
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of transient supramolecular polymers controlled by mass transfer in biphasic systems†
Shilin
Zhang
 a,
Yulian
Zhang
b,
Huiting
Wu
a,
Zhihao
Li
a,
Peichen
Shi
a,
Hang
Qu
a,
Yibin
Sun
a,
Xinchang
Wang
c,
Xiaoyu
Cao
a,
Liulin
Yang
*a and
Zhongqun
Tian
a
a,
Yulian
Zhang
b,
Huiting
Wu
a,
Zhihao
Li
a,
Peichen
Shi
a,
Hang
Qu
a,
Yibin
Sun
a,
Xinchang
Wang
c,
Xiaoyu
Cao
a,
Liulin
Yang
*a and
Zhongqun
Tian
a
aState Key Laboratory of Physical Chemistry of Solid Surface, Key Laboratory of Chemical Biology of Fujian Province, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: llyang@xmu.edu.cn
bCollege of Materials, Xiamen University, Xiamen 361005, P. R. China
cSchool of Electronic Science and Engineering (National Model Microelectronics College), Xiamen University, Xiamen 361005, P. R. China
First published on 15th November 2022
Abstract
Inspired by life assembly systems, the construction of transient assembly systems with spatiotemporal control is crucial for developing intelligent materials. A widely adopted strategy is to couple the self-assembly with chemical reaction networks. However, orchestrating the kinetics of multiple reactions and assembly/disassembly processes without crosstalk in homogeneous solutions is not an easy task. To address this challenge, we propose a generic strategy by separating components into different phases, therefore, the evolution process of the system could be easily regulated by controlling the transport of components through different phases. Interference of multiple components that are troublesome in homogeneous systems could be diminished. Meanwhile, limited experimental parameters are involved in tuning the mass transfer instead of the complex kinetic matching and harsh reaction selectivity requirements. As a proof of concept, a transient metallo-supramolecular polymer (MSP) with dynamic luminescent color was constructed in an oil–water biphasic system by controlling the diffusion of the deactivator (water molecules) from the water phase into the oil phase. The lifetime of transient MSP could be precisely regulated not only by the content of chemical fuel, but also factors that affect the efficiency of mass transfer in between phases, such as the volume of the water phase, the stirring rate, and the temperature. We believe this strategy can be further extended to multi-compartment systems with passive diffusion or active transport of components, towards life-like complex assembly systems.
Introduction
Living assembly systems featured with spatiotemporal control over structure and functions (e.g., self-adaptation, self-healing, self-replication) are normally achieved by maintaining the system out-of-equilibrium fueled by chemical energy. Inspired by nature, the study of molecular assembly systems that are out of equilibrium has aroused wide interest.1–15 One of the widely adopted strategies to construct out-of-equilibrium assembly systems is to couple the self-assembly process with chemical reactions.3,4,16–21 The precursors are converted into building blocks by chemical-fueled activation reactions, followed by the self-assembly of activated building blocks. Simultaneously, the building blocks are continuously deactivated and reverted into the original precursors, resulting in transient assembly systems. The assemblies can only be maintained when the chemical fuel is available to the system.1,22–28 An important prerequisite to obtaining the transient assemblies is that the activation rate should be faster than the deactivation rate. For the activation step, chemical fuels29 with varied amounts and feeding manners (addition in small batches or at once)30–32 are generally employed to control the activation process. For the deactivation step, the common strategies are tuning the pH,1,33 temperature,34 the content of deactivators (i.e. enzyme),22,24,35–37 and so on.Inspired by living assembly systems with reaction networks involved, more complex strategies by coupling multiple chemical reactions have been developed to regulate the activation or deactivation process.24,26,27 For instance, Walther and co-workers9,38 proposed the concept of ‘‘dormant deactivator’’—a compound that liberates active deactivator slowly through a chemical reaction—capable of creating a transient pH profile to which the assemblies respond accordingly. The lifetime of the assemblies depends on the generation rate of the active deactivator. However, coupling chemical reaction networks in a homogeneous system and making these reactions work in tandem without crosstalk is not an easy task, because of not only strict requirements for the specificity of chemical reactions, but also challenges in the kinetics matching between chemical reaction and assembly/disassembly process.
To address this issue, we propose a generic approach to constructing transient assembly systems by separating components such as chemical fuels, building blocks, and deactivators in different phases or compartments. Therefore, the kinetics can be easily regulated by controlling the transport of components with limited parameters concerned, instead of falling into the careful selection of multi-step chemical reactions and the coupling of reaction kinetics parameters. As a proof of concept, we demonstrate the construction of transient metallo-supramolecular polymers (MSP) in an oil–water biphasic system (Fig. 1). A dendron ligand T-3tpy consisting of a truxene core carrying three terpyridine branches was dissolved in chloroform. Upon the addition of chemical fuel (Mg(ClO4)2) in the oil phase, the fast coordination of Mg2+ ions with terpyridine groups in T-3tpy led to the formation of MSP. Meanwhile, water molecules as deactivators slowly diffused into the oil phase, and gradually decomposed the MSP by competitively hydrating the Mg2+ ions, making MSP transient. By controlling the water transfer into the oil phase, the lifetime of this transient MSP could be tuned from minutes to hours.
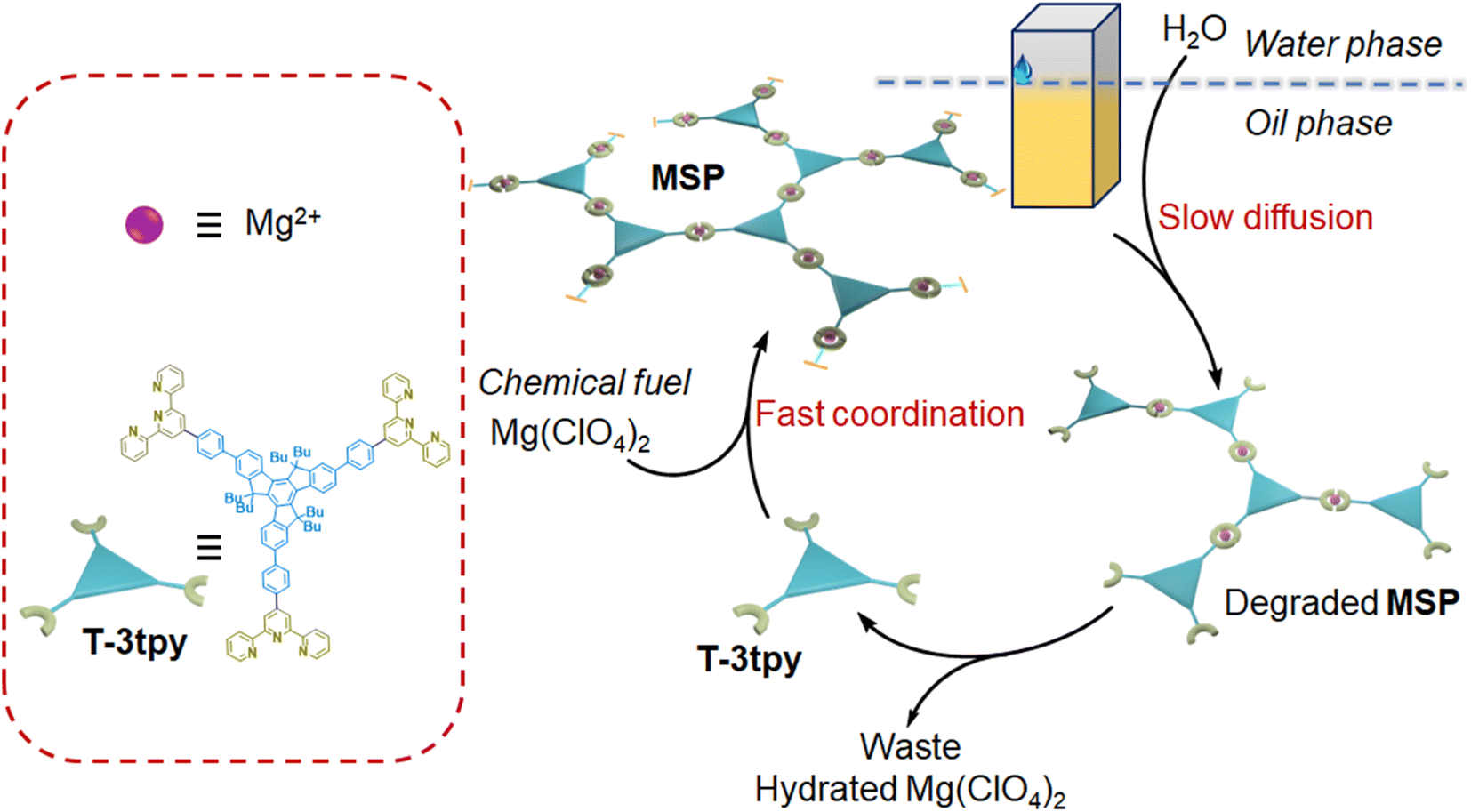 | ||
| Fig. 1 Scheme of the chemically fueled reaction cycle towards transient MSP by controlling the water diffusion in an oil–water biphasic system. | ||
Results and discussion
The coordination of T-3tpy dendron with Mg2+ was supposed to result in a hyperbranched MSP.39,40T-3tpy in CHCl3 solution has a maximum absorbance at 346 nm and a characteristic emission peak at 414 nm originated from the truxene chromophore (Fig. 2a, b and S10†). Upon titration of the T-3tpy (11 μM in CHCl3) with Mg(ClO4)2, the absorption band at 346 nm diminished accompanied by a new absorption band ca. 400 nm increased. The UV-Vis spectrum kept the same when the molar ratio of Mg(ClO4)2 and T-3tpy increased to 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating the coordination between Mg2+ and terpyridine group in a 1:2 stoichiometry.41–43 Furthermore, a broad emission peak at 525 nm rose remarkably accompanied by the decrease of the original peak at 414 nm. Accordingly, the fluorescent color of the solution when irradiated by 365 nm light switched from the initial blue to approximate white, and finally yellow-green (Fig. 2c). The formation of polymers was further confirmed by dynamic light scattering (DLS) and microscopies (Fig. 2d–f). Discrete spherical nanoparticles with diameters around 60 nm to 80 nm were clearly observed by TEM and SEM, and the measured size was in good agreement with the data (75 nm) obtained by DLS analysis. Particles with larger sizes from 75 nm to 122 nm could be obtained by increasing the equivalents of Mg(ClO4)2 from 2 to 8 eq. (Fig. S11†).
:1, indicating the coordination between Mg2+ and terpyridine group in a 1:2 stoichiometry.41–43 Furthermore, a broad emission peak at 525 nm rose remarkably accompanied by the decrease of the original peak at 414 nm. Accordingly, the fluorescent color of the solution when irradiated by 365 nm light switched from the initial blue to approximate white, and finally yellow-green (Fig. 2c). The formation of polymers was further confirmed by dynamic light scattering (DLS) and microscopies (Fig. 2d–f). Discrete spherical nanoparticles with diameters around 60 nm to 80 nm were clearly observed by TEM and SEM, and the measured size was in good agreement with the data (75 nm) obtained by DLS analysis. Particles with larger sizes from 75 nm to 122 nm could be obtained by increasing the equivalents of Mg(ClO4)2 from 2 to 8 eq. (Fig. S11†).
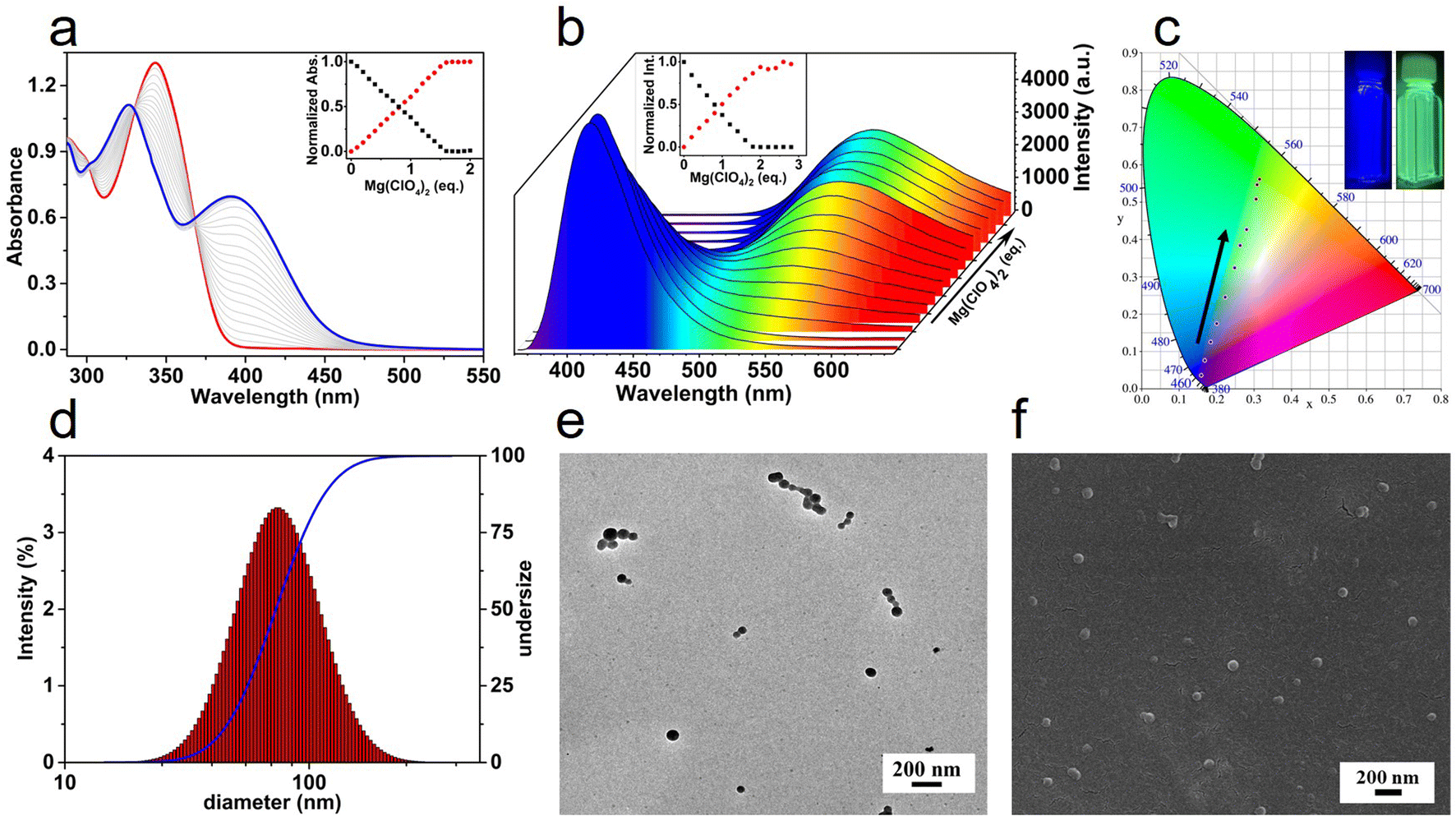 | ||
| Fig. 2 The formation of MSP was confirmed by spectroscopies, dynamic light scattering, and microscopies. (a) The UV-Vis spectra of T-3tpy in CHCl3 (11 μM) upon titration with Mg(ClO4)2 in THF (1 mM), red line: the original spectrum of T-3tpy; blue line: spectra when 2 eq. of Mg(ClO4)2 were added; (inset) normalized absorbance changes of ligand T-3tpy at 346 nm and 400 nm as a function of the molar ratio of Mg(ClO4)2. (b) The fluorescence spectra upon the addition of Mg(ClO4)2 under the same conditions as in UV-Vis titration. (Inset) Normalized intensity changes of ligand T-3tpy at 414 nm and 525 nm as a function of the molar ratio of Mg(ClO4)2. (c) Changes in the luminescent color of the solution in the 1931 CIE during titration. The inset shows the visible fluorescence of the solution (left: 0 eq. Mg(ClO4)2, right: 2 eq. Mg(ClO4)2) under the exposure of 365 nm light. (d) Size distribution of MSP determined by DLS (T-3tpy: 11 μM, Mg(ClO4)2: 2 eq.). The blue line refers to the cumulative frequency distribution of the size of MSP. (e) The TEM image and (f) the SEM image of MSP. | ||
The transient MSP system was successfully constructed in an oil–water biphasic solution. Because Mg(ClO4)2 in chloroform can be hydrated by water (Fig. S12†), it is possible to take water as a deactivator to decompose the MSP that formed in the oil phase. By controlling the diffusion of water into the oil phase, a transient MSP system could be constructed. Specifically, T-3tpy was dissolved in chloroform (2 mL, 11 μM), and the biphasic system was prepared by adding a drop of water (10 μL) on top of the chloroform solution. Upon addition of chemical fuel Mg(ClO4)2 (22 μM, 2 eq. of T-3tpy) to the chloroform phase, MSP formed instantly which was evidenced by the same transition of UV-Vis spectrum as aforementioned (Fig. 3a). Meanwhile, the water molecules slowly diffused into chloroform and decomposed the MSP, featured by the gradual decrease of the absorbance at 400 nm and the increase at 346 nm. After 24 min, the UV-Vis spectrum of the system almost recovered to the initial state, indicating the depolymerization of MSP and the regeneration of T-3tpy monomers. The polymerization and depolymerization cycle was repeated at least three times by adding Mg(ClO4)2 in batches (Fig. 3b). However, as the number of cycles increased, more Mg(ClO4)2 was required (4–6 eq.), which may owe to the increased water content in the chloroform phase over time and the interference from the accumulated waste products (hydrated Mg(ClO4)2). The transient MSP was further confirmed by tracking the morphology of polymer particles. Samples were taken at different time points (2 min, 14 min, 20 min, and 26 min) for transmission electron microscopy (TEM). As shown in Fig. 3c, spherical particles with an average size of 60 nm were observed in the sample at 2 min, which was consistent with the MSP particles at the equilibrium state. As time went on, the number and size of particles gradually decreased, accompanied by the formation of abundant tiny particles which might be ascribed to the hydrated Mg(ClO4)2. For the sample at 26 min, almost no large particles could be observed, indicating the complete decomposition of the MSP. A similar process (Fig. S13†) was also observed by scanning electron microscopy (SEM).
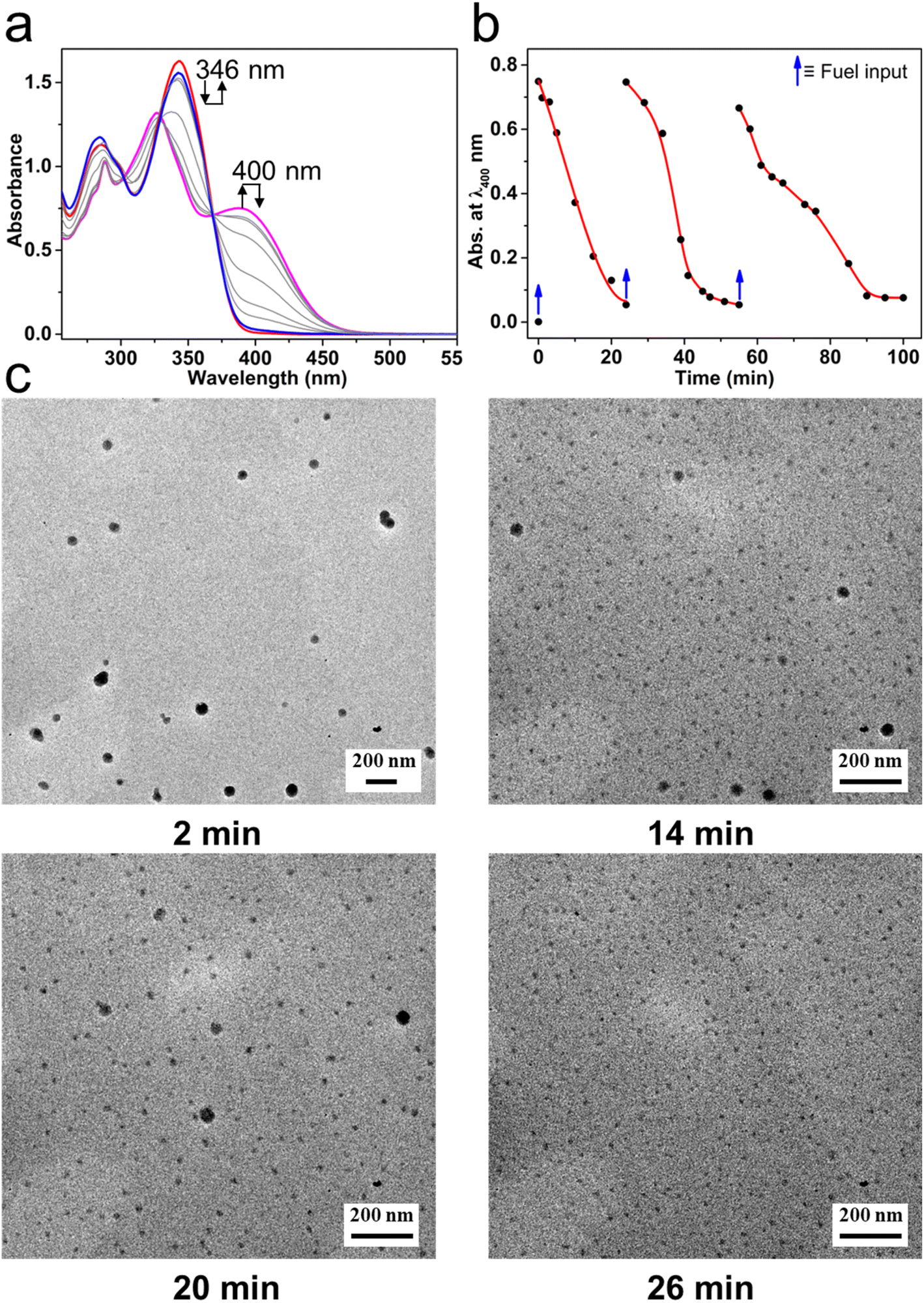 | ||
| Fig. 3 The transient behavior of MSP in the oil–water biphasic system. (a) The change of UV-Vis spectra of T-3tpy (red) after the addition of Mg(ClO4)2. The pink line indicates the formation of MSP. The spectrum recovered to the initial state (blue) over 24 min. (b) The polymerization and decomposition of MSP were repeated by adding Mg(ClO4)2 in batches. (c) Representative TEM images of MSP at different time intervals. (T-3tpy 11 μM, Mg(ClO4)2 22 μM, H2O 10 μL, stirring rate 300 rpm, 25 °C). | ||
The transient MSP system was further investigated by nuclear magnetic resonance (NMR) spectroscopy (Fig. 4). Upon addition of 2 eq. of Mg(ClO4)2 (THF-d8) to the biphasic solution (4 mM T-3tpy in 0.5 mL CDCl3 and 5 μL H2O), the NMR signal of T-3tpy monomer disappeared rapidly. Unfortunately, the 1H NMR signal of MSP could not be detected owing to the large particle size. The MSP finally decomposed indicated by the regeneration of T-3tpy signals. Meanwhile, a new signal at ca. 4.80 ppm was observed, which was identified as hydrated Mg2+ by a controlled experiment (Fig. S12†). To further confirmed the transient behavior in such a metal-coordination system, a one-armed ligand T-1tpy was taken as a model compound. The coordination of T-1tpy with Mg2+ results in a soluble dimer, which could also be transient in the same biphasic system (see ESI, Fig. S14–S18†). In this case, the transient behavior of the dimer with a similar transition process of NMR signals was observed (Fig. S14 and S15†). Therefore, we can conclude that the transient behavior of MSP should be attributed to the competitive coordination of dissolved water with Mg2+ in the oil phase.
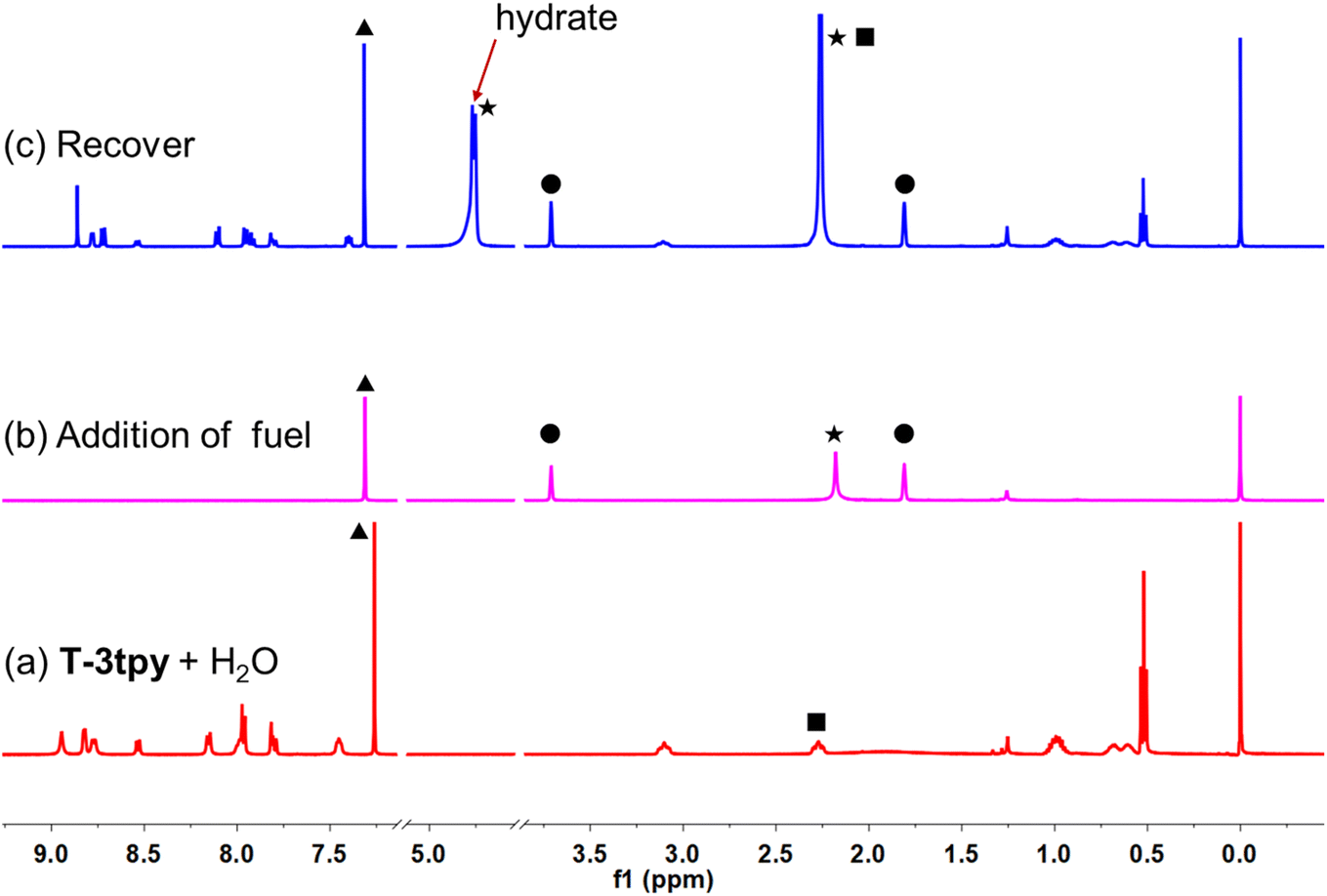 | ||
| Fig. 4 Characterization of transient MSP in the oil–water biphasic system using NMR. 1H NMR spectra of the solution containing ligand T-3tpy before (trace a) and after (trace b and c) the addition of Mg(ClO4)2. Marked signals are assigned to protons: ●, THF; ▲, CHCl3; ★, H2O in the solvent; ■, –CH2–. | ||
The lifetime of transient MSP could be easily regulated by the content of chemical fuel, i.e. Mg(ClO4)2, in the oil phase. In this study, the lifetime of MSP was defined as the period from the generation to the complete decomposition of MSP, by monitoring the absorbance at 400 nm in UV-Vis spectroscopy. As shown in Fig. 5a, the lifetime increased from 24 to 62 minutes upon increasing the equivalents of Mg(ClO4)2 from 2 to 8 while the volume of water phase was fixed at 10 μL, showing a linear proportion to the content of Mg(ClO4)2. Notably, the more Mg(ClO4)2, the longer plateau in the UV-Vis curve could be observed, suggesting the formed MSP could be protected by the excess Mg(ClO4)2 from depolymerization (Fig. S19†).
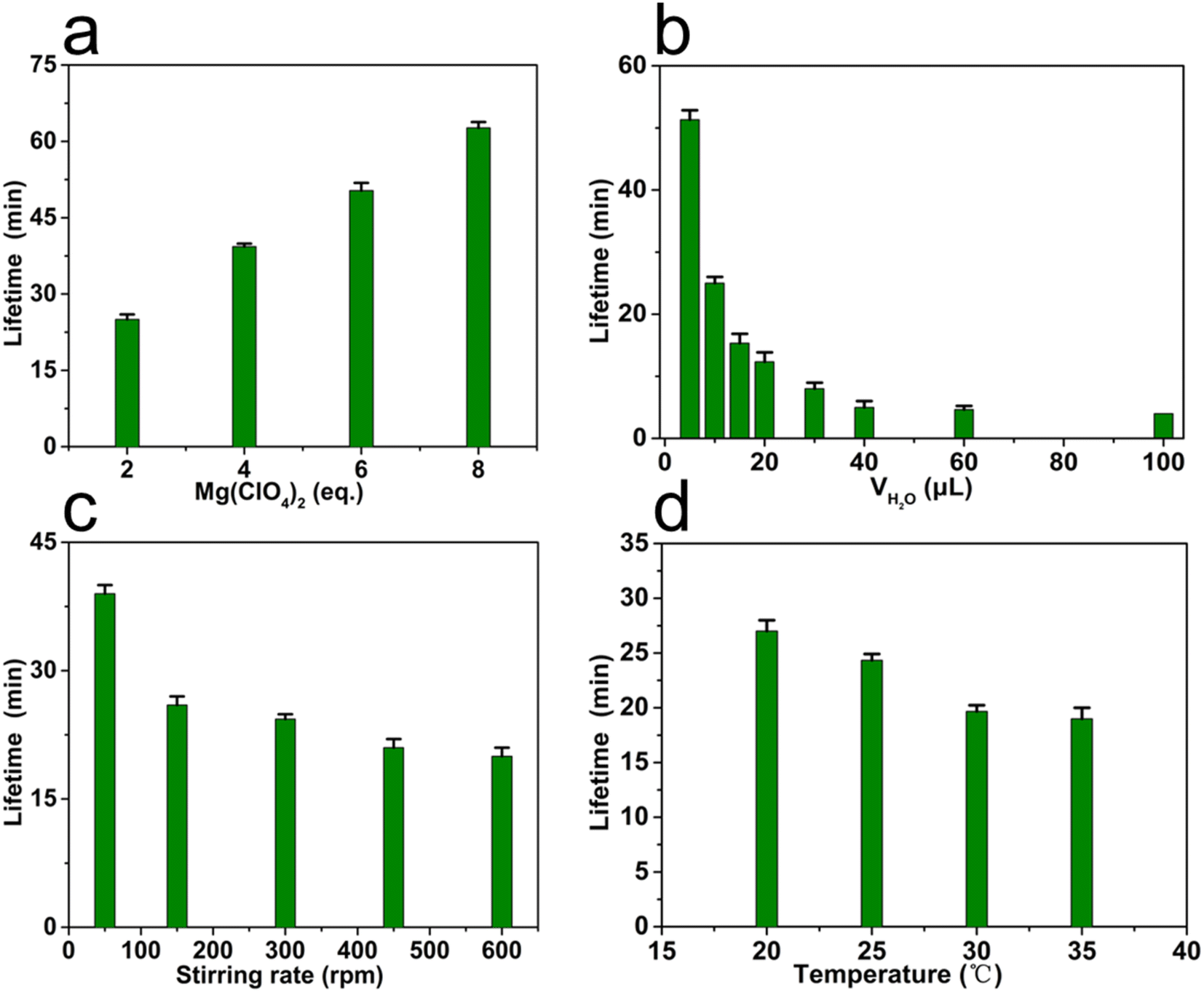 | ||
| Fig. 5 The lifetime of MSP was regulated by: (a) different equivalents of Mg(ClO4)2 (water 10 μL, 300 rpm, 25 °C), (b) volume of water (2 eq. Mg(ClO4)2, 300 rpm, 25 °C), (c) stirring rate (2 eq. Mg(ClO4)2, water 10 μL, 25 °C), and (d) temperature (2 eq. Mg(ClO4)2, water 10 μL, 300 rpm). | ||
Besides the content of fuel in the oil phase, the mass transfer44 in between phases plays a key role in tuning the lifetime of MSP. Factors that may affect the efficiency of mass transfer such as the volume of water phase, the stirring rate, and the temperature were evaluated while the content of Mg(ClO4)2 remained constant. It was found that the lifetime of MSP was highly sensitive to the volume of water and the stirring rate, but less affected by temperature. A dramatic decrease in lifetime from ∼51 min to ∼5 min was observed when increasing the volume of water from 5 μL to 40 μL (Fig. 5b), owing to the accelerated diffusion of water molecules into the oil phase (Fig. S20†). However, when the volume of water was over 40 μL, the lifetime tended to be constant, suggesting the transfer of water molecules ceased to be the rate-determining step. The stirring rate of the magnet rotor determined the speed of convection, and therefore the efficiency of mass transfer in this system (Fig. 5c and S21†). As shown in Fig. 5c, the lifetime was ∼39 min under a relatively low stirring rate (50 rpm). Increasing the stirring rate to 150 rpm resulted in a significant decline of the lifetime (∼26 min), suggesting a fast mass transfer under this condition. The results indicated that the mass transfer was dominated by molecular diffusion under a low stirring rate, while controlled by convection under a high stirring rate. The relatively low efficiency of molecular diffusion leads to a long lifetime of MSP. In an extreme case without stirring, the oil phase even turned to be inhomogeneous. Only the MSP near the oil–water interface could be decomposed while the MSP in oil bulk remained stable (Fig. S22†). Although the convection significantly shortened the lifetime of MSP, further increasing the stirring rate only resulted in a slight loss of the lifetime, similar to the case when tuning the volume of water. Considering that mass transfer can be regulated by the thermal motion of molecules, the lifetime of MSP was also investigated under different temperatures. The lifetime declined from ∼27 min to ∼19 min when the temperature was elevated from 20 °C to 35 °C (Fig. 5d), indicating a mild effect of temperature in this biphasic system.
The essential role of mass transfer was further confirmed by studying the dynamics of MSP in different solvent systems, including an oil–water biphasic system, an oil–air biphasic system, and a sealed oil solution. In the oil–water biphasic system, the water content in the oil phase increased gradually and reached a constant value of ∼1274 μg mL−1 after 30 min (Table 1). The cycle of polymerization–depolymerization was completed within 24 min in this system. Whereas in the oil–air biphasic system, the maximum water content in the oil phase was only 656.8 μg mL−1, and the MSPs were only partially degraded even after 2 h (Fig. S23†). Moreover, the volatilization of organic solvent during such a long-time exposure resulted in the precipitates of MSP, which further retarded their degradation. Furthermore, in the sealed oil phase, the water content was maintained at 265.3 μg mL−1, and the MSP could be maintained for a long time (Fig. S24†). However, if the initial water content in the sealed oil phase was too high, e.g. 1278 μg mL−1, there was no evidence for the formation of MSP after adding 2 eq. of Mg(ClO4)2 into the solution (Fig. S25†). All the results confirmed that the dynamics of MSP are indeed controlled by the transfer of water into the oil phase.
| Solutions | CHCl3/mL | THF/μL | H2Oa/μL | Water contentb/μg mL−1 | Lifetime of MSP |
|---|---|---|---|---|---|
| a The content of added water. b Water content in the oil phase measured 30 min after solution preparation. | |||||
| Oil–water system | 2 | 40 | 10 | 1274 | Within 24 min |
| Oil–air system | 2 | 40 | 0 | 656.8 | Partial degradation over 2 h |
| Sealed oil solution | 2 | 40 | 0 | 265.3 | Long live |
Benefiting from the luminescent property of the truxene moiety, the transient MSP presented a dynamic luminescence over time. Typically, the emission of initial ligand T-3tpy at 414 nm was quenched upon the addition of Mg(ClO4)2 (2 eq.), instead, a new emission band at ca. 525 nm appeared (Fig. 6a). Subsequently, the emission at 525 nm gradually decreased, accompanied by the reappearance of the emission at 414 nm within 20 min. This transient process can be traced in more detail by the ratio of fluorescence intensities I525nm/I414nm (Fig. 6b). After adding Mg(ClO4)2, the ratio rapidly increased to the maximum due to the fast coordination of the metal ion and the T-3tpy ligand. In the following 4 minutes, the ratio declined slightly, then fell sharply during the 4–10 min, and finally approached zero during 10–20 min. The transition process of the luminescent color (Movie S1†) is presented in the CIE chromaticity coordinate (Fig. 6c). Right after the addition of fuel, the luminescent color jumped from blue (0.16, 0.04) to yellow-green (0.32, 0.56), then ultimately reverted to the initial blue (0.16, 0.04). The curve of ratio metric fluorescence over time shows a plateau in the first 4 min, indicating a steady state of the MSP in this period. This might be ascribed to the protection by the slight excess of Mg(ClO4)2 from depolymerization. Increasing the concentration of ligand T-3tpy (10−4 M) and Mg(ClO4)2 (2 × 10−4 M) prolonged the lifetime of MSP to 27 min (Fig. 6e), and resulted in a more complex transition process of luminescence (Fig. 6d). A bimodal emission with continuous changes of wavelength was observed during the recovery process, resulting in a different color transition process in the order of blue–yellow green–white–blue (Movie S2†), as shown in the CIE chromaticity coordinate (Fig. 6f). These results may owe to the aggregation of MSP under high concentration, evidenced by increased particle size (∼130 nm) (Fig. S26†).
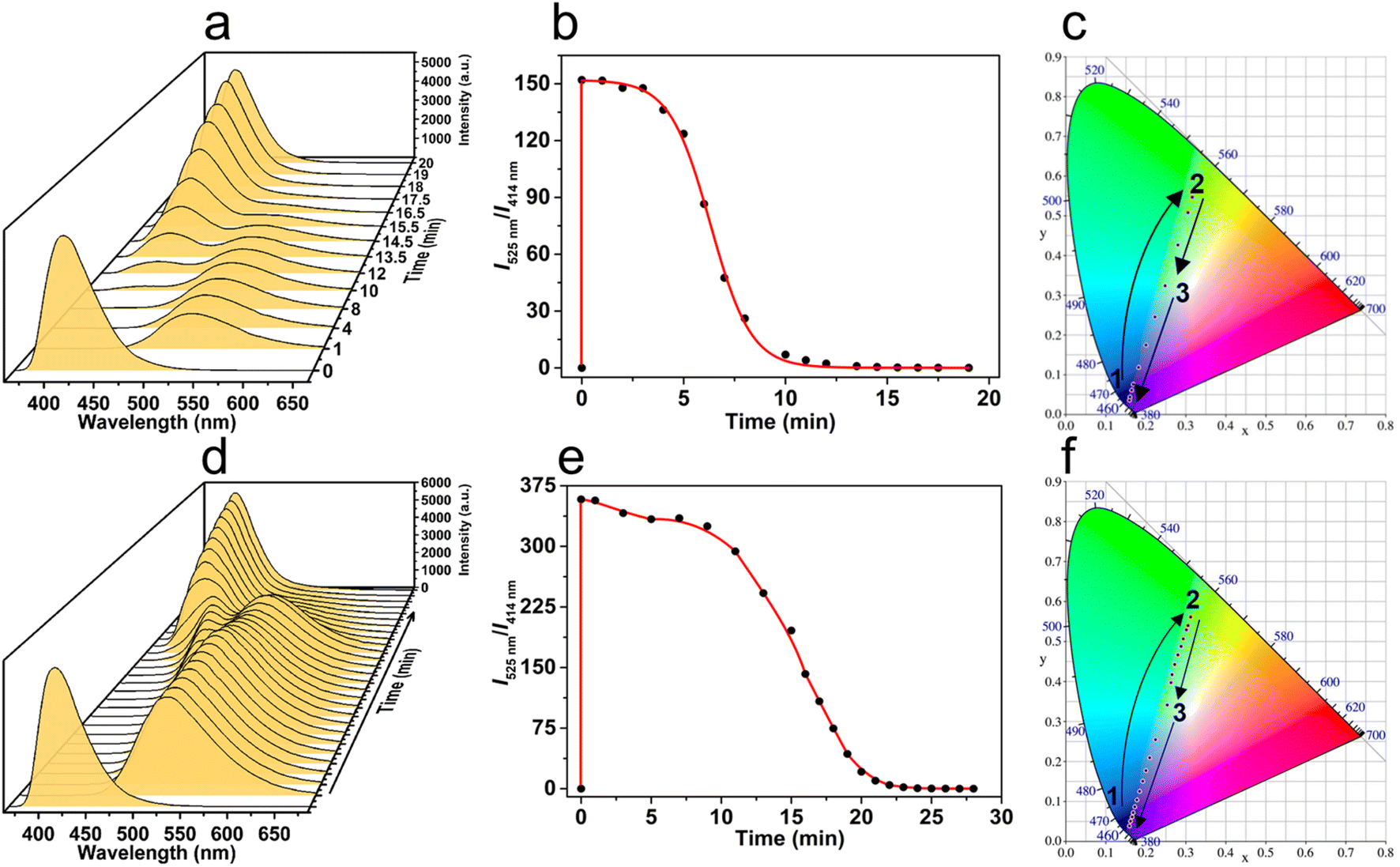 | ||
| Fig. 6 The transition process of luminescence of the transient MSP system over time. (a and d) Time-dependent fluorescence spectra. (b and e) Time-dependent relative fluorescence intensity. (c and f) Time-dependent CIE coordinate diagram showing the change trajectories of the emission color (conditions: (a–c) T-3tpy 11 μM, Mg(ClO4)2 22 μM, H2O 10 μL; (d–f) T-3tpy 10−4 M, H2O 10 μL, Mg(ClO4)2 2 × 10−4 M). In both experiments, the stirring rate was 300 rpm, and the temperature was 25 °C. | ||
Conclusions
In conclusion, we propose a facile and generic strategy for constructing transient assembly systems by coupling self-assembly, chemical reactions, and molecular diffusion in multiple phases. As a proof of concept, we have demonstrated the construction of transient MSP in an oil–water biphasic system. By controlling the water diffusion from the water phase into the oil phase, the kinetics of MSP depolymerization was facilely regulated, resulting in the transient MSP. Distinct from the strategies that regulate all the kinetic processes in a homogeneous system,24,26,27,38 this strategy introduces a new regulation dimension, i.e. the mass transfer of components through different phases, to regulate either the activation process or deactivation process. By separating components such as chemical fuels, building blocks, and deactivators in different phases, interference from the unexpected crosstalk of different components that usually troubles the homogeneous system could be effectively diminished. Meanwhile, limited experimental parameters are involved in tuning the diffusion instead of the complex kinetic matching and harsh reaction selectivity requirements in homogeneous systems. This strategy can be further extended to multi-compartment systems45,46 and liquid–liquid phase separation systems.47–50 Passive diffusion and active transport can be developed in controlling the transport of chemical fuels or deactivators between compartments. Learning more from the characteristics of out-of-equilibrium assembly in life would pave the way to move artificial molecular assembly toward life-like intelligent assembly systems.Data availability
All data supporting the findings of this study are available within the article and ESI.†Author contributions
Zhang S. L. and Zhang Y. L. synthesized the ligands and participated in all the experiments and data analysis. Wu H. T. performed the fluorescence measurements. Li Z. H. carried out the DLS characterizations and NMR experiments. Zhang S. L. and Zhang Y. L. performed the absorption spectra measurements. Shi P. C. carried out the thermodynamic and kinetic data analysis. Zhang S. L. and Yang L. L. wrote the overall manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Liubin Feng, and Jietao Hu for helpful discussion and assistance in the experiments. This work was supported by the National Natural Science Foundation of China (NSFC) (No. 21971216, 21971217, 21991130, 21991131), the Top-Notch Young Talents Program of China, and the Fundamental Research Funds for the Central Universities of China (No. 20720210007).References
- J. Boekhoven, W. E. Hendrikse, G. J. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed
.
- B. A. Grzybowski and W. T. Huck, Nat. Nanotechnol., 2016, 11, 585–592 CrossRef CAS PubMed
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC
- M. Weißenfels, J. Gemen and R. Klajn, Chem, 2021, 7, 23–37 Search PubMed
- B. Liu, J. Wu, M. Geerts, O. Markovitch, C. G. Pappas, K. Liu and S. Otto, Angew. Chem., Int. Ed., 2022, 61, e202117605 CAS
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS PubMed
- G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC
- H. S. Azevedo, S. L. Perry, P. A. Korevaar and D. Das, Nat. Chem., 2020, 12, 793–794 CrossRef CAS PubMed
- R. Merindol and A. Walther, Chem. Soc. Rev., 2017, 46, 5588–5619 RSC
- D. Jiao, F. Lossada, W. Yu, J. Guo, D. Hoenders and A. Walther, Adv. Funct. Mater., 2020, 30, 1905309 CrossRef CAS
- J. Deng and A. Walther, Nat. Commun., 2020, 11, 3658 CrossRef CAS PubMed
- J. Deng and A. Walther, Chem, 2020, 6, 3329–3343 CAS
- A. Sharko, D. Livitz, S. De Piccoli, K. J. M. Bishop and T. M. Hermans, Chem. Rev., 2022, 122, 11759–11777 CrossRef CAS PubMed
- E. Del Grosso, E. Franco, L. J. Prins and F. Ricci, Nat. Chem., 2022, 14, 600–613 CrossRef CAS PubMed
- S. Borsley, D. A. Leigh and B. M. W. Roberts, Nat. Chem., 2022, 14, 728–738 CrossRef CAS PubMed
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS PubMed
- B. Rieß, R. K. Grötsch and J. Boekhoven, Chem, 2020, 6, 552–578 Search PubMed
- L. S. Kariyawasam, M. M. Hossain and C. S. Hartley, Angew. Chem., Int. Ed., 2021, 60, 12648–12658 CrossRef CAS PubMed
- J. Rodon-Fores, M. A. Würbser, M. Kretschmer, B. Rieß, A. M. Bergmann, O. Lieleg and J. Boekhoven, Chem. Sci., 2022, 13, 11411–11421 RSC
- V. W. Liyana Gunawardana, T. J. Finnegan, C. E. Ward, C. E. Moore and J. D. Badjic, Angew. Chem., Int. Ed., 2022, 61, e202207418 CrossRef CAS PubMed
- L. Jia, L. Xu, Y. Liu, J. Hao and X. Wang, CCS Chem., 2022, 1–14, DOI:10.31635/ccschem.022.202101536
- S. Maiti, I. Fortunati, C. Ferrante, P. Scrimin and L. J. Prins, Nat. Chem., 2016, 8, 725–731 CrossRef CAS PubMed
- F. Schnitter, A. M. Bergmann, B. Winkeljann, J. Rodon Fores, O. Lieleg and J. Boekhoven, Nat. Protoc., 2021, 16, 3901–3932 CrossRef CAS PubMed
- A. Jain, S. Dhiman, A. Dhayani, P. K. Vemula and S. J. George, Nat. Commun., 2019, 10, 450 CrossRef CAS PubMed
- S. Yang, G. Schaeffer, E. Mattia, O. Markovitch, K. Liu, A. S. Hussain, J. Ottele, A. Sood and S. Otto, Angew. Chem., Int. Ed., 2021, 60, 11344–11349 CrossRef CAS PubMed
- A. H. J. Engwerda, J. Southworth, M. A. Lebedeva, R. J. H. Scanes, P. Kukura and S. P. Fletcher, Angew. Chem., Int. Ed., 2020, 59, 20361–20366 CrossRef CAS PubMed
- N. Singh, B. Lainer, G. J. M. Formon, S. De Piccoli and T. M. Hermans, J. Am. Chem. Soc., 2020, 142, 4083–4087 CrossRef CAS PubMed
- S. Bal, K. Das, S. Ahmed and D. Das, Angew. Chem., Int. Ed., 2019, 58, 244–247 CrossRef CAS PubMed
- C. Biagini, S. Albano, R. Caruso, L. Mandolini, J. A. Berrocal and S. Di Stefano, Chem. Sci., 2018, 9, 181–188 RSC
- R. K. Grotsch, C. Wanzke, M. Speckbacher, A. Angi, B. Rieger and J. Boekhoven, J. Am. Chem. Soc., 2019, 141, 9872–9878 CrossRef PubMed
- M. Tena-Solsona, J. Janssen, C. Wanzke, F. Schnitter, H. Park, B. Rieß, J. M. Gibbs, C. A. Weber and J. Boekhoven, ChemSystemsChem, 2020, 3, 1–10 Search PubMed
- S. P. Afrose, C. Mahato, P. Sharma, L. Roy and D. Das, J. Am. Chem. Soc., 2022, 144, 673–678 CrossRef CAS PubMed
- G. Wang, B. Tang, Y. Liu, Q. Gao, Z. Wang and X. Zhang, Chem. Sci., 2016, 7, 1151–1155 RSC
- C. Wanzke, M. Tena-Solsona, B. Rieß, L. Tebcharani and J. Boekhoven, Mater. Horiz., 2020, 7, 1397–1403 RSC
- K. Jalani, S. Dhiman, A. Jain and S. J. George, Chem. Sci., 2017, 8, 6030–6036 RSC
- X. Hao, H. Wang, W. Zhao, L. Wang, F. Peng and Q. Yan, CCS Chem., 2022, 4, 838–846 CrossRef CAS
- C. Pan, J. Xu, L. Wang, Y. Jia, J. Li, G. Liu, S. Zhu, B. Yang and Y. Li, CCS Chem., 2022, 1–13, DOI:10.31635/ccschem.022.202201893
- T. Heuser, A. K. Steppert, C. M. Lopez, B. Zhu and A. Walther, Nano Lett., 2015, 15, 2213–2219 CrossRef CAS PubMed
-
G. Odian, Principles of Polymerization, Wiley, Hoboken, NJ, 4th edn, 2004 Search PubMed
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed
- A. Fermi, G. Bergamini, M. Roy, M. Gingras and P. Ceroni, J. Am. Chem. Soc., 2014, 136, 6395–6400 CrossRef CAS PubMed
- E. C. Constable, J. Healy and M. G. B. Drew, Polyhedron, 1991, 10, 1883–1887 CrossRef CAS
- H. Wang, C. Guo and X. Li, CCS Chem., 2022, 4, 785–808 CrossRef CAS
- F. Al Basir and P. K. Roy, Int. J. Eng. Math., 2015, 2015, 1–9 CrossRef
- M. G. Howlett, A. H. J. Engwerda, R. J. H. Scanes and S. P. Fletcher, Nat. Chem., 2022, 14, 805–810 CrossRef CAS PubMed
- A. Chatterjee, A. Reja, S. Pal and D. Das, Chem. Soc. Rev., 2022, 51, 3047–3070 RSC
- F. Spath, C. Donau, A. M. Bergmann, M. Kranzlein, C. V. Synatschke, B. Rieger and J. Boekhoven, J. Am. Chem. Soc., 2021, 143, 4782–4789 CrossRef PubMed
- I. Myrgorodska, I. Colomer and S. P. Fletcher, ChemSystemsChem, 2020, 2, 1–5 CrossRef
- C. Donau, F. Spath, M. Sosson, B. A. K. Kriebisch, F. Schnitter, M. Tena-Solsona, H. S. Kang, E. Salibi, M. Sattler, H. Mutschler and J. Boekhoven, Nat. Commun., 2020, 11, 5167 CrossRef CAS PubMed
- K. K. Nakashima, M. A. Vibhute and E. Spruijt, Front. Mol. Biosci., 2019, 6, 1–9 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04548f |
| This journal is © The Royal Society of Chemistry 2022 |