
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iminologous epoxide ring-closure†
Chieh-Hung
Tien
 a,
Alan J.
Lough
b and
Andrei K.
Yudin
*a
a,
Alan J.
Lough
b and
Andrei K.
Yudin
*a
aDavenport Research Laboratories, Department of Chemistry, University of Toronto, Toronto, ON M5S 3H6, Canada. E-mail: andrei.yudin@utoronto.ca
bX-Ray Crystallography Laboratory, Department of Chemistry, University of Toronto, Toronto, ON M5S 3H6, Canada
First published on 10th October 2022
Abstract
The discovery of new reactions enables chemists to attain a better understanding of fundamental chemical reactivity and push the boundaries of organic synthesis. Our understanding and manipulation of high-energy states such as reactive conformations, intermediates, and transition structures contribute to this field. Herein we interrogate epoxide ring-closure by inserting the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N functionality into a well-known precursor to nucleophilic epoxide ring-closure. The synthesis of tetrasubstituted, nitrile-tethered epoxides takes place via activation of iminologous diols followed by fragmentation. Mechanistic study reveals the transformation to be stereospecific, which is consistent with the concerted nature of the epoxide ring-closure.
N functionality into a well-known precursor to nucleophilic epoxide ring-closure. The synthesis of tetrasubstituted, nitrile-tethered epoxides takes place via activation of iminologous diols followed by fragmentation. Mechanistic study reveals the transformation to be stereospecific, which is consistent with the concerted nature of the epoxide ring-closure.
Introduction
Development of new chemical transformations involves an interplay of mechanistic insights, serendipity, and unbiased screening.1 The de novo prediction of organic reactions is complicated by low-energy side processes that decrease reaction selectivity and result in low yields.2 Productive reaction path modification is evidenced in interrupted processes, which take place when a high-energy state such as a reactive intermediate or a strained conformation takes on an alternate path.3 While this appears logical, there is a lack of actionable steps that allow for the design of such transformations. Motivated by discovery of spatioenergetically-matched reaction paths, we recently initiated a program aimed at structural modification of well-established intermediates and transition state assemblies.4 Although the principle of vinylogy5 is restricted to consideration of ground states, an SN2′ pathway6 is effectively a vinylogous SN2 reaction (Scheme 1a). We felt that this way of looking at chemical reactivity might be applicable to other settings. Here we apply the principle of iminology7 to epoxide ring-closure8 and describe a hitherto unknown fragmentation reaction.9,10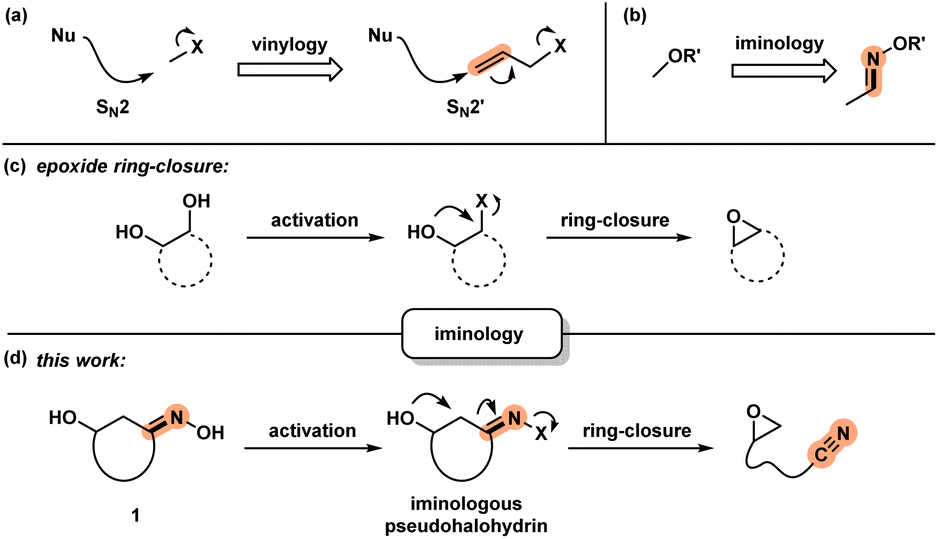 | ||
| Scheme 1 (a) SN2 and SN2′ reactions: a consideration of vinylogy; (b) application of iminology to alcohol derivatives to afford oximes; (c) ring-closure of 1,2-diols to obtain epoxides; and (d) iminologous epoxide ring-closure. | ||
Unlike the double bond in vinylogy, the utility of the imino-fragment (CN) as a two-atom relay unit has not garnered attention in the synthetic community. Structurally speaking, oximes can be considered as the iminologues of alcohols and related ethers and esters (Scheme 1b).11,12 Driven by the ongoing interest in the energy-rich N–O bond of oximes,12 we considered the iminologue of 1,2-diols (1). Doing so appeared attractive because of the known migration, ring-closing and fragmentation transformations of 1,2-diols (Scheme 1c).8,13 Insertion of an imino fragment into the C–O bond of one of the hydroxy groups would afford structures such as 1, and subsequently the iminologous pseudohalohydrin upon oxime activation (Scheme 1d).11e,14
Results and discussion
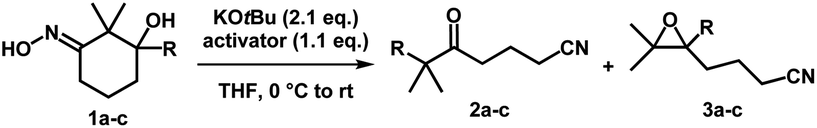
Iminologous diol 1 can be obtained from commercially available 1,3-diones by a simple 2- or 3-step sequence (see ESI†). Three initial substrates, 1a–c, were synthesized, and 1a was first reacted with 2 equivalents of KOtBu and a variety of reagents to activate the N–O bond to access the desired iminologous pseudohalohydrin in situ. The crude mixture was then analyzed by 1H and 13C NMR spectroscopy (Table 1). The reaction of 1a with TsCl yielded a nitrile-containing species as indicated by 13C NMR spectroscopy, in addition to a multitude of side products, which were difficult to separate by chromatography (entry 4). Other activating agents gave rise to the iminologous pseudohalohydrin without further reaction in conjunction with traces of other species with the characteristic nitrile 13C signals (entries 1–3, 5). Treatment of 1a with DIPEA and TsCl, followed by KOtBu provided a more manageable reaction mixture, which was subjected to column chromatography to provide ketonitrile 2a and epoxynitrile 3a in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with a 31% combined yield (entry 6).15 Iminologous diol 1b underwent a similar fragmentation process to afford a mixture of 2b and 3b in a 1:1 ratio with a 26% combined yield (entry 7). Ketonitriles 2a and 2b presumably arose from a semipinacol rearrangement pathway via a carbocation intermediate (vide infra). By switching to more electron-deficient 1c, the epoxynitrile 3c was synthesized as the sole product and isolated in 78% yield, suppressing the competing semipinacol rearrangement (entry 8). Increasing the equivalents of base and activating agent raised the yield to 88% (entry 9).
:1 ratio with a 31% combined yield (entry 6).15 Iminologous diol 1b underwent a similar fragmentation process to afford a mixture of 2b and 3b in a 1:1 ratio with a 26% combined yield (entry 7). Ketonitriles 2a and 2b presumably arose from a semipinacol rearrangement pathway via a carbocation intermediate (vide infra). By switching to more electron-deficient 1c, the epoxynitrile 3c was synthesized as the sole product and isolated in 78% yield, suppressing the competing semipinacol rearrangement (entry 8). Increasing the equivalents of base and activating agent raised the yield to 88% (entry 9).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Activator | 2 (%) | 3 (%) |
| a The crude reaction was subjected to analysis by NMR spectroscopy, but no purification was attempted. b Analysis by NMR spectroscopy indicated formation of nitrile-containing products, but no purification was attempted. c DIPEA (1.1 eq.) was added to 1a, followed by TsCl (1.1 eq.). After 1 h, KOtBu (2.1 eq.) was added. d KOtBu (2.4 eq.) and TsCl (1.4 eq.) were used. KOtBu = potassium tert-butoxide, TFAA = trifluoroacetic anhydride, MsCl = methanesulfonyl chloride, TsCl = p-toluenesulfonyl chloride, NsCl = 2-nitrosulfonyl chloride, DIPEA = diisopropylethylamine. | ||||
| 1a | 1a (R = H) | Ph2POCl | n/a | n/a |
| 2a | 1a | TFAA | n/a | n/a |
| 3a | 1a | MsCl | n/a | n/a |
| 4b | 1a | TsCl | n/a | n/a |
| 5a | 1a | NsCl | n/a | n/a |
| 6c | 1a | TsCl | 23 | 8 |
| 7 | 1b (R = Me) | TsCl | 12 | 14 |
| 8 | 1c (R = CONBn2) | TsCl | n/a | 78 |
| 9d | 1c | TsCl | n/a | 88 |
With the optimized conditions in hand, a series of iminologous diols 1d–q were synthesized, and the scope of the reaction was investigated (Scheme 2). Switching the dibenzylamide moiety to dimethylamine, derived from DMF, slightly lowered the yield to 68% (3d). The presence of an aniline on the amide substituent did not hinder the reaction and 3e was obtained in high yield. Other amides derived from cyclic amines such as pyrrolidine (3f) and morpholine (3g) were also well tolerated. Single crystals of 3f were grown and analyzed by X-ray crystallography to confirm the identity of the molecule. By switching to a less electron-rich amide derived from N-methylaniline, the reaction became less efficient, but otherwise gave 3h in synthetically useful yields. A substrate containing the antidepressant desipramine fragment was isolated in 81% yield (3i). Changing the amide fragment to an equally electron-withdrawing CF3 group afforded epoxide 3j in 24% yield. Novel [2.2]- and [2.4]-spirocyclic epoxides 3k–m were synthesized in low to good yields. Notably, the olefin group in 3m was tolerated, which could be sensitive towards other oxidant-based epoxidation conditions. Reducing ring size of the precursor to five atoms gave propionitrile-tethered epoxide 3n in 57% yield. Increasing the ring size to seven atoms gave 3o in 32% yield. Interestingly, an isobaric side product (3oa) that does not contain a nitrile functional group was isolated. Based on the 13C NMR chemical shifts of the carbonyl species, and the coupling patterns of the methylene proton (see ESI†), the side product was proposed to be in a rigid, cyclic system. Thus, the structure of 3oa was assigned to be a bicyclo[4.2.1]-dihydroisoxazole bearing an exocyclic amide and a strained bridgehead carbonyl. This product was presumed to arise from the direct intramolecular substitution on the electrophilic nitrogen atom of the respective iminologous pseudohalohydrin by the negatively charged alkoxide. Lastly, substrates with substituents on the ring can also smoothly undergo the reaction to afford 3p and 3q in moderate to good yields.
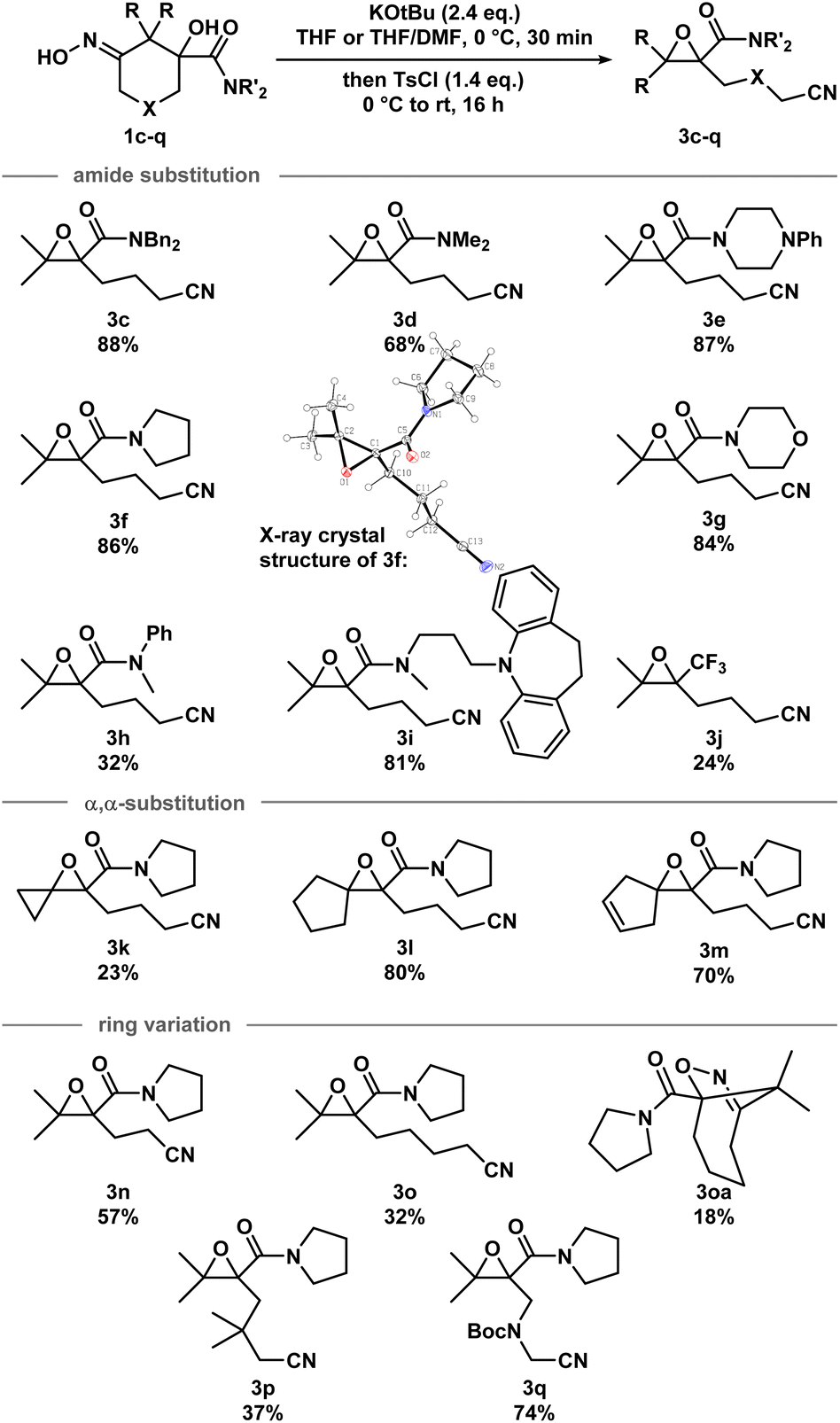 | ||
| Scheme 2 Scope of the iminologous ring-closure of iminologous diols 1c–qvia iminologous pseudohalohydrins to afford epoxynitriles 3c–q. X-ray crystal structure of 3f (displacement ellipsoid set at 30% probability level). | ||
To further expand the scope of this process, 4a and 4b, where the alcohol moiety is external to the ring system, were synthesized (Scheme 3). Treatment of 4a under standard conditions did not afford the desired epoxynitrile (Scheme 3a). To our surprise, similar to 3oa, a direct displacement of the pseudohalide by the alkoxide onto the nitrogen atom occurred to exclusively give dihydroisoxazole 5a. The identity of this species was further confirmed by single crystal X-ray crystallography. Analogous reactivity was observed when 4b was employed, and the methylated dihydrooxazines 5b was isolated in high yields (Scheme 3b). The presence of the diester substituent could lower the barrier of cyclization to favour dihydroisoxazole formation instead of the desired pathway to afford epoxides.16
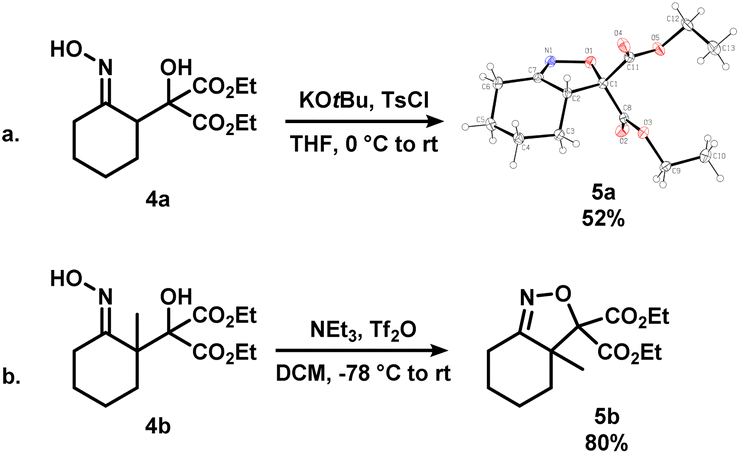 | ||
| Scheme 3 Synthesis of dihydroisoxazoles 5a (shown as X-ray crystal structure, displacement ellipsoid set at 30% probability level) and 5bvia the activation of iminologous diols 4a and 4b. | ||
The epoxynitriles can undergo further transformations to access other highly decorated molecules (Scheme 4). Treatment of 3c with SmI2 in THF effected the regioselective ring-opening of the epoxide to afford allylic alcohol 6. Lewis acid-mediated fluorination of 3g using BF3 gave fluorohydrin 7a in 37% yield. To our surprise, a significant amount of borylated heterocycle 7b did not undergo hydrolysis under reversed-phase chromatography conditions, and was isolated in 48% yield. The structure was identified by X-ray crystallography. Lastly, in the presence of a strong nucleophile such as nBuLi, the nitrile was reduced to the corresponding ketone, while maintaining the epoxide moiety, albeit in low yields.
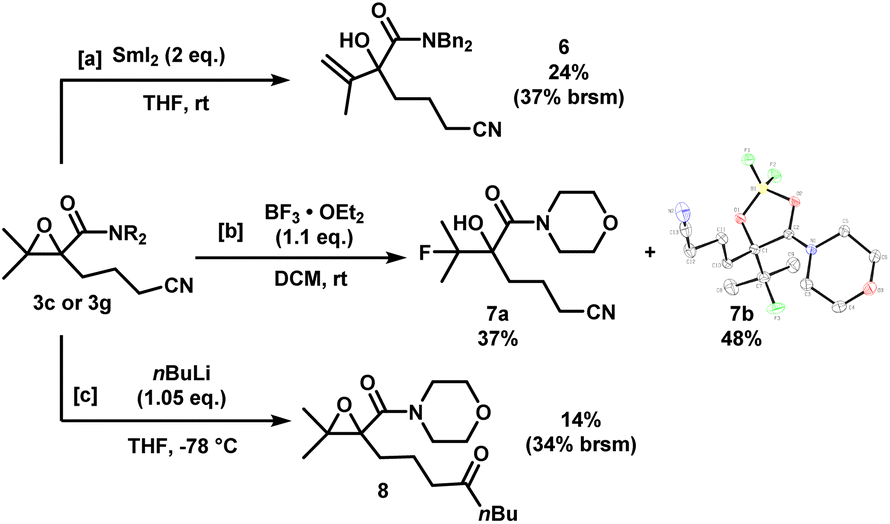 | ||
| Scheme 4 Downstream functionalization of epoxynitriles 3c and 3g. X-ray crystal structure of 7b (displacement ellipsoid set at 30% probability level). | ||
We considered the mechanism of this transformation (Scheme 5). Without TsCl, the reaction does not proceed, and 1c was recovered, indicating the intermediacy of the corresponding iminologous pseudohalohydrin (Scheme 5a). Using only one equivalent of KOtBu and TsCl gave 3c in 44% isolated yield (Scheme 5b). It was previously established that when 1a and 1b were subjected to the reaction conditions, both the ring-closing products, 3a and 3b, and the H-migration products, 2a and 2b, were isolated (Scheme 5c). The ketones are presumed to arise from the [1,2]-H migration (semipinacol rearrangement) of the intermediates that contain adjacent carbocations. To further probe the presence of carbocations in amide-tethered substrates (since no migration was observed), oximes (±)-cis-1r and (±)-trans-1r were synthesized and isolated. The major isomer was crystallized, and a crystal structure was obtained to confirm the relative stereochemistry. Both isomers, (±)-cis-1r and (±)-trans-1r, were subsequently subjected to the standard conditions, and to our surprise, the epoxide products were afforded as diastereomerically pure species (±)-trans-3r and (±)-cis-3r, respectively (Scheme 5d). The relative configuration of the two diastereomers were assigned based on NOESY NMR assignment (see ESI†). This suggests the lack of planarization of the α-carbon atom (carbon in the α-position relative to the oxime), which is inconsistent with the formation of carbocations in this process. Therefore, C–O bond formation and C–C bond cleavage must occur in a concerted or otherwise stereospecific fashion.
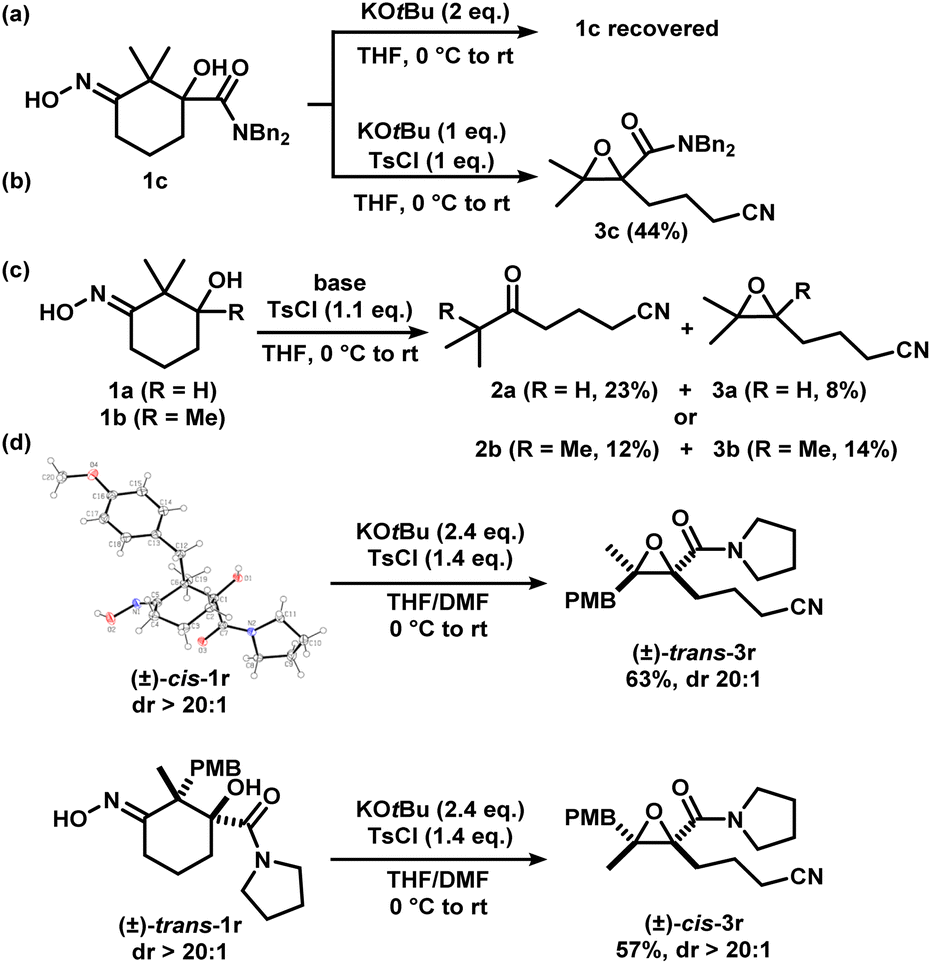 | ||
| Scheme 5 Mechanistic studies. X-ray crystal structure of (±)-cis-1r (displacement ellipsoid set at 30% probability level). | ||
Two other substrates (±)-1s and (±)-1t were synthesized and the diastereomers were separated (Scheme 6). Subjection of (±)-trans-1s and (±)-cis-1s to the optimized conditions provided epoxides (±)-cis-3s and (±)-trans-3s, respectively, with good yields in a stereospecific fashion. Ethylene ester-substituted (±)-cis-1t did not react under the reaction conditions, and the starting material was recovered upon hydrolysis during reversed-phase purification. Interestingly, diastereomeric (±)-trans-1t gave the desired epoxide (±)-cis-3t, albeit in low yields.
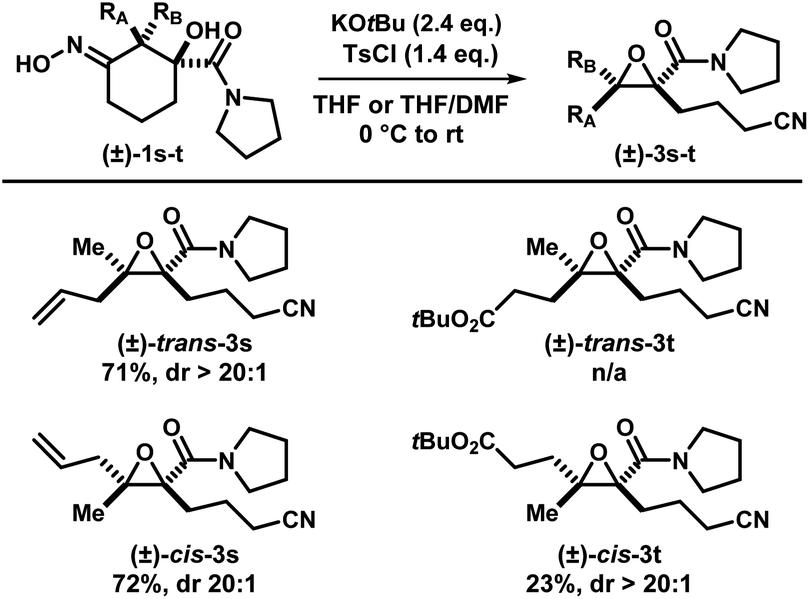 | ||
| Scheme 6 Iminologous ring-closure of diastereomerically enriched (±)-1s and (±)-1t. | ||
Considering the aforementioned results, we put forth a potential mechanism of the epoxidation process (Scheme 7a). First, the oxime is activated to form the corresponding iminologous pseudohalohydrin A. If the ring size is sufficiently large, or if the alkoxide is spatially close to the electrophilic nitrogen, a transannular or intramolecular substitution could occur to give dihydroisoxazoles B (pathway a). Otherwise, A undergoes a stereospecific C–C fragmentation, where the group antiperiplanar to the tosylate migrates onto the nitrogen atom to generate nitrilium C as in the abnormal Beckmann rearrangement.11d Facile fragmentation of C is expected to give the stabilized carbocation D, which could further undergo a semipinacol rearrangement to afford ketones 2 (pathway b). However, in amide-tethered substrates, the anionic oxygen atom in A likely undergoes direct intramolecular substitution to expel the oxime functional group by C–C cleavage in a concerted iminologous ring-closure to directly afford 3 (pathway c).17 This pathway is only possible if the oxygen atom is equatorial (Scheme 7b, green), which makes the C–O bond antiperiplanar to the breaking C–C bond as seen in the X-ray crystal structure of (±)-cis-1r. Since the equilibrium distribution of conformers is different between diastereomers, a change in reactivity would be expected, thus explaining the higher reactivity of (±)-trans-1t with respect to (±)-cis-1t.
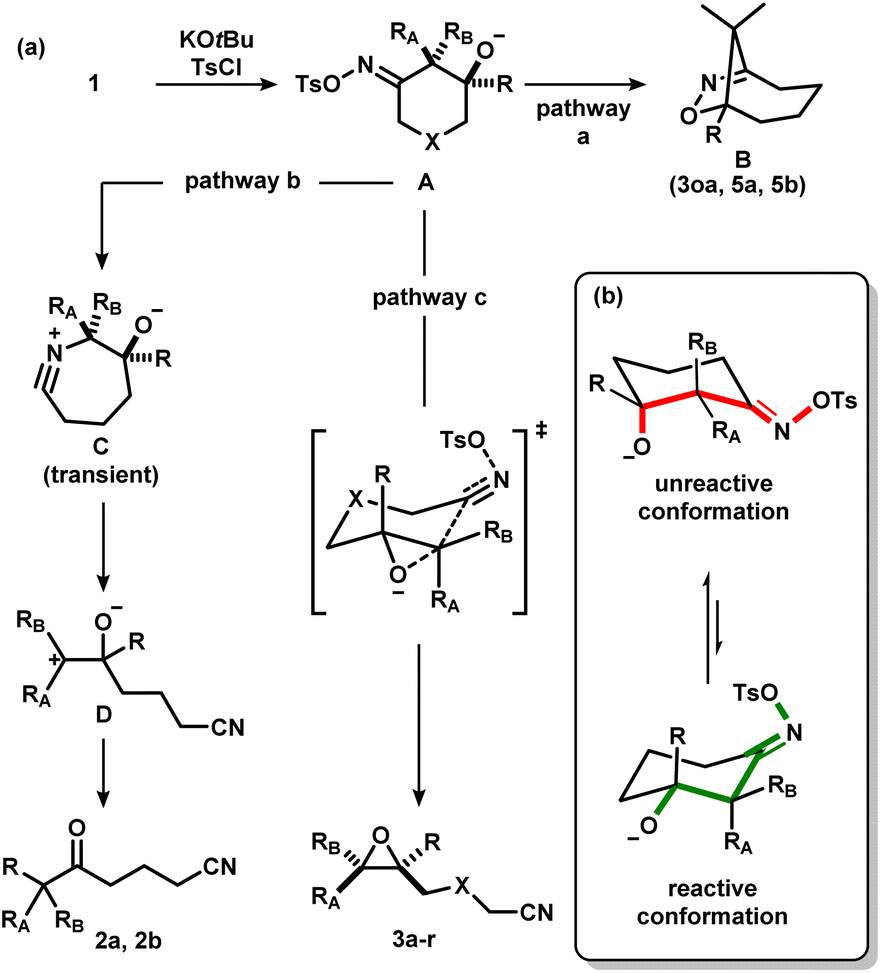 | ||
| Scheme 7 (a) Proposed mechanism for the formation of ketonitriles, epoxynitriles and dihydroisoxazoles via the activation of iminologous diols 1 with TsCl. (b) Analysis on the reactivity between different conformers. | ||
Conclusions
We have shown that insertion of the imino fragment into the key bond-breaking step of the well-established epoxide ring-closure process results in a transition state that delivers a novel iminologous fragmentation reaction to afford tetra-substituted epoxides. The mechanism has been examined, and it was proposed that the reaction is stereospecific and follows a concerted ring-closure pathway. We anticipate the application of the principle of iminology in other established processes to further demonstrate that structural modification of reactive intermediates can be used as a strategy in the development of new transformations.Author contributions
C.-H. T. and A. K. Y. conceived the project and wrote the manuscript. C.-H. T. designed and conducted the experiments. A. J. L. obtained and solved the crystal structures. All authors have given approval to the final version of the manuscript.Conflicts of interest
A. K. Y. is an associate editor of Chemical Science.Acknowledgements
NSERC is acknowledged for funding support. C.-H. T. thanks the Connaught Fund (U of T) for financial support. Discussions with the Yudin lab are gratefully acknowledged.Notes and references
- (a) K. D. Collins, T. Gensch and F. Glorius, Nat. Chem., 2014, 6, 859–871 CrossRef CAS PubMed; (b) R. M. Roberts, Serendipity: Accidental Discoveries in Science, Wiley, 1989 Search PubMed; (c) M. Shevlin, ACS Med. Chem. Lett., 2017, 8, 601–607 CrossRef CAS PubMed.
- F. Z. Dörwald, Side Reactions in Organic Synthesis, Wiley-VCH, 2005 Search PubMed.
- V. Trudel, C.-H. Tien, A. Trofimova and A. K. Yudin, Nat. Rev. Chem., 2021, 5, 604–623 CrossRef CAS.
- A. K. Yudin, Chem. Sci., 2020, 11, 12423–12427 RSC.
- For reviews, see: (a) R. C. Fuson, Chem. Rev., 1935, 16, 1–27 CrossRef CAS; (b) G. Casiraghi, F. Zanardi, G. Appendino and G. Rassu, Chem. Rev., 2000, 100, 1929–1972 CrossRef CAS PubMed; (c) G. Casiraghi, L. Battistini, C. Curti, G. Rassu and F. Zanardi, Chem. Rev., 2011, 111, 3076–3154 CrossRef CAS PubMed; (d) C. Curti, L. Battistini, A. Sartori and F. Zanardi, Chem. Rev., 2020, 120, 2448–2612 CrossRef PubMed.
- (a) R. M. Magid, Tetrahedron, 1980, 36, 1901–1930 CrossRef CAS; (b) J. A. Marshall, Chem. Rev., 1989, 89, 1503–1511 CrossRef CAS; (c) F. G. Bordwell, A. H. Clemens and J. P. Cheng, J. Am. Chem. Soc., 1987, 109, 1773–1782 CrossRef CAS.
- M. Berthelot, M. Helbert, C. Laurence, J.-Y. Le Questel, F. Anvia and R. W. Taft, J. Chem. Soc., Perkin Trans., 1993, 2, 625–627 RSC.
- (a) H. C. Kolb and K. B. Sharpless, Tetrahedron, 1992, 48, 10515–10530 CrossRef CAS; (b) S. A. Weissman, K. Rossen and P. J. Reider, Org. Lett., 2001, 3, 2513–2515 CrossRef CAS PubMed.
- (a) R. V. Kolakowski, M. Munpadi, Y. Zhang, T. J. Emge and L. J. Williams, J. Am. Chem. Soc., 2009, 131, 12910–12922 CrossRef CAS PubMed; (b) T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2010, 49, 8962–8965 CrossRef CAS PubMed; (c) M. A. Drahl, M. Manpadi and L. J. Williams, Angew. Chem., Int. Ed., 2013, 52, 11222–11251 CrossRef CAS PubMed.
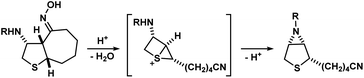
- During their synthesis of (±)-biotin, Confalone and co-workers reported an unexpected abnormal Beckmann rearrangement via a highly energetic 1-thiabicyclo[2.1.0]pentan-1-ium species as an intermediate (see scheme below), instead of a carbocation. However, no further studies were reported to support the claim:
 (a) P. T. Confalone, E. D. Lollar, G. Pizzolato and M. R. Uskoković, J. Am. Chem. Soc., 1978, 100, 6291–6292 CrossRef CAS;
(b) P. T. Confalone, G. Pizzolato, D. L. Confalone and M. R. Uskoković, J. Am. Chem. Soc., 1980, 102, 1954–1960 CrossRef CAS.
(a) P. T. Confalone, E. D. Lollar, G. Pizzolato and M. R. Uskoković, J. Am. Chem. Soc., 1978, 100, 6291–6292 CrossRef CAS;
(b) P. T. Confalone, G. Pizzolato, D. L. Confalone and M. R. Uskoković, J. Am. Chem. Soc., 1980, 102, 1954–1960 CrossRef CAS. - For reviews, see: (a) X.-Y. Yu, J. R. Chen and W. J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (b) K. A. Rykaczewski, E. R. Wearing, D. E. Blackmun and C. S. Schindler, Nat. Synth., 2022, 1, 24–36 CrossRef; (c) C. Pratley, S. Fenner and J. A. Murphy, Chem. Rev., 2022, 122, 8181–8260 CrossRef CAS PubMed; (d) B. Zhao, Y. Zheng, C. Chen, M. Wang, M. Ma and Z. Shi, Org. Chem. Front., 2021, 8, 2985–2989 RSC; (e) R. E. Gawley, Org. React., 1988, 35, 1–420 CAS.
- Selected examples: (a) P. V. Kattamuri, J. Zhao, T. Kanti Das, J. H. Siitonen, N. Morgan, D. H. Ess and L. Kürti, J. Am. Chem. Soc., 2022, 144, 10943–10949 CrossRef CAS PubMed; (b) E. R. Wearing, D. E. Blackmun, M. R. Becker and C. S. Schindler, J. Am. Chem. Soc., 2021, 143, 16235–16242 CrossRef CAS PubMed; (c) E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744–748 CrossRef CAS PubMed; (d) P. V. Kattamuri, J. Yin, S. Siriwongsup, D.-H. Kwon, D. H. Ess, Q. Li, G. Li, M. Yousufuddin, P. F. Richardson, S. C. Sutton and L. Kürti, J. Am. Chem. Soc., 2017, 139, 11184–11196 CrossRef CAS PubMed.
- (a) Z.-L. Song, C.-A. Fan and Y.-Q. Tu, Chem. Rev., 2011, 111, 7523–7556 CrossRef CAS PubMed; (b) A. S. Perlin, Glycol-cleavage oxidation, in Advances in Carbohydrate Chemistry and Biochemistry, ed. D. Horton, Elsevier, Amsterdam, 2006, vol. 60, pp. 183–250 Search PubMed.
- (a) S. J. Touchette, E. M. Dunkley, L. L. Lowder and J. Wu, Chem. Sci., 2019, 10, 7812–7815 RSC; (b) H. Fujioka, N. Matsumoto, Y. Kuboki, H. Mitsukane, R. Ohta, T. Kimura and K. Murai, Chem. Pharm. Bull., 2016, 64, 718–722 CrossRef CAS PubMed; (c) S. Kenaya, Y. Asaji, T. Yoshimura and J.-I. Matsuo, Synlett, 2020, 31, 1201–1204 CrossRef.
- Analogous competing semipinacol rearrangement and ring-closing pathways were observed in the Lewis acid-catalyzed Darzens epoxidation of carbonyl derivatives with diazoesters: D. G. Nam, S. Y. Shim, H.-M. Jeong and D. H. Ryu, Angew. Chem., Int. Ed., 2021, 60, 22236–22240 CrossRef CAS PubMed.
- M. E. Jung and G. Piizzi, Chem. Rev., 2005, 105, 1735–1766 CrossRef CAS PubMed.
- K. Prantz and J. Mulzer, Chem. Rev., 2010, 110, 3741–3766 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; mass spectrometry data; NMR spectroscopy data; X-ray crystallography data. CCDC 2194278–2194281. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04496j |
| This journal is © The Royal Society of Chemistry 2022 |