
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of a metastable uranium metal–organic framework isomer through non-equilibrium synthesis†
Sylvia L.
Hanna
 a,
Tekalign T.
Debela
c,
Austin M.
Mroz
c,
Zoha H.
Syed
a,
Kent O.
Kirlikovali
a,
Christopher H.
Hendon
*cd and
Omar K.
Farha
*ab
a,
Tekalign T.
Debela
c,
Austin M.
Mroz
c,
Zoha H.
Syed
a,
Kent O.
Kirlikovali
a,
Christopher H.
Hendon
*cd and
Omar K.
Farha
*ab
aDepartment of Chemistry and International Institute for Nanotechnology, Northwestern University, Evanston, IL 60208, USA. E-mail: o-farha@northwestern.edu
bDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA
cDepartment of Chemistry and Biochemistry, University of Oregon, Eugene, OR 97403, USA. E-mail: chendon@uoregon.edu
dMaterials Science Institute, University of Oregon, Eugene, OR 97403, USA
First published on 31st October 2022
Abstract
Since the structure of supramolecular isomers determines their performance, rational synthesis of a specific isomer hinges on understanding the energetic relationships between isomeric possibilities. To this end, we have systematically interrogated a pair of uranium-based metal–organic framework topological isomers both synthetically and through density functional theory (DFT) energetic calculations. Although synthetic and energetic data initially appeared to mismatch, we assigned this phenomenon to the appearance of a metastable isomer, driven by levers defined by Le Châtelier's principle. Identifying the relationship between structure and energetics in this study reveals how non-equilibrium synthetic conditions can be used as a strategy to target metastable MOFs. Additionally, this study demonstrates how defined MOF design rules may enable access to products within the energetic phase space which are more complex than conventional binary (e.g., kinetic vs. thermodynamic) products.
Introduction
Supramolecular isomerism occurs when more than one type of network superstructure exists for the same set of molecular building blocks.1 This phenomenon produces chemically similar materials with divergent properties, an occurrence with widespread implications. For example, protein isoforms and misfolding dramatically affect disease,2,3 polymorphism of pharmaceutical cocrystals influences drug development,4,5 and isomer selectivity in supramolecular arrays allows for engineered materials with exquisite structure–property control.6–8 Since isomer structure thus accounts for varying material performance, understanding the chemical processes that select for these structures will aid in the targeted synthesis of specific isomers.Synthetic selectivity between supramolecular isomers is broadly derived from the energetic relationships between them.9 Ideally, this phase space can be mapped on an energy landscape containing both local and global energy minima (Scheme 1). We note that for network superstructures, this phase space becomes complex and often contains more than merely binary energetic minima. For a classical non-dissipative supramolecular organization, the global energy minimum occurs under an equilibrium environment as the thermodynamic product. Conversely, local energy minima (e.g., kinetic or metastable products) arise under non-equilibrium environments and strongly depend on synthetic conditions.10,11 For instance, kinetic products are favored by rapid assembly and are accessed through kinetic control by shifting the height of existing activation energy barriers. Metastable products, however, are local energetic minima that become thermodynamically stable under the conditions of growth.12 Thus, thermodynamic control favors metastable products by shifting chemical equilibria using levers defined by Le Châtelier's principle. As an example, several studies have shown that removal of products during a reaction shifts the chemical equilibrium towards the product side and generates a metastable material.12–16
 | ||
| Scheme 1 Energy landscape of supramolecular products. Non-equilibrium non-dissipative syntheses produce metastable and kinetic products (top, pink), and equilibrium syntheses produce thermodynamic products (bottom, blue). | ||
This interplay between supramolecular structure, properties, and energetics appears in a class of self-organized network assemblies termed metal–organic frameworks (MOFs). MOFs comprise inorganic nodes and organic linkers which self-assemble into porous, crystalline materials with widespread functions.17–19 According to graph theory, one set of node and linker building blocks can connect in various arrangements to produce distinct topologies with unique underlying periodic nets.20,21 Due to this phenomenon, MOFs exhibit topological isomerism, producing various framework nets – often with unique properties – from the same set of linker and node components.22,23 However, energetic analyses of MOF isomer phase space are generally limited to binary thermodynamic and kinetic topological isomers in traditional transition metal-based MOFs.24–31 Widening the energetic understanding of MOF topological isomers involves: (1) defining what causes a certain topology to be favored, (2) determining how isomer energetics inform synthesis design rules, and (3) implementing thermodynamic control to access less explored metastable phases32,33 in non-traditional MOFs.34,35
In this study, we aim to address these three thrusts through an energetic analysis of topological isomerism in an actinide MOF system. Since structure–property relationships in actinide-based MOFs prove to be unlike those of traditional transition metal-based MOFs,36,37 we hypothesize that the energetic analysis of actinide MOF isomers will provide unique insight into their synthetic relationship. Here, we select a MOF system with a tetrahedral linker (tetrakis(4-carboxyphenyl)methane or TCPM) and a triangular, uranyl-based node (Fig. 1c). The assembly of these building blocks results in two distinct isomeric topologies: ctn (Fig. 1a) and bor (Fig. 1d).20 By systematically varying synthetic parameters, including temperature, modulator ratio, and reaction concentration, we aimed to selectively isolate one isomer as the thermodynamic product at high modulator amount, high temperature, and low concentration. Instead, we obtained bor (NU-1306) under high modulator and high temperature conditions, and we isolated ctn (NU-1305) under low reaction concentration conditions. To understand these conflicting data points, we examined the crystal structures and employed geometric strain analysis as well as density functional theory (DFT) calculations, which indicate that NU-1305 is thermodynamically favored by 8 kcal mol−1. By identifying that the synthesis of NU-1306 is in fact favored by thermodynamically controlled non-equilibrium conditions, we attribute this apparent mismatch between synthetic and energetic parameters to NU-1306 being a metastable phase. Identifying the relationship between structure and energetics in this study reveals how non-equilibrium synthetic conditions can be used as a strategy to target metastable MOFs. More generally, this investigation contributes to a broader understanding of isomeric options within the complex superstructure phase space.
 | ||
| Fig. 1 Topological isomers NU-1305 and NU-1306. Augmented topological nets for (a) ctn and (d) bor resulting from the assembly of (c) a triangular node building block and a tetrahedral linker building block. Crystal structures of (b) NU-1305, in the ctn topology, and (e) NU-1306, in the bor topology. In (b) and (e), uranium is shown in yellow, oxygen in red, and carbon in blue. Hydrogen atoms are omitted for clarity. | ||
Results and discussion
Following the synthetic procedure detailed in the ESI,† self-assembly of the triangular uranyl node and tetrahedral TCPM linker produced single crystals of ctn topology, named NU-1305 (Fig. 1b and S2†),38,39 and single crystals of bor topology, named NU-1306 (Fig. 1e and S3†). Both MOFs consist of a hexagonal bipyramidal uranyl node coordinated equatorially to three TCPM linkers through their carboxylate oxygen atoms (Fig. S5b†). Each MOF is (3,4)-connected with a formula of H+[UO2(TCPM)0.75]− or H3O+[UO2(TCPM)0.75]− (Fig. S10–S12†).NU-1305 exhibits one type of pore, comprised of eight nodes and six linkers. The largest sphere to fit in this pore possesses a radius of 19.5 Å (Fig. S4†), and further crystallographic details for NU-1305 can be found in the report by Hu et al.38 NU-1306 crystallized in the non-centrosymmetric P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m space group with a cubic unit cell of a = b = c = 20.9 Å and a solvent-accessible pore volume calculated by PLATON of 84%. Octahedral cages of 12.8 Å radii, each comprised of four nodes and six linkers, connect through vertices to form apertures with 22 Å radii (Fig. S5c and d†). Further crystallographic details can be found in Table S1.†
3m space group with a cubic unit cell of a = b = c = 20.9 Å and a solvent-accessible pore volume calculated by PLATON of 84%. Octahedral cages of 12.8 Å radii, each comprised of four nodes and six linkers, connect through vertices to form apertures with 22 Å radii (Fig. S5c and d†). Further crystallographic details can be found in Table S1.†
After confirming the formation of both topological isomers, we then aimed to determine their respective positions within the system's energy landscape. To do so, we examined which isomer required more thermodynamically favored synthetic conditions by systematically varying the modulator ratio, temperature, and reaction concentration. Since coordinating modulator is thought to compete with linkers for binding sites on metal ions, increased amounts of modulator should favor thermodynamic products by slowing down self-assembly kinetics.40,41 Published literature also indicates that higher temperatures favor thermodynamic MOF products by providing more energy to surmount higher activation energy barriers.25,27 Finally, lower reaction concentrations lead to less frequent reactant collisions, which effectively decrease reaction kinetics and afford thermodynamic MOF products.41 Thus, we anticipated that these synthetic levers would indicate which topology lies lower on the energy landscape.
Maintaining a 1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of UO2(NO3)2 (node) to TCPM (linker) in 0.8 mL N,N-dimethylformamide (DMF) at 120 °C, we first varied the formic acid (FA) modulator to DMF ratio from 0.03 FA:DMF to 0.15 FA:DMF. Powder X-ray diffraction (PXRD) revealed that samples with a FA:DMF ratio from 0.03 to 0.09 resulted in NU-1305, while samples with a FA:DMF ratio from 0.10 to 0.15 resulted in NU-1306 (Fig. 2a). Thus, decreased modulator produced NU-1305, while increased modulator produced NU-1306, pointing to NU-1306 as the thermodynamic product. We note that isomer selectivity using modulator may be influenced by the presence of missing linker defects (Fig. S18 and Table S4†).42
:1 ratio of UO2(NO3)2 (node) to TCPM (linker) in 0.8 mL N,N-dimethylformamide (DMF) at 120 °C, we first varied the formic acid (FA) modulator to DMF ratio from 0.03 FA:DMF to 0.15 FA:DMF. Powder X-ray diffraction (PXRD) revealed that samples with a FA:DMF ratio from 0.03 to 0.09 resulted in NU-1305, while samples with a FA:DMF ratio from 0.10 to 0.15 resulted in NU-1306 (Fig. 2a). Thus, decreased modulator produced NU-1305, while increased modulator produced NU-1306, pointing to NU-1306 as the thermodynamic product. We note that isomer selectivity using modulator may be influenced by the presence of missing linker defects (Fig. S18 and Table S4†).42
 | ||
| Fig. 2 Isolated isomers through tuned synthetic conditions. PXRD data of reaction products from systematically (a) increasing modulator amount while holding temperature and reaction concentration constant, (b) increasing reaction temperature while holding modulator amount and reaction concentration constant, or (c) increasing reaction concentration while holding modulator and temperature constant. Diffraction patterns are normalized. | ||
Next, we selected the conditions from above with a FA:DMF ratio of 0.09, which favored NU-1305 growth at 120 °C, as a starting point but varied the temperature of the reaction. Under identical synthetic conditions, increasing the temperature to 160 °C afforded a mixture of isomers, and heating at 170 °C favored the formation of pure NU-1306 (Fig. 2b). Similar to the requirement for increased modulator concentrations for the synthesis of NU-1306, the formation of NU-1306 at higher reaction temperatures implies that it is the thermodynamically favored topological isomer.
Finally, to test the effect of reaction concentration on isomer product, we selected the conditions above that formed NU-1305 (0.09 FA:DMF, 0.8 mL DMF, 120 °C) and diluted the reaction media, expecting to observe a transformation to the more thermodynamically stable NU-1306. Interestingly, we only observed the presence of NU-1305 and did not observe any transformation to NU-1306 by PXRD analysis (Fig. S7†). Instead, diluting the reaction conditions used to produce NU-1306 (0.10 FA:DMF, 0.8 mL DMF, 120 °C) by increasing the amount of DMF from 0.8 mL to 1.8 mL yielded PXRD patterns in which the characteristic peaks for NU-1306 decreased in intensity while those of NU-1305 grew (Fig. 2c). Contrary to previous systematic testing with modulator and temperature, these data indicate that lower reaction concentrations favor NU-1305, assigning NU-1305 as thermodynamically stable.
To gain insight into the phenomena guiding selectivity for one isomer over the other, we calculated the energetic minima of each topology using periodic hybrid Density Functional Theory (DFT) (Fig. S15, Table S3, ESI CIFs†). We further decomposed the energetic contributions by isolating the configurational differences in energy as a function of ligand and node geometry, respectively. Calculations revealed that NU-1305 is 8 kcal mol−1 more stable than NU-1306 (Fig. 3e). Through disassembly of the MOF, the NU-1305 node and linker conformations were found to be favored over those of NU-1306 by 4.8 and 3.2 kcal mol−1, respectively.
 | ||
| Fig. 3 Energetic analysis of isomer favorability. (a) Conversion of NU-1305 (left) to NU-1306 (right) and vice versa. Geometric analysis of (b) NU-1305 and (d) NU-1306 nodes. Only the immediately bound phenyl ring of one attached linker is shown for clarity (c) Standard deviation of linker dihedral angles for NU-1305 and NU-1306. In panels (a)–(d), uranium is shown in yellow, oxygen in red, and carbon in black. Hydrogen atoms are omitted for clarity. (e) Reaction coordinate diagram of NU-1305 and NU-1306 isomers. This panel is qualitative. | ||
A geometric analysis of building block components and crystal conformation also supports the energetic favorability of NU-1305 over NU-1306. The NU-1306 linker dihedral angles exhibit a higher standard deviation from the mean than those of NU-1305, showing increased distortion of the NU-1306 linker component (Fig. 3c and S14, Table S2†). The energetically unfavorable co-facial configuration of the linker in NU-1306 in turn destabilizes the node. While the node equatorial plane and the plane of the immediately-bound linker phenyl ring remain flush in NU-1305 (1.25° angle between planes, Fig. 3b), a 9.57° angle appears between these planes in NU-1306, adding strain on the node (Fig. 3d and S13†). Furthermore, the crystal density of NU-1306 lies at 0.470 g cm−3 while that of NU-1305 is 0.521 g cm−3, suggesting NU-1305 as the more energetically stable structure.24,40
In addition to the geometric analysis, conversion between topological isomers verified their respective locations within the system's energy landscape. Synthetic conditions involving high temperature and increased modulator suggest a higher activation energy barrier to form NU-1306, while reaction concentration-dependence, DFT calculations, and geometric analysis indicate that NU-1305 lies lower on the energy landscape (Fig. 3e). If both these deductions are true, then the energy required to convert from NU-1305 to NU-1306 should be higher than the energy needed to convert isomers in the opposite direction (Fig. 3e). To test this hypothesis, we heated DMF suspensions of individual MOF isomers in the presence of additional modulator and determined the least harsh conditions (lowest amount of modulator, lowest temperature) needed to observe isomer conversion. We found that NU-1305 converts to NU-1306 by submerging 5 mg of washed NU-1305 in fresh DMF with 200 μL FA at 170 °C (Fig. 3a and S8†), while conversion from NU-1306 to NU-1305 requires either 25 μL FA at 170 °C or 70 μL FA at 120 °C (Fig. 3a and S9†). From these experiments, it is apparent that converting from NU-1305 to NU-1306 requires harsher conditions (high modulator, high temperature) than converting from NU-1306 to NU-1305 (either low modulator and high temperature or high modulator and low temperature). These data reveal that NU-1305 does indeed lie lower in the energy landscape. Thus, these synthetic conversion parameters, paired with the geometric analysis of both MOF isomers, support the DFT findings of NU-1305 as the thermodynamically favored isomer.
The question then arises: how can the apparent mismatch between synthetic and energetic parameters in this system be explained? The answer to this question lies in the high temperature synthesis of NU-1306, which occurs with a starting 0.8 mL DMF volume and 0.09 FA:DMF ratio at 170 °C, well above the DMF solvent boiling point (153 °C). Because the reaction vessel used in this synthesis is not pressure-rated, evaporated solvent escapes as the reaction mixture is heated, decreasing the total reaction volume by the end of the reaction (Fig. 4b). This behavior is not observed for the reaction performed at 120 °C under otherwise analogous conditions, which produces NU-1305 (Fig. 4a).
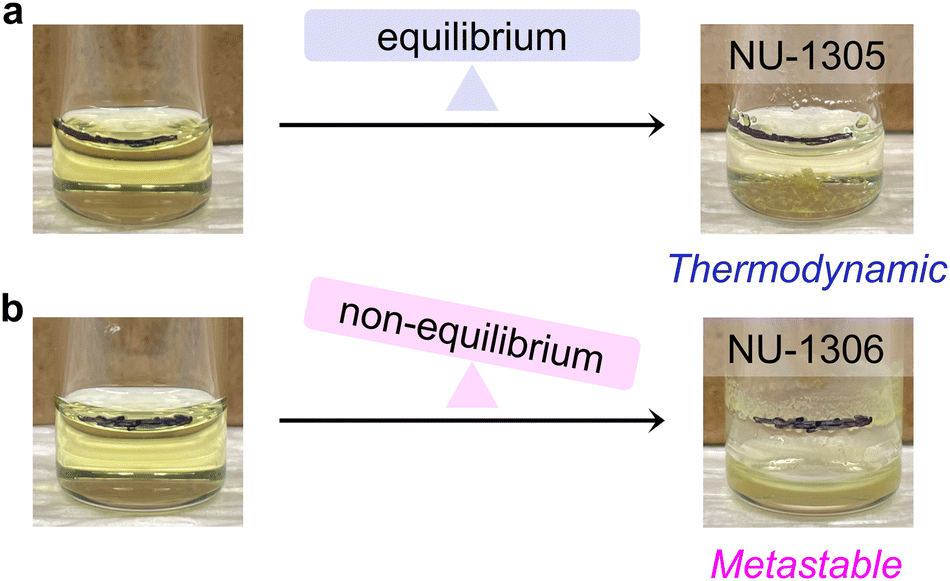 | ||
| Fig. 4 Non-equilibrium conditions for NU-1306 synthesis. Photographs of (a) NU-1305 and (b) NU-1306 before (left) and after (right) the reaction. A black horizontal line drawn on the glass vial indicates the initial solvent line before heat was added. Reaction conditions are identical (0.9 FA:DMF, 0.8 mL DMF, 1.6:1 node:linker) except that (a) was heated at 120 °C for 24 hours while (b) was heated at 170 °C for 1 hour. | ||
Thus, multiple factors may potentially influence the selection of NU-1306 over NU-1305 under these high temperature conditions, including (1) increased effective reaction concentration, (2) energy added to the system from high temperature, and (3) loss of reaction components through evaporation. If loss of material is responsible for NU-1306 growth at high temperature, it would imply that non-equilibrium conditions affect isomer selectivity, an important consideration for understanding the apparent mismatch between synthetic and energetic parameters in this system. Thus, we set out to deconvolute these three factors and determine which of them encourages NU-1306 growth at 170 °C.
If reaction concentration rather than temperature or material loss promotes the growth of NU-1306 at high temperatures, then the same reaction performed at 120 °C at high concentration should produce NU-1306. We thus performed a control experiment at 0.09 FA:DMF and 120 °C with the same total volume found at the completion of the 170 °C NU-1306 reaction. However, these conditions afford NU-1305 with very minor NU-1306 peaks observed by PXRD analysis, demonstrating that the increased reaction concentration does not account for full NU-1306 growth in this case (Fig. S7†). We note that due to the three-dimensionality of the energy landscape, higher reaction concentrations at 0.10 FA:DMF favor pure NU-1306 (Fig. 2c), while the same experiments run here at 0.09 FA:DMF do not (Fig. S7†).
Next, to deconvolute the effects of temperature and loss of material, we employed a pressure reaction vessel where solvent cannot escape. If loss of material is important for NU-1306 growth, then we expect the high temperature reaction (0.8 mL DMF, 0.09 FA:DMF, 170 °C) run in a closed pressure vessel to not produce NU-1306. However, if NU-1306 still grows under these conditions, then energy added to the system from high temperature promotes NU-1306 growth. The synthesis performed in a closed system resulted in the formation of an amorphous, black product (Fig. S6a and b†), implying that loss of material is crucial to NU-1306 formation at high temperatures. To emphasize that loss of material promotes NU-1306 growth but has no effect on NU-1305 growth, we demonstrated that the identical 120 °C synthesis of NU-1305 still proceeds when performed in a closed pressure vessel (Fig. S6c†). Thus, we deduce that loss of material over time is in fact important for the formation of NU-1306.
Analysis of this reaction provides insight into what type of material loss occurs under these conditions. While MOF synthesis is a complex phenomenon, we postulate a simplified reaction for this system in eqn (1), including components directly involved in the reaction. Here, an unquantified molar ratio (n) of formic acid decomposes to volatile products (either CO and H2O in pathway 1 or CO2 and H2 in pathway 2). Gas chromatography experiments reveal that both pathways are present for the two isomer MOF syntheses, and that pathway 1 dominates for NU-1306 synthesis while pathway 2 dominates for NU-1305 synthesis (Fig. S16 and S17†).
 | (1) |
Solvent loss in the NU-1306 synthesis indicates that volatile formic acid byproducts are also removed from the closed reaction system. Importantly, since NU-1306 only forms on the sides of the vial at the solvent/air interface (Fig. 4b), MOF product likewise is constantly syphoned out of the reaction media as the volume level decreases. Thus, multiple products leave the NU-1306 reaction as it progresses. We note that NU-1305, which forms as large crystals mostly on the bottom of the vial, still remains available for growth within the reaction media, with minimal solvent loss (Fig. 4a).
According to Le Châtelier's principle, when products are removed from a reaction, the equilibrium balance becomes tilted in favor of product formation; this can result in non-equilibrium thermodynamic control of a metastable product.12,13,16 Here, we observe a similar occurrence: products are removed from the reaction media, which stabilizes a phase that is not the thermodynamic ground state. Thus, NU-1306 becomes thermodynamically stable under the conditions of growth, even though it is a local energetic minimum. This higher temperature reaction indicates that NU-1306 is a metastable phase, which also reveals why other thermodynamic reaction parameters such as high modulator amount favor this product. Thus, from the energetic analysis of this system, we observe NU-1305 as the topological isomer at the global energy minimum (thermodynamic product) and NU-1306 at a local energy minimum (metastable product).
Conclusions
In conclusion, we demonstrate how the identification of a metastable phase resolves seemingly contradictory synthetic parameters and energetic calculations within a set of uranium MOF topological isomers. We systematically synthesized two distinct topological isomers ctn and bor, named NU-1305 and NU-1306, respectively. While NU-1306 appeared to be thermodynamically favored by synthetic trends involving modulator and temperature, reaction concentration trends, DFT calculations, geometric analysis, and isomer conversion identified NU-1305 as the thermodynamically favored isomer by 8 kcal mol−1. Since the high-temperature synthesis of NU-1306 depends on the removal of products from the reaction media over time, non-equilibrium thermodynamic control is apparent in this system, following Le Châtelier's principle. Thus, we attribute the apparent mismatch between synthetic and energetic parameters to NU-1306 being a metastable phase that becomes thermodynamically stable under the conditions of growth.The interplay between structure and energetics in this study informs design rules for topological MOF isomers by revealing that even topologies of local energetic minima may be favored thermodynamically under non-equilibrium synthesis conditions. Synthetic trends that traditionally point to the appearance of a thermodynamic MOF product, such as high modulator and high temperature, may not provide sufficient proof for the global thermodynamic product within the entire system. Instead, we encourage the use of a suite of factors for identifying MOF location within the three-dimensional energy landscape, including synthetic parameters, crystal structure examination, geometric strain analysis, and energetic calculations. Additionally, the apparent mismatch observed in this study encourages us to explore phase space outside of conventional binary (e.g., kinetic vs. thermodynamic) product options. Finally, this study also reveals how implementing thermodynamic control can access less explored metastable phases in non-traditional uranium-based MOFs. Targeting non-equilibrium synthetic conditions through Le Châtelier's principle can thus be used as a strategy to access metastable MOFs with unique properties and functions. More generally, investigating the relationship between structure and energetics broadens our understanding of phase space in both actinide materials and supramolecular isomers.
Data availability
Crystallographic data for NU-1306 has been deposited at The Cambridge Crystallographic Data Centre (CCDC) under deposition number 2203723.†Author contributions
O. K. F. and C. H. H. supervised the project. S. L. H. and O. K. F. conceived the project and led the investigation. S. L. H. designed the experiments, performed MOF synthesis and characterization, collected PXRD and SCXRD experiments, analyzed the data, and interpreted the results. Z. H. S. and O. K. F. synthesized linker. Z. H. S. assisted S. L. H. with GC measurements. T. T. D. and A. M. M. performed DFT calculations under the supervision of C. H. H. S. L. H. and O. K. F. wrote the manuscript, with the help of K. O. K., and all authors commented on and revised the manuscript.Conflicts of interest
O. K. F. has a financial interest in NuMat Technologies, a startup company that is seeking to commercialize MOFs. All other authors declare no competing interests.Acknowledgements
O. K. F. and S. L. H. acknowledge support from the U.S. Department of Energy, National Nuclear Security Administration, under award number DE-NA0003763. S. L. H. also acknowledges support from the U.S. Department of Energy National Nuclear Security Administration Stewardship Science Graduate Fellowship under award number DE-NA0003960. C. H. H., T. T. D., and A. M. M. are supported by the National Science Foundation through the Division of Materials Research under grant no. DMR-1956403 and the Camille and Henry Dreyfus Foundation. Z. H. S. is supported by the National Science Foundation Graduate Research Fellowship under grant no. DGE-1842165. K. O. K. gratefully acknowledges support from the International Institute for Nanotechnology Postdoctoral Fellowship and the Northwestern University International Institute for Nanotechnology. This work made use of the Integrated Molecular Structure Education and Research Center (IMSERC) at Northwestern University, which has received support from the State of Illinois, Northwestern University, the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), NSF CHE-1048773, and the International Institute for Nanotechnology. This work also made use of the Reactor Engineering and Catalyst Testing (REACT) core facility which acknowledges funding from the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science program (DE-SC0001329 and DE-FG02-03ER15457). S. L. H. acknowledges Timur Islamoglu for helpful conversations over the course of this project. S. L. H. also thanks Xuan Zhang, Mohammed Rasel Mian, and Charlotte Stern for assistance and advice regarding SCXRD measurements. S. L. H. and Z. H. S. thank Neil Schweitzer for assistance with GC measurements.References
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef PubMed.
- A. J. Matlin, F. Clark and C. W. J. Smith, Nat. Rev. Mol. Cell Biol., 2005, 6, 386–398 CrossRef PubMed.
- X. Yang, J. Coulombe-Huntington, S. Kang, G. M. Sheynkman, T. Hao, A. Richardson, S. Sun, F. Yang, Y. A. Shen, R. R. Murray, K. Spirohn, B. E. Begg, M. Duran-Frigola, A. MacWilliams, S. J. Pevzner, Q. Zhong, S. A. Trigg, S. Tam, L. Ghamsari, N. Sahni, S. Yi, M. D. Rodriguez, D. Balcha, G. Tan, M. Costanzo, B. Andrews, C. Boone, X. J. Zhou, K. Salehi-Ashtiani, B. Charloteaux, A. A. Chen, M. A. Calderwood, P. Aloy, F. P. Roth, D. E. Hill, L. M. Iakoucheva, Y. Xia and M. Vidal, Cell, 2016, 164, 805–817 CrossRef PubMed.
- A. Y. Lee, D. Erdemir and A. S. Myerson, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 259–280 CrossRef.
- B. Rodríguez-Spong, C. P. Price, A. Jayasankar, A. J. Matzger and N. r. Rodríguez-Hornedo, Adv. Drug Delivery Rev., 2004, 56, 241–274 CrossRef.
- A. Stannard, J. C. Russell, M. O. Blunt, C. Salesiotis, M. d. C. Giménez-López, N. Taleb, M. Schröder, N. R. Champness, J. P. Garrahan and P. H. Beton, Nat. Chem., 2012, 4, 112–117 CrossRef.
- M. Gupta and J. J. Vittal, Coord. Chem. Rev., 2021, 435, 213789–213804 CrossRef CAS.
- H. Bergeron, D. Lebedev and M. C. Hersam, Chem. Rev., 2021, 121, 2713–2775 CrossRef CAS.
- M. Aykol, S. Dwaraknath Shyam, W. Sun and A. Persson Kristin, Sci. Adv., 2018, 4, eaaq0148 CrossRef PubMed.
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
- J. R. Chamorro and T. M. McQueen, Acc. Chem. Res., 2018, 51, 2918–2925 CrossRef PubMed.
- J. P. Sheckelton, J. R. Neilson, D. G. Soltan and T. M. McQueen, Nat. Mater., 2012, 11, 493–496 CrossRef CAS.
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080–2088 RSC.
- R. D. Brown, S. A. Corcelli and S. A. Kandel, Acc. Chem. Res., 2018, 51, 465–474 CrossRef CAS.
- R. Mikita, T. Aharen, T. Yamamoto, F. Takeiri, T. Ya, W. Yoshimune, K. Fujita, S. Yoshida, K. Tanaka, D. Batuk, A. M. Abakumov, C. M. Brown, Y. Kobayashi and H. Kageyama, J. Am. Chem. Soc., 2016, 138, 3211–3217 CrossRef CAS PubMed.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- M. D. Allendorf, V. Stavila, M. Witman, C. K. Brozek and C. H. Hendon, J. Am. Chem. Soc., 2021, 143, 6705–6723 CrossRef CAS.
- J. D. Evans, V. Bon, I. Senkovska, H.-C. Lee and S. Kaskel, Nat. Commun., 2020, 11, 2690–2700 CrossRef CAS.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2018, 4, eaat9180 CrossRef CAS PubMed.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef.
- A. Karmakar, A. Paul and A. J. L. Pombeiro, CrystEngComm, 2017, 19, 4666–4695 RSC.
- T. A. Makal, A. A. Yakovenko and H.-C. Zhou, J. Phys. Chem. Lett., 2011, 2, 1682–1689 CrossRef.
- A. K. Cheetham, G. Kieslich and H. H. M. Yeung, Acc. Chem. Res., 2018, 51, 659–667 CrossRef PubMed.
- X. Gong, H. Noh, N. C. Gianneschi and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 6146–6151 CrossRef PubMed.
- W.-G. Liu and D. G. Truhlar, Chem. Mater., 2017, 29, 8073–8081 CrossRef CAS.
- D. J. Bara, C. Wilson, M. Mörtel, M. M. Khusniyarov, S. Ling, B. Slater, S. Sproules and R. S. Forgan, J. Am. Chem. Soc., 2019, 141, 8346–8357 CrossRef CAS.
- T. He, Z. Huang, S. Yuan, X.-L. Lv, X.-J. Kong, X. Zou, H.-C. Zhou and J.-R. Li, J. Am. Chem. Soc., 2020, 142, 13491–13499 CrossRef CAS.
- S.-J. Lee, J. L. Mancuso, K. N. Le, C. D. Malliakas, Y.-S. Bae, C. H. Hendon, T. Islamoglu and O. K. Farha, ACS Mater. Lett., 2020, 2, 499–504 CrossRef CAS.
- Z. Akimbekov, A. D. Katsenis, G. P. Nagabhushana, G. Ayoub, M. Arhangelskis, A. J. Morris, T. Friščić and A. Navrotsky, J. Am. Chem. Soc., 2017, 139, 7952–7957 CrossRef CAS PubMed.
- B. Karadeniz, D. Žilić, I. Huskić, L. S. Germann, A. M. Fidelli, S. Muratović, I. Lončarić, M. Etter, R. E. Dinnebier, D. Barišić, N. Cindro, T. Islamoglu, O. K. Farha, T. Friščić and K. Užarević, J. Am. Chem. Soc., 2019, 141, 19214–19220 CrossRef CAS.
- S.-H. Lo, L. Feng, K. Tan, Z. Huang, S. Yuan, K.-Y. Wang, B.-H. Li, W.-L. Liu, G. S. Day, S. Tao, C.-C. Yang, T.-T. Luo, C.-H. Lin, S.-L. Wang, S. J. L. Billinge, K.-L. Lu, Y. J. Chabal, X. Zou and H.-C. Zhou, Nat. Chem., 2020, 12, 90–97 CrossRef.
- J. Liu, B. Lukose, O. Shekhah, H. K. Arslan, P. Weidler, H. Gliemann, S. Bräse, S. Grosjean, A. Godt, X. Feng, K. Müllen, I.-B. Magdau, T. Heine and C. Wöll, Sci. Rep., 2012, 2, 921–925 CrossRef.
- Z.-J. Li, Y. Ju, Z. Zhang, H. Lu, Y. Li, N. Zhang, X.-L. Du, X. Guo, Z.-H. Zhang, Y. Qian, M.-Y. He, J.-Q. Wang and J. Lin, Chem.–Eur. J., 2021, 27, 17586–17594 CrossRef CAS PubMed.
- P. Thuery and J. Harrowfield, Cryst. Growth Des., 2018, 18, 5512–5520 CrossRef CAS.
- S. L. Hanna, S. Chheda, R. Anderson, D. Ray, C. D. Malliakas, J. G. Knapp, K.-i. Otake, P. Li, P. Li, X. Wang, M. C. Wasson, K. Zosel, A. M. Evans, L. Robison, T. Islamoglu, X. Zhang, W. R. Dichtel, J. F. Stoddart, D. A. Gomez-Gualdron, L. Gagliardi and O. K. Farha, Chem, 2022, 8, 225–242 CAS.
- P. Li, N. A. Vermeulen, C. D. Malliakas, D. A. Gómez-Gualdrón, A. J. Howarth, B. L. Mehdi, A. Dohnalkova, N. D. Browning, M. O'Keeffe and O. K. Farha, Science, 2017, 356, 624–627 CrossRef PubMed.
- K.-Q. Hu, X. Jiang, C.-Z. Wang, L. Mei, Z.-N. Xie, W.-Q. Tao, X.-L. Zhang, Z.-F. Chai and W.-Q. Shi, Chem.–Eur. J., 2017, 23, 529–532 CrossRef PubMed.
- N. R. M. C. Biology, NU-1305 was first published in ref. 38 under the name “1”. We rename it here to “NU-1305” for the sake of consistency.
- R. S. Forgan, Chem. Sci., 2020, 11, 4546–4562 RSC.
- C. R. Marshall, E. E. Timmel, S. A. Staudhammer and C. K. Brozek, Chem. Sci., 2020, 11, 11539–11547 RSC.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2203723. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04783g |
| This journal is © The Royal Society of Chemistry 2022 |