
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReassessment of N2 activation by low-valent Ti-amide complexes: a remarkable side-on bridged bis-N2 adduct is actually an arene adduct†
Daniel N.
Huh
 a,
Ross F.
Koby
a,
Zoe E.
Stuart
a,
Rachel J.
Dunscomb
a,
Nathan D.
Schley
*b and
Ian A.
Tonks
*a
a,
Ross F.
Koby
a,
Zoe E.
Stuart
a,
Rachel J.
Dunscomb
a,
Nathan D.
Schley
*b and
Ian A.
Tonks
*a
aDepartment of Chemistry, University of Minnesota – Twin Cities, Minneapolis, MN 55455, USA. E-mail: itonks@umn.edu
bDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235, USA. E-mail: nathan.schley@vanderbilt.edu
First published on 14th October 2022
Abstract
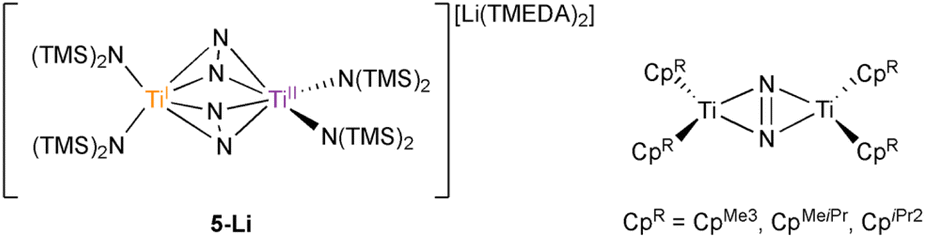
The complex {(TMEDA)2Li}{[Ti(N(TMS)2)2]2(μ-η2:η2-N2)2} (5-Li) is the only transition metal N2 complex ever reported with two side-on N2 adducts. In this report, the similarity of 5-Li to a new inverse sandwich toluene adduct {(PhMe)K}{[Ti(N(TMS)2)2]2(μ-PhMe)} (6-K) necessitated a re-examination of the structure of 5-Li. Through a reassessment of the original disordered crystal data of 5-Li and new independent syntheses brought about through revisitation of the original reaction conditions, 5-Li has been re-assigned as an inverse sandwich toluene adduct, {(TMEDA)2Li}{[Ti(N(TMS)2)2]2(μ-PhMe)} (6-Li). The original crystal data could be fitted almost equally well to structural solutions as either 5-Li or 6-Li, and this study highlights the importance of a holistic examination of modeled data and the need for secondary/complementary analytical methods in paramagnetic inorganic syntheses, especially when presenting unique and unexpected results. In addition, further examination of reduction reactions of Ti[N(TMS)2]3 and [(TMS)2N]2TiCl(THF) in the presence of KC8 revealed rich solvent- and counterion-dependent chemistry, including several degrees of N2 activation (bridging nitride complexes, terminal bridging N2 complexes) as well as ligand C–H activation.
Introduction
N2 activation by reduced metal complexes is fundamentally important in understanding how to develop molecular alternatives to the Haber–Bosch process. Myriad N2 complexes exist across the periodic table, and Ti is no exception:1 there are examples of end-on and side-on bridging N2 structures, including fully N–N cleaved nitrides from N2.2–7 In some instances, Ti mediated N2 reduction has also been used to incorporate nitrogen into organic molecules.8 Recent examples of catalytic N2 reduction with molecular Ti complexes2 motivate our renewed interest in further developing the chemistry of low-valent Ti complexes in the context of small molecule activation.Low-valent Ti complexes can also engender powerful reductive organic transformations,9–11 such as C(sp3)–H and thiophene oxidative additions,12,13 arene hydrogenation,14 and diazene disproportionation.15 Our group is interested in the synthesis and isolation of various low-valent Ti arene and (hetero)arene adducts, which have been invoked in the catalytic synthesis of pyrroles15–17via nitrene transfer through a formal TiII/TiIV redox reaction.18 To date, a variety of TiII arene complexes have been isolated (Fig. 1A).19 For example, Arnold reported a coordinated Ti–toluene complex 1 supported by a N,N′-bis(trimethylsilyl)benzamidinate ligand20 while Ozerov reported a Ti-arene complex 2 supported by a p-tert-butyl calix[4]arene ligand.21 Inverse-sandwich Ti complexes with bridging arenes have also been characterized, as demonstrated by a Cp*Ti adduct 3 reported by Mach22 and a tripyrrole Ti adduct 4 reported by Gambarotta and Budzelaar.23 Multidentate intramolecularly bound arene examples have also been reported.12,24
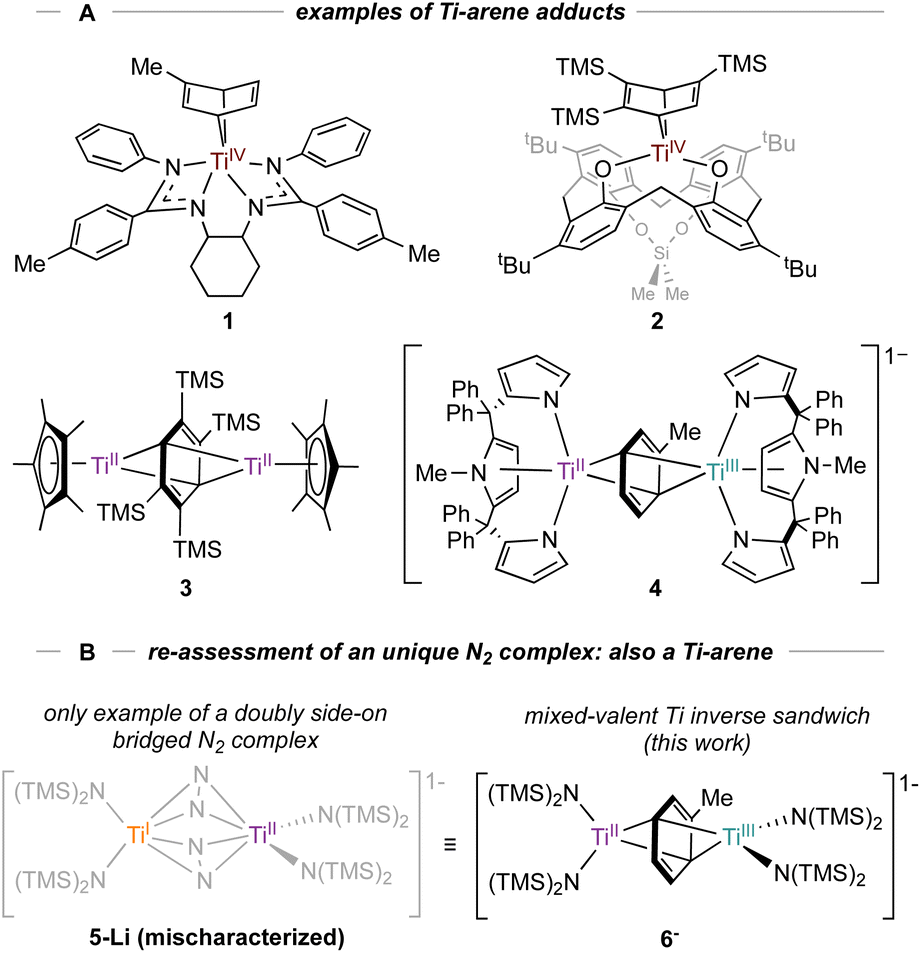 | ||
| Fig. 1 A: Examples of monometallic (1–2) and bimetallic coordinated arenes (3–4). B: New examples of inverse sandwich toluene complexes (6-K and 6-Li) indicate that a reported Ti2–(N2)2 complex 5-Li was originally mischaracterized.26 Countercations omitted for clarity. | ||
Motivated by these examples, we investigated reduction reactions involving a well-known Ti-amido complex, TiIII[N(TMS)2]3,25 envisioning that the electron-rich, sterically-encumbered (TMS)2N platform may readily bind free arenes. Herein, we report the synthesis of two inverse sandwich toluene adducts of low-valent Ti {(PhMe)K}{[Ti(N(TMS)2)2]2(μ-PhMe)} (6-K) and {(TMEDA)2Li}{[Ti(N(TMS)2)2]2(μ-PhMe)} (6-Li) (Fig. 1B, right). The structures of 6-K and 6-Li are surprisingly similar to a previous report of a remarkable example of a doubly side-on bridged N2 complex, 5-Li (Fig. 1B, left).265-Li was the landmark first report (30 years ago) of a side-on bridged Ti–N2 complex and has been the only example of a transition metal complex with two side-on bound bridging N2 ligands reported to date. Structural reassessment of 5-Li has led us to conclude that this compound was originally mischaracterized as an N2 adduct and is instead also an inverse sandwich adduct of toluene (6-Li). Further exploration of the reduction chemistry of TiIII[N(TMS)2]3 and related low-valent Ti amides under an N2 atmosphere demonstrate a suite of new N2 reduction products, although no side-on N2 adducts.
Results and discussion
TiIII[N(TMS)2]3 was reduced by KC8 in toluene in an effort to isolate a reduced Ti–toluene complex (Fig. 2). When a concentrated toluene solution was passed through a KC8 column,27 the reaction mixture immediately turned near black yielding the mixed-valent inverse sandwich complex {(PhMe)K}{[Ti(N(TMS)2)2]2(μ-PhMe)} 6-K upon crystallization at −35 °C (40% isolated yield).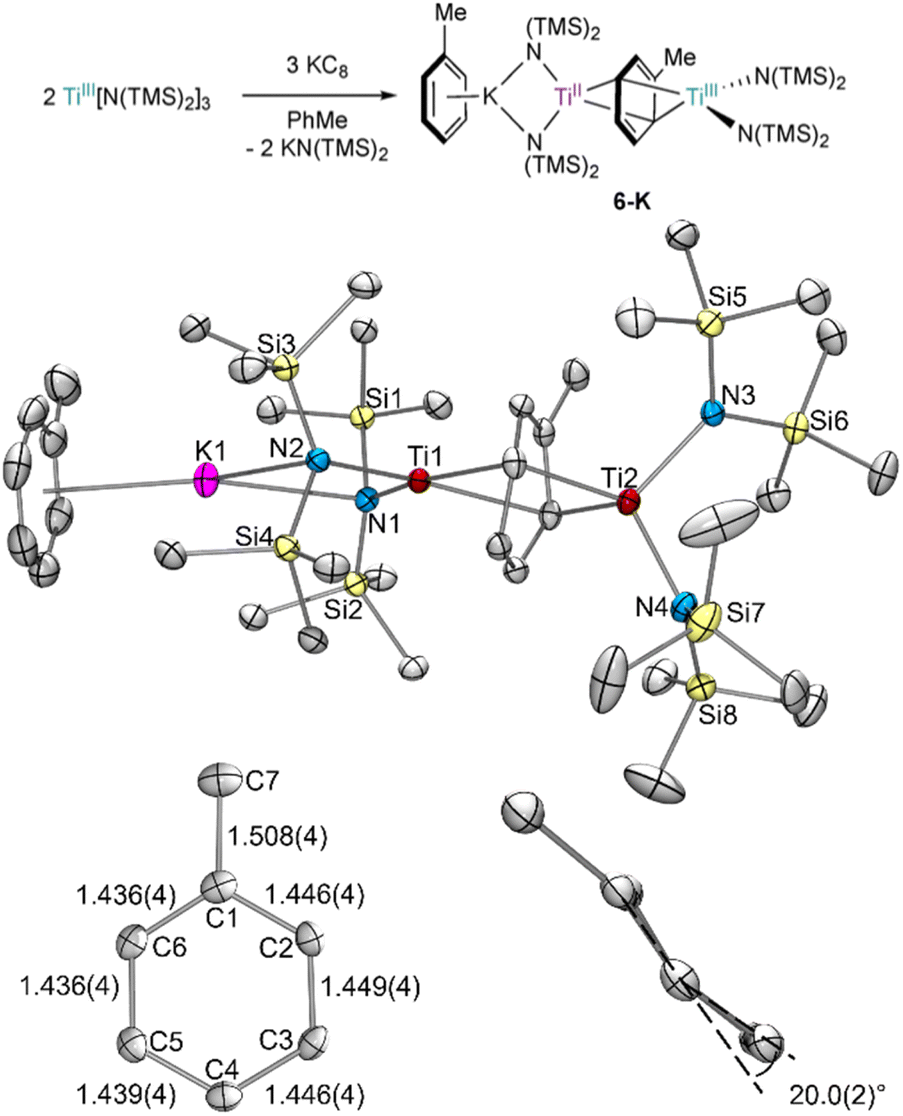 | ||
| Fig. 2 Top: synthesis of 6-K by KC8 reduction of TiIII[N(TMS)2]3 in toluene. Bottom: crystal structure of 6-K with displacement ellipsoids drawn at the 50% probability level, including zoom-in of the bridging toluene showing the dihedral angle across the C5–C6–C1–C2 and C2–C3–C4–C5 planes. Hydrogen atoms and a disordered TMS group were omitted for clarity. Selected bond distance ranges (Å) and angles (°) are shown in Table S15.† | ||
The X-ray crystal structure of 6-K is shown in Fig. 2 along with relevant toluene C–C distances and angles. A comparison of 6-K to Ti-arene complexes 1–4 is also shown in Table S15.† In the extreme electronic pictures, 6-K could be described as a TiII/TiIII complex bridged by a toluene dianion (Fig. 3, left), a TiI/TiII complex bridged by a neutral toluene (Fig. 3, right), or a TiIII/TiIV complex bridged by a toluene tetraanion;28 any of which could be consistent with the solution-state Evans method magnetic moment of 6-K 1.67μB (overall S = ½). The structural metrics indicated that 6-K is best described as a toluene dianion: the C–C bond lengths of 6-K are all remarkably elongated (1.436(4)–1.449(4) Å) and the toluene is significantly folded (dihedral angle = 20.0(2)°), indicating a loss of aromatic character, and ruling out the aromatic neutral or tetraanion toluene ligand descriptions. A picture of the formal oxidation states can also be distinguished by the Ti–N(TMS)2 bond lengths. The TiIII–N(TMS)2 moiety has Ti–N lengths of 2.028(2) and 2.030(2) Å which is consistent with Ti–N(TMS)2 bond lengths in 18 other TiIII compounds (1.929(3) to 2.057(1) Å).25,26,29–37 The TiII–N(TMS)2 bearing the (PhMe)K moiety has longer Ti–N(TMS)2 bond lengths which range from 2.129(2) to 2.146(2) Å which is expected for an ion with a lower oxidation state. A similar difference in Ti–N lengths is been found in mixed-valent TiII/TiIII inverse sandwich 4.23
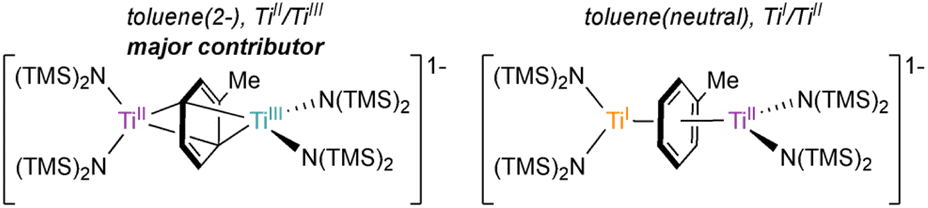 | ||
| Fig. 3 Electronic representation of 6-K as either TiII/TiIII bridged by a toluene dianion (left) or TiI/TiII bridged by a neutral toluene (right). | ||
The synthesis of 6-K caused us to re-investigate the very similar formally TiII/TiIII complex, 5-Li, which was reported to feature a bimetallic Ti–(μ-η2:η2-N2)2–Ti core containing two side-on bridging N2 moieties (Fig. 4, left).26 In the context of 6-K, several features of 5-Li are peculiar. First, the N–N bond lengths in 5-Li (1.379(21) Å) are substantially longer than 37 of 39 reported Ti2–N2 complexes (end-on range: 1.165(5)–1.315(3); side-on range: 1.216(5)–1.226(5)),1–8,26,33,38–49 although there is one other example of a highly activated end-on Ti2–N2 TrenTMS complex with an N–N distance of 1.461(8) Å.2a Second, while there are 3 other side-on Ti2–N2 complexes reported (Fig. 4, right),5,75 is the only reported doubly side-on complex, and in fact the only example of any transition metal containing 2 side-on bridging N2 ligands. These two N2 moieties were modelled as disordered over two crystallographically independent nitrogen-atom positions, with a further four positions being generated by symmetry. A close look at the six atomic sites found between the two Ti centers reveals an inverse sandwich motif that is strikingly reminiscent of the structure of 6-K, and a direct overlay of the Ti coordination spheres of 5-Li (maroon) and 6-K (teal) reveal significant overlap (Fig. 5). These features indicate that 5-Li may have been a mis-assigned arene adduct analogous to 6-K; in fact, 5-Li was synthesized in toluene.
 | ||
| Fig. 4 Reported examples of bimetallic Ti complexes bridged by a side-on N2 ligand. Left: double side-on Ti–(N2)2–Ti, 5-Li, complex.26 Right: single side-on N2 titanocene complexes.5,7 | ||
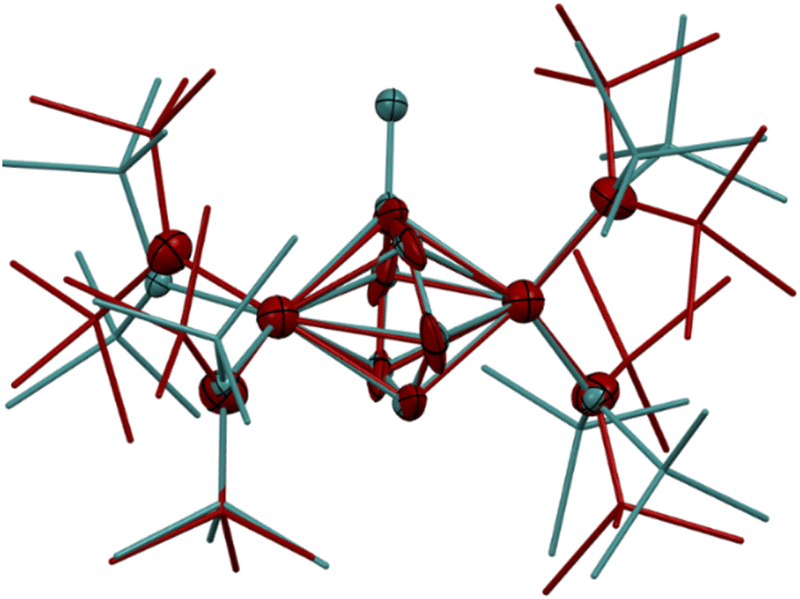 | ||
| Fig. 5 Overlay of the crystal structures of 5-Li (maroon) and 6-K (teal). Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms, NTMS2 disorder, and countercations were removed for clarity. | ||
Reinspection of the reflection intensities provided as ESI in the original publication led to the discovery of unassigned electron density surrounding the bridging N2 moieties (Fig. 6) corresponding to a largest difference peak of 0.64 e− Å−3. Remodeling of the original reflection intensities as a toluene adduct {Li(TMEDA)2}{[Ti(NR2)2]2(μ-PhMe)} (R = TMS) (6-Li) across 4 crystallographically-related toluene orientations resulted in slightly improved refinement metrics over the side-on Ti–(N2)2–Ti moiety 5-Li (Fig. 7, top). Our model for 6-Li gives unweighted and weighted R-factors of R1 = 0.0601, wR2 = 0.172, and a largest difference peak of 0.37 e− Å−3versus R1 = 0.0627, wR2 = 0.179, and 0.64 e− Å−3 for 5-Li. The relatively modest difference in model quality metrics for such different models (C7H8 = 50 e−vs. 2× N2 = 28 e−) is no-doubt influenced by the relatively poor I/σ, resolution and completeness of the original reflection data, which was not out of the norm for the era in which it was collected.
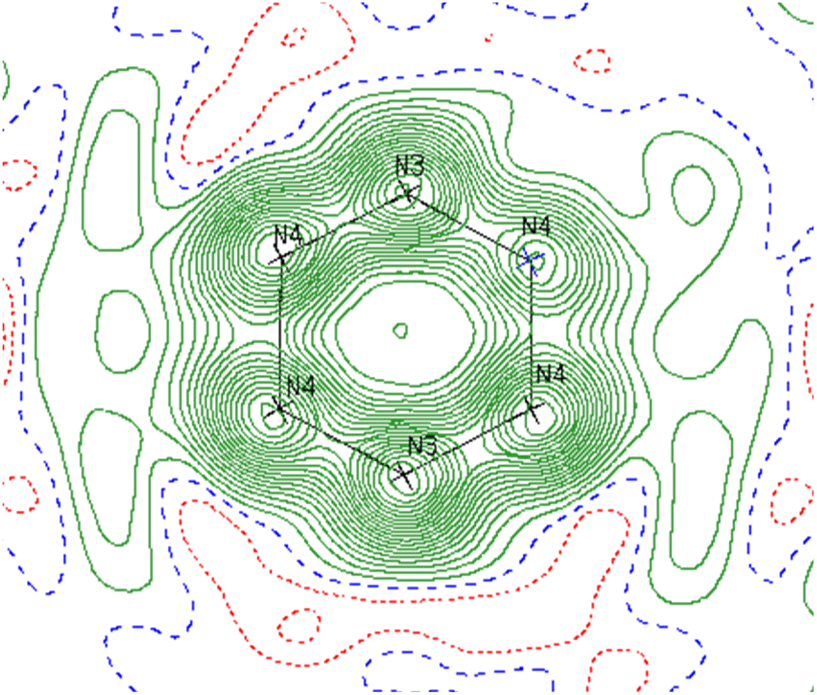 | ||
| Fig. 6 Contour map of Fobs for 5-Li showing the plane bisecting the Ti–Ti vector. Range: −0.60 to 3.80 e− Å−3 in steps of 0.20. Negative contours are shown in red and positive in green. Residual electron density outside of the N ring indicates likely partial occupancy of another atom at those positions. | ||
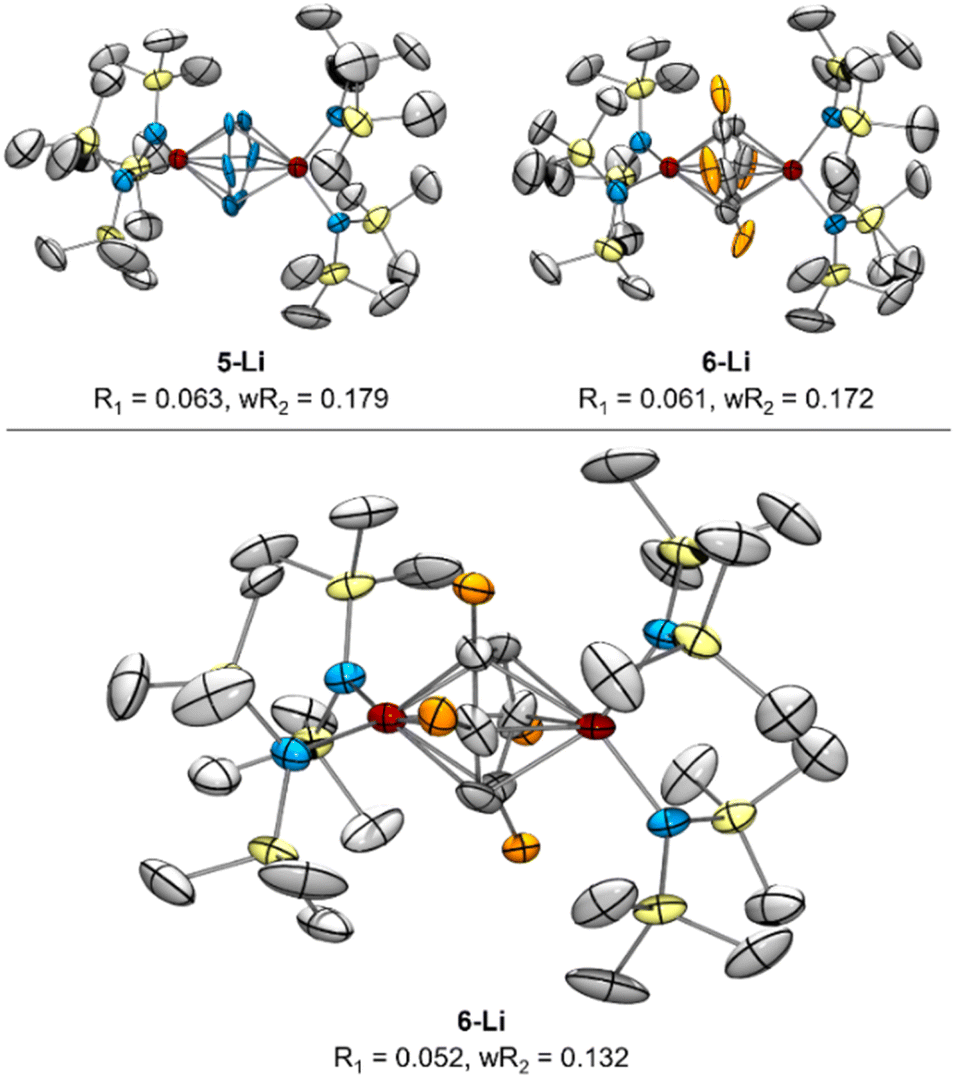 | ||
| Fig. 7 Top: re-solved SC-XRD of reported 5-Li reflection data as either 5-Li (left) or 6-Li (right) as possible solutions, with displacement ellipsoids drawn at the 50% probability level. Bottom: XRD from new reflection data of re-synthesized 6-Li following Reaction C from Fig. 8. Orange ellipsoids are disordered Me groups of the toluene dianion in 4 positions. Hydrogen atoms, NTMS2 disorder, and countercations were removed for clarity. | ||
In order to definitively establish the true identity of 5-Li, we turned our attention toward resynthesis of 5-Li in an attempt to collect higher quality X-ray data (Fig. 8). The previous report described the synthesis of both side-on 5-Li and an end-on Ti–(μ-η1:η1-N2)–Ti complex 7 (Fig. 8, Reactions A and B). Attempts to synthesize 5-Li under the reported conditions were not successful, with only the end-on bridged 7 produced instead (Fig. 8, Reaction D). In the original synthesis of 5-Li, there is a discrepancy between the conditions reported in the experimental (under Ar) and in the reaction equation (under N2). When N2 was rigorously excluded by performing the reaction on a Schlenk line under vacuum (Fig. 8, Reaction C) the toluene adduct 6-Li was isolated. This newly synthesized 6-Li has an identical unit cell to that reported for 5-Li with unweighted and weighted R-factors of R1 = 0.0519 and wR2 = 0.1320. Further, the previously published atomic coordinates for 5-Li can be refined directly against the reflection file obtained for the authentic sample of 6-Li. Modeling the new XRD data for 6-Li as 5-Li gives poorer metrics (R1 = 0.0665 and wR2 = 0.1874). Under this analysis, the largest peak in the difference map is again the unmodeled toluene methyl position which occurs at an identical position in the unit cell, being displaced by only 0.13 Å, a value well below the resolution of the experiment. Finally, hydrolysis of a crystalline sample of the newly-synthesized 6-Li yielded 57% of toluene by quantitative 1H NMR analysis (ESI, Fig. S2†).
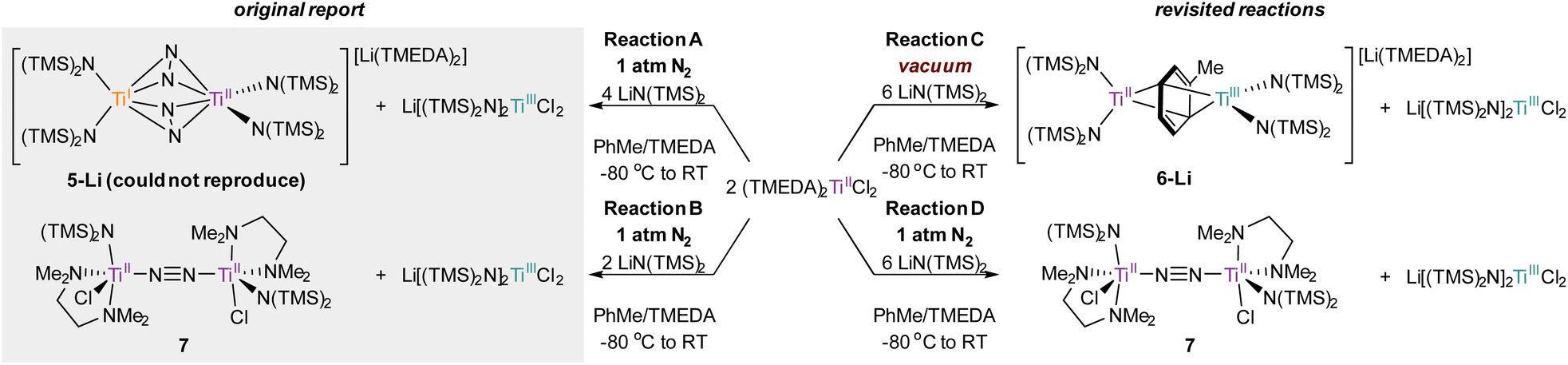 | ||
| Fig. 8 Previously reported conditions for the synthesis of 5-Li and 7 (left) and the re-examined reactivity of (TMEDA)2TiCl2 toward N2 and in vacuum conditions for the synthesis of 6-Li and 7 (right). | ||
The Raman spectra of 6-K and 6-Li are also quite similar (Fig. S26 and S27†), and lack the expected N–N stretch in the 1700 cm−1 region that is typical of bridging Ti–N2 complexes (Table S16†).2a,6,7,50 Further, attempts to calculate (m06 def2svp) optimized structures of [5]− from crystallographic coordinates of 5-Li result in either a single bridging side-on N2 for a doublet electronic state, or an η2:η1 side-on coordinated N2 (ref. 51 and 52) and a terminal end-on N2 for a quartet electronic state (ESI, Pages S26–S34†). The reorganization of the N2 core in these structures provide evidence that a doubly bridging side-on dinitrogen complex like 5 may be unstable for this system. In contrast, geometry optimization of 6-Li is facile and accurately and precisely models the experimental structure.
Thus, through reassessment of the original data combined with new synthesis and DFT analysis, we can confidently assert that the species previously assigned as the Ti–(μ-η2:η2-N2)2–Ti complex 5-Li is in fact the inverse sandwich complex Ti2–(PhMe)2−6-Li. This result further highlights the importance of looking beyond residual factors when critically analyzing crystallography models, as it is possible to have multiple reasonable solutions for the same data set. For example, Parkin and co-workers recently determined that the complex [Cd(CO)3(C6H3Cl)]4 with a reasonable R1 = 0.068 was instead [Re(CO)3(C4N2H3S)]4 with an R1 = 0.034.53
Further reduction reactions with Ti amides
The formation of toluene dianion reaction products 6-Li and 6-K provided the incentive to examine the reduction of the preformed Ti amide complexes Ti[N(TMS)2]3 (Fig. 9) and [(TMS)2N]2TiCl(THF) (Fig. 10) under N2 atmosphere.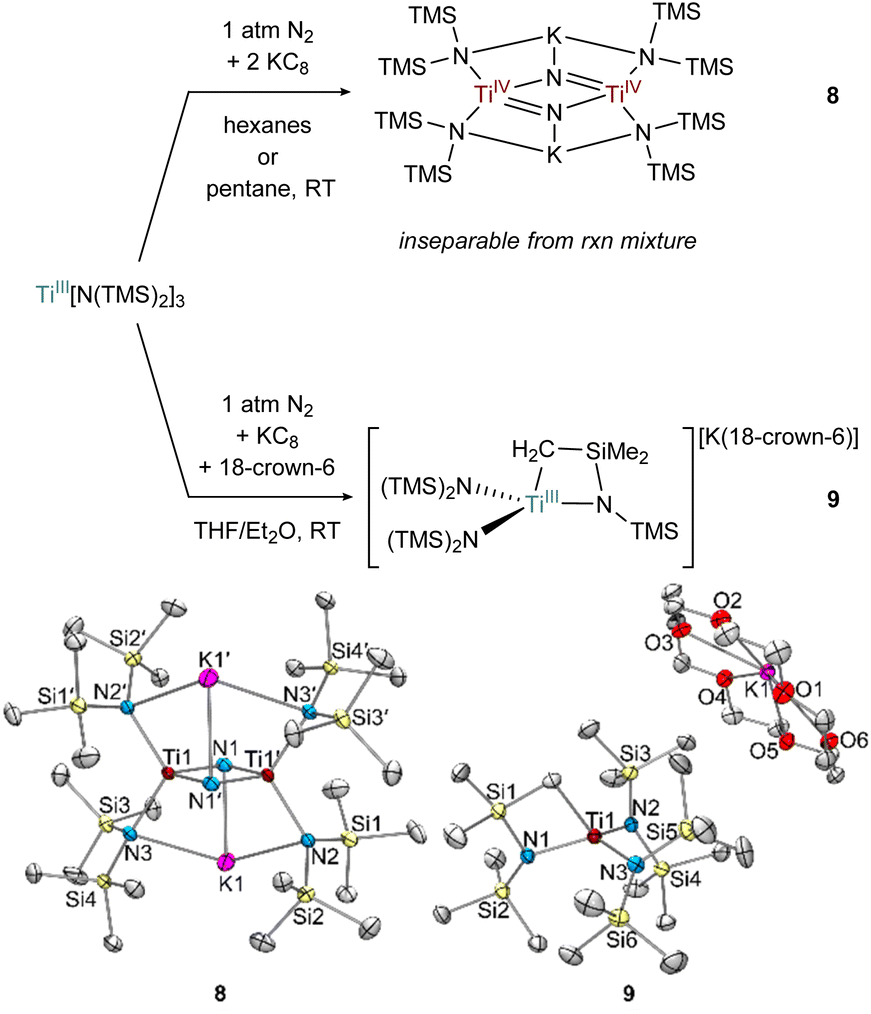 | ||
| Fig. 9 Top: condition-dependent reduction of Ti[N(TMS)2]3 forming 8 and 9 (top). Bottom: crystal structures of 8 and 9 (bottom) with displacement ellipsoids at the 50% probability level. Hydrogen atoms and were removed for clarity. | ||
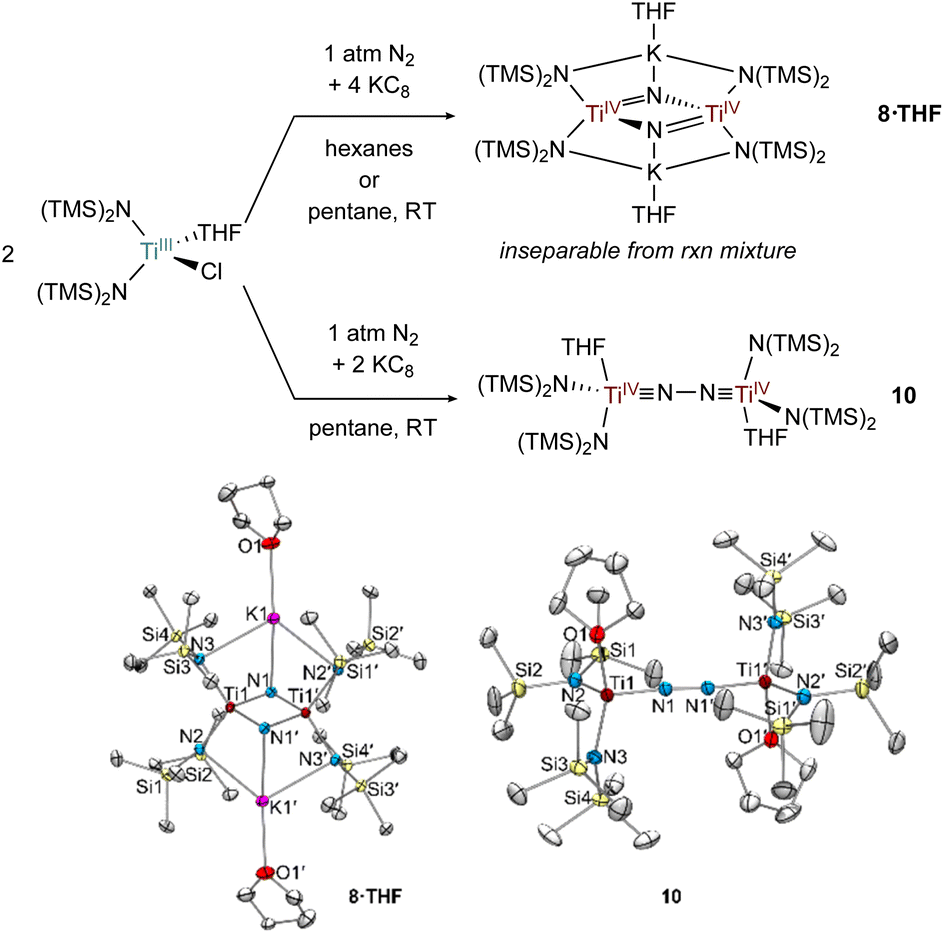 | ||
Fig. 10 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 reduction of [(TMS)2N]2TiCl(THF) using KC8 in the presence of N2 generates 8·THF and a 1:1 reduction in pentane generates 10 (top). Crystal structures of 8·THF and 10 (bottom) with displacement ellipsoids at the 50% probability level. Hydrogen atoms and disorder were removed for clarity. :2 reduction of [(TMS)2N]2TiCl(THF) using KC8 in the presence of N2 generates 8·THF and a 1:1 reduction in pentane generates 10 (top). Crystal structures of 8·THF and 10 (bottom) with displacement ellipsoids at the 50% probability level. Hydrogen atoms and disorder were removed for clarity. | ||
Preliminary reactivity studies have revealed a suite of N2 activation products, although in many cases the reaction products are inseparable. For example, reduction of Ti[N(TMS)2]3 in hexanes generates a colorless Ti bridging nitride 8, the product of full N2 cleavage, as an inseparable part of a larger mixture of unidentified products (Fig. 9). Analogous Ti-nitride formation has also been reported by Liddle and co-workers using Mg as the reducing agent.2b Interestingly, reduction of Ti[N(TMS)2]3 in THF with 18-crown-6 instead generated the TiIII C–H activated product {[(TMS)2N]2Ti[N(TMS)CH2SiMe2]}{K(18-crown-6)}, 9 in 28% isolated yield. Similar examples of cyclometallation from M[N(TMS)2]3 (M = Sc, Ti, Y, Er, Yb, Lu) complexes have been reported.54–58 In this instance, an equivalent of N(TMS)2− likely serves as a base to abstract a proton from the C–H bond, limiting the maximum reaction yield to 50%.
In the attempted reduction reactions with Ti[N(TMS)2]3 the co-product is the highly soluble KN(TMS)2 which led to challenges in cleanly separating the Ti products. To circumvent this issue, reduction reactions of [(TMS)2N]2TiCl(THF) with KC8 were attempted, envisioning that the less soluble KCl co-product would be easier to separate (Fig. 10). Reduction of [N(TMS)2]2TiCl(THF) with 2 equiv. KC8 in hexanes or pentane yielded a THF adduct of a bridging nitride {[(N(TMS)2)2Ti]2(μ-N)2}{K(THF)}2 (8·THF), analogous to the reduction of Ti[N(TMS)2]3. Unfortunately, the reaction mixture of 8·THF was similarly intractable to that of 8, and identification was only possible by picking single crystals from the mixture. However, reduction of [N(TMS)2]2TiCl(THF) with only 1 equiv. of KC8 in pentane yielded the end-on bridged N2 complex [(N(TMS)2)2Ti(THF)]2(μ-η1:η1-N2) (10). Interestingly, further reaction of 10 with excess KC8 did not result in significant formation of 8·THF.
Selected bond distances from the solid-state structures of 8, 8·THF, 7 (ref. 26) and 10 are reported in Table 1. The Ti–Nnitride distances in 8 and 8·THF have nearly identical bond metrical parameters despite containing an additional coordinated THF to each potassium cation in 8·THF. The N⋯N distances in 8 and 8·THF are >2.5 Å, consistent with complete N–N bond scission and the formation of bridging nitrides. Additionally, the Ti–Nnitride distances are symmetric, indicating resonance for the Ti![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds. The N–N bond distances in 10, (1.276(2) Å), and 7,26 (1.289(9) Å), are similar to many other reported Ti–N2–Ti bridging complexes.2–7 The short Ti–NN2 bond distances in 7 and 10 are (1.76 Å) are indicative of Ti
N double bonds. The N–N bond distances in 10, (1.276(2) Å), and 7,26 (1.289(9) Å), are similar to many other reported Ti–N2–Ti bridging complexes.2–7 The short Ti–NN2 bond distances in 7 and 10 are (1.76 Å) are indicative of Ti![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bonds, suggesting that a TiIVN–NTiIV electronic structure is predominant in both cases. DFT analysis of 10 further corroborates the TiN triple bond character, as both Ti–N p-bonds can be seen in the HOMO and HOMO − 1 (Fig. S20 and S21†). The Ti–N(TMS)2 distances in 7 and 10 similar to those in the TiIV nitride compounds 8 and 8·THF, further indicating TiIVN–NTiIV character in 7 and 10. These electronic structures are consistent with a recent framework for classifying N2 complexes based on electron counting.59
N triple bonds, suggesting that a TiIVN–NTiIV electronic structure is predominant in both cases. DFT analysis of 10 further corroborates the TiN triple bond character, as both Ti–N p-bonds can be seen in the HOMO and HOMO − 1 (Fig. S20 and S21†). The Ti–N(TMS)2 distances in 7 and 10 similar to those in the TiIV nitride compounds 8 and 8·THF, further indicating TiIVN–NTiIV character in 7 and 10. These electronic structures are consistent with a recent framework for classifying N2 complexes based on electron counting.59
| Nitride complexes | N2 complexes | |||
|---|---|---|---|---|
| 8 | 8·THF | 7 (ref. 26) | 10 | |
| Ti1–N1 | 1.825(3) | 1.824(1) | 1.762(5) | 1.763(1) |
| Ti1–N1′ | 1.836(3) | 1.835(2) | ||
| Ti1–N2 | 2.073(3) | 2.075(1) | 2.023(5) | 1.995(2) |
| Ti1–N3 | 2.092(3) | 2.094(1) | — | 2.011(1) |
| N1⋯N1′ | 2.505(4) | 2.503(2) | — | — |
| N1–N1′ | — | — | 1.289(9) | 1.276(2) |
Conclusion
In summary, the bis-N2 side-on complex, 5-Li is in fact a bridging toluene complex, 6-Li. This misassignment arises from the modeling of the disorder of the bridging atoms. Despite the reasonable difference in models (C7H8 = 50 e−vs. 2× N2 = 28 e−) the poor I/σ, resolution and completeness of the original reflection data limited proper interpretation. New experimental and computational analysis of data confirmed the formation of the bridging toluene complex: synthesis under vacuum in toluene yields the same crystals as reported for the incorrectly-assigned N2 adduct, and NMR analysis of the hydrolyzed crystals reveals the presence of toluene. The reassignment of 5-Li means that there are zero examples of a double-side-on bridging mode of N2 activation, and we posit that this particular bonding motif may not be possible.Experimental section
General considerations
All syntheses and manipulations described below were conducted under nitrogen with exclusion of air using glovebox, Schlenk-line, and high-vacuum techniques. Ti[N(TMS)2]3,25 (TMEDA)2TiCl2,60 and KC8 (ref. 61) were prepared using previously published procedures. TMEDA was vacuum transferred, passed through activated alumina, stored over 4 Å molecular sieves in the glovebox prior to use. 18-Crown-6 was sublimed prior to use. NMR solvent C6D6 was dried over Na0/Ph2CO and vacuum transferred before passing through activated alumina in the glovebox. Pentane, hexanes, and toluene were dried on a Pure Process Technology solvent purification system, passed through activated alumina, and stored over activated 4 Å molecular sieves prior to use. 1H NMR spectra were obtained on a Bruker Avance 400 MHz spectrometer at 298 K. Elemental analysis was shipped to and performed by Midwest Microlab. Attempts to collect elemental analysis on samples of the newly-reported 6-K, 6-Li, 9, and 10 were unsuccessful as these sensitive complexes decomposed during shipment. SC-XRD data of 6-K, 7, 8·THF, 9 and 10 were collected on a Bruker-AXS Venture Photon-III using a Mo source, 6-Li on a Bruker-AXS Venture Photon-III using a Cu source, and 8 was collected on a Bruker-AXS Smart Apex-II using a Mo source. Crystal and refinement data are available in ESI.† UV-visible absorption spectra were recorded on a HP8453A diode array spectrometer from Unisoku, Scientific Instruments (Osaka, Japan). Raman spectra were obtained at room temperature with excitation at 457 nm (500 mW at source, Cobolt Lasers) through the sample in a J. Young NMR tube using a 135° backscattering arrangement. The collimated Raman scattering was collected using two plano convex lenses (f = 12 cm, placed at an appropriate distance) through appropriate long pass edge filters (Semrock) into an Acton AM-506M3 monochromator equipped with a Princeton Instruments ACTON PyLON LN/CCD-1340 × 400 detector. The detector was cooled to −120 °C prior to the experiments. Spectral calibration was performed using the Raman spectrum of acetonitrile/toluene 50:50 (v/v).62 Each spectrum was acquired 60 times with 1 s acquisition time, resulting in a total acquisition time of 1 min per spectrum. The collected data was processed using Spectragryph63 and a multi-point baseline correction was performed for all spectra.
Synthesis of {(PhMe)K}{[Ti(N(TMS)2)2]2(μ-PhMe)} 6-K
In an N2-filled glovebox, a blue toluene (1.5 mL) solution of Ti[N(TMS)2]3 (196 mg, 0.37 mmol) was passed through a KC8 filled 7 mm diameter pipette column (5 cm of KC8) yielding a darkly coloured eluent. An additional 1.5 mL of PhMe was used to wash the KC8 column. The resulting near black solution was dried in vacuo to yield an oil. This oil was extracted with −78 °C pentane (3 × 1.5 mL) and the pentane solution was filtered through glass wool. The resulting pentane solution was then dried in vacuo to yield a dark navy-blue powder with colorless crystals (identified as KN(TMS)2via SC-XRD). The powder was then extracted and filtered two additional times with −78 °C pentane (3 × 1.5 mL) until colorless solids were no longer observed. Upon final drying, near black microcrystalline solids of {(PhMe)K}{[Ti(N(TMS)2)2]2(μ-PhMe)} formed (71 mg, 40%). Single crystal X-ray quality crystals were obtained from a concentrated PhMe solution at −35 °C. Evans method: 1.67μB.Synthesis of {(TMEDA)2Li}{[Ti(N(TMS)2)2]2(μ-PhMe)}, 6-Li
In an N2-filled glovebox, purple (TMEDA)2TiCl2 (300 mg, 0.84 mmol) and colorless LiN(TMS)2 (422 mg, 2.52 mmol) were added to a Teflon screw cap 100 mL flask. To another 100 mL Teflon screw cap flask, toluene (10 mL) and TMEDA (0.5 mL) were added. Both flasks were sealed, removed from the glovebox, and placed on a high-vac line. The flask containing the solid (TMEDA)2TiCl2 and LiN(TMS)2 were placed under vacuum (10−4 torr) for 3 h. The PhMe/TMEDA solution was degassed via three 1 h freeze–pump–thaw cycles. The PhMe/TMEDA solution was vacuum transferred to the flask containing solids using a −78 °C bath. The flask was then sealed and the purple mixture was stirred and warmed to room temperature. After 30 min of stirring, the purple mixture turned brown. After 1 d, degassed Et2O (via three 1 h, freeze–pump–thaw cycles) was vacuum transferred to the frozen brown mixture. Upon thawing, an Et2O layer on the brown solution formed. The sealed flask was stored in a −30 °C freezer. After 1 d, both colorless and brown crystals formed. The colorless crystals were identified as [Li(TMEDA)2]{[(TMS)2N]2TiCl2} via unit-cell analysis (CSD Refcode: JOHBAJ). A full dataset of the brown crystals were collected and identified as {(TMEDA)2Li}{[Ti(N(TMS)2)2]2(μ-PhMe)}, 6-Li.Synthesis of [(TMEDA)Ti(N(TMS)2)Cl]2(μ-N2), 7
In an N2-filled glovebox, purple (TMEDA)2TiCl2 (300 mg, 0.84 mmol) and colorless LiN(TMS)2 (421 mg, 2.52 mmol) were added to a 100 mL flask and chilled to −78 °C. To this flask, −78 °C toluene (10 mL) and TMEDA (0.5 mL) were added. The purple mixture was stirred and warmed to room temperature. The purple mixture turned brown within minutes. After 1 d, the brown solution was layered with Et2O and placed in a −30 °C freezer. After 1 d, colorless and brown crystals formed. The colorless crystals were identified as [Li(TMEDA)2]{[(TMS)2N]2TiCl2} via unit-cell analysis (CSD Refcode: JOHBAJ). A full dataset of the brown crystals were collected and identified as [(TMEDA)Ti(N(TMS)2)Cl]2(μ-N2), 7.Synthesis of reaction mixture containing 8
In an N2-filled glovebox, a blue Ti[N(TMS)2]3 (100 mg, 0.19 mmol) solution in hexanes (5 mL) was added to KC8 (54 mg, 0.38 mmol) forming a darkly coloured mixture. After stirring for 3 d, the near black mixture was filtered through Celite to remove graphite. The resulting near black solution was placed in a −30 °C freezer. After 1 d, colourless crystals covered in tar formed from the near black solution and were identified as the bridging nitride complex 8. Crystals of K[N(TMS)2] were also identified in the mixture by matching the unit-cell (CSD Refcode: VETFOP).Synthesis of reaction mixture containing 8·THF
In an N2-filled glovebox, a light blue [(TMS)2N]2TiCl(THF) (100 mg, 0.21 mmol) solution in hexanes (5 mL) was added to KC8 (57 mg, 0.42 mmol) forming a darkly coloured mixture. After stirring for 3 d, the black mixture was filtered through Celite to remove graphite. The resulting solution was placed in a −30 °C freezer. After 1 d, colorless crystals covered in tar formed from the near black solution and were identified as the bridging nitride complex 8·THF.Synthesis of [K(18-crown-6)]{[(TMS)2N]2Ti[N(TMS)(SiMe2CH2)]}, 9
In an N2-filled glovebox, a blue 3:1 Et2O:THF (4 mL) solution of Ti[N(TMS)2]3 (100 mg, 0.19 mmol) was added dropwise to a dark blue 3:1 Et2O:THF (10 mL) mixture of 18-crown-6 (52 mg, 0.19 mmol) and KC8 (27 mg, 0.19 mmol) forming a darkly coloured mixture. After 4 d of stirring, the dark green mixture was filtered though Celite to remove graphite and the dark green solution was placed under vacuum. The resulting green oil was redissolved in Et2O (1 mL) and layered into pentane (10 mL) and placed in a −30 °C freezer. After 1 d, green single-crystals formed and were identified as [K(18-crown-6)]{[(TMS)2N]2Ti[N(TMS)(SiMe2CH2)]}, 9, via SC-XRD (44 mg, 28%).
Modified synthesis of [(TMS)2N]2TiCl(THF)36
In an N2-filled glovebox, TiCl3(THF)3 (1006 mg, 2.7 mmol) and LiN(TMS)2 (918 mg, 5.4 mmol) were stirred in pentane (50 mL) for 1 h. The resulting light blue mixture was filtered through Celite twice to remove LiCl. The clear blue solution was placed in a −30 °C freezer. After 1 d, light blue crystals formed in a dark green solution. The green solution as decanted away and the light blue crystals were washed with −78 °C pentane (3 × 2 mL) and isolated (983 mg, 76%).Synthesis of [(TMS)2N2Ti(THF)]2(μ-N2), 10
In an N2-filled glovebox, light blue pentane (5 mL) solution of [(TMS)2N]2TiCl(THF) (200 mg, 0.42 mmol) was added to KC8 (57 mg, 0.42 mmol) forming a darkly coloured mixture. After stirring for 1 d, the mixture was filtered through Celite to remove graphite and KCl. The resulting brown solution was concentrated to ∼0.5 mL of pentane and placed in a −30 °C freezer. After 1 d, brown single-crystals formed and were identified as [(TMS)2N2Ti(THF)]2(μ-N2), 10. The crystals were washed with −78 °C pentane and isolated (34 mg, 18%). Crystalline yields are low due to the high solubility of this compound. 1H (400 MHz, C6D6, 25 °C, δ, ppm) 4.17 (s, 8H, THF) 1.46 (m, 8H, THF), 0.43 (s, 72H, SiMe3).Data availability
All experimental and computational data is available as a part of the ESI.†Author contributions
Daniel N. Huh: conceptualization, investigation, formal analysis, writing – original draft, writing – review & editing, funding acquisition. Ross F. Koby: investigation, formal analysis, writing – original draft, writing – review & editing. Zoe E. Stuart: investigation. Rachel J. Dunscomb: investigation. Nathan D. Schley: investigation, formal analysis, writing – original draft, writing – review & editing. Ian A. Tonks: conceptualization, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Institutes of Health (R35GM119457). Additional support was provided by the National Institute of General Medicine Sciences of the National Institutes of Health to D. N. H. from the Ruth L. Kirschstein NRSA Postdoctoral Fellowship (F32GM137547). Instrumentation for the University of Minnesota Chemistry NMR facility was supported from a grant through the National Institutes of Health (S10OD011952). X-ray diffraction experiments were performed with a diffractometer purchased through a grant from NSF/MRI (1229400) and the University of Minnesota. We thank Dr Victor G. Young Jr and Margaret C. Clapham of the University of Minnesota X-ray Crystallographic Laboratory for their assistance with SC-XRD. We also thank Chase Abelson and Prof. Larry Que for assistance with UV-vis and Raman spectroscopy.References
- D. Singh, W. R. Buratto, J. F. Torres and L. J. Murray, Chem. Rev., 2020, 120, 5517–5581 CrossRef CAS PubMed.
- (a) L. R. Doyle, A. J. Wooles, L. C. Jenkins, F. Tuna, E. J. L. McInnes and S. T. Liddle, Angew. Chem., Int. Ed., 2018, 57, 6314–6318 CrossRef CAS PubMed; (b) L. R. Doyle, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed., 2019, 58, 6674–6677 CrossRef CAS PubMed.
- T. E. Hanna, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 14688–14689 CrossRef CAS PubMed.
- T. E. Hanna, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 6018–6019 CrossRef CAS PubMed.
- T. E. Hanna, W. H. Bernskoetter, M. W. Bouwkamp, E. Lobkovsky and P. J. Chirik, Organometallics, 2007, 26, 2431–2438 CrossRef CAS.
- T. E. Hanna, E. Lobkovsky and P. J. Chirik, Organometallics, 2009, 28, 4079–4088 CrossRef CAS.
- S. P. Semproni, C. Milsmann and P. J. Chirik, Organometallics, 2012, 31, 3672–3682 CrossRef CAS.
- Y. Nakanishi, Y. Ishida and H. Kawaguchi, Angew. Chem., Int. Ed., 2017, 56, 9193–9197 CrossRef CAS PubMed.
- S. Fortier and A. Gomez-Torres, Chem. Commun., 2021, 57, 10292–10316 RSC.
- A. Gomez-Torres, C. Saucedo and S. Fortier, Arene Complexes of Group 4 Metals. Comprehensive Organometallic Chemistry IV, ed. K. Meyer, D. O'Hare and G. Parkin, Elsevier Ltd, 2022, pp. 502–549 Search PubMed.
- G. B. Wijeratne, E. M. Zolnhofer, S. Fortier, L. N. Grant, P. J. Carroll, C.-H. Chen, K. Meyer, J. Krzystek, A. Ozarowski, T. A. Jackson, D. J. Mindiola and J. Telser, Inorg. Chem., 2015, 54, 10380–10397 CrossRef CAS.
- J. R. Aguilar-Calderón, A. J. Metta-Magaña, B. Noll and S. Fortier, Angew. Chem., Int. Ed., 2016, 55, 14101–14105 CrossRef PubMed.
- A. Gómez-Torres, J. R. Aguilar-Calderón, C. Saucedo, A. Jordan, A. Metta-Magaña, B. Pinter and S. Fortier, Chem. Commun., 2020, 56, 1545–1548 RSC.
- A. Gómez-Torres, J. R. Aguilar-Calderón, A. M. Encerrado-Manriquez, M. Pink, A. J. Metta-Magaña, W.-Y. Lee and S. Fortier, Chem.–Eur. J., 2020, 26, 2803–2807 CrossRef PubMed.
- Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2016, 8, 63–68 CrossRef CAS.
- Z. W. Davis-Gilbert, X. Wen, J. D. Goodpaster and I. A. Tonks, J. Am. Chem. Soc., 2018, 140, 7267–7281 CrossRef CAS PubMed.
- J. Guo, X. Deng, C. Song, Y. Lu, S. Qu, Y. Dang and Z.-X. Wang, Chem. Sci., 2017, 8, 2413–2425 RSC.
- I. A. Tonks, Acc. Chem. Res., 2021, 54, 3476–3490 CrossRef CAS PubMed.
- S. Fortier, A. Gomez-Torres and C. Saucedo, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2021, DOI:10.1016/B978-0-12-820206-7.00012-3.
- J. R. Hagadorn and J. Arnold, Angew. Chem., Int. Ed., 1998, 37, 1729–1731 CrossRef CAS PubMed.
- O. V. Ozerov, B. O. Patrick and F. T. Ladipo, J. Am. Chem. Soc., 2000, 122, 6423–6431 CrossRef CAS.
- R. Gyepes, J. Pinkas, I. Císařová, J. Kubišta, M. Horáček and K. Mach, RSC Adv., 2016, 6, 94149–94159 RSC.
- G. B. Nikiforov, P. Crewdson, S. Gambarotta, I. Korobkov and P. H. M. Budzelaar, Organometallics, 2007, 26, 48–55 CrossRef CAS.
- J. N. Boynton, J.-D. Guo, F. Grandjean, J. C. Fettinger, S. Nagase, G. J. Long and P. P. Power, Inorg. Chem., 2013, 52, 14216–14223 CrossRef CAS PubMed.
- M. A. Putzer, J. Magull, H. Goesmann, B. Neumüller and K. Dehnicke, Chem. Ber., 1996, 129, 1401–1405 CrossRef CAS.
- R. Duchateau, S. Gambarotta, N. Beydoun and C. Bensimon, J. Am. Chem. Soc., 1991, 113, 8986–8988 CrossRef CAS.
- W. J. Evans, Organometallics, 2016, 35, 3088–3100 CrossRef CAS.
- D. Patel, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2013, 42, 5224–5227 RSC.
- C. R. Stennett, J. C. Fettinger and P. P. Power, Inorg. Chem., 2020, 59, 1871–1882 CrossRef CAS PubMed.
- C. R. Stennett and P. P. Power, Inorg. Chem., 2021, 60, 18503–18511 CrossRef CAS PubMed.
- M. A. Putzer, R. J. Lachicotte and G. C. Bazan, Inorg. Chem. Commun., 1999, 2, 319–322 CrossRef CAS.
- J. J. H. Edema, R. Duchateau, S. Gambarotta, R. Hynes and E. Gabe, Inorg. Chem., 1991, 30, 154–156 CrossRef CAS.
- N. Beydoun, R. Duchateau and S. Gambarotta, J. Chem. Soc., Chem. Commun., 1992, 244–246, 10.1039/C39920000244.
- B. C. Bailey, F. Basuli, J. C. Huffman and D. J. Mindiola, Organometallics, 2006, 25, 2725–2728 CrossRef CAS.
- C. C. Quadri, K. W. Tornroos and E. Le Roux, IUCrData, 2017, 2, x171488 CrossRef CAS.
- L. Scoles and S. Gambarotta, Inorg. Chim. Acta, 1995, 235, 375–380 CrossRef CAS.
- R. K. Minhas, L. Scoles, S. Wong and S. Gambarotta, Organometallics, 1996, 15, 1113–1121 CrossRef CAS.
- R. D. Sanner, D. M. Duggan, T. C. McKenzie, R. E. Marsh and J. E. Bercaw, J. Am. Chem. Soc., 1976, 98, 8358–8365 CrossRef CAS.
- J. M. de Wolf, R. Blaauw, A. Meetsma, J. H. Teuben, R. Gyepes, V. Varga, K. Mach, N. Veldman and A. L. Spek, Organometallics, 1996, 15, 4977–4983 CrossRef CAS.
- A. Scherer, K. Kollak, A. Lützen, M. Friedemann, D. Haase, W. Saak and R. Beckhaus, Eur. J. Inorg. Chem., 2005, 2005, 1003–1010 CrossRef.
- P. P. Fontaine, B. L. Yonke, P. Y. Zavalij and L. R. Sita, J. Am. Chem. Soc., 2010, 132, 12273–12285 CrossRef CAS.
- J. R. Hagadorn and J. Arnold, J. Am. Chem. Soc., 1996, 118, 893–894 CrossRef CAS.
- S. M. Mullins, A. P. Duncan, R. G. Bergman and J. Arnold, Inorg. Chem., 2001, 40, 6952–6963 CrossRef CAS PubMed.
- G. Bai, P. Wei and D. W. Stephan, Organometallics, 2006, 25, 2649–2655 CrossRef CAS.
- Y. Sekiguchi, F. Meng, H. Tanaka, A. Eizawa, K. Arashiba, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Dalton Trans., 2018, 47, 11322–11326 RSC.
- B. Wang, G. Luo, M. Nishiura, S. Hu, T. Shima, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2017, 139, 1818–1821 CrossRef CAS PubMed.
- W. A. Chomitz and J. Arnold, Chem. Commun., 2007, 4797–4799, 10.1039/B709763H.
- R. Baumann, R. Stumpf, W. M. Davis, L.-C. Liang and R. R. Schrock, J. Am. Chem. Soc., 1999, 121, 7822–7836 CrossRef CAS.
- T. Kurogi, Y. Ishida and H. Kawaguchi, Chem. Commun., 2013, 49, 11755–11757 RSC.
- F. Studt, N. Lehnert, B. E. Wiesler, A. Scherer, R. Beckhaus and F. Tuczek, Eur. J. Inorg. Chem., 2006, 291–297 CrossRef CAS.
- M. D. Fryzuk, S. A. Johnson and S. J. Rettig, J. Am. Chem. Soc., 1998, 120, 11024–11025 CrossRef CAS.
- D. Pun, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2008, 130, 6047–6054 CrossRef CAS PubMed.
- E. Amemiya, A. Loo, D. G. Shlian and G. Parkin, Chem. Sci., 2020, 11, 11763–11776 RSC.
- M. Karl, K. Harms, G. Seybert, W. Massa, S. Fau, G. Frenking and K. Dehnicke, Z. Anorg. Allg. Chem., 1999, 625, 2055–2063 CrossRef CAS.
- M. A. Putzer, B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 1998, 624, 1087–1088 CrossRef CAS.
- F. Han, J. Zhang, W. Yi, Z. Zhang, J. Yu, L. Weng and X. Zhou, Inorg. Chem., 2010, 49, 2793–2798 CrossRef CAS PubMed.
- M. Niemeyer, Inorg. Chem., 2006, 45, 9085–9095 CrossRef CAS PubMed.
- M. Fang, J. E. Bates, S. E. Lorenz, D. S. Lee, D. B. Rego, J. W. Ziller, F. Furche and W. J. Evans, Inorg. Chem., 2011, 50, 1459–1469 CrossRef CAS PubMed.
- L. S. Yamout, M. Ataya, F. Hasanayn, P. L. Holland, A. J. M. Miller and A. S. Goldman, J. Am. Chem. Soc., 2021, 143, 9744–9757 CrossRef CAS PubMed.
- E. M. Zolnhofer, G. B. Wijeratne, T. A. Jackson, S. Fortier, F. W. Heinemann, K. Meyer, J. Krzystek, A. Ozarowski, D. J. Mindiola and J. Telser, Inorg. Chem., 2020, 59, 6187–6201 CrossRef CAS PubMed.
- D. E. Bergbreiter and J. M. Killough, J. Am. Chem. Soc., 1978, 100, 2126–2134 CrossRef CAS.
- Standard Guide for Raman Shift Standards for Spectrometer Calibration, ASTM E1840-96(2007), ASTM International, West Conshohocken, PA, 2007 Search PubMed.
- F. Menges, Spectragryph – optical spectroscopy software, version 1.2.15, 2021, http://www.effemm2.de/spectragryph/ Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 218344–218346, 2176319, 2183462, 2183463 and 2183467. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04368h |
| This journal is © The Royal Society of Chemistry 2022 |