
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemplate-assisted alloying of atom-precise silver nanoclusters: a new approach to generate cluster functionality†
Sourav
Biswas‡
a,
Anish Kumar
Das‡
a,
Surya Sekhar
Manna
 b,
Biswarup
Pathak
b and
Sukhendu
Mandal
*a
b,
Biswarup
Pathak
b and
Sukhendu
Mandal
*a
aSchool of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Kerala 69551, India. E-mail: sukhendu@iisertvm.ac.in
bDepartment of Chemistry, Indian Institute of Technology Indore, Madhya Pradesh 453552, India
First published on 6th September 2022
Abstract
To acquire the atomic design of new functional Ag(I) nanoclusters (NCs), a new synthetic approach of site-specific alloying has been unveiled, by which the neutral CO2 templated Ag20 core is confined through Cu containing two peripheral motif units. The impact of surface charge, size and shape of the template on the self-assembly of Ag(I) has been precisely controlled here for the first time and as a result, a similar pentagonal gyrobicupola-like Ag20 core is formed while varying the templates (S2−, CO32− and CO2). However, the surface charge generated on the Ag(I) core due to the presence of a neutral template opens up the possibility of this novel alloying process. The introduction of strongly interacted peripheral motif units (DMA-CuS−) on the Ag20 core enforces more rigidity in the skeleton that reduces the probability of non-radiative transition in the excited state by lowering the intramolecular vibration. In addition to this, the incorporation of electron-donating peripheral motif units modulates the frontier molecular arrangement that helps in attaining the synergy which would ultimately turn on the room-temperature emission properties. The electron-donating effect of the peripheral motif units further leads to a sharp reduction of the bandgap and the symmetric position of the heterometal in the cluster minimizes the intercluster distances which further influences the intercluster charge carrier transport. So, the precise structure–property correlation with this novel synthetic approach will pave the way for a well-functioning NC design.
Introduction
Metal nanoclusters (NCs) with atomic precision have drawn immense research interest owing to their capability of atomic-level control over the structure and properties.1–3 In contrast to their surface plasmon absorption-dominated nanoparticle counterparts, the quantized electronic states of NCs in their ultra-small size regime bear the opportunity to explore many new molecular-like properties.4–8 To date, a number of noble metal NCs have been synthesized by modulating the metal–metal and metal–ligand bonding.1,2 Among these, thiolate-protected Ag NCs show outstanding luminescence properties and attract application-oriented research interest.3,9–12 The extensive research progress on designing thiolate-protected Ag NCs is divided into two distinct directions.13–15 The Ag NCs containing a metallic Ag(0) core are synthesized by using a reducing agent; however the other congeners, Ag(I)-based NCs are mostly synthesized in an uncontrolled self-assembly process without any reducing agent and contain no free valence electrons as the former.2 The high reactivity of metal precursors with thiolate ligands along with the associated fast kinetics of nucleation and growth make this self-assembly process the most challenging for rational structure design.15The introduction of structure directing agents in the self-assembly of Ag(I) can control the precise structure design. Since then anions have been mostly utilized as structure directing agents in Ag(I)-based NCs synthesis which controls the size, shape and overall charge distribution of the synthesized NCs.16,17 Due to the variable ligation number of the different anions, the preferences of Ag(I) coordination along with the argentophilic interaction introduce an extra contribution to the structural stability.16,18 To date F−,19 Cl−,19–21 CO32−,22–24 S2−,25,26 MO42− (M = S, Se, Mo, Cr)27 and polyoxometalates28 have been utilized as templates for thiolate-protected Ag(I) NCs synthesis. The size, shape and structural stability of thiolated Ag(I) NCs also depend on the bulkiness of the thiolate ligands. Sun et al. first reported the StBu protected Ag20 NC in a DMF medium where CO32− serves as a template.22 Later, various StBu protected Ag20 NCs were discovered by simply changing the auxiliary ligands; however in all cases, CO32− is present as the specific template formed by in situ fixation of CO2 indicating the importance of a specific template anion in the assembly of Ag(I) atoms.23,24 Afterward, Alhilaly et al. synthesized StBu protected Ag16 NCs by using Cl− as a template.20 So, the reduction in the size, shape and charge of the template anion decreases the number of assembled Ag(I) atoms in the cluster node. In our previous work, we observed the dominant role of the Cl− template in S-Adm protected Ag16 NC design over the bulkiness of the adamantanethiolate ligand.21 It has been observed in the literature that the physical properties can be brought into the cluster system by the incorporation of the functional anion template.16 However, the alteration in the cluster node restricts the proper understanding of such influence in cluster property determination by varying the templates. So, it will be more effective if any synthesis process could be designed and executed where the number of self-assembled Ag(I) atoms will not depend on the size, shape and charge of the template.
Although Ag NCs have attracted us with their outstanding luminescence properties, most of the Ag(I) NCs are unable to exhibit their luminescence properties at room temperature due to their thermally activated dominant non-radiative transition.20–24 So, the complex synthesis process of making Ag(I) NCs would not be beneficial due to the absence of their most alluring properties. Very recently, Li et al. showed the effect of post-synthetic modification through ligand exchange on a structurally defined Ag20 NC in photoluminescence (PL) generation by achieving more rigidity.29 However, such post-synthetic modification will not be favorable all the time as it involves much more approximation in retaining the actual core size. So, any in situ modification for confining the Ag(I) core to mitigate the dominant non-radiative transition may be more favorable.
One such method could be alloying or bimetallic cluster formation as it can generate cluster luminescence.30–34 In the alloying process of Ag(I) NCs, the incorporation of more reactive foreign atoms is mainly focused on replacing one or more Ag(I) atoms from the core by doping.35,36 This alters the number of Ag(I) ions in the core and electronic structure which hinders the correlation of the role of foreign atoms for a specific core. Thus, for tracking the alteration in the electronic structure of heterometallic NCs more precisely, we need to keep the position of the parent Ag(I) atoms in their original place instead of replacing them with other chemically-different d10 metal atoms. Until now no such synthetic procedure has been implemented where the structurally intact Ag(I) core has been probed with foreign metal atoms. The effect of templates on stabilizing such alloy Ag(I)-based heterometallic NCs synthesis is still unknown.
The structural integrity in long-range order and the unique charge carrier transport of the NCs bear an immense opportunity for electronic materials design.37,38 The charge carrier transport between the two-cluster units depends on the size of the cluster nodes and the intercluster distances.39–41 However, the presence of a highly resistive surface protecting ligand environment at the outer surface of the cluster nodes and the absence of interconnectivity through linker molecules generally reduce the intercluster charge transportation in molecular Ag(I) NCs. For this reason, researchers have so far shown little interest in exploring the charge transportation of Ag(I) NCs. To improve the charge carrier transport in Ag(I) NCs, the size of the Ag(I) cluster node is the primary choice that could be increased. However, this is not possible by keeping the total number of Ag(I) atoms constant. On the other hand, the intercluster distance would be intact if the size of the cluster nodes is the same. So, incorporating any additional metal atom at the peripheral position of the cluster node will alleviate the short-coming of the finite size distribution between the two cluster nodes for efficient charge carrier transport. However, such approaches are still not available for Ag(I) NCs.
Considering the above-mentioned facts, here we present a strategy for controlling the template-assisted growth of functional Ag(I) NCs in a different solvent medium and explore the functionalities with structural correlation at the atomic level. At first, our challenge is in the direction of overcoming the excessive dependence of the template on the self-assembly of the Ag(I) atoms. To be specific, we report three new crystal structures containing the same Ag20 as the cluster core which accommodates different in situ generated templates, namely S2−, CO32− and CO2 at the center. The primary origin of all in situ generated templates differs from the precursor to the aerial sources which are generated by altering the basicity of the reaction medium. Under these particular reaction conditions, the associated charge on each template is proved to be essential for the fast assembly of Ag(I) atoms; however, this is not at all decisive. Due to the neutral charge of CO2, the sluggish assembly of twenty Ag(I) atoms along with ten-thiolate surface protecting ligands and eight auxiliary ligands provide the scope of alloying and accommodating two foreign DMA-CuS− units as peripheral staple motifs in specific positions. The growth mechanism of each NC is discussed here. Although the structural anatomy of these three NCs shows notable similarities, the presence of the peripheral staple motifs confines the intracluster molecular motion of the cluster node which turns on the room-temperature luminescence properties. An alteration in the charge carrier transport properties is also detected after alloying, as the cluster radius is increased and the associated intercluster distance is decreased, thus resulting in a ∼104 order of enhancement in single-crystal conductivity. So, our strategic approaches represent a new method of template-assisted alloying through the incorporation of peripheral motifs on the Ag(I) cluster nodes to modulate the optical and charge carrier transport properties. With improved control of the growth of the NCs and proper correlation with the functionalities, we assume that this study will be a paradigm for future exploration of alloy NCs.
Results and discussion
Synthesis and crystallization of Ag(I) NCs
A typical synthesis of Ag(I) NCs involves the chemical reactions between the metal precursors in the solvent medium at room temperature. Here, three different Ag(I) NCs were synthesized in a facile one-pot synthetic procedure by changing the solvent medium. The strategical approach relies on the formation of different templates by varying the solvent medium that governs the formation and growth of the NCs although we haven't added any templating agent during the reactions. For homometal NC synthesis initially, the reaction occurs between the AgStBu complex and AgCF3COO in the solvent medium. Then the nature of the in situ generated templates in different solvent media plays a critical role in the formation of the Ag(I) NCs. However, for the synthesis of heterometallic NCs the reaction conditions are the same as those for the former but the Cu(I) precursor was added to incorporate the heterometal atom. High-quality crystals were obtained from the resulting clear solution by slow evaporation at room temperature. Obtained crystals are structurally elucidated by single-crystal X-ray diffraction (SCXRD) analysis.Structural anatomy of synthesized Ag(I) NCs
The structural elucidation of the obtained three NCs via SCXRD analysis shows that all three NCs crystallized in the identical crystal system of monoclinic (Tables S1–S3†). However, these exhibit different sizes, shapes, and compositions. The crystallographic data infers the overall composition of the Ag(I)-based thiolated NCs that are formulated as [S@Ag20(StBu)10(CF3COO)8(CH3CN)4]. (EtOH) (NC 1), [CO3@Ag20(StBu)10(CF3COO)8(DMF)3(H2O)]. (DMF) (NC 2), and [CO2@Ag20Cu2S2(StBu)10(CF3COO)8(DMA)4]. (DMA) (NC 3) (Fig. 1). In demonstrating the structural comparison among NC 1, NC 2, and NC 3, it was found that all the NCs feature the doughnut-like Ag20 core geometry (Fig. S1†), where the twenty Ag(I) atoms are held together by argentophilic interactions.42,43 The Bader's charge analysis between the two adjacent Ag atoms in each Ag20 core indicates the reality of non-covalent argentophilic interaction (Fig. S2†).44 The geometry of the core belongs to the Johnson solid category, which is classified as a pentagonal gyrobicupola (J31).45 According to the arrangement of the Ag atoms, the skeleton of Ag20 can be divided into three planes, (1) an upper pentagonal plane (constructed from five Ag atoms), (2) a middle decagonal plane (constructed from ten Ag atoms), and (3) a lower pentagonal plane (constructed from five Ag atoms) (Fig. S3†). This middle plane is connected with the other two planes through ten square and ten triangular facets (Fig. S4†). Interestingly, the different templates are accommodated at the center of each NC core, as revealed by the X-ray crystal data, and that is one of the parts of structural variation which make all the NCs distinguishable from each other. Hence, NC 1, NC 2, and NC 3 are described as the S2−, CO32−, and CO2 templated NC, respectively (Fig. 2a–c). However, all the NCs accommodate the same number of thiolate ligands (−StBu), which is ten as a main protecting ligand, and the same number of trifluroacetate (CF3COO−), which is eight as an auxiliary ligand. Moreover, the positions of the attached thiolate ligands and the trifluroacetates are exactly the same for all the NCs including the bridging style with the core. More precisely, each of the S atoms of the −StBu ligand is attached to each square facet of the pentagonal gyrobicupola through μ4 bridging mode, adopting the square pyramid geometry by the Ag4S unit, and the −StBu ligands are directed out from the core by making an angle of 60° with respect to the decagonal plane (Fig. 2d–f). But among the eight CF3COO−, six CF3COO− are bonded via μ2 bridging mode with the middle plane of the Ag20 skeleton, and the remaining two CF3COO− adopted μ1 bridging mode with the other two pentagonal planes (Fig. 2g–i). Furthermore, to elucidate the attachment of the solvent molecules with the core, we have noticed that there is also a structural difference among the NCs. NC 1 and NC 2 contain the respective solvent molecules (i.e., CH3CN for NC 1 and DMF for NC 2) in the same place, though in the case of NC 2, one of the desired solvent molecules, i.e., DMF, is replaced by the H2O molecule (Fig. 2j and k). The attachment of the H2O molecules instead of DMF gives us a clue about the specificity of the attachment of solvent molecules to the Ag20 core. Interestingly, in the case of NC 3, instead of attaching four DMA molecules to the core, two of the opposite solvent molecules are swapped by the two DMA-CuS− units, which results in the formation of alloy NCs in the DMA medium (Fig. 2l). The attachment of one DMA-CuS− unit specifically took place exactly where the H2O molecule is attached to NC 2 and the other one is just placed at the opposite position to keep the symmetry unchanged. In contrast, we synthesized NC 3 by adding an excess of Cu(I) salt that did not give rise to any product containing more than two Cu atoms. So, we believe that the attachment of the two DMA-CuS− units is highly site-specific and it is quite easy to replace the less coordinated solvent molecules through highly interacted Ag–S coordination in a controlled manner. Finally, each of the NCs contains one lattice solvent molecule, depending on the solvent system used in the synthesis (Fig. 1).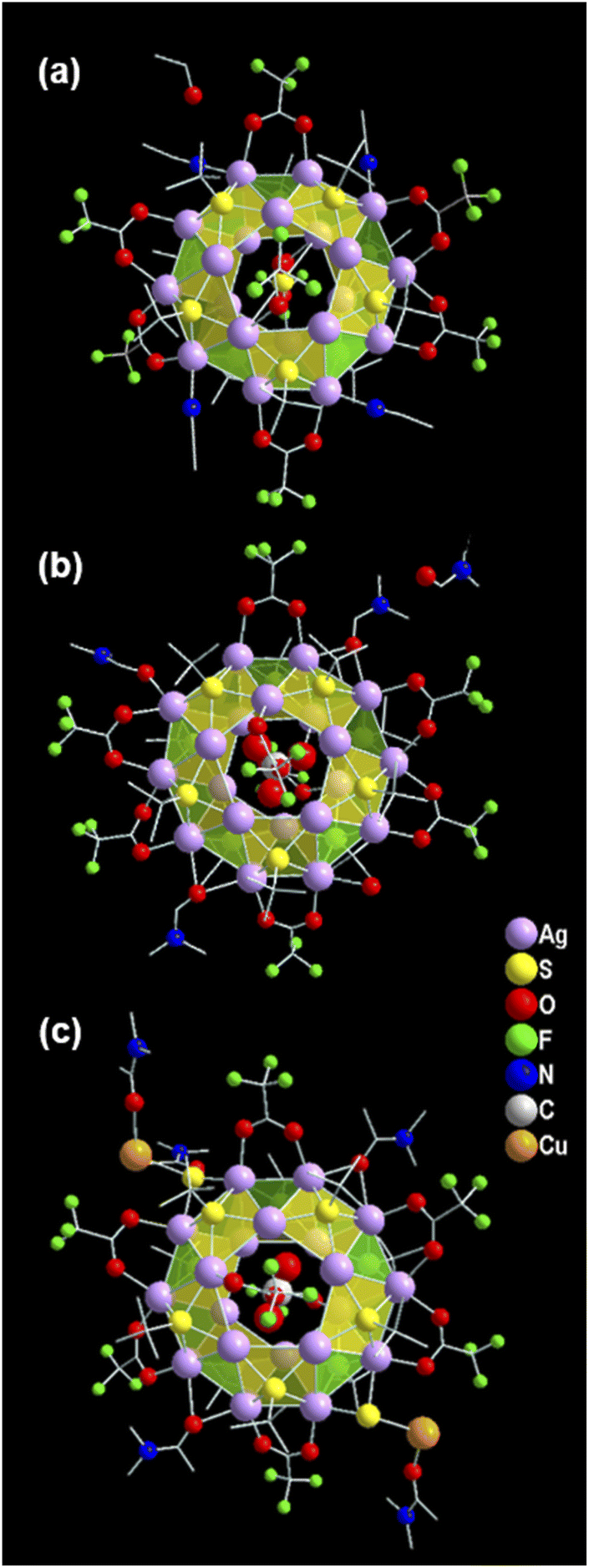 | ||
| Fig. 1 Crystal structural illustration of (a) NC 1, (b) NC 2 and (c) NC 3. | ||
 | ||
| Fig. 2 Detailed structural anatomy of NC 1, NC 2 and NC 3, respectively; (a), (b), (c) the pentagonal gyrobicupola geometry of the Ag20 skeleton with different templates inside; (d), (e), (f) connections of −StBu units on the square facets of each skeleton; (g), (h), (i) linking of CF3COO− as auxiliary ligands on the skeleton; (j), (k), (l) linking of respective solvents on the skeleton. In the case of NC 3 the two solvents are swapped by the two DMA-CuS− units. H atoms are omitted for clarity. | ||
Growth mechanism of the synthesized Ag(I) NCs
In spite of the synthesized NCs having a similar number of Ag(I) atoms and surface protecting ligands, they accommodate different templates in their core. As discussed earlier we haven't added any such reagents during the reactions, so the templates are only generated during the course of the reaction. The basicity of the solvent medium is important here for the selective formation of templates. Due to the least basic nature of the ethanol/acetonitrile (E/ACN) mixture, the scission of the C–S bond of the thiolate ligand is more facile which releases the excessive amount of S2− anions. The highly polarizable in situ generated S2− anions are then quickly encapsulated by the available Ag(I) ions in the solution to form Ag NCs that grow quickly with the surface protecting ligands covered on the outer surface, resulting in a neutral NC 1 structure. Most interestingly, the other two crystals grow with CO32− anions and CO2 as templates although there are no starting materials involved which can generate these two templates. As the reactions are performed in an ambient atmosphere, the incorporation of such species could be due to the presence of aerial CO2. In the case of CO32− templation, the preferable basic nature of the DMF solvent favours the conversion of CO2 to CO32−via hydration followed by deprotonation that allows prompt encapsulation of the Ag(I) ions with surface protecting ligands to form the neutral NCs. However, in the case of CO2 templated Ag(I) NCs, the highest basic nature of DMA (among the solvents used in these cases) favors the templation without any conversion of CO2. The slow encapsulation of neutral CO2 by the twenty Ag(I) atoms along with the eighteen surface protecting ligands results in a cationic cluster unit in this medium which is further stabilized by the addition of two DMA-CuS− units at the peripheral positions as a peripheral motif (–S–Cu-DMA) as described in Scheme 1. The successful identification of the unstable intermediate cationic cluster unit [CO2@Ag20(StBu)10(CF3COO)8(DMA)2]2+ through electrospray ionization mass spectrometry (ESI-MS) measurement when the Cu(I) precursor is absent, gives further leverage to explain the growth mechanism (Fig. S5†). The formation of this intermediate indicates that the addition of extra Ag–S at the peripheral position of the Ag20 core is not facile which may be due to the confined size of the highly steric outer surface. However, our strategical approach offsets such issues by selecting the smallest congener of group 11, which prefer to be at the peripheral position. In spite of the deliberate addition of Cu(I) during the NC 1 and NC 2 synthesis, we find a similar crystal that excludes the possibility of any other parameter being involved in linking the peripheral motifs other than charge neutrality. So, after the formation of this cationic cluster unit, the attachment of the specific peripheral motif units is necessary for neutralizing the overall cluster unit. However, such incorporation of peripheral motifs maintains the overall symmetry as all these three NCs have the same crystal system. | ||
| Scheme 1 Growth mechanism of the template-assisted alloy NC starts from the neutral template and self-assembly of silver atoms with the association of surface protecting groups. The intermediate exhibits a 2+ charge which is neutralized by the incorporation of two peripheral motif units at the site-specific symmetric position and NC 3 is finally obtained. | ||
So, it is clear that the formation of the Cu(I) incorporated Ag(I) NCs is also completely governed by the template. Generally, anionic templates are advantageous in assembling cationic metal atoms via balancing the local positive charges. We are hereby reporting for the first time that the neutral template also imposes similar types of assembling forces to control the size and shape of the Ag(I) NCs but at a slower rate as the synthesis of NC 3 takes the longest time in comparison to the synthesis of NC 1 and NC 2. So, the slow aggregation of the Ag(I) ions with the surface protecting ligands on the outer surface of CO2 is governed by argentophilic interactions and the coordination specificity. The formation of the Cu–S bond again leads to the slow breaking of some C–S bonds in the DMA medium at room temperature, facilitating the CO2 templation. The outer surface of the Cu(I) in Cu–S− is protected by DMA and gives the ultimate stability to the alloy NC. To prove the fixation of aerial CO2 in these two Ag(I) NCs i.e., NC 2 and NC 3, we have performed controlled reactions inside a desiccator under a N2 atmosphere that resulted in a yellowish solution mixture without any brown coloration or crystals even after three weeks which indicates the importance of the template source for the crystallization of the desired product. We have also deliberately added Na2CO3 as a source of CO32− which resulted in the quick precipitation of Ag2CO3 instead of the desired product. This observation clearly indicates the potential of the slow introduction of CO2 during the course of the reaction.
Various spectroscopic and microscopic characterization studies of the synthesized Ag(I) NCs
To verify the chemical composition of these as-synthesized NCs, ESI-MS was performed in positive mode (Fig. 3). Four prominent peaks are identified in the spectrum of NC 1. The peak corresponding to the m/z value at 4196.58 which is well-matched with the simulated isotropic patterns, is associated with [S@Ag20(StBu)10(CF3COO)8(CH3CN)4. EtOH + H+] and justifies the cluster molecular formula and its neutral state. All other associated fragments and the simulated isotropic patterns of these fragments are depicted in Fig. S6.† Similarly, NC 2 also exhibited four prominent peaks and the peak corresponding to the m/z value at 4324.77, is associated with [(CO3)@Ag20(StBu)10(CF3COO)8(DMF)3(H2O). DMF + H+] and also justifies the cluster molecular formula and its neutral state. All other associated fragments and the simulated isotropic patterns of these fragments are depicted in Fig. S7.† However, NC 3 exhibits three prominent peaks and among these the highest intense m/z peak at 4625 corresponds to [(CO2)@Ag20Cu2S2(StBu)10(CF3COO)8(DMA)4. DMA + H+] and validates the cluster molecular formula and its neutral state. All other associated fragments and the simulated isotropic patterns of these fragments are depicted in Fig. S8.† Some associated fragments of each NC directly confirm the presence of S2−, CO32− and CO2 which we also found as templates from the crystallographic data. The existence of the respective elements and their oxidation states in these three NCs is confirmed by X-ray photoelectron spectroscopic (XPS) survey spectra (Fig. S9†). The binding energy spectra of individual elements confirm that all the metallic atoms are in the +1 state (Fig. S10–S12†). The existence of Cu(I) in only NC 3 is further verified by XPS. The resulting XPS data determined the amount of Cu present in NC 3 showing an Ag/Cu ratio of 9.48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.16 which is consistent with the obtained crystal structure. The irreversible cyclic voltammogram of NC 3 features one oxidation event at E = 0.19 (vs. Ag wire) for Cu(I) → Cu(II) transformation (Fig. S13†). So, this oxidation event further indicates the presence of Cu(I) in NC 3. The energy dispersive X-ray spectra of each NC also corroborate with the XPS elemental analysis (Fig. S14†). The microscopic characterization of these single-crystals confirms their block shape (Fig. S15†). The elemental mapping of NC 3 using a transmission electron microscope confirms the peripheral position of the Cu atoms (Fig. S16†). The stability of these three NCs in the solution medium is verified by 1H and 19F NMR studies in CDCl3 resembling the reported individual components (Fig. S17–S20†).46
:1.16 which is consistent with the obtained crystal structure. The irreversible cyclic voltammogram of NC 3 features one oxidation event at E = 0.19 (vs. Ag wire) for Cu(I) → Cu(II) transformation (Fig. S13†). So, this oxidation event further indicates the presence of Cu(I) in NC 3. The energy dispersive X-ray spectra of each NC also corroborate with the XPS elemental analysis (Fig. S14†). The microscopic characterization of these single-crystals confirms their block shape (Fig. S15†). The elemental mapping of NC 3 using a transmission electron microscope confirms the peripheral position of the Cu atoms (Fig. S16†). The stability of these three NCs in the solution medium is verified by 1H and 19F NMR studies in CDCl3 resembling the reported individual components (Fig. S17–S20†).46
 | ||
| Fig. 3 ESI-MS spectra of NC 1, NC 2 and NC 3. (Inset) Experimental and simulated patterns (red color) of the parent NCs. | ||
Effect of alloying on optical absorption
To interpret the photophysical properties of these three NCs, we recorded UV-vis absorbance spectra of these three NCs in a DCM solution medium at room temperature. NC 1 and NC 2 exhibit similar types of monotonic decrease in absorbance data indicating a very similar electronic transition (Fig. 4a), whereas a prominent shoulder peak is observed at 350 nm for NC 3. Such a difference is mainly generated due to the alteration in the electronic structure induced by the alloying atoms. The TDDFT calculations reveal the influence of peripheral motif units in the frontier molecular orbital arrangements (FMOs) of NC 3 in the most prominent optical transition. The band at 3.51 eV (∼353 nm) of NC 3 refers to the most intense transition which originates from HOMO−7 to LUMO+1 (Fig. 4b). However, the most intense optical transitions originate from HOMO−14 to LUMO for NC 1 and HOMO−10 to LUMO for NC 2 (Fig. S21†). According to the frontier Kohn–Sham orbital population of NC 3, the HOMO−7 is constructed by the main contribution of the s and p-like states of the ligands along with s, p and d-like states of the peripheral motif whereas the unoccupied orbitals are constructed by the contribution from the s, p and d-like states of the core (Fig. 4c). In contrast, the occupied orbitals are constructed only by the major contributions of the s and p-like states of the ligands and the unoccupied orbitals are constructed by the major contribution that forms the core for NC 1 and NC 2. So, the incorporation of peripheral motif units directly influences the FMO through the hybridization of different states. It is also identified that with the attachment of electron-donating peripheral motif units to the core, the occupied energy levels of the alloy NC elevate in an upward direction, while unoccupied energy levels remain the same. Hence, the ligand to metal charge transition process in NC 3 is directly induced by the significant difference in the orbital arrangement in contrast to others. Thus, the appearance of a prominent shoulder peak at a lower energy in the enlarged orbital separation of NC 1 and NC 2 is not facile. This large red-shift in the UV-vis absorbance of NC 3 is also consistent with its size parameter. Due to the incorporation of the peripheral motif units on the Ag20 core, NC 3 exhibits the highest cluster size (metal–metal distance, Fig. S22†) and contains the highest number of metal atoms inside the cluster in comparison with NC 1 and NC 2.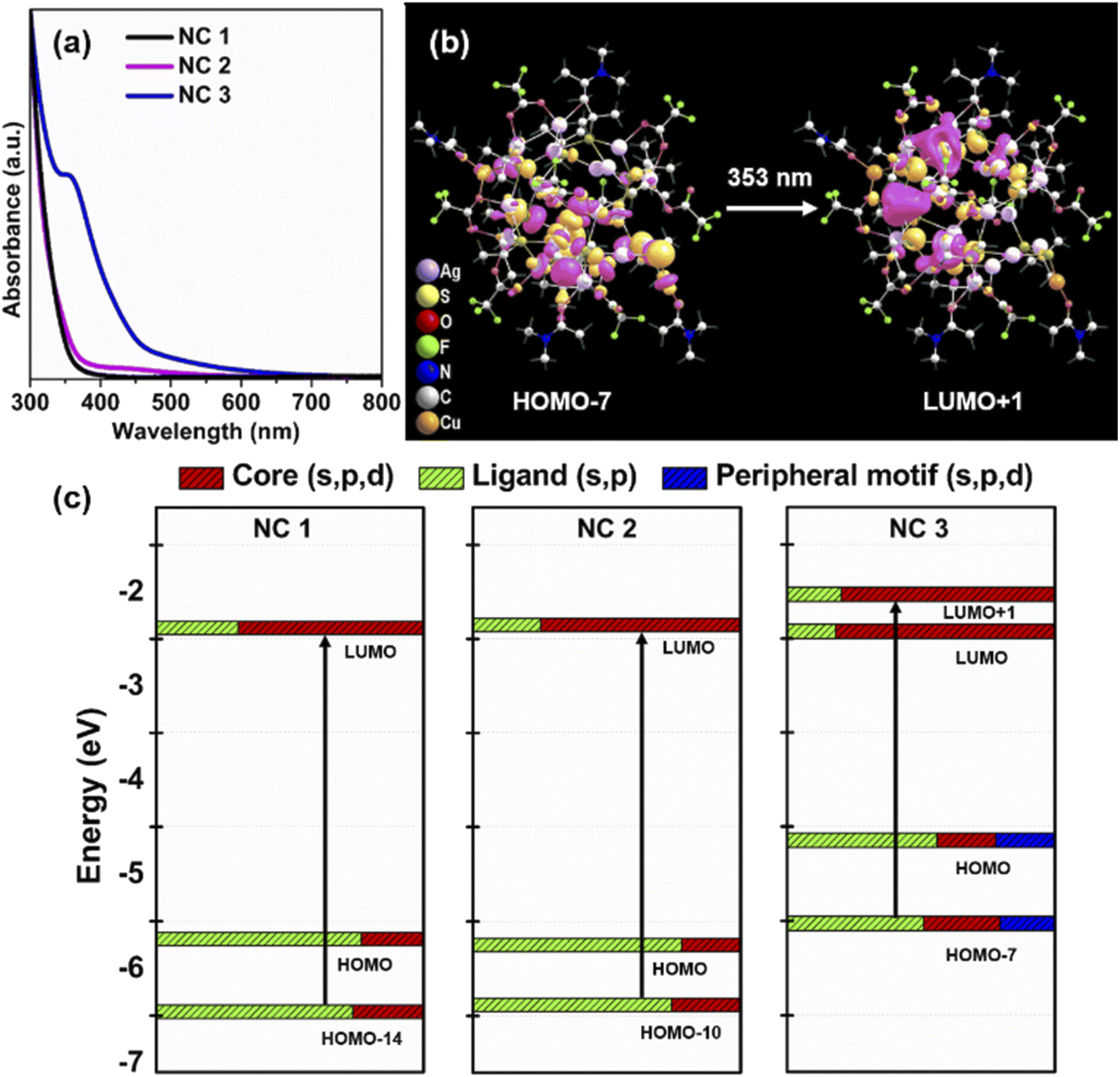 | ||
| Fig. 4 (a) UV-vis absorbance spectra of NC 1, NC 2 and NC 3 dissolved in DCM, (b) the associated frontier-molecular orbitals of NC 3 corresponding to the transition at 353 nm and (c) Kohn–Sham diagram of each NC and the associated transitions. | ||
Effect of alloying on luminescence
The luminescence properties of NCs are largely affected by the electronic structure.2 Therefore, we further compare the luminescence properties of these three NCs in the DCM solution medium at room temperature. In general anion template mono-layered Ag(I) core-based NCs are not emissive in both solution and solid-states at room-temperature (298 K).20–24 Here also we could not identify the steady-state PL properties of NC 1 and NC 2 in the DCM medium. However, after irradiation at 353 nm, strong photoluminescence is detected for NC 3 with an emission maximum at 402 nm (Fig. 5a) and an impressive relative quantum yield of 0.21. We identify that the mono exponentially fitted emission lifetime of NC 3 is 12 ns (Fig. S23†). TDDFT analysis suggests that this violet emission is mainly attributed to the cluster-centered charge transfer between the Ag(I) core and the combination of Cu(I) incorporated peripheral motif along with the ligand system. However, in the case of NC 1 and NC 2 the involvement of such a Ag(I) core and ligand environment without peripheral motif units doesn't generate such emission. So, it is evident that the incorporation of peripheral motifs in the FMO arrangement plays a crucial role in generating such violet emission in NC 3. It is worth noting that previously reported Ag(I)-based NCs only exhibited PL emission at very low temperature which is attributed to the restricted intramolecular motion through the confined vibration of the cluster node.22,23 But at room temperature, the dominative thermally activated non-radiative decay stimulates the molecular vibrations that result in the disappearance of photoluminescence.47 In contrast, the intense violet emission of NC 3 at room temperature appears due to the additional rigidity of the structure imposed by the two DMA-CuS− units. The introduction of DMA-CuS− peripheral motifs resulted in stronger Ag–S interaction in the Ag20 core that enforces more rigidity in the skeleton and reduces the probability of non-radiative transition in the excited state by lowering the intramolecular vibration. The confinement of the intramolecular vibrations is envisioned by Bader's quantum theory of atoms in molecules (QTAIM) calculations.48,49 The highest bond critical points (ρ(rc) = 0.016, and 0.056) and Laplacian of electron density [∇2ρ(rc) = 0.051, and 0.148] in between Ag⋯Ag and Ag⋯S in NC 3 suggest very strong non-covalent interactions (Fig. 5b and S24†) and this may confine the Cu atoms in the peripheral positions instead of the Ag sites in the Ag20 core. As a result of this non-covalent interaction, the resultant bonding character and the rigidity of the cluster node increase that suppresses the non-radiative transition pathway. Therefore, the template-assisted alloying process imposes optimum rigidity in the Ag(I)-based NC skeleton along with the different FMO arrangements which could effectively turn on its room-temperature luminescence. A monotonic decrease in steady-state PL intensities is observed after increasing the temperature starting from 273 K without any shift in the emission peak position (Fig. S25†). The reduction in the quantum yield and associated emission lifetime at 308 K suggest the enhancement of thermally activated non-radiative pathways due to the enhancement in the surface molecular vibration of NC 3 at a higher temperature (Table S4†). We have calculated the radiative and non-radiative rates and identified the changes (Table S4†). The perfect linear relationship between the PL intensities and temperature will provide the scope of this NC 3 for luminescent thermometer design in the near future.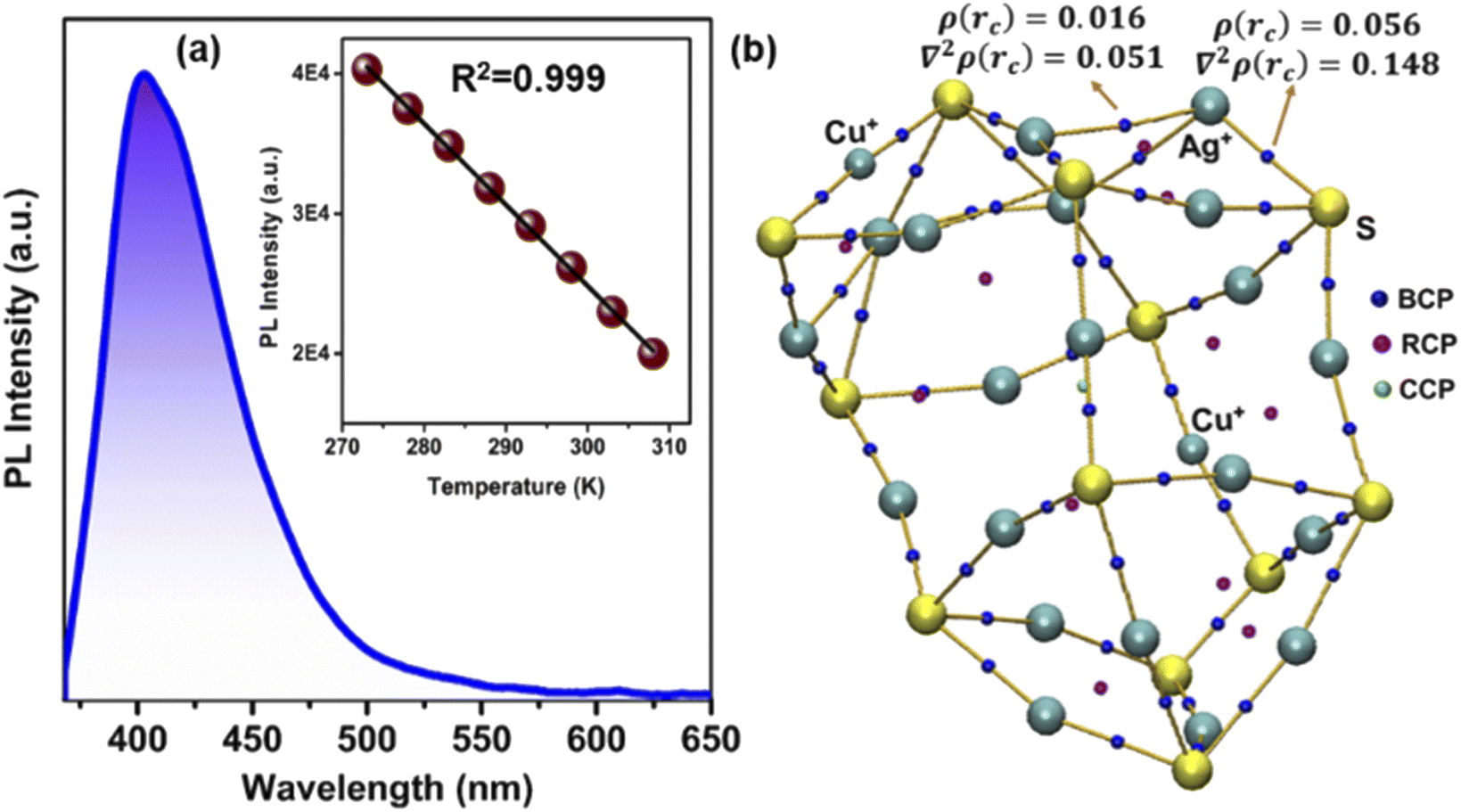 | ||
| Fig. 5 (a) The PL spectrum of NC 3; inset showing the linear relationship between the changes in PL intensities with respect to temperature and (b) QTAIM molecular plot of the bond critical point (BCP), ring critical point (RCP) and cage critical point (CCP) of NC 3. For clarity, ligands were removed from the structure. | ||
Effect of alloying on single-crystal conductivity
The solid-state optical diffuse reflectance spectra of each NC are also measured here. The estimated bandgap of NC 3 (1.26 eV) is lower than that of NC 1 (1.89 eV) and NC 2 (1.87 eV), respectively (Fig. S26†). Interestingly, after 1 month of synthesis we obtained exactly the same bandgap of NC 3 but a deviation was observed for NC 2 and NC 1 confirming the higher stability of NC 3 (Fig. S27†). The projected density of state (PDOS) calculations of each NC reveal that the incorporation of the peripheral motifs into the cluster significantly influences the spatial separation of the electron density distribution (Fig. 6a). The electron-donating properties of the peripheral motif units extend the valence band region of NC 3 towards the Fermi energy level while keeping the conduction band unaffected which results in the reduction of the bandgap. The calculated bandgaps of the NCs are 2.08 eV (NC 1), 2.05 eV (NC 2) and 1.18 eV (NC 3) which are well-matched with the experimental values. Now, this sharp decrease in the bandgap with the shift of the valence band edge may have profound effects on the charge transportation characteristics.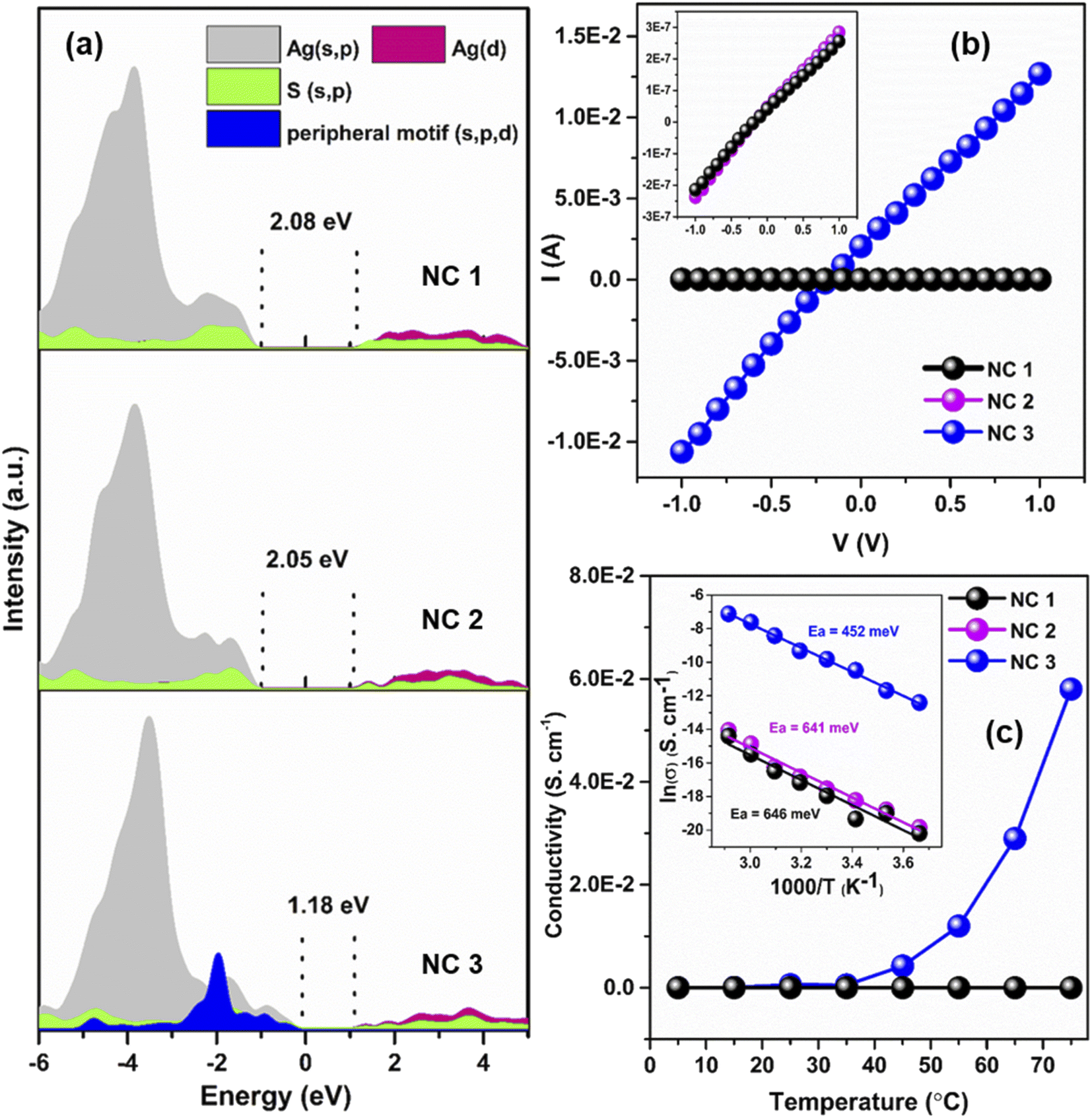 | ||
| Fig. 6 (a) PDOS analysis of each NC, (b) I–V plot of each NC, inset showing the zoomed version of NC 1 and NC 2 and (c) temperature-dependent conductivity of each NC, inset showing the activation energy of each NC. | ||
To evaluate the charge transport properties in these NCs, the single-crystal conductivity of each crystal is measured here. The representative I–V characteristic curves are shown in Fig. 6b. NC 3 shows the highest single-crystal conductivity of 6.1 × 10−4 S cm−1 which is ∼104 times higher than that of NC 1 and NC 2, respectively (Table S5†). So, the incorporation of electron-donating peripheral motif units influences the band structure of the NC and decreases the bandgap value which can ensure better charge carrier transport in NC 3. To get a more detailed picture we have identified the difference in intercluster distances. The incorporation of Cu atoms precisely at the peripheral position of the Ag20 moiety leads to a lowering of the intercluster distances between the adjacent NCs. Fig. S28† displays the superior intercluster distances of NC 3 in both end-to-end and face-to-face directions in comparison to NC 1, which results in faster charge transportation.
The magnitude of the bandgap of each NC indicates its semiconducting nature. The semiconducting properties are further verified by temperature-dependent (T = 278–348 K) conductivity measurement. The results show that conductivity increases with the increase of temperature as thermal energy increases the number of charge carriers and this is consistent with all three NCs (Fig. 6c). The highest single-crystal conductivity of 5.8 × 10−2 S cm−1 is achieved by NC 3 at 348 K. The measured temperature reliance of these three NCs is followed by an Arrhenius-type temperature-activated hopping pathway. The presence of the insulating surface protecting ligands and the absence of any linker between the two cluster units inevitably invites a hopping charge transport mechanism. The fitting of electrical conductivity as a function of the reciprocal of temperature and the activation energies of each NC are estimated. The results suggest that NC 3 requires ∼30% less activation energy to transport the charge carriers in comparison to NC 1 and NC 2 (Fig. 6c inset). Temperature-activated charge carrier transport in single-crystals solely depends on the Coulomb charging energy as the associated energetic disorders are negligible for single-crystals.34,50,51 The Coulomb charging energy signifies the energy required for the addition or removal of an additional charge carrier to the NC. However, this energy is inversely proportional to the radius of the cluster. As the Cu⋯Cu distance in NC 3 is much higher than the Ag⋯Ag distances of the other two NCs (Fig. S22†), it indicates the lowest Coulomb charging energy. Now due to this lowest Coulomb charging energy, the temperature-activated charge carrier transport will be more favorable for NC 3 which coordinates with the lower activation energy where the nearest neighbor hopping is prevailing. Thus, as a combination of the above-mentioned factors, NC 3 shows an enhancement in electronic coupling which is directly associated with the incorporation of Cu atoms in the Ag(I) cluster node through the template-assisted alloying process. These findings correlate with the structure and electronic properties of atomically precise NCs that would surely direct any future endeavors to further increase electronic coupling through decreasing intercluster distance.
Conclusion
The current research provides a paradigm to counteract the bottleneck in the field of atomically precise anion template Ag(I) NC synthesis. The excessive dependence of the size, shape and the charge of the template on the Ag(I) core design is now becoming obsolete as the current synthetic protocols provide a great control on the self-assembly of Ag(I) atoms. However, the results clearly elucidate the different roles of the template charge on total cluster neutrality and provide the scope for alloying on the peripheral surface of the Ag(I) core. This novel strategy of attaching peripheral motifs turns on the room-temperature emission by confining the thermally activated vibrations of the Ag(I) core through rigid structural architecture and different FMO arrangements. The interactive non-covalent interactions are more prominent here for rigidifying the Ag20 core. Furthermore, the symmetric peripheral position of the alloying metal atoms and the modified bandgap between the hybridized orbital states facilitate the intercluster charge carrier transport properties. The new synthetic strategy for the preparation of NC 3 involving both solvent and surface reconstruction provides a new pathway for the synthesis of other NCs with fascinating structures and property design. This can create more attraction to design new synthetic procedures, property exploration, and application of alloy/bimetallic clusters with a neutral CO2 template.Experimental section
Synthesis of NC 1
0.1 mmol of the [AgStBu]n complex and 0.1 mmol of CF3COOAg were treated in 4 mL (1:1) ethanol/acetonitrile at room temperature until a clear solution was obtained. After three days block-shaped yellow-colored crystals were obtained from the clear solution mixture with a yield of 62% (based on Ag) in a dark environment.
Synthesis of NC 2
A similar procedure was used for NC 2 synthesis to that previously mentioned only by replacing the solvent with DMF. The block-shaped orange-colored crystals were obtained after five days with a yield of 61% (based on Ag).Synthesis of NC 3
In a similar way, the 0.1 mmol of [AgStBu]n complex and 0.1 mmol of CF3COOAg were treated in 4 mL DMA at room temperature until a clear solution was obtained. After this, 0.05 mmol of Cu(CH3CN)4BF4 was added and kept stirring for another 30 min. The resultant clear solution was then kept for crystallization in a similar manner. After ten days, block-shaped deep red-colored crystals were obtained from the resultant brown-colored solution with a yield of 54% (based on Ag).It is important to note that without the addition of Cu(CH3CN)4BF4 in the NC 3 synthesis protocol, we obtained a yellowish colored solution. Even after three weeks no crystals were obtained from this solution.
We have also deliberately added Cu(CH3CN)4BF4 during the synthesis of NC 1 and NC 2 but ultimately we have obtained similar types of crystals having similar cell parameters.
The precursor, AgStBu complex was synthesized by treating equimolar amounts of AgNO3 and HStBu with Et3N in methanol.46
Data availability
The ESI† contains experimental details, computational details, crystal structure parameters, luminescence parameters, electrical conductivity and associated parameters, doughnut-like Ag20 core, reduced density gradient (RDG) isosurface of argentophilic interaction of the three NCs, three planes of the Ag20 skeleton, connection among the three planes, ESI-MS data of the intermediate, ESI-MS fragments of the three NCs, XPS survey spectra of three NCs, deconvoluted XPS spectra of the three NCs, cyclic-voltammetry plot of NC 3, EDS spectra of the three NCs, SEM micrographs and optical microscope image of the three NCs, TEM image of NC 3 and EDS analysis, 1H NMR of the three NCs, 19F NMR of the three NCs, FMO of NC 1 and NC 2, metal–metal distances in the three NCs, emission lifetime of NC 3, QTAIM molecular plots of the three NCs, temperature-dependent PL emission of NC 3, solid-state band gap of each NC, edge-to-edge and face-to-face distance of NC 1 and NC 3 and references.Author contributions
A. K. D. and S. B. performed the synthesis, characterization and data interpretation. S. S. M. and B. P. performed the theoretical calculations. S. M conceived the project, analysed the data and was involved in manuscript preparation. All authors discussed the results and were involved in manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. B. acknowledges the NPDF fund from SERB (PDF/2020/001085). We are thankful to Mrs Saniya Gratious for her help in ESI-MS measurement.References
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2019, 120, 526–622 CrossRef.
- S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2014, 47, 816–824 CrossRef CAS PubMed.
- S. Maity, D. Bain and A. Patra, Nanoscale, 2019, 11, 22685–22723 RSC.
- J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS.
- J. Yang and R. Jin, ACS Mater. Lett., 2019, 1, 482–489 CrossRef CAS.
- J. Yang and R. Jin, J. Phys. Chem. C, 2020, 125, 2619–2625 CrossRef.
- T. Kawawaki, A. Ebina, Y. Hosokawa, S. Ozaki, D. Suzuki, S. Hossain and Y. Negishi, Small, 2021, 17, 2005328 CrossRef CAS PubMed.
- H. Hirai, S. Ito, S. Takano, K. Koyasu and T. Tsukuda, Chem. Sci., 2020, 11, 12233–12248 RSC.
- Q. Yao, Z. Wu, Z. Liu, Y. Lin, X. Yuan and J. Xie, Chem. Sci., 2021, 12, 99–127 RSC.
- C. Sun, B. K. Teo, C. Deng, J. Lin, G.-G. Luo, C.-H. Tung and D. Sun, Coord. Chem. Rev., 2021, 427, 213576 CrossRef CAS.
- Y. Jin, C. Zhang, X.-Y. Dong, S.-Q. Zang and T. C. Mak, Chem. Soc. Rev., 2021, 50, 2297–2319 RSC.
- Q.-M. Wang, Y.-M. Lin and K.-G. Liu, Acc. Chem. Res., 2015, 48, 1570–1579 CrossRef CAS PubMed.
- R. Vilar, Angew. Chem., Int. Ed., 2003, 42, 1460–1477 CrossRef CAS PubMed.
- K. Bowman-James, Acc. Chem. Res., 2005, 38, 671–678 CrossRef CAS PubMed.
- C. Liu, H.-W. Chang, C.-S. Fang, B. Sarkar and J.-C. Wang, Chem. Commun., 2010, 46, 4571–4573 RSC.
- M. J. Alhilaly, R.-W. Huang, R. Naphade, B. Alamer, M. N. Hedhili, A.-H. Emwas, P. Maity, J. Yin, A. Shkurenko, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2019, 141, 9585–9592 CrossRef CAS PubMed.
- A. K. Das, S. Biswas, S. S. Manna, B. Pathak and S. Mandal, Chem. Sci., 2022, 13, 8355–8364 RSC.
- D. Sun, H. Wang, H.-F. Lu, S.-Y. Feng, Z.-W. Zhang, G.-X. Sun and D.-F. Sun, Dalton Trans., 2013, 42, 6281–6284 RSC.
- S. Yuan, Y.-K. Deng, X.-P. Wang and D. Sun, New J. Chem., 2013, 37, 2973–2977 RSC.
- K. Zhou, C. Qin, X.-L. Wang, K.-Z. Shao, L.-K. Yan and Z.-M. Su, CrystEngComm, 2014, 16, 7860–7864 RSC.
- G. Li, Z. Lei and Q.-M. Wang, J. Am. Chem. Soc., 2010, 132, 17678–17679 CrossRef CAS PubMed.
- S. Biswas, A. K. Das, A. C. Reber, S. Biswas, S. Bhandary, V. B. Kamble, S. N. Khanna and S. Mandal, Nano Lett., 2022, 22, 3721–3727 CrossRef CAS PubMed.
- J.-H. Liao, H.-W. Chang, H.-C. You, C.-S. Fang and C. Liu, Inorg. Chem., 2011, 50, 2070–2072 CrossRef CAS PubMed.
- K. Zhou, C. Qin, H.-B. Li, L.-K. Yan, X.-L. Wang, G.-G. Shan, Z.-M. Su, C. Xu and X.-L. Wang, Chem. Commun., 2012, 48, 5844–5846 RSC.
- S. Li, X.-S. Du, B. Li, J.-Y. Wang, G.-P. Li, G.-G. Gao and S.-Q. Zang, J. Am. Chem. Soc., 2018, 140, 594–597 CrossRef CAS PubMed.
- T. Kawawaki, Y. Imai, D. Suzuki, S. Kato, I. Kobayashi, T. Suzuki, R. Kaneko, S. Hossain and Y. Negishi, Chem.–Eur. J., 2020, 26, 16150–16193 CrossRef CAS PubMed.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- W.-Q. Shi, Z.-J. Guan, J.-J. Li, X.-S. Han and Q.-M. Wang, Chem. Sci., 2022, 13, 5148–5154 RSC.
- M. Van Der Linden, A. J. Van Bunningen, L. Amidani, M. Bransen, H. Elnaggar, P. Glatzel, A. Meijerink and F. M. De Groot, ACS Nano, 2018, 12, 12751–12760 CrossRef CAS.
- S. Wang, Q. Li, X. Kang and M. Zhu, Acc. Chem. Res., 2018, 51, 2784–2792 CrossRef CAS PubMed.
- T. U. Connell, S. Sandanayake, G. N. Khairallah, J. M. White, A. Richard, P. S. Donnelly and S. J. Williams, Dalton Trans., 2013, 42, 4903–4907 RSC.
- X. Kang, M. Zhou, S. Wang, S. Jin, G. Sun, M. Zhu and R. Jin, Chem. Sci., 2017, 8, 2581–2587 RSC.
- F. Fetzer, A. Maier, M. Hodas, O. Geladari, K. Braun, A. J. Meixner, F. Schreiber, A. Schnepf and M. Scheele, Nat. Commun., 2020, 11, 6188 CrossRef CAS PubMed.
- P. Yuan, R. Zhang, E. Selenius, P. Ruan, Y. Yao, Y. Zhou, S. Malola, H. Häkkinen, B. K. Teo and Y. Cao, Nat. Commun., 2020, 11, 2229 CrossRef CAS.
- C. Bansal, S. G. Praveen, J. T. T. Kumaran and A. Chatterjee, Sci. Rep., 2015, 5, 7685 CrossRef PubMed.
- J.-P. Choi and R. W. Murray, J. Am. Chem. Soc., 2006, 128, 10496–10502 CrossRef CAS PubMed.
- M. Galchenko, A. Black, L. Heymann and C. Klinke, Adv. Mater., 2019, 31, 1900684 CrossRef.
- A. K. Das, S. Biswas, S. S. Manna, B. Pathak and S. Mandal, Inorg. Chem., 2021, 60, 18234–18241 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- N. W. Johnson, Can. J. Math., 1966, 18, 169–200 CrossRef.
- A. K. Das, S. Biswas, A. Thomas, S. Paul, A. S. Nair, B. Pathak, M. S. Singh and S. Mandal, Mater. Chem. Front., 2021, 5, 8380–8386 RSC.
- J.-S. Yang, Z. Han, X.-Y. Dong, P. Luo, H.-L. Mo and S.-Q. Zang, Angew. Chem., Int. Ed., 2020, 59, 11898–11902 CrossRef CAS PubMed.
- T. Lu and F. Chen, Acta Chim. Sin., 2011, 69, 2393–2406 CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef CAS.
- G. Schmid and U. Simon, Chem. Commun., 2005, 697–710 RSC.
- A. Zabet-Khosousi and A.-A. Dhirani, Chem. Rev., 2008, 108, 4072–4124 CrossRef CAS PubMed.
Footnotes |
| † CCDC 2183686–2183688. For crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc04390d |
| ‡ S. B. and A. K. D. contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |