
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular structure design of planar zwitterionic polymer electrode materials for all-organic symmetric batteries†
Jun
Wang
a,
Haichao
Liu
 c,
Chunya
Du
c,
Yu
Liu
d,
Bing
Liu
a,
Haoran
Guan
a,
Shaowei
Guan
*a,
Zhenhua
Sun
*b and
Hongyan
Yao
*a
c,
Chunya
Du
c,
Yu
Liu
d,
Bing
Liu
a,
Haoran
Guan
a,
Shaowei
Guan
*a,
Zhenhua
Sun
*b and
Hongyan
Yao
*a
aNational & Local Joint Engineering Laboratory for Synthesis Technology of High Performance Polymer, Key Laboratory of High Performance Plastics, Ministry of Education, College of Chemistry, Jilin University, Qianjin Street 2699, Changchun, 130012, P. R. China. E-mail: yaohy@jlu.edu.cn; guansw@jlu.edu.cn
bShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, Shenyang 110016, P. R. China. E-mail: zhsun@imr.ac.cn
cState Key Lab of Supramolecular Structure and Materials, Jilin University, 2699 Qianjin Avenue, Changchun, 130012, P. R. China
dCollege of Sciences, Shenyang University of Chemical Technology, Shenyang, 110142, P. R. China
First published on 16th September 2022
Abstract
All-organic symmetric lithium-ion batteries (LIBs) show promising prospects in sustainable energy storage systems, due to their environmental friendliness, structural diversity and low cost. Nevertheless, it remains a great challenge to explore suitable electrode materials and achieve excellent battery performance for all-organic symmetric LIBs. Herein, a squaraine-anthraquinone polymer (PSQ) electrode material was designed through rational molecular engineering. The well-designed extended π-conjugated system, donor–acceptor structure, abundant redox-active sites and rational manipulation of weak inter-/intramolecular interactions endow the PSQ electrode with outstanding electrochemical performance. The capacity of the PSQ cathode can be optimized to 311.5 mA h g−1 by in situ carbon-template polymerization. Impressively, PSQ-based all-organic symmetric LIBs displayed high reversible capacity (170.8 mA h g−1 at 50 mA g−1), excellent rate performance (64.9% capacity retention at 4000 mA g−1vs. 50 mA g−1), ultralong cycle life up to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 2000 mA g−1 and 97% capacity retention after 2500 cycles at 500 mA g−1, which is one of the best comprehensive battery performances among the all-organic LIBs reported thus far.
000 cycles at 2000 mA g−1 and 97% capacity retention after 2500 cycles at 500 mA g−1, which is one of the best comprehensive battery performances among the all-organic LIBs reported thus far.
Introduction
Rechargeable lithium-ion batteries (LIBs) have been commercialized for decades and currently play a crucial role as the main power supply in mobile electronics and electrical vehicles.1–4 However, concerns of limited resources, vast energy consumption during the fabrication process, and serious environmental pollution are inevitably caused by using conventional inorganic electrode materials.5,6 As an alternative, organic electrode materials have gained widespread attention for their inherent merits of abundant resources, environmental friendliness, and high structural designability.7–9 To date, tremendous advances have been made to explore novel organic electrode materials (cathodes or anodes) and optimize their electrochemical performance in organic/inorganic hybrid half-battery systems.10–14 Nevertheless, studies of all-organic batteries for developing sustainable energy storage systems are still heavily lagging far behind.15–18All-organic batteries are assembled with organic cathode and anode materials. They are classified into three cell configurations according to the type of electrode materials used in the cathode and anode (n, p and bipolar type), namely, cationic rocking-chair, anionic rocking-chair and dual-ion batteries.15,19,20 It is usually beneficial to simplify the fabrication process and reduce manufacturing costs to construct all-organic symmetric batteries using the same material as electrodes.21–23 The chemical structures of electrode materials significantly affect the electrochemical performance of all-organic symmetric batteries. The molecular design for electrode materials of all-organic symmetric batteries needs to rationally balance the working voltage, specific capacity, and cycling stability of both the cathode and anode.22,24 First, reversible redox reactions of electrode materials are supposed to take place at both higher voltage platforms (>2 V) and lower voltage platforms (<1 V) for all-organic symmetric batteries. The organic electrode materials containing cathode-active groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN, etc.) and anode-active groups (CC, etc.) may match the redox potential needs in all-organic symmetric batteries.17,25 Additionally, rationally increasing the loading of redox-active sites (or reducing the redox-inert units) has been proven to be a universal strategy to improve the theoretical capacity for both organic cathode and anode materials.26–29 For instance, Chen et al. reported a polymer with a high loading of redox-active groups (CO and CN), which exhibited an impressive specific capacity of 502.4 mA h g−1 at 0.05C.30 Moreover, methods used in organic half batteries, such as manipulating weak intermolecular or intramolecular interactions (π–π interactions, hydrogen bonds, ion–dipole interactions, etc.)31–34 as well as constructing an extended π-conjugated system35,36 might be helpful to improve the cycling stability of all-organic symmetric batteries. Although some advances have been made in developing all-organic batteries, they still face many challenges and problems, such as unsatisfactory rate performance, inferior capacity and fast capacity fading, due to the inferior electronic conductivity, introduction of inactive linkages, and dissolution of active materials in organic electrolytes.15,21 Hence, the exploration of novel electrode materials and the achievement of excellent comprehensive electrochemical performance for all-organic symmetric batteries remain highly challenging and urgent.
O, CN, etc.) and anode-active groups (CC, etc.) may match the redox potential needs in all-organic symmetric batteries.17,25 Additionally, rationally increasing the loading of redox-active sites (or reducing the redox-inert units) has been proven to be a universal strategy to improve the theoretical capacity for both organic cathode and anode materials.26–29 For instance, Chen et al. reported a polymer with a high loading of redox-active groups (CO and CN), which exhibited an impressive specific capacity of 502.4 mA h g−1 at 0.05C.30 Moreover, methods used in organic half batteries, such as manipulating weak intermolecular or intramolecular interactions (π–π interactions, hydrogen bonds, ion–dipole interactions, etc.)31–34 as well as constructing an extended π-conjugated system35,36 might be helpful to improve the cycling stability of all-organic symmetric batteries. Although some advances have been made in developing all-organic batteries, they still face many challenges and problems, such as unsatisfactory rate performance, inferior capacity and fast capacity fading, due to the inferior electronic conductivity, introduction of inactive linkages, and dissolution of active materials in organic electrolytes.15,21 Hence, the exploration of novel electrode materials and the achievement of excellent comprehensive electrochemical performance for all-organic symmetric batteries remain highly challenging and urgent.
Squaraines (SQ), typical donor–acceptor type organic dye semiconductive molecules, are extensively applied in the fields of nonlinear optics, photovoltaics, photodynamic therapy, etc.37–39 Benefiting from the planar zigzagged zwitterionic resonance structure, some SQs are expected to have extended π-conjugated systems and low energy gaps (Eg),40,41 which make them potential promising electrode materials in the field of LIBs. In this work, we designed and proposed a squaraine-anthraquinone polymer (PSQ) that can be used simultaneously as cathode and anode materials in all-organic batteries. From the perspective of molecular engineering, PSQ was designed and applied as an electrode material based on the following advantages and considerations: (1) the high loading of active sites (CO) guarantees the high theoretical capacity of the PSQ cathode in a relatively high voltage range, (2) the abundant unsaturated carbon in the aromatic ring is possibly helpful for the PSQ anode to achieve high capacity in a low voltage range,8,42,43 and (3) the planar and π-conjugated structure of monomers is beneficial to reduce the solubility and promote charge transfer in the polymer electrode. As a result, the PSQ cathode exhibited a high reversible capacity (210.7 mA h g−1 at 50 mA g−1), excellent rate performance (75.5% capacity retention at 2000 mA g−1vs. 50 mA g−1), and outstanding cycling stability (95% capacity retention after 6000 cycles at 2000 mA g−1). The capacity of the PSQ cathode can be optimized to 311.5 mA h g−1 by in situ carbon-template polymerization. However, the cycling stability of the PSQ/carbon composite cathode shows obvious degradation of performance. Meanwhile, the PSQ anode also displayed a high capacity of 723.2 mA h g−1 at 100 mA g−1 after 10 cycles and a long cycle life of 2000 cycles. More impressively, the PSQ-based all-organic symmetric LIBs assembled with the PSQ cathode and anode showed extraordinary battery performance. It exhibited a high reversible capacity (170.8 mA h g−1 at 50 mA g−1), excellent rate performance (64.9% capacity retention at 4000 mA g−1vs. 50 mA g−1), and ultralong cycle life up to 30000 cycles with a low capacity fading rate of 0.0017% and 97% capacity retention after 2500 cycles at 500 mA g−1. This work provides an efficient molecular design strategy for exploring polymeric electrode materials for all-organic symmetric LIBs with high comprehensive performance.
Results and discussion
The structural and electronic features of organic electrodes are inherent reasons for their diverse electrochemical behavior,28,44 which prompted us to explore and evaluate the suitability for PSQ being applied as an organic electrode material at the molecular level via theoretical calculations. The optimized configuration of two monomers (squaraine (SQ) and 2,6-diaminoanthraquinone (2,6-DAQ)) and PSQ is planar (Fig. 1a and Table S1†). The planar configuration of PSQ is beneficial for the extension of π-conjugation and enhancing molecular rigidity, which could reduce its solubility in organic electrolytes and improve its long-term cycling stability.35,44 The top view of the optimized structure (repeat units n = 2) illustrates a planar zigzagged zwitterionic configuration of PSQ (Fig. 1b). The distance between the O atoms in squaraine moieties and the adjacent H atoms in the aromatic ring of anthraquinone is ca. 2.16 Å, which is much shorter than the sum of the van der Waals radii of the O and H atoms (2.72 Å). This result indicates the existence of intramolecular hydrogen bonds that make PSQ show a planar configuration.32,33 The extended π-conjugated system and planar structure of PSQ can facilitate intermolecular π–π interactions (Fig. 1c),32,35 which could be conducive to reducing the solubility of PSQ and enhancing the cycling stability of PSQ-based batteries.35 The molecular electrostatic potential (MESP) of PSQ was determined using one (n = 1) and two (n = 2) repeat units based on the optimized configurations to predict the possible redox-active sites and Li binding sequence of PSQ (Fig. 1d).45–47 In principle, the atoms with lower MESP values on the van der Waals surface have a higher binding affinity to Li+, which is favorable for serving as redox-active sites. All the CO and C–O− in squaraine moieties and the CO in anthraquinone moieties show low MESP values, and the MESP values of the former are lower than those of the latter. These results indicate that the squaraine and anthraquinone moieties can bind with Li+ in sequence during the reduction process of the PSQ electrode.
 | ||
| Fig. 1 (a) Synthesis routes for PSQ (above) and side view of the optimized configuration for monomers and PSQ (below). Schematic diagrams of (b) intramolecular hydrogen bonds and (c) intermolecular π–π interactions of PSQ based on the optimized molecular structure (C: gray; H: white; O: red; N: blue). (d) The molecular electrostatic potential (MESP) of PSQ (repeat unit n = 1 and 2). (e) LUMO and HOMO energy diagrams for PSQ (repeat unit n = 2) and monomers. | ||
PSQ displays a lower lowest unoccupied molecular orbital (LUMO) level than the two monomers (Fig. 1e (n = 2), Fig. S1†(n = 1)). The low LUMO level endows PSQ with larger electron affinity, higher oxidizing ability, and higher discharging voltage than those of its monomers (2,6-DAQ and SQ). PSQ (n = 2) also has a much smaller energy gap (2.61 eV) than monomers, mainly due to its donor–acceptor structure, i.e., the SQ moiety as the donor and the 2,6-DAQ moiety as the acceptor.48,49 It is worth noting that the energy gap of PSQ (n = 2, 2.61 eV) is smaller than that of PSQ (n = 1, 2.83 eV), suggesting that extending the π-conjugation length is helpful for lowering the energy gap.50,51 The weak intermolecular and intramolecular interactions, extended π-conjugated system, and donor–acceptor structure are expected to significantly facilitate the charge transfer rate and stabilize the cycling process of the PSQ electrode.
PSQ was synthesized from SQ and 2,6-DAQ via a facile one-pot polymerization with a high production yield beyond 90%. The monomers are obtained from commercial sources at low prices, and the synthesis of PSQ is scalable, allowing multi-dozen gram-scale production (Fig. S2†). The PSQ is insoluble in common organic solvents and is expected to have good stability in the electrolyte. The chemical structure, composition, aggregate structure, microscopic morphology, and thermal stability of PSQ were characterized by Fourier transform infrared (FT-IR), solid-state 13C nuclear magnetic resonance (13C NMR), elemental analysis (EA), X-ray photoelectron spectroscopy (XPS), X-ray diffraction analysis (XRD), field-emission scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM), and thermogravimetric analysis (TGA). The characteristic bands of N–H (3252 cm−1) and CN (1795 cm−1) in the FT-IR spectrum of PSQ (Fig. 2a) indicate the successful polymerization of SQ and 2,6-DAQ.52 The absorption bands at 1619 cm−1 and 1388 cm−1 are assigned to CO and C–O in 1,3-squarate moieties of PSQ,53 respectively, while the absorption band at 1657 cm−1 is ascribed to CO in anthraquinone moieties. In the solid-state 13C NMR spectrum of PSQ (Fig. 2b), the peak at 178 ppm, the peaks at 165 and 152 ppm, and the peaks at 115–141 ppm are attributed to the carbons of CO in anthraquinone moieties, the carbons of CO and C–O in 1,3-squarate moieties, and the carbons of the benzene ring in anthraquinone moieties and four-membered ring of 1,3-squarate moieties, respectively.52,53 The elements in XPS (Fig. S3†) and element contents obtained from EA measurement (Table S2†) are in accordance with the PSQ and its theoretical value. The above characterizations confirm the successful synthesis of PSQ. The PSQ has satisfactory thermal stability (Fig. S4†). The strong diffusion peak at ca. 27.5° in the XRD pattern indicates strong π–π intermolecular interactions in PSQ (Fig. S5†)35,54 The SEM (Fig. 2c and S6†) and TEM images (Fig. 2d and S6†) show that PSQ is a lamellar structure with a size below 1 μm. The layer-by-layer morphology of PSQ originates from the intermolecular π–π interactions, which is consistent with the XRD results and their optimized configuration.
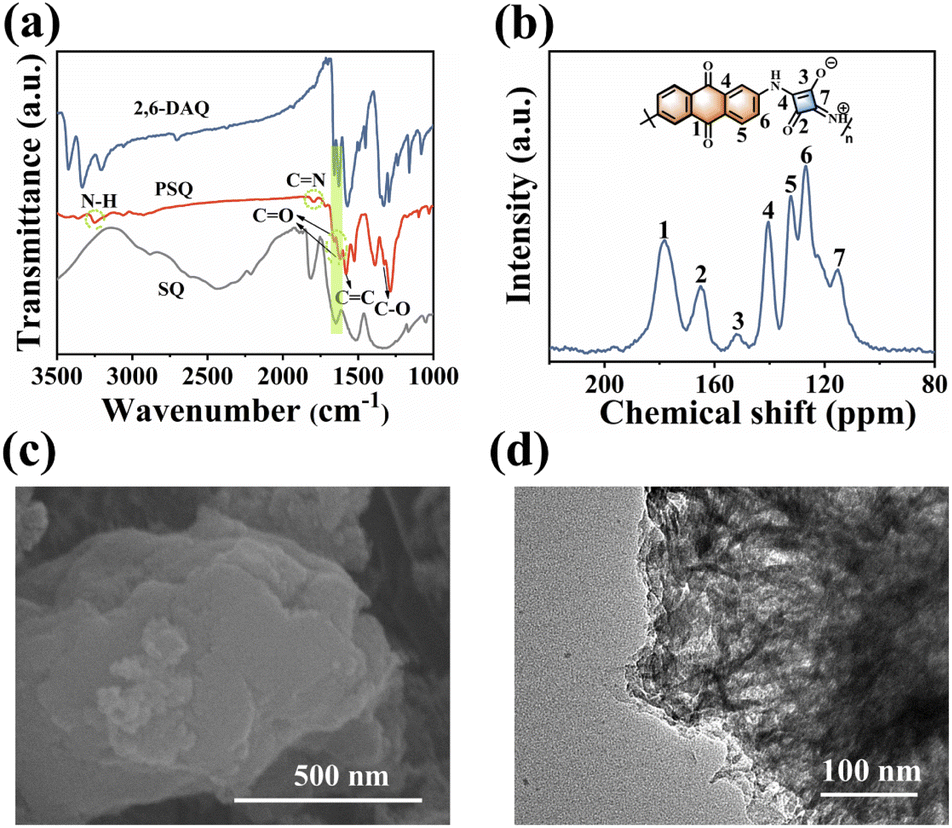 | ||
| Fig. 2 (a) FT-IR spectra of PSQ and monomers. (b) Solid-state 13C NMR spectrum of PSQ. (c) SEM image and (d) TEM image of PSQ. | ||
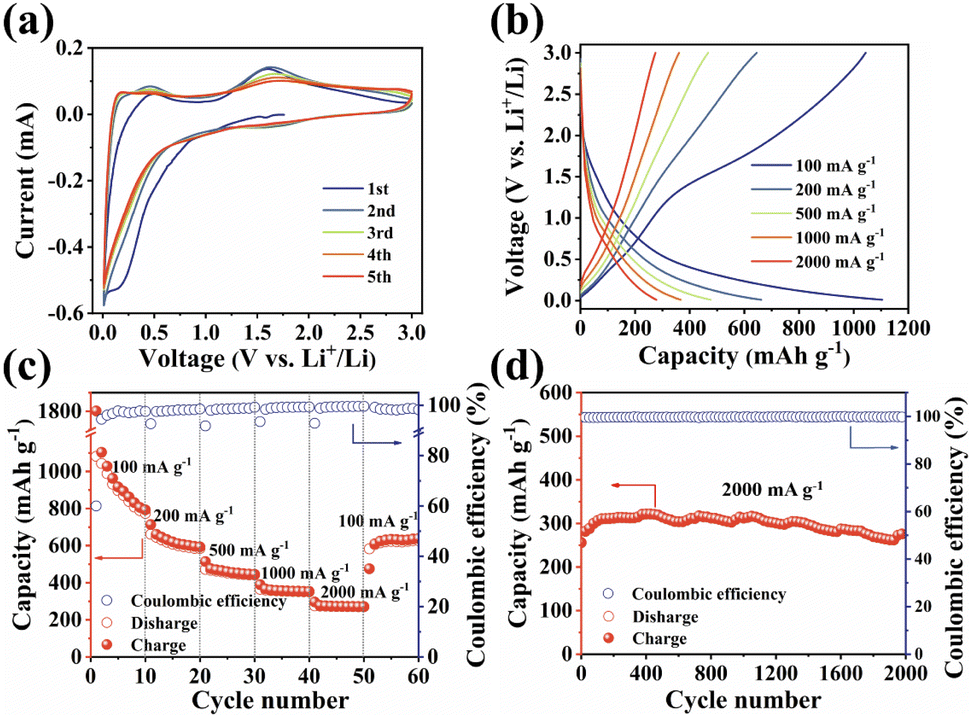
The unique structural characteristics including the extended π-conjugated system, zigzagged zwitterionic resonance structure, low energy gap, and abundant redox-active sites make PSQ promising electrode materials in organic LIB applications.55 The electrochemical performance of the PSQ cathode was tested in a voltage range of 1.0–3.5 V in a half-cell using lithium as an anode. In the cyclic voltammetry (CV) curves of PSQ at 0.2 mV s−1 (Fig. 3a), a pair of closely overlapped and broad peaks situated at 1.96 V (cathodic) and 2.22 V (anodic) are observed, which are attributed to the reversible insertion/desertion process of Li+ in the CO of anthraquinone and 1,3-squarate moieties.53,56 In the charge–discharge curves, PSQ exhibits long sloping discharge curves from 2.5 V to 1.75 V and a small sloping discharge region in the range of 1.2–1.0 V, which is consistent with the results of the CV curves. The PSQ cathode shows an initial capacity of 134.2 mA h g−1, and the capacity gradually increases to 182.8 mA h g−1 at the 500th cycle (Fig. 3b) (as shown in Fig. S7,† the capacity contribution of conductive carbon in the PSQ cathode is negligible). The CV of PSQ at 2 mV s−1 for 30 cycles was tested to explore the gradually increasing capacity (activation process) of PSQ during the electrochemical test (Fig. S8†). The CV area gradually increases with the scans, suggesting the improved utilization of the active sites. The voltage peak separations are smaller by degrees, which means that the electrochemical polarization in the PSQ cathode is decreased with the scans. The activation process of PSQ is also observed in the electrochemical impedance spectra (EIS) (Fig. S9†). The electrochemical impedance decreases with the increasing cycle number. The above results suggest the facilitated charge transfer process when the electrolytes penetrate the densely packed and crystal structure of PSQ, which contributes to the occurrence of the activation process.42,53
 | ||
| Fig. 3 (a) CV curves of the PSQ cathode at 1–3.5 V (vs. Li+/Li). (b) The charge–discharge curves of the PSQ cathode at 100 mA g−1. (c) Rate performance of the PSQ and 2,6-DAQ cathodes. (d) Plots of peak current versus scan rate and the b-values obtained according to i = avb. (e) Separation of capacitive and diffusion currents at different scan rates. (f) The Li+ diffusion coefficients (DLi+) of the PSQ cathode in the corresponding voltage range (calculated from GITT tests). (g) Long-term cycling performance of the PSQ cathode at 2000 mA g−1. | ||
The PSQ cathode exhibits reversible capacities of 151.2, 145.4, 138.7, 127.6, 119.3 and 114.1 mA h g−1 at 50, 100, 200, 500, 1000 and 2000 mA h g−1, respectively (Fig. 3c). The capacity recovers to 152.7 mA h g−1 when the current density is reset to 50 mA g−1. The capacity keeps increasing and the value stabilizes beyond 210.7 mA h g−1 after the 120th cycle at 50 mA g−1. The rate capacity retention is as high as 75.5% (2000 mA g−1vs. 50 mA g−1). The good rate performance is attributed to the extended π-conjugated structure and the narrow energy gap of PSQ.12,35 The fast reaction kinetics of PSQ were explored via CV curves at various sweep rates from 0.2 to 2 mV s−1. The CV curves display similar shapes with gradually magnified currents (Fig. S10†). In principle, the peak current (i) obeys a power-law relationship with the scan rate (v), according to the equation: i = avb. The b value is determined by plotting log(i) vs. log(ν), which is an indicator of reaction kinetics. The b values of the PSQ cathode are 0.93, 0.99, 0.88 and 0.95, suggesting fast kinetics dominated by a surface capacitive process for the PSQ cathode (Fig. 3d).57 The capacitive contribution at various scan rates is quantified according to the equation i = k1v + k2v1/2. The capacitive contributions are beyond 82.1% at various scan rates and increase progressively with the scan rates (Fig. 3e). The highest capacitive ratio of 94.2% is observed at 2 mV s−1. Additionally, the Li+ diffusion coefficients (DLi+) of the PSQ cathode were obtained via the galvanostatic intermittent titration technique (GITT) test (Fig. 3f and S11†). The average DLi+ values determined according to equation 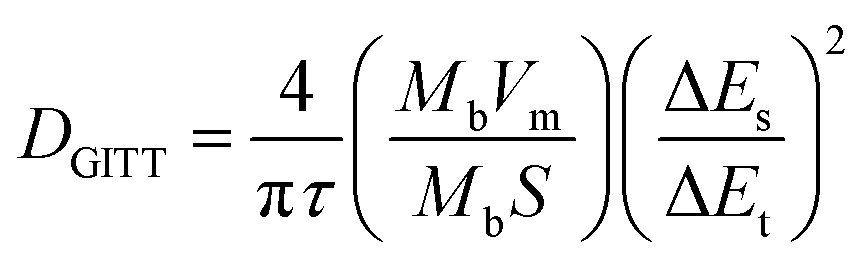 are 1.03 × 10−9 and 1.01 × 10−9 cm s−1 for discharge and charge processes, respectively. The DLi+ of PSQ is at a high level among organic electrode materials, indicating the fast charge transfer rate for the PSQ cathode. The above results indicate that the redox reaction of the PSQ cathode is dominated by the surface-induced capacitive and fast charge-transfer process, which contributes to the high-power capability of PSQ.
are 1.03 × 10−9 and 1.01 × 10−9 cm s−1 for discharge and charge processes, respectively. The DLi+ of PSQ is at a high level among organic electrode materials, indicating the fast charge transfer rate for the PSQ cathode. The above results indicate that the redox reaction of the PSQ cathode is dominated by the surface-induced capacitive and fast charge-transfer process, which contributes to the high-power capability of PSQ.
The long-term cycling stability of the PSQ cathode was evaluated. The PSQ cathode displays an activation process at 100 mA g−1 and shows a high capacity of 182.8 mA h g−1 after 500 cycles during the long-term cycling test (Fig. S12†). At current densities of 500 and 1000 mA g−1, the PSQ cathode shows high capacity retention of 97.9% and 92.6% after 1000 and 2000 cycles, respectively. Remarkably, the PSQ cathode also displays an ultralong cycle life at 2000 mA g−1. The capacity retention is 95% after 6000 cycles with an extremely low capacity decay rate of 0.00089% (Fig. 3g), which is among the best results in organic cathode materials.14,26 The excellent cycling stability and long cycle life of PSQ are attributed to its insolubility and high structural stability, which is enabled by weak intermolecular or intramolecular interactions (i.e., π–π interactions, hydrogen bonds, etc.), extended π-conjugated system and planar rigid structure.
The abundant unsaturated carbon is beneficial for PSQ to achieve high capacity in a low voltage range and make it a potential candidate as an organic anode material. The PSQ anode half-cell was assembled using PSQ and a lithium reference electrode and tested in a low voltage range of 0.01–3 V. The CV curves of the PSQ anode display two pairs of redox peaks at a voltage of approximately 1.6 V and 0.2 V (Fig. 4a), which are assigned to the reversible lithiation/delithiation process of CO and CC, respectively.58 The high overlap of CV curves demonstrates the good redox reversibility of the PSQ anode. The PSQ anode exhibits typical long slopes in charge–discharge curves (Fig. 4b), corresponding to the successive redox reactions of CO and CC with Li+ in the PSQ anode. The discharge capacity of the PSQ anode at the 2nd cycle is 1020.3 mA h g−1 at 100 mA g−1 with a CE of 94.5%, and the value becomes 723.2 mA h g−1 with a high CE of 97.8% after 10 cycles. (The capacities are obtained after deducting the capacity contribution of conductive carbon and shown in Fig. S13†). The rapid capacity degradation in initial cycles might be attributed to partial destruction of PSQ structure upon the intercalation of Li+ into per unsaturated CC. It is also worth noting that the inferior reversibility, especially in the first several cycles, is also usually observed in the reported organic electrodes with the so-called “superlithium” phenomenon, which prompts us to pay more attention to the in-depth mechanism and causes of the “superlithium” phenomenon.58,61 Besides, the capacity of the PSQ anode is 793.2, 593.6, 446, 353.7 and 271.7 mA h g−1 at current densities of 100, 200, 500, 1000 and 2000 mA g−1, respectively (Fig. 4c). A high capacity is retained when the current density is reset to 100 mA g−1, indicating the good rate performance of the PSQ anode. Additionally, the PSQ anode exhibits outstanding cycling stability. High capacity retention of 86.7% is achieved after 2000 cycles at 2000 mA g−1 (Fig. 4d).
 | ||
| Fig. 4 Electrochemical performance for the PSQ anode. (a) CV curves. (b) Rate performance. (c) The charge–discharge curves. (d) Cycling performance at 2000 mA g−1. | ||
The storage mechanism of the PSQ cathode was studied by the evolution of ex situ FT-IR and XPS spectra for the PSQ cathode at different discharged and charged states. When the battery is discharged to 1 V, the decreased intensity of the CO band and the increased intensity of the C–O band in the high-resolution C 1s XPS spectra indicate Li+ intercalation into the CO of PSQ (Fig. 5a). The opposite changes occur in the C 1s XPS spectrum when the battery is charged to 3.5 V, which corresponds to Li+ extraction from the PSQ cathode.30,53 The high-resolution O 1s XPS spectra (Fig. 5b) also show the same change trend during the charge–discharge process and confirm the high redox reversibility of the PSQ cathode. The ex situ FT-IR spectra were also recorded from the open-circuit voltage (2.7 V) to the fully charged state (3.5 V) (Fig. 5c). The intensity of the characteristic CO band (1615 cm−1) in 1,3-squarate moieties decreases when the battery is discharged to 2.2 V. During the following discharge process, the intensity of the CO characteristic band in anthraquinone moieties which is located at 1653 cm−1 decreases in the voltage range of 2.2–1.0 V. These results demonstrate that the CO in 1,3-squarate moieties is discharged in priority and the CO in anthraquinone moieties is discharged later because the former CO has a lower MESP value and higher binding affinity to Li+.45,46 The opposite intensity change trend of the CO and the C–O band is observed in the charge process, suggesting the Li+ desertion process in redox-active sites. Additionally, the reversible intensity changes of C–N (1347 cm−1), CN (1323 cm−1) and Li–O (1086 cm−1) also confirm the high reversibility of the redox reaction for the PSQ cathode. Based on the above results, the storage mechanism for the PSQ cathode is proposed (Fig. 5d). During discharge process, the CO in 1,3-squarate and anthraquinone moieties are reduced by Li in sequence, and the opposite oxidization process occurs in the charge process. The whole charge–discharge process involves a four e− redox reaction for the PSQ cathode.
 | ||
| Fig. 5 Ex situ (a) C 1s and (b) O 1s XPS spectra and (c) FT-IR spectra of the PSQ cathode. (d) Schematic diagram of the redox mechanism for the PSQ cathode. Ex situ (e) C 1s and (f) O 1s XPS spectra and (g) FT-IR spectra of the PSQ anode. (h) Schematic diagram of the redox mechanism for the PSQ anode. | ||
Moreover, the LUMO and HOMO distribution and the energy gap of PSQ in different lithiated states were obtained via DFT calculations (Fig. S14†). The pristine and lithiated PSQ display similar donor–acceptor structures and low Eg, indicating the good conductivity of the electrode materials during the charge–discharge process.
The lithium storage behavior of the PSQ anode was also explored via ex situ FT-IR, XPS and Raman tests. In the XPS C 1s spectra (Fig. 5e), reversible intensity changes of CO, C–O and C–Li are observed in the charge–discharge process of the PSQ anode, corresponding to the reversible insertion/desertion process of Li+ in the CO and CC groups. The XPS O 1s spectra also show reversible intensity changes of CO, C–O and O–Li (Fig. 5f), which is in accordance with that in C 1s spectra. The results of ex situ FT-IR also confirm the reversible redox process in CC and CO of the PSQ anode (Fig. 5g). Reversible intensity changes occur in the characteristic bands of CO (1614 cm−1 and 1654 cm−1) and CC (1586 cm−1). In addition, the reversible band changes of disordered C (D band, C–C) and CO in the ex situ Raman spectra (Fig. S15†) also support that the redox mechanism of PSQ anode involves the intercalation/deintercalation of Li+ in CO and CC groups.22 Based on the above results, the redox mechanism of the PSQ anode is illustrated as a possibly maximum of 20 e− reversible redox processes in an ideal case (Fig. 5h). The SEM images of the PSQ cathode and anode in different charge–discharge states (Fig. S16–S18†) and the TEM images of the cycled PSQ anode (Fig. S19†) show the compact electrode surface, which might be attributed to the development of a solid electrolyte interface (SEI).58–60 The SEI film might be helpful for inhibiting the dissolution of organic materials in the electrolyte and protecting them from being affected by electrochemical byproducts such as HF, thus being conducive to their long-term cycling stability.
It is known that the PSQ cathode has an ideal 4 e− redox process during the discharge–charge process from the storage mechanism analysis and a high theoretical capacity of 339 mA h g−1. In contrast, the PSQ shows a moderate reversible capacity of 210.7 mA h g−1 which corresponds to ca. 62% active site utilization. To fully realize the electrochemical capacity of the PSQ cathode, PSQ-K was prepared with a Ketjen black template by in situ polymerization. Different from the dense lamellar structure of PSQ, PSQ-K shows a uniformly distributed microstructure (Fig. S20†), which enables the active sites in PSQ-K to be highly accessible. As a result, PSQ-K exhibits an outstanding reversible capacity of 311.5 mA h g−1 at 50 mA g−1, corresponding to a high active site utilization of ca. 92% (Fig. S21†). The PSQ-K also shows a high rate performance that is comparable with that of the PSQ. However, the cycling stability (71.1% retention after 2000 cycles at 2000 mA g−1) of PSQ-K is much inferior to that of PSQ, which might be attributed to the improved solubility of PSQ-K caused by the reduced intermolecular interactions after the in situ polymerization process. Thus, considering the simplified production process as well as the long cycle life of pristine PSQ, which is more favorable for potential practical applications, PSQ rather than PSQ-K was selected as the main research object in this work. Nevertheless, the above results suggest the great potential of PSQ/carbon composites are high-energy and high-power electrode materials, and the systematic optimization of PSQ/carbon composite electrodes will be performed in future work.
The electrode materials in all-organic symmetric batteries need to balance the voltage, specific capacity and cycling stability of the cathode and anode.15 Benefiting from the outstanding electrochemical performance and suitable voltage of the PSQ cathode and anode, PSQ is expected to be a promising cathode and anode material for all-organic symmetric LIBs. The PSQ-based all-organic symmetric LIBs were assembled by using the PSQ cathode and fully lithiated PSQ (discharged to 0.01 V) anode. In the cationic rocking chair configuration for the PSQ-based all-organic symmetric battery, Li+ shuttles back and forth between the cathode and anode during the charge–discharge process (Fig. 6a). The electrolyte constituent was optimized (the electrochemical tests using the other electrolyte were shown in Fig. S22†), and a mass ratio of the anode and cathode (1:3) was selected. The electrochemical performance of the PSQ-based all-organic symmetric battery was comprehensively evaluated. In the CV curves (Fig. 6b), two pairs of redox peaks appear at approximately 1.1 V and 1.72 V which are ascribed to the reversible redox reaction of CO in PSQ. The long and continuous sloping voltage profiles in charge–discharge curves (Fig. S23†) are in accordance with the broad redox peaks in the voltage range of 0.2–2.2 V in the CV curves. The PSQ-based all-organic symmetric battery displays a satisfactory discharge voltage of ca. 1.4 V. It is worth noting that the working voltage of the PSQ-based all-organic symmetric battery is moderate among reported all-organic batteries. It is far from comparable to commercial batteries which show a high average working voltage. There is still much room for improving the working voltage of the full battery via molecular design strategies. In addition, the PSQ-based all-organic symmetric battery also exhibits excellent rate performance (Fig. 6c and d). They exhibit high capacities of 156.4, 152.4, 134.4, 116, 102.1, and 98.6 mA h g−1 at 100, 200, 500, 1000, 2000 and 4000 mA g−1, respectively. When the current rate is reset to 50 mA g−1 and keeps running for another 50 cycles, a high discharge capacity of 170.8 mA h g−1 is observed. The excellent rate capability is attributed to the fast reaction kinetics (Fig. 6e and S24†), which are facilitated by the low energy gap. Moreover, the PSQ-based all-organic symmetric battery exhibits outstanding long-term cycling stability. They exhibit an excellent capacity retention of 97% at 500 mA g−1 after 2500 cycles (Fig. 6f). Moreover, they show high capacity retentions of 68.4% and 46.2% after 10000 and 30000 cycles at a high current density of 2000 mA g−1, respectively, with ultra-low capacity fading rates of 0.0032% and 0.0017%, respectively (Fig. 6g). The corresponding coulombic efficiency is close to 100% during the whole cycling test. Consequently, the high reversible specific capacity, excellent rate performance, satisfactory working voltage and ultrahigh cycling stability of the PSQ-based all-organic symmetric battery make it one of the best high-performance all-organic batteries (Fig. 6d and Table S3†).
 | ||
| Fig. 6 Electrochemical performance of PSQ-based all-organic symmetric battery. (a) Schematic diagram of the charge–discharge process. (b) CV curves at 2 mV s−1. (c) Rate performance at various current densities. (d) Comprehensive electrochemical performance comparison radar chart for reported all-organic batteries (the details are shown in Table S3†). (e) Surface and diffusion-controlled contributions of the PSQ cathode at 2 mV s−1. (f) Cycling performance at 500 mA g−1. (g) Cycling performance at 2000 mA g−1. | ||
Conclusions
A planar zwitterionic squaraine-anthraquinone polymer (PSQ) was designed and synthesized as an electrode material for all-organic symmetric LIBs. The PSQ electrode for half-batteries exhibited high capacities (210.7 mA h g−1 at 50 mA g−1 for the cathode and 311.5 mA h g−1 at 50 mA g−1 for the PSQ-K cathode prepared by in situ polymerization, 723.2 mA h g−1 at 100 mA g−1 for the anode), excellent rate performance (114.1 mA h g−1 at 2000 mA g−1 for the cathode, 271.7 mA h g−1 at 2000 mA g−1 for the anode), and outstanding cycling stability (95% capacity retention after 6000 cycles at 2000 mA g−1 for the cathode, 86.7% after 2000 cycles at 2000 mA g−1 for the anode). More impressively, the all-organic symmetric battery assembled using PSQ as both the cathode and anode showed outstanding comprehensive battery performance including high reversible capacity (170.8 mA h g−1 at 50 mA g−1), excellent rate performance (98.6 mA h g−1 at 4000 mA g−1) and ultralong cycle life up to 30000 cycle life at 2000 mA g−1 with a low capacity fading rate of 0.0017%, which are among the best results of reported all-organic batteries. The excellent comprehensive electrochemical performance of PSQ is attributed to the well-designed extended π-conjugated system, donor–acceptor structure, abundant redox-active sites, and the rational manipulation of weak inter-/intramolecular interactions. Our results demonstrate the promoting effects of rational manipulation of weak inter-/intramolecular interactions as well as constructing planar and donor–acceptor structures in polymers for addressing the intrinsic dissolution and low conductivity problems of organic electrode materials. This work provides new insights into the molecular design of high-performance and low-cost all-organic symmetric battery electrode materials, which is meaningful for the practical applications of organic electrode materials in future sustainable energy storage systems.
Data availability
Detailed experimental procedures are included in the ESI.†Author contributions
Conceptualization: J. W., Y. H. Y., S. W. G.; funding acquisition: Y. H. Y. and S. W. G.; formal analysis: H. C. L. and C. Y. D.; investigation: J. W., H. C. L., C. Y. D., Y. L., B. L., and H. R. G., methodology: J. W., Y. H. Y., and Z. H. S.; validation: B. L. and H. R. G.; supervision: Y. H. Y. and S. W. G.; project administration: Y. H. Y. and S. W. G.; writing-original draft: J. W., Y. H. Y., and S. W. G.; writing-review and editing: J. W., Y. H. Y., Z. H. S. and S. W. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is dedicated to the 70th anniversary of the chemistry of Jilin University. The authors thank the financial support from the National Natural Science Foundation of China (No. 51903100) and the Science and Technology Development Plan of Jilin Province, P. R. China (No. 20210402060GH).Notes and references
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef.
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS.
- H. K. Bruce Dunn, Jean-Marie Tarascon, Science, 2011, 334, 928–935 CrossRef.
- T. B. Schon, B. T. McAllister, P. F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 14, 6490–6557 CrossRef PubMed.
- Y. Liang and Y. Yao, Joule, 2018, 2, 1690–1706 CrossRef CAS.
- K. Nakashima, T. Shimizu, Y. Kamakura, A. Hinokimoto, Y. Kitagawa, H. Yoshikawa and D. Tanaka, Chem. Sci., 2020, 11, 37–43 RSC.
- B. Lee, Y. Ko, G. Kwon, S. Lee, K. Ku, J. Kim and K. Kang, Joule, 2018, 2, 61–75 CrossRef CAS.
- Q. Zhang, X. Cui, S. Hao, Q. Zhang, Z. Guo, H. Li, Z. Lin and Y. Yang, Mater. Today, 2021, 50, 170–198 CrossRef CAS.
- T. P. Nguyen, A. D. Easley, N. Kang, S. Khan, S. M. Lim, Y. H. Rezenom, S. Wang, D. K. Tran, J. Fan, R. A. Letteri, X. He, L. Su, C. H. Yu, J. L. Lutkenhaus and K. L. Wooley, Nature, 2021, 593, 61–66 CrossRef CAS PubMed.
- M. Lee, J. Hong, J. Lopez, Y. Sun, D. Feng, K. Lim, W. C. Chueh, M. F. Toney, Y. Cui and Z. Bao, Nat. Energy, 2017, 2, 861–868 CrossRef CAS.
- C. Peng, G.-H. Ning, J. Su, G. Zhong, W. Tang, B. Tian, C. Su, D. Yu, L. Zu, J. Yang, M.-F. Ng, Y.-S. Hu, Y. Yang, M. Armand and K. P. Loh, Nat. Energy, 2017, 2, 17074 CrossRef CAS.
- F. Zhang, W. Zhang, D. Wexler and Z. Guo, Adv. Mater., 2022, 34, 2107965 CrossRef CAS PubMed.
- N. Goujon, N. Casado, N. Patil, R. Marcilla and D. Mecerreyes, Prog. Polym. Sci., 2021, 122, 101449 CrossRef CAS.
- X. Dong, Z. Guo, Z. Guo, Y. Wang and Y. Xia, Joule, 2018, 2, 902–913 CrossRef CAS.
- J. Xie, Z. Wang, Z. J. Xu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1703509 CrossRef.
- A. Wild, M. Strumpf, B. Häupler, M. D. Hager and U. S. Schubert, Adv. Energy Mater., 2017, 7, 1601415 CrossRef.
- W. Yan, C. Wang, J. Tian, G. Zhu, L. Ma, Y. Wang, R. Chen, Y. Hu, L. Wang, T. Chen, J. Ma and Z. Jin, Nat. Commun., 2019, 10, 2513 CrossRef.
- L. Fan, R. Ma, J. Wang, H. Yang and B. Lu, Adv. Mater., 2018, 30, 1805486 CrossRef PubMed.
- G. Dai, Y. He, Z. Niu, P. He, C. Zhang, Y. Zhao, X. Zhang and H. Zhou, Angew. Chem., Int. Ed., 2019, 58, 9902–9906 CrossRef CAS PubMed.
- Y. Zhao, M. Wu, H. Chen, J. Zhu, J. Liu, Z. Ye, Y. Zhang, H. Zhang, Y. Ma, C. Li and Y. Chen, Nano Energy, 2021, 86, 106055 CrossRef CAS.
- S. Wang, L. Wang, Z. Zhu, Z. Hu, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2014, 126, 6002–6006 CrossRef.
- C. Friebe, A. Lex-Balducci and U. S. Schubert, ChemSusChem, 2019, 12, 4093–4115 CrossRef CAS PubMed.
- Y. Zhang, Z. Sun, X. Kong, Y. Lin and W. Huang, J. Mater. Chem. A, 2021, 9, 26208–26215 RSC.
- Y. Liang, P. Zhang and J. Chen, Chem. Sci., 2013, 4, 1330 RSC.
- S. T. M. Tan, T. J. Quill, M. Moser, G. LeCroy, X. Chen, Y. Wu, C. J. Takacs, A. Salleo and A. Giovannitti, ACS Energy Lett., 2021, 6, 3450–3457 CrossRef CAS.
- Y. Lu, Y. Cai, Q. Zhang and J. Chen, Adv. Mater., 2021, 34, 2104150 CrossRef.
- G. Dai, Y. Liu, Z. Niu, P. He, Y. Zhao, X. Zhang and H. Zhou, Matter, 2019, 1, 945–958 CrossRef.
- M. Wu, Y. Zhao, B. Sun, Z. Sun, C. Li, Y. Han, L. Xu, Z. Ge, Y. Ren, M. Zhang, Q. Zhang, Y. Lu, W. Wang, Y. Ma and Y. Chen, Nano Energy, 2020, 70, 104498 CrossRef CAS.
- C. Wang, Energy Environ. Mater., 2020, 3, 441–452 CrossRef CAS.
- L. Sieuw, A. Jouhara, E. Quarez, C. Auger, J. F. Gohy, P. Poizot and A. Vlad, Chem. Sci., 2019, 10, 418–426 RSC.
- A. Halder, M. Ghosh, M. A. Khayum, S. Bera, M. Addicoat, H. S. Sasmal, S. Karak, S. Kurungot and R. Banerjee, J. Am. Chem. Soc., 2018, 140, 10941–10945 CrossRef CAS PubMed.
- Y. Zu, Y. Xu, L. Ma, Q. Kang, J. Wang, H. Yao, J. Hou, Y. Zu, Y. Xu, L. Ma, Q. Kang, J. Wang, H. Yao and J. Hou, Energy Storage Mater., 2021, 41, 240–247 CrossRef.
- M. Tang, S. Zhu, Z. Liu, C. Jiang, Y. Wu, H. Li, B. Wang, E. Wang, J. Ma and C. Wang, Chem, 2018, 4, 2600–2614 CAS.
- Z. Jin, Q. Cheng, A. M. Evans, J. Gray, R. Zhang, S. T. Bao, F. Wei, L. Venkataraman, Y. Yang and C. Nuckolls, Chem. Sci., 2022, 13, 3533–3538 RSC.
- Q. Xiao, J. Tian, Q. Xue, J. Wang, B. Xiong, M. Han, Z. Li, Z. Zhu, H. L. Yip and Z. Li, Angew. Chem., Int. Ed., 2019, 58, 17724–17730 CrossRef CAS PubMed.
- M. Gsanger, E. Kirchner, M. Stolte, C. Burschka, V. Stepanenko, J. Pflaum and F. Wurthner, J. Am. Chem. Soc., 2014, 136, 2351–2362 CrossRef PubMed.
- G. Chen, H. Sasabe, T. Igarashi, Z. Hong and J. Kido, J. Mater. Chem. A, 2015, 3, 14517–14534 RSC.
- A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 3770–3774 CrossRef CAS PubMed.
- W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS PubMed.
- J. Wang, H. Yao, C. Du and S. Guan, J. Power Sources, 2021, 482, 228931 CrossRef CAS.
- X. Han, G. Qing, J. Sun and T. Sun, Angew. Chem., Int. Ed., 2012, 51, 5147–5151 CrossRef CAS PubMed.
- J. Yang, P. Xiong, Y. Shi, P. Sun, Z. Wang, Z. Chen and Y. Xu, Adv. Funct. Mater., 2020, 30, 1909597 CrossRef CAS.
- L. Liu, L. Miao, L. Li, F. Li, Y. Lu, Z. Shang and J. Chen, J. Phys. Chem. Lett., 2018, 9, 3573–3579 CrossRef CAS PubMed.
- A. A. Franco, A. Rucci, D. Brandell, C. Frayret, M. Gaberscek, P. Jankowski and P. Johansson, Chem. Rev., 2019, 119, 4569–4627 CrossRef CAS PubMed.
- Z. Tie, S. Deng, H. Cao, M. Yao, Z. Niu and J. Chen, Angew. Chem., Int. Ed., 2021, 61, e202115180 Search PubMed.
- Z. Song, H. Zhan and Y. Zhou, Angew. Chem., Int. Ed., 2010, 49, 8444–8448 CrossRef CAS PubMed.
- W. Ma, L. W. Luo, P. Dong, P. Zheng, X. Huang, C. Zhang, J. X. Jiang and Y. Cao, Adv. Funct. Mater., 2021, 31, 2105027 CrossRef CAS.
- H. Liu, Y. Gu, Y. Dai, K. Wang, S. Zhang, G. Chen, B. Zou and B. Yang, J. Am. Chem. Soc., 2020, 142, 1153–1158 CrossRef CAS PubMed.
- H. Liu, Q. Bai, L. Yao, H. Zhang, H. Xu, S. Zhang, W. Li, Y. Gao, J. Li, P. Lu, H. Wang, B. Yang and Y. Ma, Chem. Sci., 2015, 6, 3797–3804 RSC.
- J. Liu, H. Li, J. Wang, Y. Zhang, D. Luo, Y. Zhao, Y. Li, A. Yu, X. Wang and Z. Chen, Adv. Energy Mater., 2021, 11, 2101926 CrossRef CAS.
- S. Zhuo, M. Tang, Y. Wu, Y. Chen, S. Zhu, Q. Wang, C. Xia and C. Wang, Nanoscale Horiz., 2019, 4, 1092–1098 RSC.
- J. Liu, P. Lyu, Y. Zhang, P. Nachtigall and Y. Xu, Adv. Mater., 2018, 30, 1705401 CrossRef.
- A. Ajayaghosh, Chem. Soc. Rev., 2003, 32, 181–191 RSC.
- Y. Hu, Y. Gao, L. Fan, Y. Zhang, B. Wang, Z. Qin, J. Zhou and B. Lu, Adv. Energy Mater., 2020, 10, 2002780 CrossRef CAS.
- G. Wang, N. Chandrasekhar, B. P. Biswal, D. Becker, S. Paasch, E. Brunner, M. Addicoat, M. Yu, R. Berger and X. Feng, Adv. Mater., 2019, 31, 1901478 CrossRef PubMed.
- Y. Wang, Y. Deng, Q. Qu, X. Zheng, J. Zhang, G. Liu, V. S. Battaglia and H. Zheng, ACS Energy Lett., 2017, 2, 2140–2148 CrossRef CAS.
- Z. Tong, S. Tian, H. Wang, D. Shen, R. Yang and C. S. Lee, Adv. Funct. Mater., 2020, 30, 1907656 CrossRef CAS.
- D. Wang, X. You, M. Wu, H. Huang, L. Chen, D. Wu and J. Xia, ACS Appl. Mater. Interfaces, 2021, 13, 16396–16406 CrossRef CAS PubMed.
- A. E. Lakraychi, F. Dolhem, A. Vlad and M. Becuwe, Adv. Energy Mater., 2021, 2101562 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, summary of previous studies, TGA curves, SEM and TEM images. See https://doi.org/10.1039/d2sc04508g |
| This journal is © The Royal Society of Chemistry 2022 |