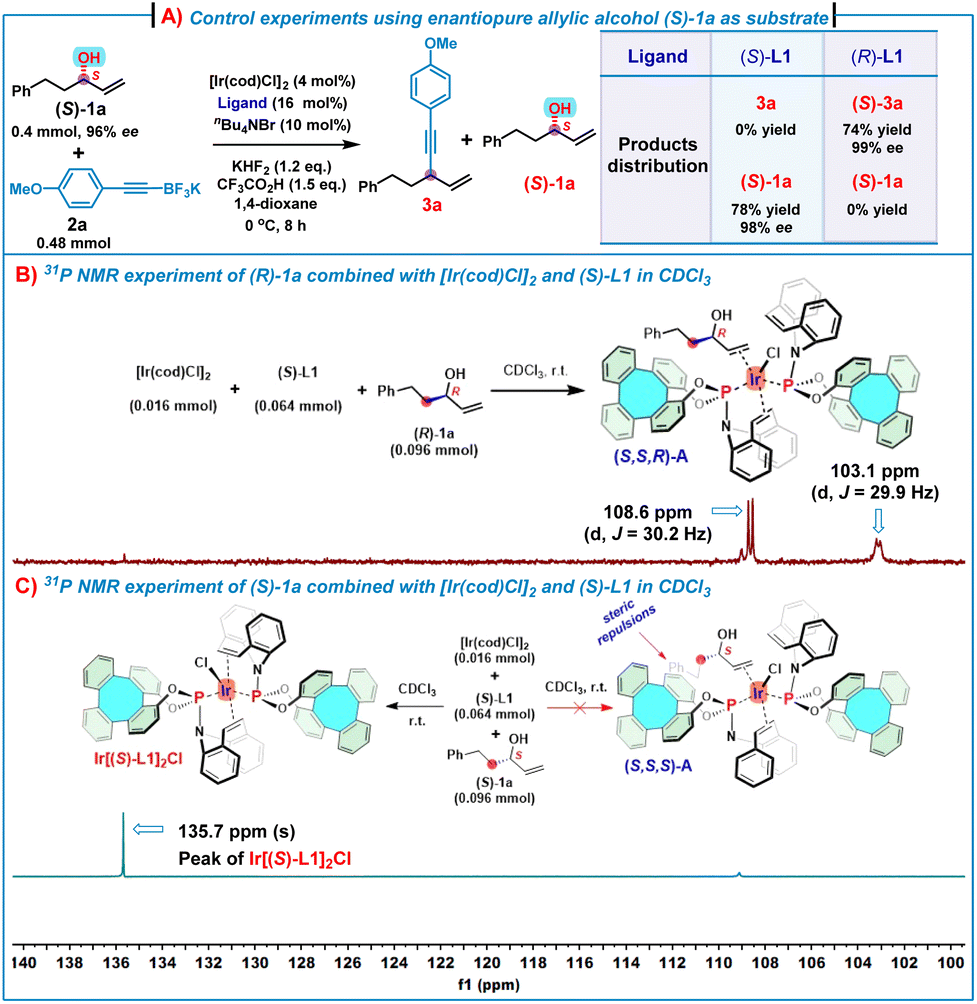 | ||
| Scheme 4 Control experiments and NMR experiments for mechanistic studies. | ||
On the basis of the experimental results and mechanistic studies on the iridium/(phosphoramidite, olefin) complexes,7h a plausible catalytic reaction cycle was proposed (Scheme 5). In principle, on mixing of the allylic alcohol (±)-1 with [Ir(cod)Cl]2 and (S)-L1, the η2-coordination of the olefin moiety to the iridium center should result in the formation of two diastereomers A1 and A2. However, the coordination of (R)-1 with the chiral Ir/(S)-L1 catalyst likely forms the favored intermediate A1, because the intermediate A2 is disfavored due to the severe steric repulsions between the alkyl moiety of (S)-1 and the phenyl moiety of (S)-L1. Subsequently, the favorable intermediate A1 undergoes an acid-promoted oxidative addition to furnish (η3-allyl)iridium(III) species B, which leads to olefin–iridium complex C through the nucleophilic attack by 2 on the allyl fragment. Finally, a ligand exchange occurs with (R)-allylic alcohol to complete the catalytic cycle. As a result, (R)-1 is competitively consumed by an enantioselective allylic alkynylation, allowing an efficient kinetic resolution to generate the alkynylation product (R)-3, as well as the unreacted enantioenriched (S)-1.
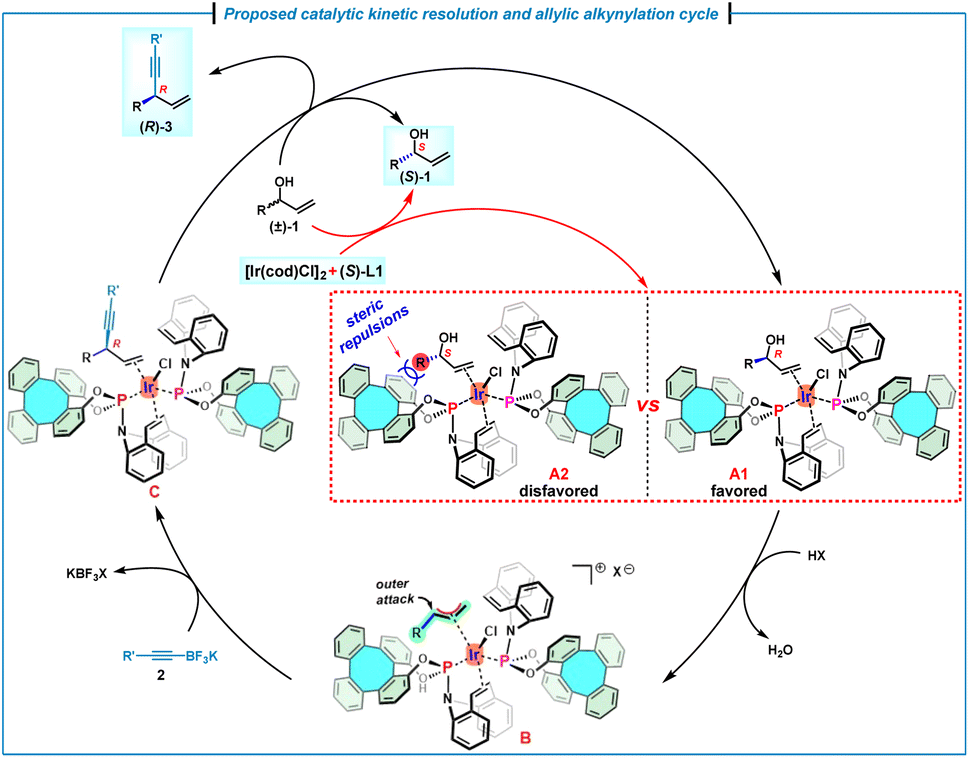 | ||
| Scheme 5 Proposed catalytic kinetic resolution and the allylic alkynylation cycle. | ||
Conclusions
In summary, we have disclosed an efficient method for the kinetic resolution/enantioselective alkynylation of readily available alkyl-substituted non-activated allylic alcohols enabled by an iridium/(phosphoramidite, olefin) catalyst system. A broad range of racemic alkyl allylic alcohols and potassium alkynyltrifluoroborates are compatible under the optimized conditions, affording both chiral 1,4-enynes and unreacted alkyl allylic alcohols with excellent enantiopurities and with an s-factor up to 922. In addition, the facile and scalable access towards useful synthons of prostaglandins and their analogs as well as naturally occurring (R)- and (S)-matsutakeols demonstrated aptly the potential of this kinetic resolution process in asymmetric synthesis. Mechanistic studies by means of control experiments revealed that the origin of enantio-recognition and efficient kinetic resolution might be due to the significant distinction of the η2-coordination between the (R)- and (S)-allylic alcohols with the Ir/(S)-L1 complex.Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.‡ Crystallographic data for compound (S)-1a′ has been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC 2159810.Author contributions
J. G. performed the experimental work. H.-R. M., W.-B. X. and L. F. synthesized some starting materials and collected and analysed the spectroscopic data. J.-F. C., H. N. C. W. and Y.-Y. Z. conceived the project and wrote the manuscript. All authors discussed the results and contributed to the preparation of the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge with thanks financial support from the Research Start-up Fund of Southern University of Science and Technology, Guangdong Provincial Key Laboratory of Catalysis (2020B121201002), the Innovation and Technology Commission (Hong Kong) in the form of subsidy to the State Key Laboratory of Synthetic Chemistry, and the University Development Fund Grants from the Chinese University of Hong Kong (Shenzhen). We thank Dr Xiaoyong Chang for the X-ray crystallographic analysis. H. N. C. W. acknowledges with gratitude his appointment as a Long-term Distinguished Visiting Professor by Southern University of Science and Technology.Notes and references
- (a) T. P. Heffron and T. F. Jamison, Org. Lett., 2003, 5, 2339–2342 CrossRef CAS PubMed; (b) A. Buzas and F. Gagosz, J. Am. Chem. Soc., 2006, 128, 12614–12615 CrossRef CAS PubMed; (c) B. A. Bhanu Prasad, F. K. Yoshimoto and R. Sarpong, J. Am. Chem. Soc., 2005, 127, 12468–12469 CrossRef CAS PubMed; (d) X. Shi, D. J. Gorin and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 5802–5803 CrossRef CAS PubMed; (e) T. Sato, T. Onuma, I. Nakamura and M. Terada, Org. Lett., 2011, 13, 4992–4995 CrossRef CAS PubMed; (f) D. Vasu, A. Das and R.-S. Liu, Chem. Commun., 2010, 46, 4115–4117 RSC; (g) G. Stork and M. Isobe, J. Am. Chem. Soc., 1975, 97, 4745–4746 CrossRef CAS PubMed; (h) K. Iida and M. Hirama, J. Am. Chem. Soc., 1994, 116, 10310–10311 CrossRef CAS; (i) V. Hickmann, M. Alcarazo and A. Fürstner, J. Am. Chem. Soc., 2010, 132, 11042–11044 CrossRef CAS PubMed; (j) X.-F. Wei, X.-W. Xie, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2017, 139, 4647–4650 CrossRef CAS PubMed; (k) S. Gao, H. Liu, C. Yang, Z. Fu, H. Yao and A. Lin, Org. Lett., 2017, 19, 4710–4713 CrossRef CAS PubMed; (l) Y.-L. Su, L.-L. Li, X.-L. Zhou, Z.-Y. Dai, P.-S. Wang and L.-Z. Gong, Org. Lett., 2018, 20, 2403–2406 CrossRef CAS PubMed; (m) X. Fang, Q. Li, R. Shi, H. Yao and A. Lin, Org. Lett., 2018, 20, 6084–6088 CrossRef CAS PubMed; (n) X.-F. Wei, T. Wakaki, T. Itoh, H.-L. Li, T. Yoshimura, A. Miyazaki, K. Oisaki, M. Hatanaka, Y. Shimizu and M. Kanai, Chem, 2019, 5, 585–599 CrossRef CAS; (o) M. Jacobson, R. E. Redfern, W. A. Jones and M. H. Aldridge, Science, 1970, 170, 542–544 CrossRef CAS PubMed.
- (a) Y. Huang, C. Ma, S. Liu, L.-C. Yang, Y. Lan and Y. Zhao, Chem, 2021, 7, 812–826 CrossRef CAS; (b) X.-Y. He and Z.-X. Wang, Chem. Commun., 2021, 57, 11988–11991 RSC; (c) Q. Yang, Y. Zhou, J. Chen, X. He, J. Xu, F. Y. Kwong and B. Fan, Eur. J. Org. Chem., 2015, 2015, 5330–5333 CrossRef CAS; (d) V. V. Grushin and H. Alper, J. Org. Chem., 1992, 57, 2188–2192 CrossRef CAS; (e) Y.-X. Li, Q.-Q. Xuan, L. Liu, D. Wang, Y.-J. Chen and C.-J. Li, J. Am. Chem. Soc., 2013, 135, 12536–12539 CrossRef CAS PubMed; (f) P. Xie, Z. Sun, S. Li, L. Zhang, X. Cai, W. Fu, X. Yang, Y. Liu, X. Wo and T.-P. Loh, Org. Lett., 2020, 22, 1599–1604 CrossRef CAS PubMed; (g) Q. Wang and Y. Kobayashi, Tetrahedron Lett., 2010, 51, 5592–5595 CrossRef CAS; (h) Z. Yang, R. K. Kumar, P. Liao, Z. Liu, X. Li and X. Bi, Chem. Commun., 2016, 52, 5936–5939 RSC; (i) S. Mondal, T. Pinkert, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2021, 60, 5688–5692 CrossRef CAS PubMed; (j) H. Li and A. Alexakis, Angew. Chem., Int. Ed., 2012, 51, 1055–1058 CrossRef CAS PubMed; (k) C. P. Grugel and B. Breit, Org. Lett., 2018, 20, 1066–1069 CrossRef CAS PubMed; (l) B. M. Trost, S. Hildbrand and K. Dogra, J. Am. Chem. Soc., 1999, 121, 10416–10417 CrossRef CAS; (m) M. Shirakura and M. Suginome, Angew. Chem., Int. Ed., 2010, 49, 3827–3829 CrossRef CAS PubMed; (n) S. Okusu, H. Okazaki, E. Tokunaga, V. A. Soloshonok and N. Shibata, Angew. Chem., Int. Ed., 2016, 55, 6744–6748 CrossRef CAS PubMed; (o) J.-F. Bai, K. Yasumoto, T. Kano and K. Maruoka, Angew. Chem., Int. Ed., 2019, 58, 8898–8901 CrossRef CAS PubMed.
- J. A. Dabrowski, F. Gao and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 4778–4781 CrossRef CAS PubMed.
- (a) Y. Makida, Y. Takayama, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2013, 52, 5350–5354 CrossRef CAS PubMed; (b) A. Harada, Y. Makida, T. Sato, H. Ohmiya and M. Sawamura, J. Am. Chem. Soc., 2014, 136, 13932–13939 CrossRef CAS PubMed.
- X.-Y. Cui, Y. Ge, S. M. Tan, H. Jiang, D. Tan, Y. Lu, R. Lee and C.-H. Tan, J. Am. Chem. Soc., 2018, 140, 8448–8455 CrossRef CAS PubMed.
- W.-Y. Huang, C.-H. Lu, S. Ghorai, B. Li and C. Li, J. Am. Chem. Soc., 2020, 142, 15276–15281 CrossRef CAS PubMed.
- (a) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS PubMed; (b) J. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539–2555 CrossRef CAS PubMed; (c) Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed; (d) S. L. Rössler, D. A. Petrone and E. M. Carreira, Acc. Chem. Res., 2019, 52, 2657–2672 CrossRef PubMed; (e) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef CAS PubMed; (f) Y.-N. Yu and M.-H. Xu, Chin. J. Chem., 2017, 75, 655–670 CAS; (g) H.-F. Tu, P. Yang, Z.-H. Lin, C. Zheng and S.-L. You, Nat. Chem., 2020, 12, 838–844 CrossRef CAS PubMed; (h) S. L. Rössler, S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 3603–3606 CrossRef PubMed; (i) C. Defieber, M. A. Ariger, P. Moriel and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 3139–3143 CrossRef CAS PubMed; (j) T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 15164–15165 CrossRef CAS PubMed; (k) Z.-T. He and J. F. Hartwig, Nat. Chem., 2019, 11, 177–183 CrossRef CAS PubMed; (l) X. Jiang and J. F. Hartwig, Angew. Chem., Int. Ed., 2017, 56, 8887–8891 CrossRef CAS PubMed; (m) G. Lipowsky, N. Miller and G. Helmchen, Angew. Chem., Int. Ed., 2004, 43, 4595–4597 CrossRef CAS PubMed; (n) M. Gärtner, S. Mader, K. Seehafer and G. Helmchen, J. Am. Chem. Soc., 2011, 133, 2072–2075 CrossRef PubMed; (o) J. Y. Hamilton, N. Hauser, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2014, 53, 10759–10762 CrossRef CAS PubMed; (p) T. Sandmeier and E. M. Carreira, Angew. Chem., Int. Ed., 2021, 60, 9913–9918 CrossRef CAS PubMed; (q) S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Nat. Catal., 2020, 3, 48–54 CrossRef CAS; (r) R. Jiang, L. Ding, C. Zheng and S.-L. You, Science, 2021, 371, 380–386 CrossRef CAS PubMed; (s) Q.-F. Wu, C. Zheng, C.-X. Zhuo and S.-L. You, Chem. Sci., 2016, 7, 4453–4459 RSC; (t) Q.-L. Xu, L.-X. Dai and S.-L. You, Chem. Sci., 2013, 4, 97–102 RSC; (u) X. Zhang, W.-B. Liu, H.-F. Tu and S.-L. You, Chem. Sci., 2015, 6, 4525–4529 RSC.
- J. Y. Hamilton, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 7532–7535 CrossRef CAS PubMed.
- (a) J.-W. Han, J.-X. Chen, X. Li, X.-S. Peng and H. N. C. Wong, Synlett, 2013, 24, 2188–2198 CrossRef CAS; (b) J.-W. Han, X. Li and H. N. C. Wong, Chem. Rec., 2015, 15, 107–131 CrossRef CAS PubMed; (c) J.-W. Han, X.-S. Peng and H. N. C. Wong, Natl. Sci. Rev., 2017, 4, 892–916 CrossRef CAS; (d) H.-Y. Peng, C.-K. Lam, T. C. W. Mak, Z. Cai, W.-T. Ma, Y.-X. Li and H. N. C. Wong, J. Am. Chem. Soc., 2005, 127, 9603–9611 CrossRef CAS PubMed; (e) C.-K. Hau, H. He, A. W. M. Lee, D. T. W. Chik, Z. Cai and H. N. C. Wong, Tetrahedron, 2010, 66, 9860–9874 CrossRef CAS; (f) G.-L. Chai, J.-W. Han and H. N. C. Wong, Synthesis, 2017, 49, 181–187 CAS; (g) G.-L. Chai, A. Q. Sun, D. Zhai, J. Wang, W.-Q. Deng, H. N. C. Wong and J. Chang, Org. Lett., 2019, 21, 5040–5045 CrossRef CAS PubMed; (h) J. Guo, W.-B. Xiong, H.-R. Ma, L. Fan, Y.-Y. Zhou, H. N. C. Wong and J.-F. Cui, Chem. Sci., 2022, 13, 4608–4615 RSC.
- (a) G.-L. Chai, B. Zhu and J. Chang, J. Org. Chem., 2019, 84, 120–127 CrossRef CAS PubMed; (b) G.-L. Chai, Y. Qiao, P. Zhang, R. Guo, J. Wang and J. Chang, Org. Lett., 2020, 22, 8023–8027 CrossRef CAS PubMed; (c) J.-F. Wen, W. Hong, K. Yuan, T. C. W. Mak and H. N. C. Wong, J. Org. Chem., 2003, 68, 8918–8931 CrossRef CAS PubMed; (d) J.-F. Cui, H. Huang and H. N. C. Wong, Synlett, 2011, 2011, 1018–1022 CrossRef; (e) P. Zhang, G.-L. Chai, E.-Z. Yao, L.-X. Guo, X.-Y. Liu and J. Chang, Org. Chem. Front., 2021, 8, 1575–1580 RSC; (f) E.-Z. Yao, G.-L. Chai, P. Zhang, B. Zhu and J. Chang, Org. Chem. Front., 2022, 9, 2375–2381 RSC.
- (a) A. T. Meza, T. Wurm, L. Smith, S. W. Kim, J. R. Zbieg, C. E. Stivala and M. J. Krische, J. Am. Chem. Soc., 2018, 140, 1275–1279 CrossRef CAS PubMed; (b) S. W. Kim, T. T. Schempp, J. R. Zbieg, C. E. Stivala and M. J. Krische, Angew. Chem., Int. Ed., 2019, 58, 7762–7766 CrossRef CAS PubMed; (c) W.-O. Jung, B. K. Mai, B. J. Spinello, Z. J. Dubey, S. W. Kim, C. E. Stivala, J. R. Zbieg, P. Liu and M. J. Krische, J. Am. Chem. Soc., 2021, 143, 9343–9349 CrossRef CAS PubMed.
- (a) E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974–4001 CrossRef CAS PubMed; (b) H. Pellissier, in Separation of Enantiomers, ed. M. Todd, 2014, ch. 3, pp. 75–122, DOI:10.1002/9783527650880.ch3; (c) V. S. Martin, S. S. Woodard, T. Katsuki, Y. Yamada, M. Ikeda and K. B. Sharpless, J. Am. Chem. Soc., 1981, 103, 6237–6240 CrossRef CAS; (d) M. Y. Jin, Y. Zhou, D. Xiao, Y. You, Q. Zhen, G. Tao, P. Yu and X. Xing, Angew. Chem., Int. Ed., 2022, 61, e202112993 CAS; (e) H. Wu, C. Margarita, J. Jongcharoenkamol, M. D. Nolan, T. Singh and P. G. Andersson, Chem. Sci., 2021, 12, 1937–1943 RSC; (f) C.-H. Zhang, Q. Gao, M. Li, J.-F. Wang, C.-M. Yu and B. Mao, Org. Lett., 2021, 23, 3949–3954 CrossRef CAS PubMed; (g) T. Sandmeier and E. M. Carreira, Org. Lett., 2021, 23, 2643–2647 CrossRef PubMed; (h) Y. Liu, S. Liu, D. Li, N. Zhang, L. Peng, J. Ao, C. E. Song, Y. Lan and H. Yan, J. Am. Chem. Soc., 2019, 141, 1150–1159 CrossRef CAS PubMed; (i) F. Severin, G. M. Fusi, C. Wartmann, J.-M. Neudörfl and A. Berkessel, Angew. Chem., Int. Ed., 2022, 61, e202201790 CrossRef CAS PubMed.
- N. Kanbayashi and K. Onitsuka, Angew. Chem., Int. Ed., 2011, 50, 5197–5199 CrossRef CAS PubMed.
- F. Zhang, J. Zeng, M. Gao, L. Wang, G.-Q. Chen, Y. Lu and X. Zhang, Nat. Chem., 2021, 13, 692–697 CrossRef CAS PubMed.
- (a) J. Liu, H. Li, C. Zheng, S. Lu, X. Guo, X. Yin, R. Na, B. Yu and M. Wang, Molecules, 2017, 22, 364 CrossRef PubMed; (b) W. H. Lee, I. H. Bae, B. M. Kim and Y.-B. Seu, Bull. Korean Chem. Soc., 2016, 37, 1910–1911 CrossRef CAS; (c) R. K. Rej, R. Kumar and S. Nanda, Tetrahedron, 2015, 71, 3185–3194 CrossRef CAS; (d) S. Kusuda, Y. Ueno and T. Toru, Tetrahedron, 1994, 50, 1045–1062 CrossRef CAS; (e) F. R. Bisogno, A. Rioz-Martínez, C. Rodríguez, I. Lavandera, G. de Gonzalo, D. E. Torres Pazmiño, M. W. Fraaije and V. Gotor, ChemCatChem, 2010, 2, 946–949 CrossRef CAS; (f) R. K. Rej and S. Nanda, Eur. J. Org. Chem., 2014, 2014, 860–871 CrossRef CAS.
- CCDC 2159810 contains the supplementary crystallographic data of (S)-1a′ for this paper.‡.
- (a) J. Chen, H.-M. Guo, Q.-Q. Zhao, J.-R. Chen and W.-J. Xiao, Chem. Commun., 2018, 54, 6780–6783 RSC; (b) Z. Liu and B. Breit, Org. Lett., 2018, 20, 300–303 CrossRef CAS PubMed.
- (a) D. C. Ramb, L. Kost and G. Haufe, Chimia, 2014, 68, 436–441 CrossRef CAS PubMed; (b) T. J. Donohoe, P. D. Johnson, R. J. Pye and M. Keenan, Org. Lett., 2004, 6, 2583–2585 CrossRef CAS PubMed; (c) M. Zhang, X. Zhao and S. Zheng, Chem. Commun., 2014, 50, 4455–4458 RSC.
- R. Larouche-Gauthier, C. J. Fletcher, I. Couto and V. K. Aggarwal, Chem. Commun., 2011, 47, 12592–12594 RSC.
- (a) H. Wang, R. Zhang, Q. Zhang and W. Zi, J. Am. Chem. Soc., 2021, 143, 10948–10962 CrossRef CAS PubMed; (b) S. Lauzon and T. Ollevier, Chem. Commun., 2021, 57, 11025–11028 RSC.
Footnotes |
| † Dedicated to Professor Keiji Maruoka of Kyoto University on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2159810. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04892b |
| This journal is © The Royal Society of Chemistry 2022 |