
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom peptides to proteins: coiled-coil tetramers to single-chain 4-helix bundles†
Elise A.
Naudin
 a,
Katherine I.
Albanese
ab,
Abigail J.
Smith
c,
Bram
Mylemans
ab,
Emily G.
Baker
ac,
Orion D.
Weiner
d,
David M.
Andrews
e,
Natalie
Tigue
f,
Nigel J.
Savery
*cg and
Derek N.
Woolfson
*abcg
a,
Katherine I.
Albanese
ab,
Abigail J.
Smith
c,
Bram
Mylemans
ab,
Emily G.
Baker
ac,
Orion D.
Weiner
d,
David M.
Andrews
e,
Natalie
Tigue
f,
Nigel J.
Savery
*cg and
Derek N.
Woolfson
*abcg
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: d.n.woolfson@bristol.ac.uk
bMax Planck-Bristol Centre for Minimal Biology, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK
cSchool of Biochemistry, University of Bristol, Medical Sciences Building, University Walk, Bristol BS8 1TD, UK. E-mail: n.j.savery@bristol.ac.uk
dCardiovascular Research Institute, Department of Biochemistry and Biophysics, University of California, 555 Mission Bay Blvd. South, San Francisco, CA 94158, USA
eOncology R&D, AstraZeneca, Cambridge Science Park, Darwin Building, Cambridge CB4 0WG, UK
fBioPharmaceuticals R&D, AstraZeneca, Granta Park, Cambridge CB21 6GH, UK
gBrisEngBio, School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK
First published on 20th September 2022
Abstract
The design of completely synthetic proteins from first principles—de novo protein design—is challenging. This is because, despite recent advances in computational protein–structure prediction and design, we do not understand fully the sequence-to-structure relationships for protein folding, assembly, and stabilization. Antiparallel 4-helix bundles are amongst the most studied scaffolds for de novo protein design. We set out to re-examine this target, and to determine clear sequence-to-structure relationships, or design rules, for the structure. Our aim was to determine a common and robust sequence background for designing multiple de novo 4-helix bundles. In turn, this could be used in chemical and synthetic biology to direct protein–protein interactions and as scaffolds for functional protein design. Our approach starts by analyzing known antiparallel 4-helix coiled-coil structures to deduce design rules. In terms of the heptad repeat, abcdefg—i.e., the sequence signature of many helical bundles—the key features that we identify are: a = Leu, d = Ile, e = Ala, g = Gln, and the use of complementary charged residues at b and c. Next, we implement these rules in the rational design of synthetic peptides to form antiparallel homo- and heterotetramers. Finally, we use the sequence of the homotetramer to derive in one step a single-chain 4-helix-bundle protein for recombinant production in E. coli. All of the assembled designs are confirmed in aqueous solution using biophysical methods, and ultimately by determining high-resolution X-ray crystal structures. Our route from peptides to proteins provides an understanding of the role of each residue in each design.
Introduction
De novo protein design is advancing rapidly;1,2 indeed our ability to design proteins from scratch is said to have come of age.3 Protein-design processes have evolved over the past four decades from minimal design that uses straightforward chemical principles, through rational design that incorporates sequence-to-structure relationships learnt from natural proteins, to computational design that builds proteins from fragments or parametric templates and scores them using statistical or physical forcefields.4 Today, the field has also progressed to include state-of-the-art computational methods such as artificial intelligence and machine learning that allow protein designers to “hallucinate” proteins in the computer ahead of making and characterizing in the laboratory.5,6 A possible downside of these computational innovations is that we no longer understand what we are—that is, what the computer is—designing. Consequently, one of the original motivations for the field could be lost; namely, the idea that protein design tests our understanding of the chemical and physical principles of protein folding, assembly, and stability.4 That aside, the field has also matured from being structure-centric to one committed to developing synthetic proteins with useful functions that mimic or augment natural protein functions both in vitro and in biological contexts.6–11 Whilst many challenges remain,2,4 these are truly exciting and promising times for de novo protein design.Over the past 4 decades, 4-helix bundles (4HBs) have been one of the go-to targets for de novo peptide and protein design.1,12–14 Historically, 4HB design began with minimal approaches employing patterns of hydrophobic (e.g., leucine) and polar (e.g., glutamate and lysine) residues to design single short amphipathic α helices that self-associate due to the hydrophobic effect, or to program libraries of single-chain 4-helix proteins that fold through hydrophobic collapse.15–18 Again, these design approaches have evolved by incorporating biological information and rational design, which have led more readily to high-resolution X-ray crystal structures.4,19–22 Most recently, interest has shifted to using computational methods that use backbone and fold parametrization, optimization of core packing, and specific interaction networks between core residues.23–28 Furthermore, 4HBs present a variety of assembly modes for protein designers to target, including: single peptides that associate to tetramers, helix-loop-helix constructs that can dimerize, and self-contained single-chain proteins.17,29–31 However, they also present pitfalls—or alternate states—that designers must learn to navigate away from using negative-design principles.22,32,33 For instance, for the tetramers, adjacent helices can have all-parallel, antiparallel, or mixed arrangements; and for helix-loop-helix and single-chain systems various topologies are possible.31,34,35 These different architectures, the relatively large hydrophobic cores, and the apparent robustness to modification, have been exploited to functionalize 4HBs and introduce small-molecule binding,36 catalysis,37–39 allostery,40 and the control of protein–protein interaction including regulation of gene expression.41,42
One specific type of 4HBs form α-helical coiled coils (CCs). In CCs, tight and regular packing between side chains of neighbouring helices—known as knobs-into-holes (KIH) packing—specifies the structure, including defining oligomer state, partner preferences, and helix orientation. This has proved extremely powerful in rational and computational design of CCs.43–45 In more detail, CCs are supercoiled assemblies of amphipathic α helices. Generally, CC assembly is programmed by sequence repeats of hydrophobic (h) and polar (p) residues, hpphppp, often called heptads and denoted abcdefg (Fig. 1A).44,46 Many sequence-to-structure relationships, especially at the hydrophobic a/d interface, have come from analyses of natural structures and empirical studies.19,47 In turn, these have been used to deliver a wide range of structured and increasingly functional CC designs.4,43,44 For example, our own basis set of de novo CCs currently comprises parallel assemblies from dimer to nonamer,20,48,49 and these are being used increasing by us and others in various applications.41,50–55 That all said, designing antiparallel CC assemblies from first principles has been more challenging.33,56–58 Moreover, subtle changes in primary sequence or even experimental conditions can induce switches from energetically close parallel assemblies to antiparallel conformations.33,59–61 For example, recently, we reported the rational redesign of an antiparallel CC tetramer, apCC-Tet, following the serendipitous discovery of up–down–up–down tetramers adopted by point mutations in our original parallel hexamer, CC-Hex.33
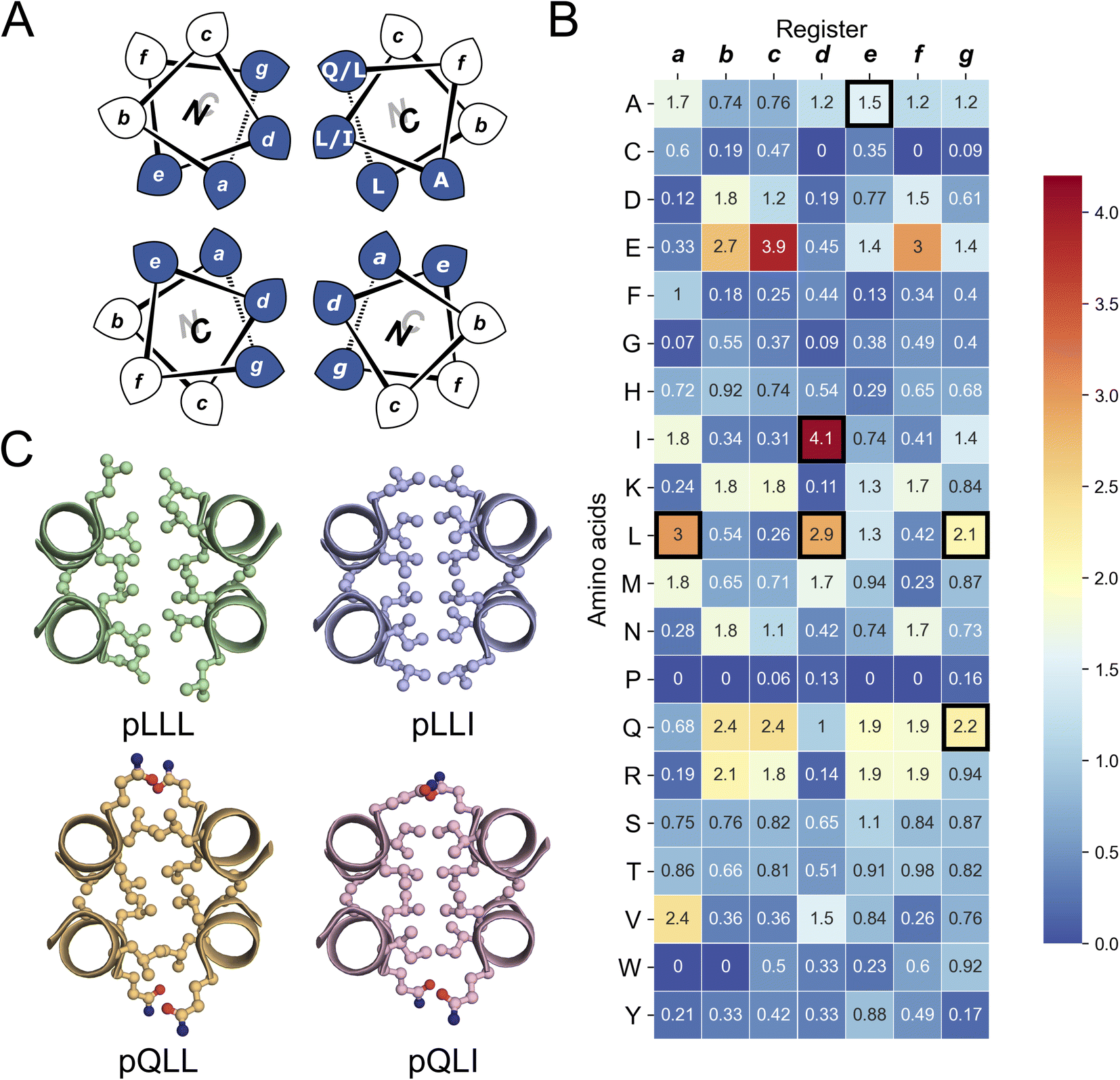 | ||
| Fig. 1 The rational design of new sequences to form antiparallel CC tetramers. (A) Helical-wheel representation of an antiparallel four-helix CC. Sequences have heptad repeats, abcdefg. The interfacial positions, a, d, e, and g, where our designs focused are highlighted in blue. Selected residues in our designed sequences are shown on the top-right helix. The N-to-C-terminal directions of the helices are indicated with the ‘N’ or ‘C’ with the darker font indicating that end is closer to the viewer. (B) Propensity table of residues for each amino acid at each position of the heptad repeat for antiparallel 4-helix CCs found in CC+.65 Raw counts (Table S1†) were normalized using the amino-acid frequencies in SWISS-PROT to give the propensity scale shown as a heat map (high, red; low, blue). A propensity of 0 indicates that no examples of that amino acid were found at that position in the database. Residues identified for the design of the new antiparallel tetramer sequences are highlighted with dark square boxes. (C) Heptad-repeat slices through the AlphaFold2-multimer67–69 models for each designed sequence: pLLL (top left), pLLI (top right), pQLL (bottom left), and pQLI (bottom right). Images for panel C were generated in PyMOL (https://www.pymol.org). | ||
Establishing clear principles for de novo design, such as sequence-to-relationships for a given target, would help navigate the complex energy landscape of helical assemblies. Moreover, it would deliver design rules to direct the assembly of different helical states to improve and expand toolkits such as the CC basis set and similar sets from others.62–64 In turn, these would provide platforms for protein redesign and applications where the impact of modifications required for functionalization could be anticipated.
Here, we elaborate a set of sequence-to-structure relationships for designing CC-based antiparallel 4HBs. By inspecting the structural database of CCs (CC+),65 we deduce clear design rules for this target. In turn, these are used to deliver three de novo structures: an antiparallel homotetramer, apCC-Tet*, a heterotetramer, apCC-Tet*3-A2B2, and a single-chain 4HB, sc-apCC-4. All three designs are characterized fully in solution, and to high-resolution by determining X-ray crystal structures. The designs are hyperstable with respect to thermal and chemical denaturation, and they fold, assemble, and function in E. coli. These properties make them ideal scaffolds to functionalize for future in vitro and subcellular applications.
Results and discussion
Rational designs based on analysis of known coiled-coil structures
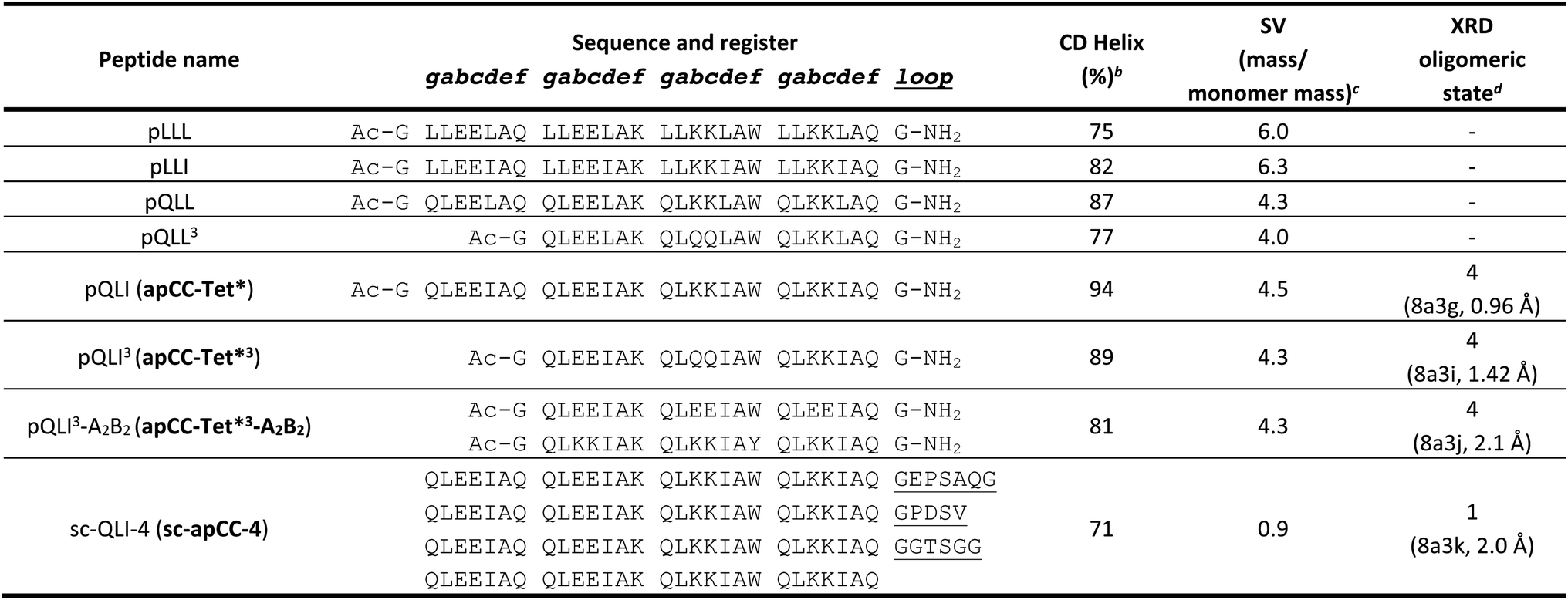
To garner sequence-to-structure relationships to design the target antiparallel CC tetramers and bundles, we analysed relevant structures in the CC+ database.65 We selected sequences for all antiparallel, four-helix, homo- and heteromeric CC assemblies with ≤50% sequence redundancy. These were used to compile an amino-acid profile for the heptad repeats, abcdefg, of these structures (Fig. 1A);44,46 the raw counts are available in Table S1.† The profile was normalized using amino-acid frequencies from SWISS-PROT as the expected values to give propensities for each residue at each position of the a–g repeat (Fig. 1B). Next, we used these propensities to deduce new de novo repeat sequences. We focused on the interfacial positions, g, a, d and e, as these contribute most to CC folding, stability, and oligomer state specification (Fig. 1A).44 For the a site, we selected leucine (Leu, L) as this was overwhelmingly preferred in the profile with a propensity of 3; i.e., it occurred three times more frequently than expected by chance at this site (Fig. 1B). We did not consider the next most prevalent residues at a, the β-branched isoleucine (Ile, I) and valine (Val, V), as these are known to favour both parallel and antiparallel dimers when placed at this site.19 Two residues, Ile and Leu, had high propensities for d, occurring at ≈4× and ≈3× the expected frequency, respectively, so we considered both in our initial designs. At e, no residues appeared above twice the expected frequency. Therefore, we opted for Ala at this site, as it occurred frequently in the dataset (Table S1†) and is known to promote antiparallel tetramers via Alacoil formation.33,66 Finally, Leu and glutamine (Gln, Q) had propensity values exceeding 2 for the g position. Therefore, both residues were investigated at g in our initial designs.Consequently, our analysis led to four distinct sequence combinations with the potential to form antiparallel tetramers: namely, L/Q-L-b-c-I/L-A-f in g → f repeats. For stable CC designs,20,33,48,49 we concatenated 4 copies of each repeat into each of 4 designed homomeric peptide sequences. We used the unspecified b and c sites to direct antiparallel assemblies further, specifically in homomers. Our rationale was to create a ‘bar-magnet’ charge pattern in the sequences by placing negatively charged glutamic acid (Glu, E) at the b and c sites of the first two heptad repeats, and positively charged lysine (Lys, K) at these sites in the two C-terminal repeats.33 The sequences were completed with the remaining 4 f sites filled with Gln, Lys, tryptophan (Trp, W), and Gln, respectively. The final sequences were capped with glycine (Gly, G) at both ends and N-terminally acetylated and C-terminally amidated (Table 1). Initially, we named the sequences after the residues at the g, a and d sites, i.e., pLLL, pLLI, pQLL, and pQLI.
|
Ahead of experiments, we modelled the four new sequences using the AlphaFold2-multimer predictor (Fig. 1C and S1–S4†).67–69 Encouragingly, the AlphaFold2 predictions for both Q@g sequences, pQLL and pQLI, gave antiparallel tetramers as designed and with high confidence (Fig. 1C) even when an oligomeric state larger than 4 was provided as a target to AlphaFold2 (Fig. S3 and S4†). By contrast, although the L@g sequences, pLLL and pLLI, could be predicted to form antiparallel 4HBs by AlphaFold2 (Fig. 1C), this was not consistently observed when higher chain numbers were used; in these cases, higher-order α-helical assemblies were predicted (Fig. S1 and S2†).
Experimental characterization of a robust antiparallel coiled-coil tetramer, apCC-Tet*
The four sequences pLLL, pLLI, pQLL and pQLI (Table 1) were synthesized by solid-phase peptide synthesis (SPPS) and confirmed by MALDI-TOF mass spectrometry (Fig. S5–S8†). First, circular dichroism (CD) spectroscopy was used to assess the secondary structure and stability of the designs in aqueous buffer near neutral pH. All four peptides were highly α helical with characteristic minima at 208 and 222 nm (Fig. 2A, S11 and S12†). Furthermore, the structures resisted thermal denaturation up to 95 °C (Fig. 2B, S11 and S12†). Next, sedimentation-velocity (SV) experiments using analytical ultracentrifugation (AUC) revealed monodisperse oligomers in all four cases (Fig. 2C and S13–S16†). Interestingly, and consistent with the AlphaFold2 modelling, both L@g peptides, pLLL and pLLI, formed hexamers in solution (Fig. S13 and S14†). This was despite the amino-acid profiles, and the precedence of L@g in other 4HB designs, notably from computational design.27,70 With hindsight, the hexamers that we observed might have been anticipated, as other de novo peptides with predominantly hydrophobic residues at g, a, d and e are Type-II CC sequences, which often form oligomers of >4, including α-helical barrels.44,48,49 Therefore, these two peptides, pLLL and pLLI, were not investigated further in this study, which aimed to deliver antiparallel 4HBs. By contrast, and consistent with the design target, both Q@g peptides, pQLL and pQLI, returned tetrameric molecular weights in AUC by both sedimentation equilibrium (SE) and SV experiments (Fig. 2C, S15 and S16†). Both of these designs were taken forward.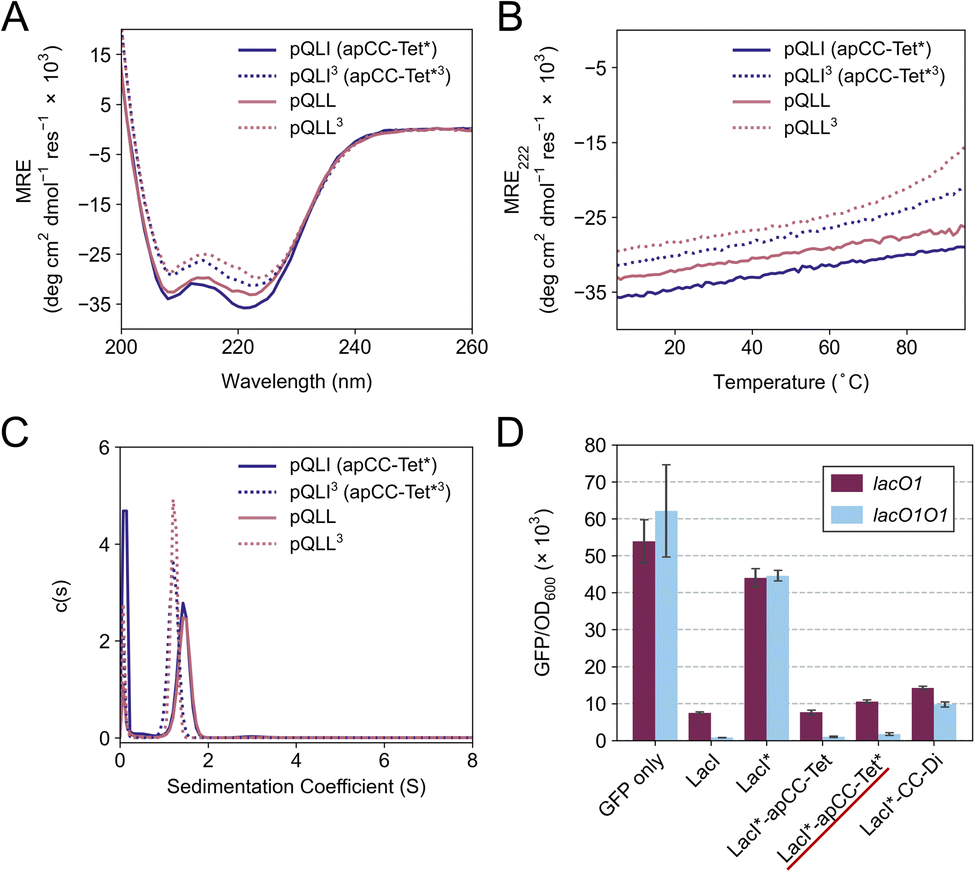 | ||
| Fig. 2 Biophysical and in-cell characterization of the homotetrameric peptides pQLI (apCC-Tet*) and pQLL. (A) CD spectra at 5 °C and (B) thermal responses of the CD signals at 222 nm (ramping up from 5 to 95 °C) of pQLI (apCC-Tet*), pQLI3 (apCC-Tet*3), pQLL, and pQLL3. Conditions: 50 μM peptide, PBS, pH 7.4. (C) Sedimentation-velocity data from AUC of pQLI, pQLI3, pQLL, and pQLL3. Fits returned weights of 4.5, 4.3, 4.3 and 4.0× monomer mass, respectively. Conditions: 150 μM peptide, PBS, pH 7.4. (D) Transcription repression assay in E. coli. CC peptides were fused to LacI*, a destabilized variant of the Lac repressor. The reporter gene, GFP, was expressed from the lacUV5 promoter with or without an additional lac O1 operator placed upstream of the lacUV5 O1 operator. Results are shown for LacI*-apCC-Tet* (underlined in red) and for controls LacI*-apCC-Tet and LacI*-CC-Di. GFP fluorescence was normalized to the OD600 of the cell culture and is an average of three repeats shown with standard error. | ||
In an attempt to access an unfolding transition for one of the Q@g designs, we measured CD spectra of the pQLI peptide in guanidinium hydrochloride, Gn·HCl. Surprisingly, neither the equilibrium spectra recorded at 5 °C nor the mean residue ellipticity at 222 nm (MRE222) signal recorded over 5–95 °C changed appreciably in the range of 0–6 M Gn·HCl (Fig. S17 and S18†). Thus, pQLI is another hyperstable de novo peptide assembly. To probe this further, we truncated both pQLL and pQLI to 3-heptad repeats, yielding pQLL3 and pQLI3, respectively (Table 1). The overall charge pattern was preserved, though only the first and the last heptads had charged residues at b and c positions and the central repeat had Gln at these sites. Both truncated designs retained stable α-helical structures by equilibrium and variable-temperature CD measurements (Fig. 2A and B). However, reducing the peptides concentrations to 5 μM accessed reversible thermal unfolding transitions, which were sigmoidal indicative of cooperativity, with estimated midpoints of 91 °C and 76 °C for pQLI and pQLL, respectively (Fig. S19†). Moreover, tetrameric assemblies for both peptides were confirmed by SV and SE experiments in AUC consistent with the target assemblies (Fig. 2C, S20 and S21†).
Next, we screened the 3- and 4-heptad variants of pQLL and pQLI for crystallization. Interestingly, only the pQLI peptides yielded crystals (Table S3†). Both peptides gave good-quality X-ray diffraction data. These allowed structures to be determined by molecular replacement using ideal α helices implemented in Fragon71 for pQLI, or using the AlphaFold267–69 model for pQLI3 to resolutions of 0.96 and 1.42 Å, respectively (Fig. 3A, B and Table S4†). The solved structures confirmed the pQLI designs as antiparallel CC tetramers with knobs-into-holes packing identified by SOCKET2 (Table S5†).72,73 Inspection of a one-heptad slice through either structure (Fig. 3C) illustrates this packing and immediately highlights the selection rules used in the design, namely: (i) a core of Leu@a that pack into holes on neighbouring helices; (ii) a wide helix–helix interface formed by the bulky Ile@d residues and flanked by Gln@g; and a narrow helix–helix interface with Ala@e allowing close helical contacts consistent with Alacoils33,66,95 and flanked by Glu@b → Lys@b′ salt bridges. In the two narrow interfaces of pQLI, 4 of such salt bridges are made with Cδ → Nζ distances of 3.5 Å. Finally, the new X-ray crystal structures aligned closely with AlphaFold2 model for both pQLI analogues (RMSDall-atom = 0.359 Å and 0.584 Å for pQLI and pQLI3, respectively, Fig. S22†).
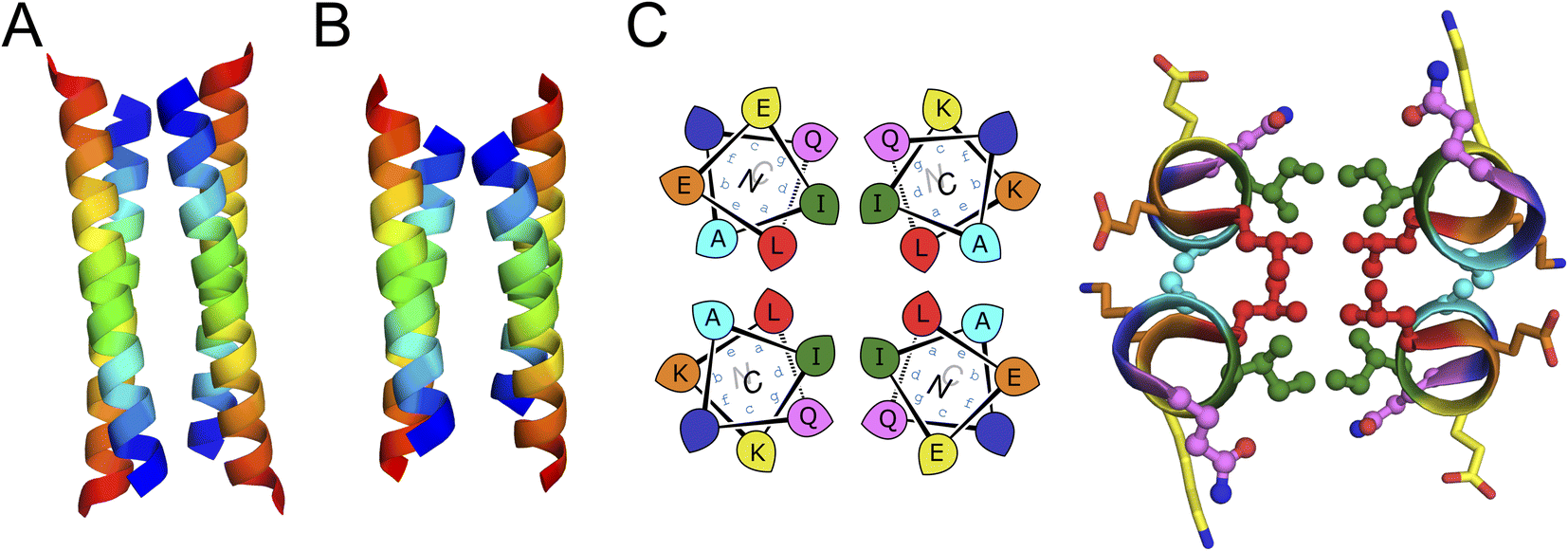 | ||
| Fig. 3 X-ray crystal structures for the antiparallel homotetrameric assemblies of pQLI analogues. (A) pQLI (apCC-Tet*, PDB ID: 8a3g) with 4 heptad repeats. (B) The shorter pQLI3 (apCC-Tet*3, PDB ID: 8a3i). The chains of both structures are coloured in chainbow from the N (blue) to the C termini (red). (C) (Left) Helical wheels for the heptad repeats of pQLI. (Right) Slice through a heptad of the X-ray crystal structures for pQLI. Each position of the heptad is depicted in different color following the SOCKET2 scheme.73 Amino acids that compose the design rules are depicted in ball-and-stick representation. | ||
We propose that the new designs with their clear and interpretable sequence-to-structure relationships offer stable modules for future applications in protein design and for chemical and synthetic biology. Therefore, we rename pQLI as apCC-Tet* to add to our basis set of robust and fully characterized de novo CCs. To demonstrate its potential utility, next we developed the design in a number of different assemblies as described below. The pQLL sequences were not taken forward from this point.
apCC-Tet* assembles efficiently and functions in E. coli
To investigate the portability of apCC-Tet* into cells, we tested it as a component in an established transcriptional assay based on the oligomeric Lac repressor, LacI, in Escherichia coli (E. coli).53 In this assay, the repressor targets the lac promoter to control expression of a GFP reporter gene introduced on a plasmid: competent LacI complexes bind the promoter and repress the GFP gene (Fig. 2D). We used a monomeric Lac repressor variant, LacI*, in which the wild-type (WT) tetramerization domain is removed, and the LacI dimer interface is disrupted.74,75 We have demonstrated previously that de novo designed CCs can substitute for the natural oligomerization domain, which is an antiparallel homotetramer.41,53 LacI* does not repress GFP production when expressed at low levels. However, when apCC-Tet* was fused to the C terminus of LacI*, repression was restored (Fig. 2D). Moreover, the level of repression achieved was comparable to the parent LacI and the previous apCC-Tet design.33,41 As WT LacI is a dimer of dimers, it can contact one or two lacO1 operator sites in the promoter and, with the latter, repression is extremely tight. Importantly, we found that the level of repression induced by LacI*-apCC-Tet* was much greater with two lacO1 operators in the reporter plasmid than with a single copy, indicating that tetramerization of the LacI* fusion protein occurred (Fig. 2D). This contrasts with a control, where LacI* is fused to the dimeric CC-Di,20 which gave similar levels of GFP repression with one or two operators present. Overall, these data indicate that the designed apCC-Tet* efficiently tetramerizes in E. coli to restore the fully active LacI* complex and its DNA-binding function.apCC-Tet* can be adapted to build antiparallel heterotetramers
Breaking the symmetry of de novo CC homo-oligomers to make heteromeric systems has many advantages and potential applications.37,52,76 We sought to expand the utility of apCC-Tet* by redesigning it to make an A2B2 heterotetramer, apCC-Tet*-A2B2. For this, we maintained the g, a, d and e sites as Gln, Leu, Ile, and Ala, respectively, and made two potentially complementary peptides: an acidic peptide, apCC-Tet*-A, with Glu at all b and c positions; and a basic peptide, apCC-Tet*-B, with Lys at those sites. The only other change was the subtle use of Trp or Tyr, respectively, at an f position to further distinguish the A and B peptides. These designs were made in two lengths of 3 and 4 heptads to give two potential pairings, apCC-Tet*3-A2B2 and apCC-Tet*-A2B2, respectively (Tables 1 and S2†). The four peptides were synthesized by SPPS, purified, verified by mass spectrometry (Fig. S23–S26†), and characterized alone and as equimolar paired mixtures as follows.Equilibrium and variable-temperature CD spectra revealed that the individual 4-heptad acidic and basic peptides were both folded and stable in PBS (Fig. S27†), and AUC-SV experiments showed that these isolated peptides formed tetramers like the parent homo-assembly despite the lack of complementary charges (Fig. S28†). An equimolar mixture of apCC-Tet*-A and apCC-Tet*-B spontaneously aggregated. Annealing the sample by heating up to 90 °C and then slowly cooling at room temperature resulted in soluble complexes, which were characterized as a folded and stable heterotetramer (Fig. S27 and S28†). However, the annealed mixture had a lower α-helical content than the respective isolated peptides. Overall, these properties are far from ideal for a de novo designed module that can be used in other contexts and applications. Therefore, we turned to the 3-heptad pair, apCC-Tet*3-A plus apCC-Tet*3-B. Although fully or partly folded (Fig. 4A), the individual acidic and basic peptides had accessible thermal unfolding transitions with midpoints of 61 °C and 42 °C, respectively (Fig. 4B). (N.B. The helicity of the basic peptide increased upon cooling back to below 20 °C.) When mixed at 20 °C, the acidic and basic peptides formed a partly helical assembly (Fig. 4B). Moreover, upon heating between ≈40–55 °C, the mixture folded to a more-helical and hyperthermally stable assembly without an observable melting transition up to 95 °C (Fig. 4B). AUC-SV experiments of annealed samples confirmed the presence of monodispersed tetramers in solution, consistent with an apCC-Tet*3-A2B2 design (Fig. S29†).
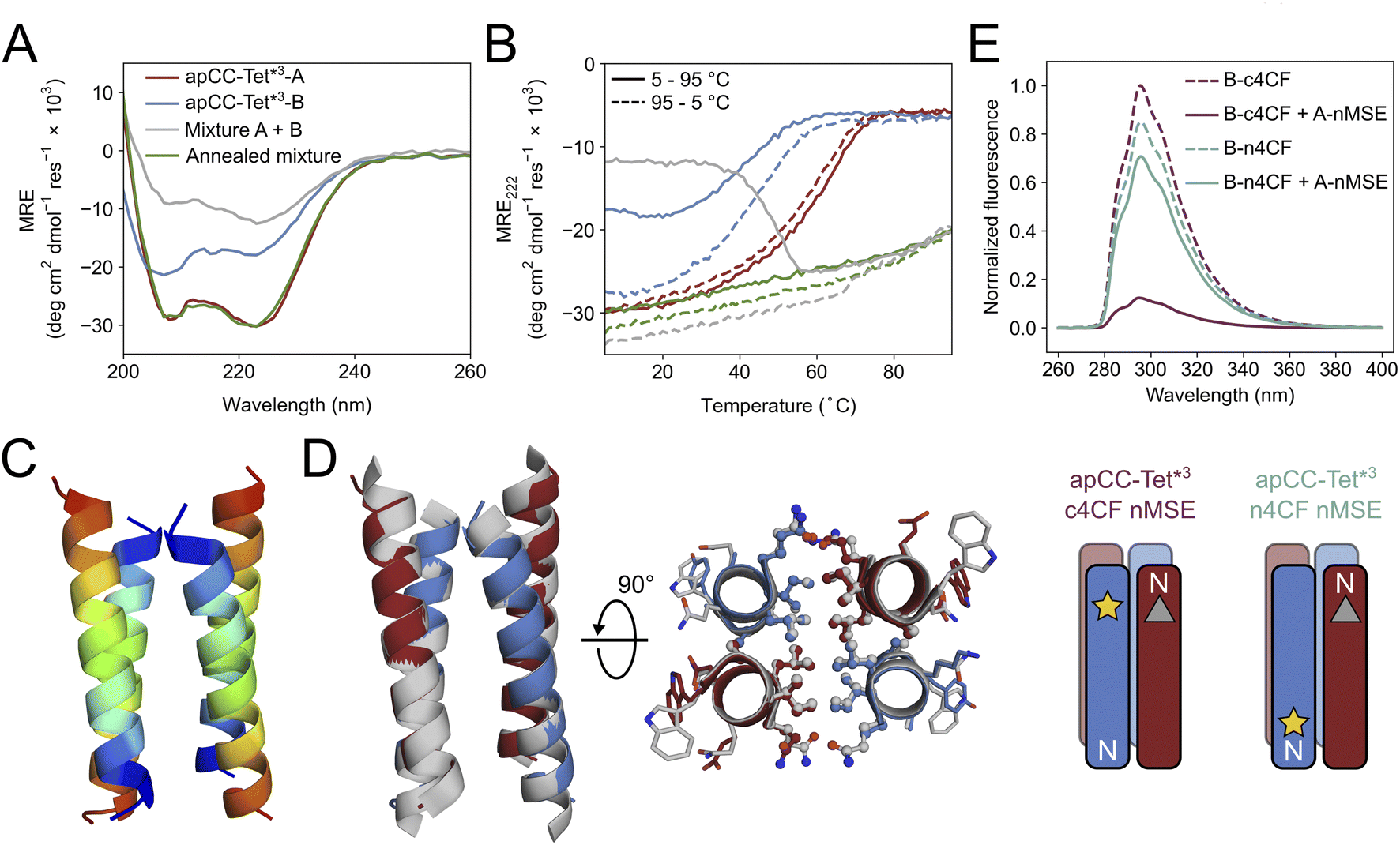 | ||
| Fig. 4 Biophysical and structural characterization of the heterotetrametric complex apCC-Tet*3-A2B2. (A) CD spectra at 5 °C and (B) thermal response curves (ramping up, solid lines; and ramping down, dashed line) for apCC-Tet*3-A (red), apCC-Tet*3-B (blue), the pre-annealed mixture apCCTet*3-A2B2 (grey), and the annealed mixture apCCTet*3-A2B2 (green). Conditions: 50 μM peptide, PBS, pH 7.4. (C) X-ray crystal structure of the heteromeric assembly apCCTet*3-A2B2 (PDB ID: 8a3j) with the chains coloured from the N (blue) to the C termini (red). (D) Alignment of the crystal structures of apCCTet*3-A2B2 (apCC-Tet*3-A, red; apCC-Tet*3-B, blue) and the related homotetramer apCC-Tet*3 (grey). (E) Fluorescence-quenching assay for labelled apCCTet*3-A2B2 peptides. 4CF is the 4-cyano-L-phenylalanine fluorophore (yellow star) and MSE is the L-selenomethionine fluorescence quencher (grey triangle). ‘n’ and ‘c’ indicate mutations near the N and C termini, respectively. In this panel only, peptide names are shortened for clarity. Conditions: 50 μM concentration of each peptide in phosphate buffer (8.2 mM sodium phosphate dibasic, 1.8 mM potassium phosphate monobasic), pH 7.4. | ||
We crystallized a mixture of apCC-Tet*3-A and apCC-Tet*3-B near neutral pH and obtained X-ray diffraction data out to 2.1 Å resolution (Tables S3 and S4†). The resulting crystal structure revealed an antiparallel hetero-tetramer confirming the target apCC-Tet*3-A2B2 complex (Fig. 4C). Like those for apCC-Tet* and apCC-Tet*3, the structure of apCC-Tet*3-A2B2 had a well-packed hydrophobic core with narrow and wide interfaces. Indeed, the heterotetramer overlaid well with the 3-heptad homotetramer (RMSDall-atom = 0.390 Å, Fig. 4D). Again, the design rules—a = Leu, d = Ile, e = Ala, and g = Gln—are readily identifiable from visual inspection of the structure (Fig. 4D).
Despite this experimental structure revealing an antiparallel orientation, there is one potential issue in moving from the ‘bar-magnet’ charge pattern of the homomeric system to the all-acidic plus all-basic design of the hetero-tetramer: the latter opens the possibility of accessing a parallel arrangement of helices in solution. To test this, we probed the arrangement of the assembled helices in solution using fluorescence-quenching experiments introduced by Raleigh.77 Guided by the X-ray crystal structure, we inserted the fluorescent 4-cyanophenylalanine (4CF) at the C-terminal e site of the B peptide to give apCC-Tet*3-B-c4CF (Table S2 and Fig. S30†); and we added a quencher, selenomethionine (MSE), at the N-terminal b position of the A peptide (apCC-Tet*3-A-nMSE, Table S2 and Fig. S31†). As a control, we placed the 4CF residue at the N-terminal c position of the B peptide (apCC-Tet*3-B-n4CF, Table S2 and Fig. S32†), which should be too distant from the MSE residue for quenching in an antiparallel assembly with apCC-Tet*3-A-nMSE. Indeed, this control combination fluoresced comparably to the apCC-Tet*3-B-n4CF peptide alone (Fig. 4E). Conversely, fluorescence was substantially quenched when apCC-Tet*3-B-c4CF was mixed with apCC-Tet*3-A-nMSE, indicating that the 4CF and the MSE groups were proximal, and confirming the assembly of antiparallel helices in solution (Fig. 4E).
Constructing a single-chain de novo proteins from apCC-Tet*
As a final demonstration of the utility of the apCC-Tet* system in protein design, we targeted the construction of a single-chain protein that could be expressed from a synthetic gene in E. coli. This route of taking peptides into full-length proteins – from peptides to proteins – has been pursued before14,30,78 and discussed recently.79–81 The advantages of revisiting this approach are: (i) that well-understood de novo peptide assemblies like apCC-Tet* should provide a strong basis for constructing robust de novo proteins; and (ii) that the resulting proteins should be functionalizable through mutations to break symmetry whilst maintaining the majority of the design rules and, therefore, the design specification. Encouraged by the consistency of the apCC-Tet* peptides and the clear rules underpinning them, we targeted a single-chain 4-helix protein by looping together the peptides of the apCC-Tet* tetramer.We hypothesized that helix packing would drive folding of the single-chain protein with only minor influences from the loops, and that no extensive design of the latter should be required. Therefore, we searched for loop sequences of reasonable composition that matched distances between the termini of the helices in the apCC-Tet* structure, while avoiding extended structures that can have unfavourable entropy contribution in the folding.82,83 From the apCC-Tet* structure, we calculated end-to-end inter-helix distances of 17.4–18.5 Å and 12.5–15.0 Å for the wide and narrow faces, respectively. We treated these distances similarly to find loops in the PDB and from the literature to span both interfaces. The selected loops70,82,84 were arbitrarily incorporated into apCC-Tet*. Alphafold2 predictions indicated that the resulting sequence (Table 1) should form the desired single-chain 4HB (Fig. S41†). We called this single-chain protein sc-apCC-4.
A synthetic gene for sc-apCC-4 was expressed in E. coli, and the protein product was purified in sodium phosphate buffer (Fig. S42 and S43†). Biophysical characterization by CD spectroscopy showed a highly α-helical structure that was fully resistant to thermal denaturation like the parent apCC-Tet* peptide (Fig. 5A and B). Moreover, sc-apCC-4 was hyperstable to chemical denaturation, i.e., up 6 M Gn·HCl (Fig. S44 and S45†). AUC-SV and SE experiments indicated that the de novo protein was a monodispersed monomer in solution (Fig. 5C and S46†). Finally, an X-ray crystal structure for sc-apCC-4 was obtained at 2.0 Å resolution. The structure was solved by molecular replacement using apCC-Tet* as starting model. It confirmed a monomeric four-helix CC bundle with an antiparallel (up-down-up-down) topology (Fig. 5D). The sc-apCC-4 structure is consistent with all of our designs in this series: it has a well-packed hydrophobic core, wide and narrow faces, and the sequence-to-structure relationships are clear from visual inspection (Fig. 5E). Moreover, and interestingly, it overlaid extremely well with the AlphaFold2 prediction with all all-atom RMSD of 0.475 Å (Fig. S47†). This suggests that core packing drives the folding over the loops demonstrating that design rules for apCC-Tet* are robust and transposable to build larger and well-defined proteins with analogous biophysical and structural properties.
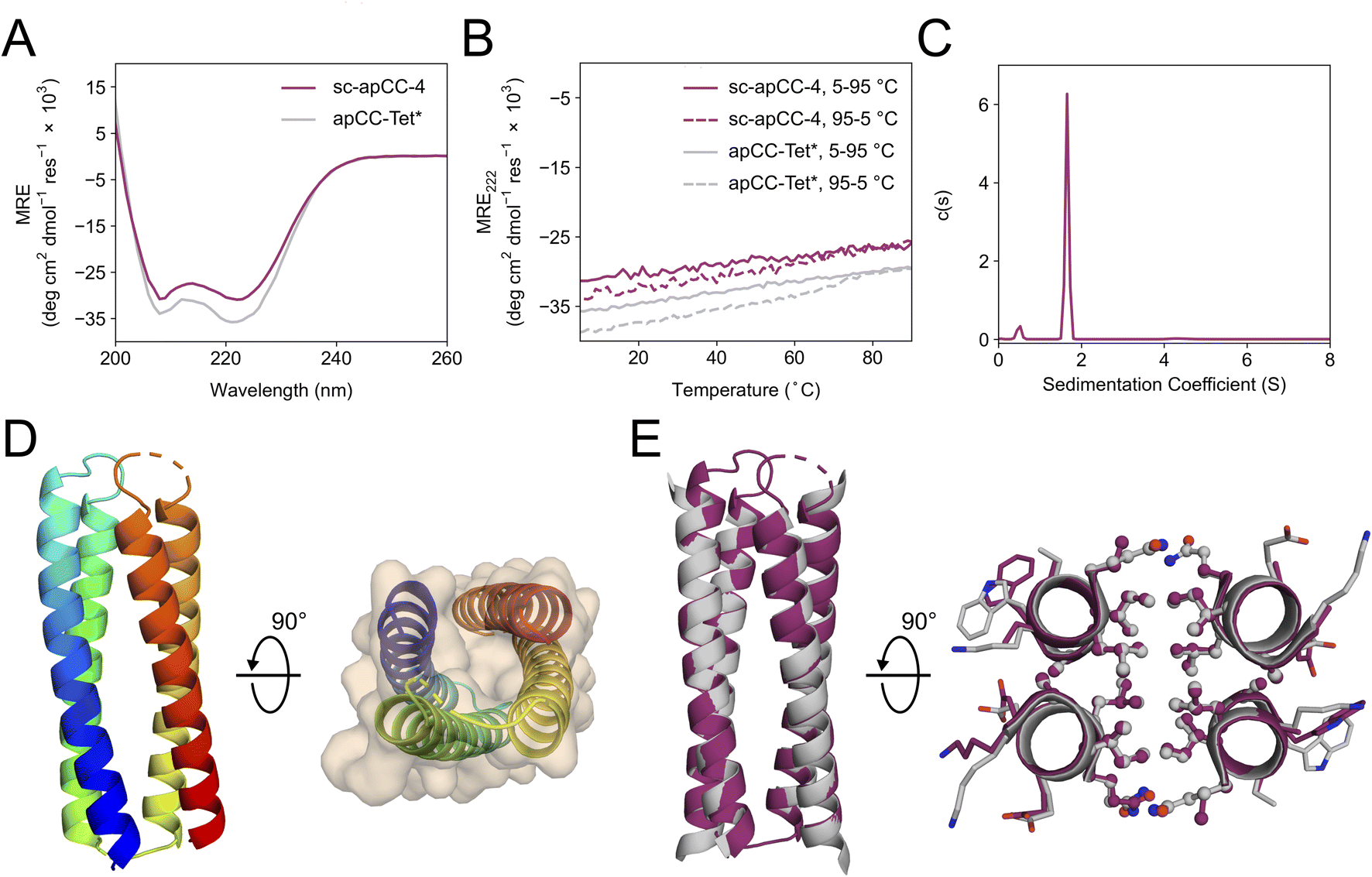 | ||
| Fig. 5 Characterization of the single-chain de novo protein, sc-apCC-4. (A) CD spectra at 5 °C and (B) thermal response curves (ramping up, solid lines; and ramping down, dashed line) for sc-apCC-4 (purple) in comparison with apCC-Tet* peptide (grey). Conditions: 25 μM protein in 50 mM sodium phosphate, 150 mM NaCl, pH 7.4 for the single-chain analogue; and 50 μM peptide, PBS, pH 7.4 for apCC-Tet*. (C) Sedimentation-velocity data from AUC for sc-apCC-4. The fit returned a weight of 0.9× monomer mass. Conditions: 25 μM protein in 50 mM sodium phosphate, 150 mM NaCl, pH 7.4. (D) (Left) X-ray crystal structure of sc-apCC-4 (PDB ID: 8a3k) coloured chainbow from the N (blue) to the C terminus (red). (Right) sc-apCC-4 structure viewed from the termini with chainbow colouring and surface representations. (E) Orthogonal views of the overlay between the structures of sc-apCC-4 (purple) and apCC-Tet* (grey) with a RMSDall-atom of 0.447 Å. | ||
We would like to note that this de novo protein design was achieved in one step from the successful apCC-Tet* design and, thus, without any computational or experimental iterations.
Conclusions
We have combined bioinformatic analysis and rational protein design to determine a set of rules for the design of antiparallel four-helix coiled-coil bundles. Specifically, the rules are Gln@g, Leu@a, Ile@d, and Ala@e in the heptad repeats, abcdefg, of coiled-coil sequences. Using these rules, we have built a new homotetramer, apCC-Tet*, with ‘bar-magnet’ patterning of charged residues at b and c to help direct antiparallel helices. apCC-Tet* is hyperstable with respect to heat and chemical denaturation, and to truncation down to 3 heptad repeats. We have also used the rules to design heterotetramers comprising two different peptide chains, one with completely acidic residues and the other with basic residues at the b and c sites. Thus, in apCC-Tet*-A2B2, only the g, a, d and e sites are needed to direct antiparallel assembly. Finally, we show that the apCC-Tet* sequence can be concatenated to construct a single-chain 4-helix coiled-coil protein, sc-apCC-4, with loops taken from the PDB or the literature, and which can be expressed recombinantly in E. coli. sc-apCC-4 was achieved in a single step without the need for design iterations. All of the designs are fully characterized experimentally in solution and to atomic resolution by X-ray crystallography. Simple visual inspection of the resulting structures reveals that the design rules can be read straight from these structures. Thus, the rules are interpretable, robust, and transferable.From the success of this rational approach, we contend that we now understood the contribution made by each amino acid in our designed sequences for 4-helix bundles. In turn, we anticipate that the newly designed peptides and protein will provide robust modules for further protein design to introduce function; and in chemical and synthetic biology as synthetic oligomerization domains. Such studies will be facilitated by the biophysical and structural characterizations that we provide here. Moreover, the different designs—of homo- and hetero-tetrameric peptides, and a monomeric protein—present opportunities to target and fine-tune different functions and uses. As an example of this potential, the relatively large and well-defined hydrophobic cores of tetrameric coiled coils and 4-helix bundles have been exploited by others to introduce cavities, small-molecule-binding pockets, and catalytic functionalities.37,39,85–87 Moreover, because our designed peptides and protein assemble efficiently in cells, such as E. coli, we anticipate applications to intervene in and to augment natural sub-cellular processes.10,53,58,62,88,89
In short, we posit that our work adds fundamental understanding of the structural principles and sequence-to-structure relationships for coiled coils generally and 4-helix bundles specifically; and that our new designs provide platforms for future de novo design, and chemical and synthetic biology programs.
Of course, many others have designed de novo antiparallel 4-helix bundles and coiled coils over the past four decades.1,4 These have been achieved by modifying natural protein domains (e.g., the GNC4 leucine zipper, and the tetramerization domain of the Lac repressor),19,90,91 through rational approaches that focus on designing amphipathic helices,18,29,30 and by taking computational approaches.26,27,70 This has led to many different sequences for similar design targets. Therefore, to place our work in this broader context and to explore the sequence variations used for these target, we examined other engineered and de novo designed sequences that (i) have been confirmed with high-resolution structures, and (ii) contain knobs-into-holes packing as detected by SOCKET2 (Table S6†).73 Interestingly, we found that most of the foregoing sequences have no clear residue fingerprints at the g, a, d and e sites that we have focused on. Indeed, there was no discernible consensus from these sequences. Those with the most regular hydrophobic cores and most similarity to our own designs are based on Harbury's GCN4-pLI sequence.19 These have Leu@a and Ile@d, but less regularity at the flanking e and g positions, which can be Leu, charged, or other residues (Table S6†).60 Clearly, these and the other sequences ‘work’ and are solutions to the 4-helix-bundle design problem. However, we suggest that the heterogeneity in sequences and the lack of pinpointable sequence-to-structure relationships may make them less attractive as robust and mutable modules for future redesign and design studies.
Finally, it is interesting to speculate on the broader implications and applications of the approach of transforming self-assembling peptides to single-chain proteins as we demonstrate here in one step, and others have done elsewhere.18,30,78 This can be likened to a possible evolutionary process in which primitive proteins might have assembled from the association and subsequent concatenation of smaller peptides,80 similar to the oligomerization of apCC-Tet* peptide to form robust tetramer and then the single-chain protein. The ease of looping the four helices together while maintaining the core folding provides some support to such a mechanism.92 Our future research aims to apply this approach to transform other well-understood multi-chain de novo coiled-coil peptides20,48,49 into single-chain proteins with clear sequence-to-structure features. We anticipate that the resulting synthetic proteins will be robust and stable, and, therefore, highly mutable to allow the incorporation of residues for binding, catalysis, and other functions.36,51,52,78,88,93,94
Data availability
Details of the computational and experimental methods and procedures, additional experimental data, and computational models are given in the ESI.†Author contributions
EAN, EGB, NJS and DNW conceived the study and contributed to experimental design. EAN designed the peptide sequences. EAN and KIA synthesized and characterized the polypeptides. AJS conducted the in-cell experiments. EAN, KIA, and BM determined the protein X-ray crystal structures. EAN and DNW wrote the paper. All authors have read and contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
EAN, AJS, NJS and DNW are supported by a Biotechnology and Biological Sciences Research Council (BBSRC) grant![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (BB/S002820/1). KIA, ODW and DNW are supported by a BBSRC-NSF grant (BB/V004220/1 and 2019598). BM and DNW are supported by a BBSRC grant (BB/V006231/1). We are also grateful to the Max Planck-Bristol Centre for Minimal Biology, which supports KIA, BM, and DNW. DNW was also supported by BrisEngBio, a BBSRC-funded Engineering Biology Research Centre (BB/L01386X/1), and a Royal Society Wolfson Research Merit Award (WM140008). ODW is grateful for a National Institutes of Health grant (GM-118167). We thank the University of Bristol, School of Chemistry, Mass Spectrometry Facility for access to the EPSRC-funded Bruker Ultraflex MALDI-TOF instrument (EP/K03927X/1) and to the Synapt G2S nanospray instrument. We would like to thank Diamond Light Source for access to beamlines I04 and I24 (Proposal mx23269) and the European Synchrotron Radiation Facility (ESRF) for access to beamline ID30B (Proposal mx2373). We thank Will Dawson, Prasun Kumar, Freddie Martin, and members of the Woolfson laboratory for helpful discussions.
(BB/S002820/1). KIA, ODW and DNW are supported by a BBSRC-NSF grant (BB/V004220/1 and 2019598). BM and DNW are supported by a BBSRC grant (BB/V006231/1). We are also grateful to the Max Planck-Bristol Centre for Minimal Biology, which supports KIA, BM, and DNW. DNW was also supported by BrisEngBio, a BBSRC-funded Engineering Biology Research Centre (BB/L01386X/1), and a Royal Society Wolfson Research Merit Award (WM140008). ODW is grateful for a National Institutes of Health grant (GM-118167). We thank the University of Bristol, School of Chemistry, Mass Spectrometry Facility for access to the EPSRC-funded Bruker Ultraflex MALDI-TOF instrument (EP/K03927X/1) and to the Synapt G2S nanospray instrument. We would like to thank Diamond Light Source for access to beamlines I04 and I24 (Proposal mx23269) and the European Synchrotron Radiation Facility (ESRF) for access to beamline ID30B (Proposal mx2373). We thank Will Dawson, Prasun Kumar, Freddie Martin, and members of the Woolfson laboratory for helpful discussions.
Notes and references
- I. V. Korendovych and W. F. DeGrado, Q. Rev. Biophys., 2020, 53, e3 CrossRef CAS.
- X. Pan and T. Kortemme, J. Biol. Chem., 2021, 296, 100558 CrossRef CAS PubMed.
- P.-S. Huang, S. E. Boyken and D. Baker, Nature, 2016, 537, 320–327 CrossRef CAS.
- D. N. Woolfson, J. Mol. Biol., 2021, 433, 167160 CrossRef CAS PubMed.
- I. Anishchenko, S. J. Pellock, T. M. Chidyausiku, T. A. Ramelot, S. Ovchinnikov, J. Hao, K. Bafna, C. Norn, A. Kang, A. K. Bera, F. DiMaio, L. Carter, C. M. Chow, G. T. Montelione and D. Baker, Nature, 2021, 600, 547–552 CrossRef CAS PubMed.
- J. Wang, S. Lisanza, D. Juergens, D. Tischer, J. L. Watson, K. M. Castro, R. Ragotte, A. Saragovi, L. F. Milles, M. Baek, I. Anishchenko, W. Yang, D. R. Hicks, M. Expòsit, T. Schlichthaerle, J.-H. Chun, J. Dauparas, N. Bennett, B. I. M. Wicky, A. Muenks, F. DiMaio, B. Correia, S. Ovchinnikov and D. Baker, Science, 2022, 377, 387–394 CrossRef CAS PubMed.
- K. J. Grayson and J. L. R. Anderson, J. R. Soc., Interface, 2018, 15, 20180472 CrossRef PubMed.
- W. M. Dawson, G. G. Rhys and D. N. Woolfson, Curr. Opin. Chem. Biol., 2019, 52, 102–111 CrossRef CAS.
- A. Marchand, A. K. Van Hall-Beauvais and B. E. Correia, Curr. Opin. Struct. Biol., 2022, 74, 102370 CrossRef CAS PubMed.
- W. Zhou, T. Šmidlehner and R. Jerala, FEBS Lett., 2020, 594, 2199–2212 CrossRef CAS.
- S. L. Lovelock, R. Crawshaw, S. Basler, C. Levy, D. Baker, D. Hilvert and A. P. Green, Nature, 2022, 606, 49–58 CrossRef CAS PubMed.
- R. B. Hill, D. P. Raleigh, A. Lombardi and W. F. DeGrado, Acc. Chem. Res., 2000, 33, 745–754 CrossRef CAS PubMed.
- A. Lombardi, F. Pirro, O. Maglio, M. Chino and W. F. DeGrado, Acc. Chem. Res., 2019, 52, 1148–1159 CrossRef CAS PubMed.
- K. J. Grayson and J. L. R. Anderson, Curr. Opin. Struct. Biol., 2018, 51, 149–155 CrossRef CAS PubMed.
- M. H. Hecht, J. S. Richardson, D. C. Richardson and R. C. Ogden, Science, 1990, 249, 884–891 CrossRef CAS PubMed.
- S. Kamtekar, J. M. Schiffer, H. Xiong, J. M. Babik and M. H. Hecht, Science, 1993, 262, 1680–1685 CrossRef CAS.
- S. P. Ho and W. F. DeGrado, J. Am. Chem. Soc., 1987, 109, 6751–6758 CrossRef CAS.
- R. L. Koder, J. L. R. Anderson, L. A. Solomon, K. S. Reddy, C. C. Moser and P. L. Dutton, Nature, 2009, 458, 305–309 CrossRef CAS PubMed.
- P. B. Harbury, T. Zhang, P. S. Kim and T. Alber, Science, 1993, 262, 1401–1407 CrossRef CAS PubMed.
- J. M. Fletcher, A. L. Boyle, M. Bruning, G. J. Bartlett, T. L. Vincent, N. R. Zaccai, C. T. Armstrong, E. H. Bromley, P. J. Booth, R. L. Brady, A. R. Thomson and D. N. Woolfson, ACS Synth. Biol., 2012, 1, 240–250 CrossRef CAS PubMed.
- A. Lombardi, C. M. Summa, S. Geremia, L. Randaccio, V. Pavone and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6298–6305 CrossRef CAS PubMed.
- Y. Deng, J. Liu, Q. Zheng, D. Eliezer, N. R. Kallenbach and M. Lu, Structure, 2006, 14, 247–255 CrossRef CAS.
- G. Grigoryan and W. F. DeGrado, J. Mol. Biol., 2011, 405, 1079–1100 CrossRef CAS.
- S. Dunin-Horkawicz and A. N. Lupas, J. Struct. Biol., 2010, 170, 226–235 CrossRef CAS PubMed.
- G. S. Murphy, B. Sathyamoorthy, B. S. Der, M. C. Machius, S. V. Pulavarti, T. Szyperski and B. Kuhlman, Protein Sci., 2015, 24, 434–445 CrossRef CAS PubMed.
- P.-S. Huang, G. Oberdorfer, C. Xu, X. Y. Pei, B. L. Nannenga, J. M. Rogers, F. DiMaio, T. Gonen, B. Luisi and D. Baker, Science, 2014, 346, 481–485 CrossRef CAS PubMed.
- S. E. Boyken, Z. Chen, B. Groves, R. A. Langan, G. Oberdorfer, A. Ford, J. M. Gilmore, C. Xu, F. DiMaio, J. H. Pereira, B. Sankaran, G. Seelig, P. H. Zwart and D. Baker, Science, 2016, 352, 680–687 CrossRef CAS PubMed.
- M. ElGamacy, M. Coles and A. Lupas, J. Struct. Biol., 2018, 204, 380–387 CrossRef CAS PubMed.
- D. Eisenberg, W. Wilcox, S. M. Eshita, P. M. Pryciak, S. P. Ho and W. F. DeGrado, Proteins, 1986, 1, 16–22 CrossRef CAS PubMed.
- L. Regan and W. F. DeGrado, Science, 1988, 241, 976–978 CrossRef CAS PubMed.
- R. B. Hill and W. F. DeGrado, J. Am. Chem. Soc., 1998, 120, 1138–1145 CrossRef CAS.
- S. F. Betz and W. F. DeGrado, Biochemistry, 1996, 35, 6955–6962 CrossRef CAS.
- G. G. Rhys, C. W. Wood, J. L. Beesley, N. R. Zaccai, A. J. Burton, R. L. Brady, A. R. Thomson and D. N. Woolfson, J. Am. Chem. Soc., 2019, 141, 8787–8797 CrossRef CAS PubMed.
- S. R. Presnell and F. E. Cohen, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 6592–6596 CrossRef CAS PubMed.
- N. L. Harris, S. R. Presnell and F. E. Cohen, J. Mol. Biol., 1994, 236, 1356–1368 CrossRef CAS PubMed.
- N. F. Polizzi and W. F. DeGrado, Science, 2020, 369, 1227–1233 CrossRef CAS.
- J. Kaplan and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11566–11570 CrossRef CAS PubMed.
- A. E. Donnelly, G. S. Murphy, K. M. Digianantonio and M. H. Hecht, Nat. Chem. Biol., 2018, 14, 253–255 CrossRef CAS PubMed.
- D. W. Watkins, J. M. X. Jenkins, K. J. Grayson, N. Wood, J. W. Steventon, K. K. Le Vay, M. I. Goodwin, A. S. Mullen, H. J. Bailey, M. P. Crump, F. MacMillan, A. J. Mulholland, G. Cameron, R. B. Sessions, S. Mann and J. L. R. Anderson, Nat. Commun., 2017, 8, 358 CrossRef PubMed.
- F. Pirro, N. Schmidt, J. Lincoff, Z. X. Widel, N. F. Polizzi, L. Liu, M. J. Therien, M. Grabe, M. Chino, A. Lombardi and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 33246–33253 CrossRef CAS PubMed.
- C. L. Edgell, A. J. Smith, J. L. Beesley, N. J. Savery and D. N. Woolfson, ACS Synth. Biol., 2020, 9, 427–436 CrossRef CAS PubMed.
- T. H. Nguyen, G. Dods, M. Gómez-Schiavon, M. Wu, Z. Chen, R. Kibler, D. Baker, H. El-Samad and A. H. Ng, GEN Biotechnology, 2022, 1, 91–100 CrossRef.
- D. N. Woolfson, Adv. Protein Chem., 2005, 70, 79–112 CrossRef CAS PubMed.
- D. N. Woolfson, in Subcell. Biochem., ed. D. A. D. Parry and J. M. Squire, Springer International Publishing, Cham, 2017, vol. 82, pp. 35–61 Search PubMed.
- A. N. Lupas and J. Bassler, Trends Biochem. Sci., 2017, 42, 130–140 CrossRef CAS PubMed.
- A. N. Lupas and M. Gruber, Adv. Protein Chem., 2005, 70, 37–38 CrossRef CAS.
- D. N. Woolfson and T. Alber, Protein Sci., 1995, 4, 1596–1607 CrossRef CAS PubMed.
- A. R. Thomson, C. W. Wood, A. J. Burton, G. J. Bartlett, R. B. Sessions, R. L. Brady and D. N. Woolfson, Science, 2014, 346, 485–488 CrossRef CAS PubMed.
- W. M. Dawson, F. J. O. Martin, G. G. Rhys, K. L. Shelley, R. L. Brady and D. N. Woolfson, Chem. Sci., 2021, 12, 6923–6928 RSC.
- F. Thomas, W. M. Dawson, E. J. M. Lang, A. J. Burton, G. J. Bartlett, G. G. Rhys, A. J. Mulholland and D. N. Woolfson, ACS Synth. Biol., 2018, 7, 1808–1816 CrossRef CAS PubMed.
- A. J. Burton, A. R. Thomson, W. M. Dawson, R. L. Brady and D. N. Woolfson, Nat. Chem., 2016, 8, 837–844 CrossRef CAS PubMed.
- J. M. Fletcher, K. A. Horner, G. J. Bartlett, G. G. Rhys, A. J. Wilson and D. N. Woolfson, Chem. Sci., 2018, 9, 7656–7665 RSC.
- A. J. Smith, F. Thomas, D. Shoemark, D. N. Woolfson and N. J. Savery, ACS Synth. Biol., 2019, 8, 1284–1293 CrossRef CAS PubMed.
- N. H. Joh, T. Wang, M. P. Bhate, R. Acharya, Y. Wu, M. Grabe, M. Hong, G. Grigoryan and W. F. DeGrado, Science, 2014, 346, 1520–1524 CrossRef CAS PubMed.
- S. A. Farhadi, R. Liu, M. W. Becker, E. A. Phelps and G. A. Hudalla, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2024117118 CrossRef CAS PubMed.
- M. G. Oakley and J. J. Hollenbeck, Curr. Opin. Struct. Biol., 2001, 11, 450–457 CrossRef CAS.
- C. Negron and A. E. Keating, J. Am. Chem. Soc., 2014, 136, 16544–16556 CrossRef CAS PubMed.
- G. G. Rhys, J. A. Cross, W. M. Dawson, H. F. Thompson, S. Shanmugaratnam, N. J. Savery, M. P. Dodding, B. Höcker and D. N. Woolfson, Nat. Chem. Biol., 2022, 18, 999–1004 CrossRef CAS PubMed.
- O. D. Monera, N. E. Zhou, P. Lavigne, C. M. Kay and R. S. Hodges, J. Biol. Chem., 1996, 271, 3995–4001 CrossRef CAS PubMed.
- M. K. Yadav, L. J. Leman, D. J. Price, C. L. Brooks, C. D. Stout and M. R. Ghadiri, Biochemistry, 2006, 45, 4463–4473 CrossRef CAS PubMed.
- R. Lizatovic, O. Aurelius, O. Stenstrom, T. Drakenberg, M. Akke, D. T. Logan and I. Andre, Structure, 2016, 24, 946–955 CrossRef CAS PubMed.
- K. E. Thompson, C. J. Bashor, W. A. Lim and A. E. Keating, ACS Synth. Biol., 2012, 1, 118–129 CrossRef CAS PubMed.
- R. O. Crooks, A. Lathbridge, A. S. Panek and J. M. Mason, Biochemistry, 2017, 56, 1573–1584 CrossRef CAS.
- T. Lebar, D. Lainšček, E. Merljak, J. Aupič and R. Jerala, Nat. Chem. Biol., 2020, 16, 513–519 CrossRef CAS PubMed.
- O. D. Testa, E. Moutevelis and D. N. Woolfson, Nucleic Acids Res., 2009, 37, D315–D322 CrossRef CAS.
- K. M. Gernert, M. C. Surles, T. H. Labean, J. S. Richardson and D. C. Richardson, Protein Sci., 1995, 4, 2252–2260 CrossRef CAS.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- R. Evans, M. O’Neill, A. Pritzel, N. Antropova, A. Senior, T. Green, A. Žídek, R. Bates, S. Blackwell, J. Yim, O. Ronneberger, S. Bodenstein, M. Zielinski, A. Bridgland, A. Potapenko, A. Cowie, K. Tunyasuvunakool, R. Jain, E. Clancy, P. Kohli, J. Jumper and D. Hassabis, bioRxiv, preprint, 2021, 2021.2010.2004.463034 Search PubMed.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS.
- Z. Chen, S. E. Boyken, M. Jia, F. Busch, D. Flores-Solis, M. J. Bick, P. Lu, Z. L. VanAernum, A. Sahasrabuddhe, R. A. Langan, S. Bermeo, T. J. Brunette, V. K. Mulligan, L. P. Carter, F. DiMaio, N. G. Sgourakis, V. H. Wysocki and D. Baker, Nature, 2019, 565, 106–111 CrossRef CAS.
- H. Jenkins, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 205–214 CrossRef CAS PubMed.
- J. Walshaw and D. N. Woolfson, J. Mol. Biol., 2001, 307, 1427–1450 CrossRef CAS PubMed.
- P. Kumar and D. N. Woolfson, Bioinformatics, 2021, 37, 4575–4577 CrossRef CAS.
- J. Chen and K. S. Matthews, J. Biol. Chem., 1992, 267, 13843–13850 CrossRef CAS PubMed.
- F. Dong, S. Spott, O. Zimmermann, B. Kisters-Woike, B. Müller-Hill and A. Barker, J. Mol. Biol., 1999, 290, 653–666 CrossRef CAS PubMed.
- E. A. Naudin, A. G. McEwen, S. K. Tan, P. Poussin-Courmontagne, J.-L. Schmitt, C. Birck, W. F. DeGrado and V. Torbeev, J. Am. Chem. Soc., 2021, 143, 3330–3339 CrossRef CAS.
- M. D. Watson, I. Peran and D. P. Raleigh, Biochemistry, 2016, 55, 3685–3691 CrossRef CAS.
- J. L. R. Anderson, C. T. Armstrong, G. Kodali, B. R. Lichtenstein, D. W. Watkins, J. A. Mancini, A. L. Boyle, T. A. Farid, M. P. Crump, C. C. Moser and P. L. Dutton, Chem. Sci., 2014, 5, 507–514 RSC.
- M. L. Romero Romero, A. Rabin and D. S. Tawfik, Angew. Chem., Int. Ed., 2016, 55, 15966–15971 CrossRef CAS PubMed.
- V. Alva and A. N. Lupas, Curr. Opin. Struct. Biol., 2018, 48, 103–109 CrossRef CAS.
- M. ElGamacy and B. Hernandez Alvarez, Curr. Opin. Struct. Biol., 2021, 68, 224–234 CrossRef CAS.
- Y. Yu and S. Lutz, Trends Biotechnol., 2011, 29, 18–25 CrossRef CAS PubMed.
- A. D. Nagi and L. Regan, Folding Des., 1997, 2, 67–75 CrossRef CAS PubMed.
- R. G. Garces, W. Gillon and E. F. Pai, Protein Sci., 2007, 16, 176–188 CrossRef CAS PubMed.
- N. F. Polizzi, Y. Wu, T. Lemmin, A. M. Maxwell, S. Q. Zhang, J. Rawson, D. N. Beratan, M. J. Therien and W. F. DeGrado, Nat. Chem., 2017, 9, 1157–1164 CrossRef CAS.
- M. K. Yadav, J. E. Redman, L. J. Leman, J. M. Alvarez-Gutiérrez, Y. Zhang, C. D. Stout and M. R. Ghadiri, Biochemistry, 2005, 44, 9723–9732 CrossRef CAS PubMed.
- C. Karas and M. Hecht, Life, 2020, 10, 9 CrossRef CAS PubMed.
- Z. Chen, R. D. Kibler, A. Hunt, F. Busch, J. Pearl, M. Jia, Z. L. VanAernum, B. I. M. Wicky, G. Dods, H. Liao, M. S. Wilken, C. Ciarlo, S. Green, H. El-Samad, J. Stamatoyannopoulos, V. H. Wysocki, M. C. Jewett, S. E. Boyken and D. Baker, Science, 2020, 368, 78–84 CrossRef CAS PubMed.
- S. Shui, P. Gainza, L. Scheller, C. Yang, Y. Kurumida, S. Rosset, S. Georgeon, R. B. Di Roberto, R. Castellanos-Rueda, S. T. Reddy and B. E. Correia, Nat. Commun., 2021, 12, 5754 CrossRef CAS PubMed.
- B. C. Root, L. D. Pellegrino, E. D. Crawford, B. Kokona and R. Fairman, Protein Sci., 2009, 18, 329–336 CrossRef CAS PubMed.
- R. Fairman, H.-G. Chao, T. B. Lavoie, J. J. Villafranca, G. R. Matsueda and J. Novotny, Biochemistry, 1996, 35, 2824–2829 CrossRef CAS PubMed.
- R. G. Smock, I. Yadid, O. Dym, J. Clarke and D. S. Tawfik, Cell, 2016, 164, 476–486 CrossRef CAS PubMed.
- A. J. Scott, A. Niitsu, H. T. Kratochvil, E. J. M. Lang, J. T. Sengel, W. M. Dawson, K. R. Mahendran, M. Mravic, A. R. Thomson, R. L. Brady, L. Liu, A. J. Mulholland, H. Bayley, W. F. DeGrado, M. I. Wallace and D. N. Woolfson, Nat. Chem., 2021, 13, 643–650 CrossRef CAS PubMed.
- C. Yang, F. Sesterhenn, J. Bonet, E. A. van Aalen, L. Scheller, L. A. Abriata, J. T. Cramer, X. Wen, S. Rosset, S. Georgeon, T. Jardetzky, T. Krey, M. Fussenegger, M. Merkx and B. E. Correia, Nat. Chem. Biol., 2021, 17, 492–500 CrossRef CAS PubMed.
- K. Szczepaniak, G. Lach, J. M. Bujnicki and S. Dunin-Horkawicz, J. Struct. Biol., 2014, 188, 123–133 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods and ESI data. See https://doi.org/10.1039/d2sc04479j |
| This journal is © The Royal Society of Chemistry 2022 |