
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium nanoparticles canopied by heptagon-containing saddle-shaped nanographenes as efficient aromatic hydrogenation catalysts†
Christian
Cerezo-Navarrete
 a,
Arthur H. G.
David
b,
Adrián
García-Zaragoza
a,
Marcos D.
Codesal
b,
Pascual
Oña-Burgos
a,
Iker
del Rosal
c,
Romuald
Poteau
c,
Araceli G.
Campaña
b and
Luis M.
Martínez-Prieto
*ad
a,
Arthur H. G.
David
b,
Adrián
García-Zaragoza
a,
Marcos D.
Codesal
b,
Pascual
Oña-Burgos
a,
Iker
del Rosal
c,
Romuald
Poteau
c,
Araceli G.
Campaña
b and
Luis M.
Martínez-Prieto
*ad
aITQ, Instituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Av. de los Naranjos 46022, Valencia, Spain. E-mail: luismiguel.martinez@csic.es
bDepartamento Química Orgánica, Universidad de Granada (UGR), C. U. Fuentenueva, 18071 Granada, Spain
cLPCNO; Laboratoire de Physique et Chimie des Nano-Objets, INSA-CNRS (UMR 5215)-UPS, Institut National des Sciences Appliquées, 135, Avenue de Rangueil, F-31077 Toulouse, France
dDepartamento de Química Inorgánica, Universidad de Sevilla (US) – IIQ, Instituto de Investigaciones Químicas (CSIC-US), Avda. Americo Vespucio 49, 41092 Seville, Spain
First published on 17th October 2022
Abstract
The search for new ligands capable of modifying the metal nanoparticle (MNP) catalytic behavior is of increasing interest. Herein we present the first example of RuNPs stabilized with non-planar heptagon-containing saddle-shaped nanographenes (Ru@1 and Ru@2). The resemblance to graphene-supported MNPs makes these non-planar nanographene-stabilized RuNPs very attractive systems to further investigate graphene–metal interactions. A combined theoretical/experimental study allowed us to explore the coordination modes and dynamics of these nanographenes at the Ru surface. The curvature of these saddle-shaped nanographenes makes them efficient MNP stabilizers. The resulting RuNPs were found to be highly active catalysts for the hydrogenation of aromatics, including platform molecules derived from biomass (i.e. HMF) or liquid organic hydrogen carriers (i.e. N-indole). A significant ligand effect was observed since a minor modification on the hept-HBC structure (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 instead of CO) was reflected in a substantial increase in the MNP activity. Finally, the stability of these canopied RuNPs was investigated by multiple addition experiments, proving to be stable catalysts for at least 96 h.
CH2 instead of CO) was reflected in a substantial increase in the MNP activity. Finally, the stability of these canopied RuNPs was investigated by multiple addition experiments, proving to be stable catalysts for at least 96 h.
Introduction
The use of metal nanoparticles (MNPs) in catalysis has attracted huge attention in the last few years.1 The reason for this massive interest is the great potential of MNPs as catalysts. Their particular electronic configuration, small size (1–100 nm) and high active surface area make them exceptionally active catalysts. The catalytic properties of MNPs are greatly influenced by ligands used as stabilizers (i.e. nitrogen-, phosphorus- carbon- or oxygen-containing ligands),2 which, in the same way as in organometallic chemistry, are able to modify the electronic and steric properties of active metal centers.3 Ligand–substrate interactions can also enhance or hinder the reactivity of certain functional groups, and therefore influence the reactivity of ligand-stabilized MNPs.4 In this context, the search for potential ligands capable of modifying the MNP catalytic behavior is always a challenge. For example, some of us have recently developed a new class of ligands based on zwitterionic adducts of N-heterocyclic carbenes (NHCs) and carbodiimides as efficient stabilizers able to modify MNP reactivity,5 where small variations in the N-substituents lead to significant changes in the catalytic activity of MNPs.6The pursuit of new families of ligands led us to explore for the first time the use of non-planar polycyclic aromatic hydrocarbons (PAHs) or distorted nanographenes in MNP stabilization. Non-planar carbon-based aromatic systems have been gaining great attention due to their special geometry and optoelectronic properties compared to their planar counterparts.7 In addition, these non-planar nanographenes can be suitable models for MNP–graphene interaction studies, since graphene materials normally contain non-hexagonal rings (pentagons or heptagons) that curve these 2D structures, modifying their original properties.7a,8 It is very well known that these defect sites act as excellent anchoring points for MNPs.9 However, most of the interaction studies between MNPs and graphene materials have been performed by DFT calculations.10 In general, these computational studies predict that the interaction between MNPs and defect-free graphenes is weak, and although there is an overlapping between the π orbitals of the graphene and the d electrons of the MNPs, the presence of defect sites (i.e. curvature, doping elements or functional groups) is necessary for a strong MNP–graphene interaction. The lack of experimental interaction studies, together with the possibility of synthesizing distorted nanographenes doped or functionalized, makes non-planar PAHs potentially ideal molecules for fundamental metal–graphene interaction investigations. Thus, non-planar PAHs with a five-membered-ring and a positive curvature have been reported to interact with multi-metallic complexes on account of their adaptability and shape.11 It has also been shown that the inclusion of seven-membered rings in nanographenes leads to a saddle-shaped curvature and induces changes in optoelectronic responses and increases solubility.12 Particular supramolecular interactions of those saddle-shaped nanographenes have been scarcely reported,13 which might have an impact on the interaction with MNPs and the subsequent catalytic activity. Besides, to the best or our knowledge, their use as ligands for MNPs has no precedents.
Spectroscopic techniques, such as NMR, XPS or FT-IR, have been demonstrated as appropriate techniques to investigate the binding mode and dynamics of coordinating ligands on MNP surfaces.14 This, together with the great importance of theoretical calculations to clarify experimental data, makes joint theoretical/experimental studies ideal to determine the coordination mode of stabilizing ligands6a and better understand the chemical processes at the MNP surface.15 Recently, the coordination of carboxylic acids to ruthenium nanoparticles (RuNPs) was investigated through a combined experimental and theoretical study.16 The interaction of ligands at the MNP surface can occur through different coordination modes, what ultimately is going to define the catalytic properties of the catalyst. Therefore, coordination and ligand effect studies are essential to design more efficient nanocatalysts. In this regard, ligand-stabilized RuNPs are candidates of choice to be applied in catalytic processes as they can be easily characterized by NMR due to the absence of magnetic perturbations (knight shift, paramagnetism, etc.), and they are highly active catalysts in hydrogenation reactions, including aromatic reductions.
Today, the hydrogenation of aromatics is an active research area.17 The formation of substituted cyclohexanes from the corresponding aromatic substrates is extensively applied in the chemical industry (e.g. hydrogenation of benzene into cyclohexane/cyclohexene for nylon production).18 Aromatic hydrogenation is also an important process in the valorization of platform molecules derived from biomass such as hydroxymethylfurfural (HMF) or phenol.19 Furthermore, arene hydrogenation/dehydrogenation is of key importance in hydrogen storage by using liquid organic hydrogen carriers (LOHCs).20 In comparison with other simpler olefins, the reduction of aromatics is much more difficult due to their aromaticity.21 Thus, the hydrogenation of arenes has been traditionally carried out using heterogenous systems under harsh conditions (high temperature and/or pressure).22 Although there are some recent examples about supported Ru NPs operating under milder conditions,23 soluble ligand-stabilized RuNPs have also demonstrated to be efficient materials for mild aromatic hydrogenation reactions.24
Herein, we have generated stable RuNPs canopied with distorted nanographenes for the first time. In particular, we used saddle-shaped heptagon-containing hexa-peri-hexabenzocoronene analogues (hept-HBC) as stabilizing ligands. These novel RuNPs were characterized by the state-of-the-art techniques (TEM, HRTEM, XPS and TGA) and their surface chemistry was studied by using CO as a probe molecule (solid state NMR and DRIFT). The interaction of distorted nanographenes with the ruthenium surface was also investigated by solution NMR and DFT calculations. The presence of the saddle curvature within the structure of the nanographene is crucial for MNP stabilization. In addition, the catalytic activity of these canopy RuNPs has been evaluated in the hydrogenation of various aromatic substrates, finding interesting ligand effects in terms of activity and selectivity.
Results and discussion
Synthesis, characterization and surface studies
Heptagon-containing saddle-shaped nanographenes were prepared according to a previously reported procedure, via a Co-catalyzed alkyne cyclotrimerization followed by a Scholl cyclodehydrogenation.25 In particular, two different hept-HBC nanographenes were synthesized and used as stabilizers to prepare RuNPs through an organometallic approach: one functionalized with a carbonyl group (1), and another one an all-carbon analogue with a methylene unit (2) (Scheme 1). Comparison of both ligands would give information about the influence of the carbonyl moiety or the curved aromatic surface on the stabilization of the MNPs. More specifically, RuNPs were obtained by reduction of Ru(COD)(COT) (COD = cyclooctadiene; COT = cyclooctatriene) in THF under 3 bar H2 in the presence of 0.1 equivalents (equiv.) of the corresponding hept-HBCs (Scheme 1 and see ESI Section S1, Experimental part†). After purification by washing with pentane, the resulting RuNPs were characterized by Transmission Electronic Microscopy (TEM) and High Resolution TEM (HRTEM) (Fig. 1). Microscopy analysis of Ru@1 and Ru@2 revealed the formation of spherical, crystalline and well-dispersed NPs with a mean diameter of 1.6 ± 0.4 and 1.6 ± 0.5 nm, respectively. Both nanomaterials presented the hexagonal close packed (hcp) structure characteristic of bulk ruthenium. Size and crystalline structure were confirmed by X-ray powder diffraction (XRD) (see ESI Section S2, Fig. S1†). | ||
| Scheme 1 Synthesis of Ru@1 and Ru@2 by using heptagon-containing saddle-shaped nanographenes (hept-HBCs) 1 and 2 as stabilizers. | ||
 | ||
| Fig. 1 TEM images and size histograms of (a) of Ru@1 and (c) Ru@2. (b) Fourier analysis applied to a HRTEM micrograph of Ru@1, which displays reflections to the (101), (102) and (100) atomic planes. (d) HRTEM image of Ru@2 showing a lattice fringe spacing of 2.32 Å that corresponds to the Ru (001) crystal plane of metallic Ru. Both HRTEM images reveal the presence of crystalline RuNPs retaining the hcp structure. | ||
The metal percentage of RuNPs was determined by thermogravimetric analysis (TGA), giving ruthenium contents of 58.8 and 57.6 wt% for Ru@1 and Ru@2, respectively (see ESI Section S3, Fig. S2 and S3†).26 These metal content values, similar to theoretical ones (∼60%), suggest the coordination of all hept-HBC molecules added during the synthesis (0.1 equiv.). However, the Ru(s)/L ratio for Ru@1 and Ru@2 (ca. ∼6) is not large enough to accommodate all the nanographene molecules on the ruthenium surface (see ESI Section S3, Table S1†). Thus, these remaining molecules may be organized in a second sphere of coordination, probably by π–π stacking between the aromatic rings of the hept-HBCs.13
The surface chemistry of Ru@1 and Ru@2 was investigated by solid-state 13C MAS-NMR with and without 1H–13C cross-polarization (CP). Most of the hept-HBC signals can be identified in the MAS NMR spectra of RuNPs recorded on purified samples (Fig. 2a and S4, see ESI Section S4†). In addition to the intense signals corresponding to the tBu groups at ca. 27 ppm, a broad resonance between 120 and 130 ppm for the aromatic rings is clearly observed. The presence of this wide peak indicates that aromatic moieties of 1 and 2 were not hydrogenated during the nanoparticle synthesis, being able to stabilize the RuNPs through aromatic π-to-metal surface interactions. Moreover, in the case of Ru@1, the peak corresponding to CO is not visible (normally observed at ca. 200 ppm), suggesting that 1 also coordinates to the MNP through the carbonyl group by σ-donation of the oxygen lone pair to Ru. This lack of visibility is attributed to a line broadening produced by the coordination of a ligand to the metal surface.5,27 Coordination of CO has been demonstrated to be an ideal tool to investigate the MNP surface through the location of the active sites. While the CO molecules located at the faces of the MNPs are normally coordinated in a bridging mode (COb), the COs placed on their edges or apexes are coordinated in a terminal mode (COt).28 The 13C MAS NMR spectra of solid samples of Ru@1 and Ru@2 NPs exposed to 1 bar of 13CO (Fig. 2b) show two new signals attributed to adsorbed CO molecules. The broad peak at about δ ∼ 230 ppm can be assigned to COb, while the sharp resonance at δ = 200 ppm is attributed to COt. Comparing the intensities of these two peaks, we can deduce that in both cases most of the CO molecules are coordinated in a terminal way. This is probably because the faces of the particle are not easily available due to the coordination of the saddle-shaped nanographenes through π-interactions between the aromatic rings and MNP faces. However, this interaction does not poison the RuNP catalysts, since under catalytic conditions these canopy RuNPs are highly active in the hydrogenation of aromatic compounds (reaction that requires MNPs with available faces on their surface) (vide infra). Furthermore, the presence of spinning side bands (*) indicates that COt are static on the MNP surface, which can be a consequence of the close coordination of hept-HBCs, thus restricting their mobility. In the 13C CP-MAS NMR spectra of Ru@1 and Ru@2 (see ESI Section S4, Fig. S5†) the intensity of the COt signals slightly increases compared to the COb ones. This means that bridging COs are less affected by cross polarization than terminal CO molecules, which are closer to the hydrogen carrier molecules, namely hept-HBCs.
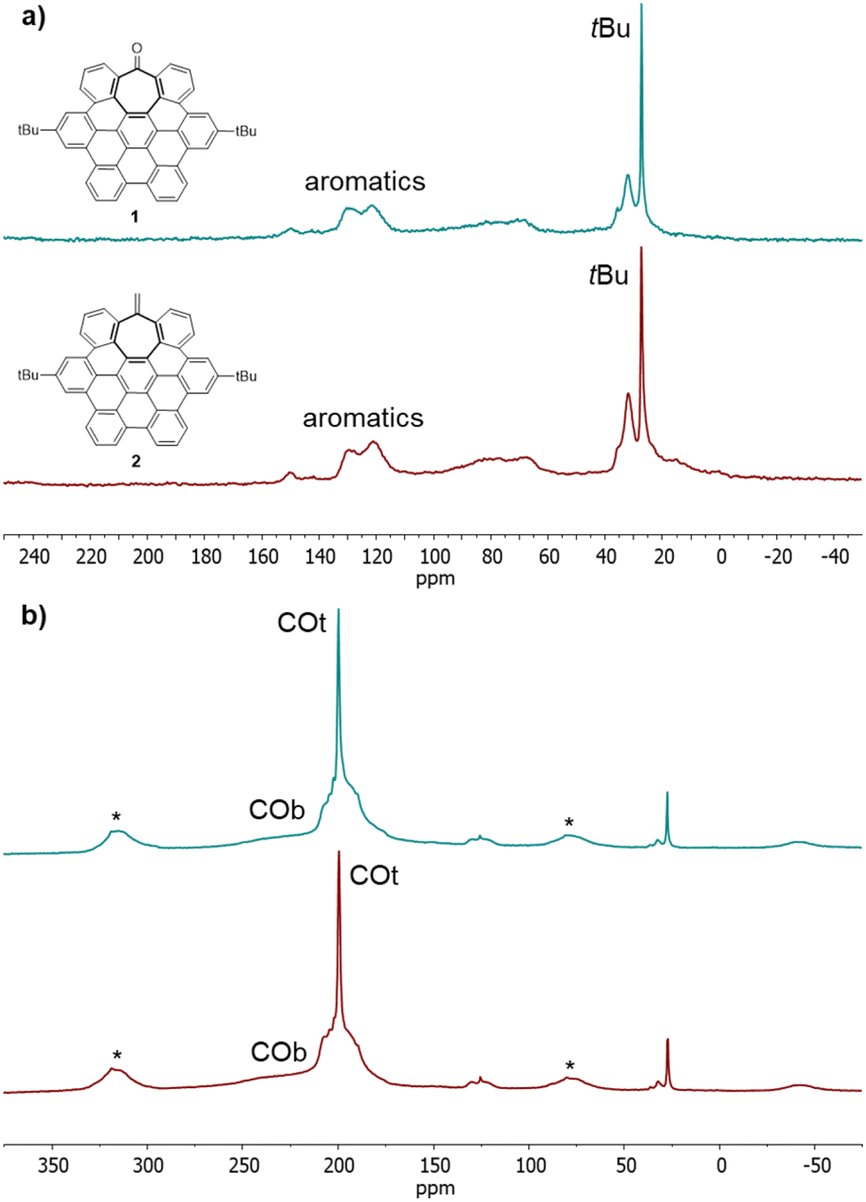 | ||
| Fig. 2 (a) 13C CP-MAS NMR spectra of Ru@1 (blue) and Ru@2 (red). (b) 13C MAS NMR spectra of Ru@1 (blue) and Ru@2 (red) after exposure to 13CO (1 bar, 20 h, r.t.). The signals with asterisks correspond to spinning side bands. | ||
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was also used to investigate the surface chemistry of Ru@1 and Ru@2. To continue with the location of the free sites present on the MNP surface, CO was bubbled for 5 min into a THF solution of Ru@1/Ru@2. Interestingly, DRIFT spectra of purified Ru@1 and Ru@2 NPs already showed the characteristic band of CO absorption at ca. 2000 cm−1 before the reaction with carbon monoxide (see ESI Section S5, Fig. S6 and S7†). This small amount of CO coordinated to the Ru surface is due to the partial decarbonylation of THF used during the synthesis, as previously reported.5 This is an indication of the high reactivity of these RuNPs. After bubbling CO, the aforementioned band showed an increase in intensity and shift to higher frequency (2019–2024 cm−1), evidencing the coordination of CO, and the presence of a large number of available active sites in these canopy RuNPs.
X-ray photoelectron spectroscopy (XPS) is a well-known technique to analyze the metal composition and oxidation states of surface catalysts. In addition, it has been recently reported to investigate the coordination mode of surface ligands on the MNP surface.6,14e,f A decrease in the binding energy of coordinating atoms (i.e. N-atoms) indicates a loss of electron density and the coordination of the ligand to the metal surface through these electron donor atoms. Unfortunately, the O 1s signal for Ru@1 presents identical binding energy (BE) to the free hept-HBC 1 used as a stabilizer (532.4 eV; see ESI Section S6, Fig. S8†). Thus, we cannot evidence the coordination of 1 through the oxygen of the carbonyl group to the Ru surface by XPS. In spite of this, a shoulder at ca. 530 eV can be clearly observed, which can be attributed to RuO2 and indicates that the Ru surface could be partially oxidized. The oxidation states of the as-synthetized RuNPs have been studied by XPS analysis of the Ru 3p region, since the overlapping of the Ru 3d and C 1s signals makes the deconvolution and interpretation difficult. Fig. S9a and b (see ESI Section S6†) show a Ru 3p5/2 peak at a BE of ca. 462 eV which can be deconvoluted in two components. The main contribution located at 461.8–461.3 eV is assigned to Ru(0),29 which suggests that Ru@1 and Ru@2 are mainly composed of metallic ruthenium. Another contribution at 463.7–463.8 eV corresponds to some RuO2 (26–28%),29 probably formed during the preparation of the samples for XPS in air.
DFT and NMR coordination studies
The solubility of Ru@1 and Ru@2 in organic solvents such as THF allowed us to characterize them by liquid NMR, employing 1D and 2D experiments to carry out a complete assignation of the signals, and obtaining interesting findings about the interaction between the hept-HBC nanographenes and the RuNP surface. The 1H NMR peak assignation of the free 1 and 2 ligands was also secured by DFT calculations (see ESI Section S7, Fig. S11†). The 1H NMR spectrum of Ru@1 in THF(d8) showed that the resonances of the aromatic rings are significantly displaced with respect to those of the free nanographene (Fig. 3), and full-assignation was completed with 2D 1H–1H gCOSY (see ESI Section S8, Fig. S18†). The signals affected by the proximity of the ruthenium surface, due to aromatic π-to-metal surface interactions, are normally deshielded and thus displaced to lower field. For example, the peak attributed to the aromatic protons closest to the carbonyl group (protons a) is considerably shifted to downfield (from 7.87 to 8.26 ppm). In particular, this remarkable displacement suggests that hept-HBC 1 is also coordinated to the ruthenium surface through the carbonyl group. The absence of the carbonyl signal due to a line broadening in the 1H–13C gHMBC spectrum of Ru@1 supports the σ-interaction between the CO group and the metal surface (see ESI Section S8, Fig. S19†). This 2D experiment has been selected due to its sensitivity which is 20 times better than that of a simple 13C experiment. Other aromatic peaks shifted to downfield, thus interacting with the ruthenium surface, are those belonging to protons f, g and h (Fig. 3). On the other hand, aromatic resonances influenced by the presence of another polycyclic aromatic molecule coordinated through π–π stacking (located in a second coordination sphere) are more shielded and thus shifted to high field (b and c). The upfield shift of the aromatic resonances due to a self-association by π–π stacking between different molecules of 1 was previously observed.12 Finally, resonances corresponding to protons d, e and those of the tBu groups are not essentially affected, which means that these protons are far away from both the ruthenium surface and nearest-neighbor nanographenes (Fig. 3 and S20, see ESI Section S8†). These results confirm the presence of 1 at the ruthenium surface, probably coordinated through the carbonyl group (σ-donation) and the aromatics (π-interactions) at the same time, and the existence of a second sphere of coordination through π–π stacking between the aromatic rings of the hept-HBC nanographenes, which was previously deduced by thermogravimetric analysis (see ESI Section S3, Table S1†).
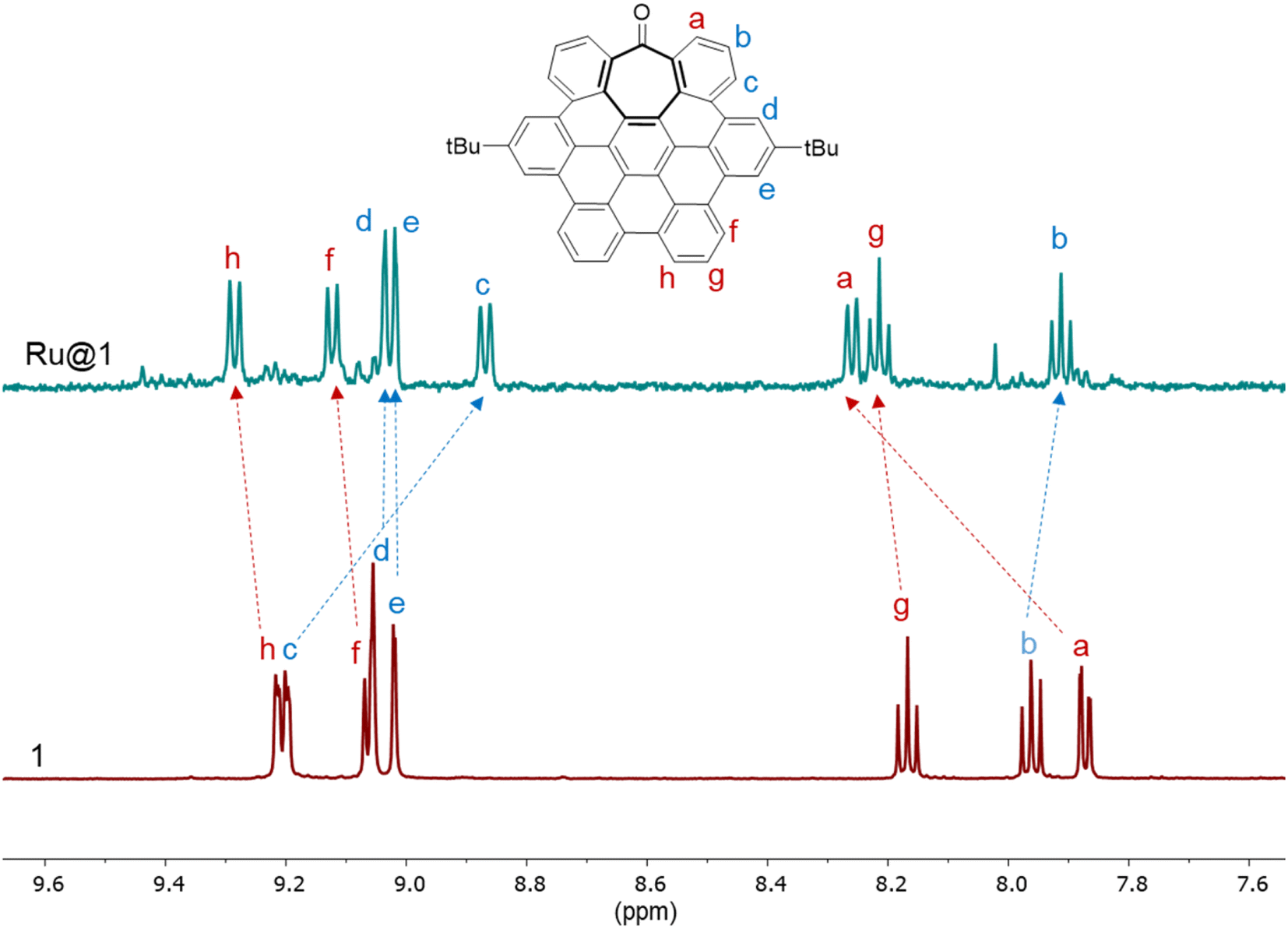 | ||
| Fig. 3 Aromatic region of 1H NMR spectra of 1 (red) and Ru@1 (blue) in THF(d8). | ||
The 1H NMR spectrum of Ru@2 in THF(d8) also showed a noticeable displacement of the aromatic resonances with respect to the signals of the nanographene 2. Again, we can obtain valuable insights about ruthenium–nanographene interactions by analyzing their chemical shifts by 1H and 2D 1H–1H gCOSY NMR (Fig. 4, S21 and S22, see ESI Section S8†). However, here the assignment is somewhat more complex since some signals are split in two. Again, protons a next to the vinyl group are close to the ruthenium surface and thus shifted to downfield. This, together with the invisibility of the characteristic signal of the vinyl group at 5.03 ppm, points that hept-HBC nanographene 2 interacts with the metal surface through π–metal interactions of this aromatic ring. Interestingly, protons f, g and h are also shifted to high field, which means that these protons are also near to the ruthenium surface. In contrast, the signal for protons c is not only displaced to upfield, which means that these protons are mostly interacting with another hept-HBC molecule through π–π stacking, but it is also split in two. This double set of signals is probably due to two different coordination modes of this nanographene on the NP surface or to a loss of symmetry in the coordinated nanographene. The same signal-split happens to the resonances corresponding to protons f and b (in the case of b one of the two signals is shifted to downfield and the other one to upfield). As in the 1H NMR spectrum of Ru@1, protons d and e, together with the tBu peak (see ESI Section S8, Fig. S22†), are practically in the same position, and therefore we can assume that they are almost not influenced neither by the ruthenium surface, nor by other nanographene molecules, due to the bulkiness of these tBu groups. Again, these results confirm the coordination of 2 to the nanoparticle surface through π-interactions and the existence of a second coordination sphere (π–π stacking). Altogether these data corroborate that hept-HBC molecules 1 and 2 interact in different manners with the Ru surface, and thus we should expect an important ligand effect in catalysis (vide infra).
 | ||
| Fig. 4 Aromatic region of 1H NMR spectra of 2 (red) and Ru@2 (blue) in THF(d8). | ||
To check the coordination mode and stability of the ligands attached at the ruthenium NP surface we have carried out DFT calculations at the DFT-PBE level of theory. These calculations were performed on an hcp spherical model containing 57 ruthenium atoms (13 in the core and 44 on the surface) with a diameter of 1 nm close to the aforementioned mean diameter of Ru@1 and Ru@2. Two hydrogen coverages, 0.84 or 1.07H/Rusurf, were also considered which correspond to respectively 37 or 47 hydrogen atoms coordinated on the RuNP surface. On these two hydrogenated NPs the coordination of three 1 or 2 ligands at the RuNP surface can occur through two coordination modes involving: (i) the methylene carbon of the heptafulvene group or the oxygen atom of the tropone group and the HBC moiety (Fig. 5a) or (ii) only the HBC moiety (Fig. 5b). Interestingly, when the three 1 or 2 ligands interact with the Ru55H37 surface only through the HBC moiety we observe a cupola-type coordination in which the CO or CH2 groups point outwards. In this case, the nanographenes are μ-η2:η2 coordinated to the atoms of the edges creating a cavity between the center of the nanographene and the surface of the NP. From a thermodynamic point of view, this coordination mode is between 22 and 24 kcal per mol per ligand less stable than the second one in which the nanographene ligand is coordinated by both the methylene carbon of the heptafulvene group or the oxygen atom of the tropone group and the HBC moiety. In this case, we observe the σ/π-donation of the oxygen atom in 1 or the π-donation of the methylene group in 2 and an aromatic π-to-metal surface interaction, with at least 3 aromatic cycles of each ligand exhibiting a typical μ3-η2:η2:η2 face-capping coordination mode. When adsorbed on an edge, 1 or 2 involves a nice canopy-like coordination thanks to the curvature of the ligands. Small species, such as hydrides, can coordinate below this protection without any steric discomfort (Fig. 6b).
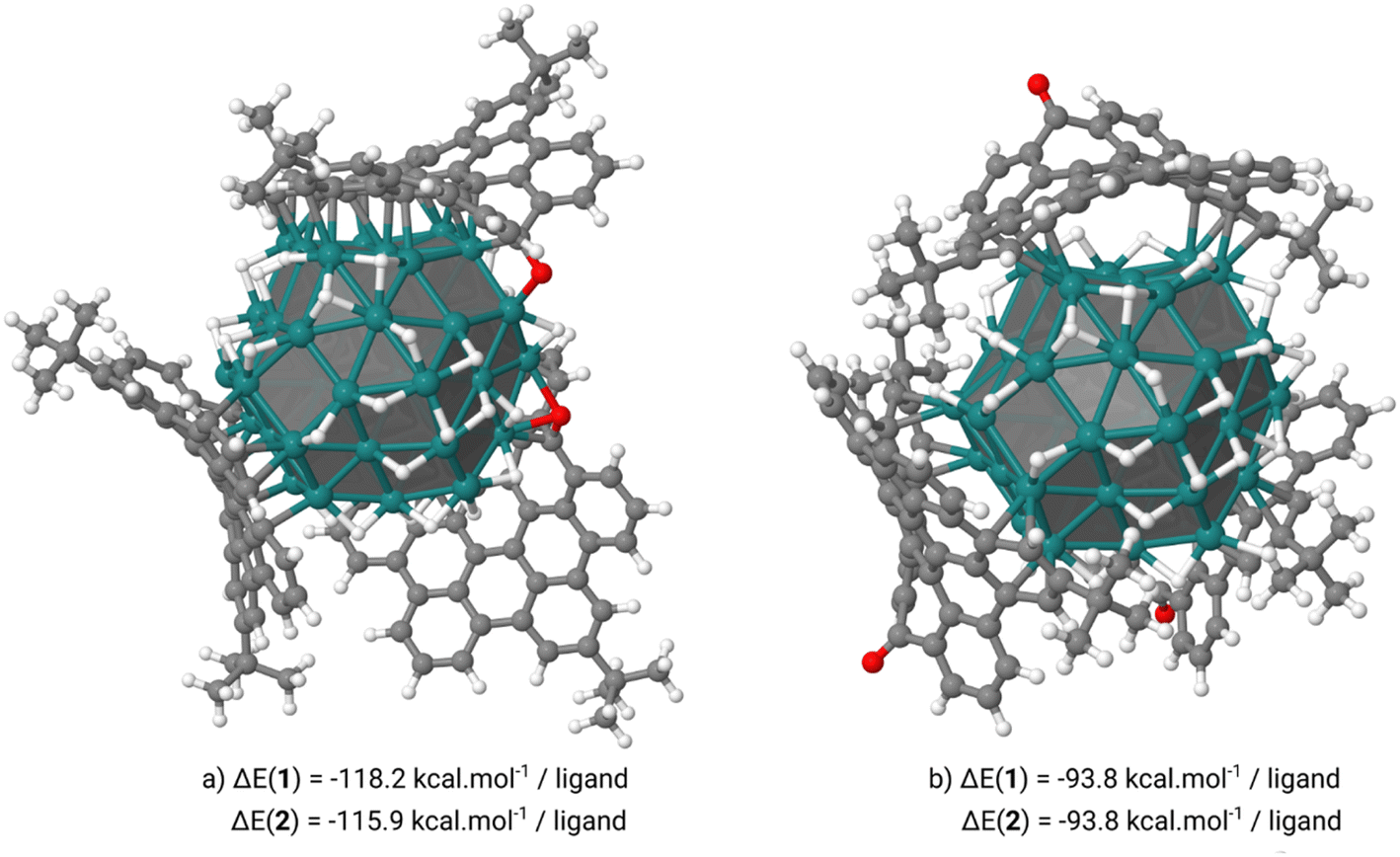 | ||
| Fig. 5 Comparison of the two possible coordination modes of 1 and 2 on the Ru55H37 NP, through: (a) the oxygen atom of the tropone group and the HBC moiety (canopy mode) and (b) only the HBC moiety (cupola mode). The very similar structures obtained for the Ru@2 model are not shown. Average adsorption energies are also given below each 3D structure. | ||
 | ||
| Fig. 6 Drift effect on the canopy coordination mode of 1 on Ru55H37 NP. Only adsorption energies are shown for the Ru@2 model [ΔE(2)], given the structural similarity to the Ru@1 model. On the left (a), there is only one wide canopy-protected zone, whereas in (b) two canopy-protected zones are available. | ||
For comparison (see ESI Section S7, Fig. S12†), in a higher hydrogenated model (Ru57H47) the cupola-type adsorption strength is slightly affected by the increase of the number of surface H atoms (−93.8 kcal mol−1 on Ru55H37vs. −77.1 kcal mol−1 on Ru57H47) due to the migration of the hydrogen atoms within the aforementioned cavity. The σ/π-CO/π-HBC coordination is much more strongly affected (−113.4 kcal mol−1 on Ru57H37vs. −88.4 kcal mol−1 on Ru57H47) but remains more favorable than the cupola one. Another interesting point is the possible mobility of the ligands once coordinated to the RuNP surface. As we can see in Fig. 6 the drift of the ligands inducing the coordination of 1 or 2 by the outermost and not central parts is a thermodynamically facile process in view of the energy difference between these two coordination modes (+4.8 kcal mol−1 for Ru57H37@1 and +3.8 kcal mol−1 for Ru57H37@2). Thus, it is possible that this drift motion takes place dynamically on the surface of such canopied RuNPs.
Density of states (DOS), crystal orbital Hamilton population (COHP) and charge analysis30 studies have also been undertaken for the Ru57H37@1 and Ru57H37@2 models (see ESI Section S7, Fig. S13 and S14†). It appears that the strong coordination of 1 and 2 on the Ru surface stems from a mixing of some d states of the metal core with the molecular orbitals (MOs) of the ligands as well as from a significant metal-to-ligand charge transfer (MLCT). Some typical MOs of the Ru57H37@1 model are reported in Fig. 7. Bonding MOs close to the Fermi energy involve d states of the metal core and π MOs of the surface ligands. The three most stable MOs shown in this figure lie below the bottom of the d-band of the metal core. These MLCT MOs partly account for the electron withdrawing ability of 1. Two of them also involve a weak contribution of the 5s band of the Ru core. The Ru57H37@2 model has similar MOs (see ESI Fig. S13†). As a result of the complex metal–ligand orbital electronic interaction, each ligand 1 withdraws on average 1.77|e| to the metal core, a value slightly higher than that found for 2 (1.59|e|). The COHP and DOS profiles for the Ru@1 and Ru@2 models are nevertheless very similar, with almost identical surface and core d-band center31 values (surface: 2.92 eV vs. 2.91 eV; core: 3.41 eV vs. 3.43 eV). On the basis of these basic descriptors, the catalytic activity of these species should not differ much.
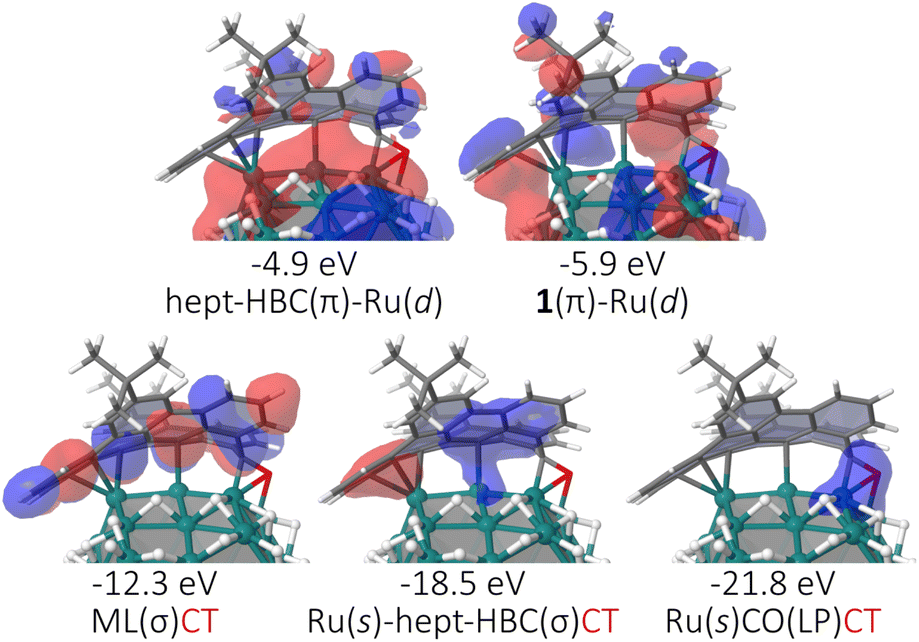 | ||
| Fig. 7 Selection of MOs typical of the metal–ligand interaction in the Ru@1 model. Similar MOs are found for Ru@2 (see Fig. S13†). Energies are given w.r.t. the Fermi energy. | ||
Finally, the possible accommodation of a fourth 1 or 2 ligand in the second coordination sphere of the RuNP was investigated (see ESI Section S7, Fig. S15†). The results show that the formation of a π–π stacking interaction between one of the three ligands coordinated to the RuNP surface and the fourth ligand is thermodynamically stable regardless of the orientation of the latter (between −26.5 and −32.1 kcal mol−1 for 1 and between −19.2 and −37.1 kcal mol−1 for 2). In all three cases, the average distance between the two ligands is around 4.3 Å, with a difference in stability governed by the steric hindrance of tBu groups. Indeed, in the most stable form (see ESI Section S7 and Fig. S15a†) the CO or CH2 groups point outwards and the two ligands are rotated by 90° with respect to each other, thus maximizing the π–π stacking interaction by avoiding steric clashes between tBu ligands.
Additional coordination studies
RuNPs have been generated on a large variety of carbon materials, including graphite,32 activated carbon,33 carbon nanotubes (CNTs),34 fullerenes,35 carbon nanofibers (CNFs)36 and graphenes.37 However, studies about the interaction of MNPs with carbon derived supports are practically restricted to DFT calculations. Thanks to the possibility of preparing hept-HBC nanographenes with different functional groups and the soluble nature of Ru@1 and Ru@2 in organic solvents (i.e. THF), coordination studies could be performed, obtaining useful information about MNP–nanographene interactions. To understand better the role of the curvature and the carbonyl group of these hept-HBC nanographenes in the stabilization of ruthenium nanoparticles a series of control experiments were carried out. First, we tried to generate RuNPs following the same organometallic approach that is described in Scheme 1, but using coronene (Fig. 8a) as stabilizer. However, after the reduction of Ru(COD)(COT) under 3 bar H2 in the presence of 0.5 equiv. of coronene, we did not observe the formation of RuNPs. TEM pictures only showed the presence of large-sized Ru agglomerates (see ESI Section S9, Fig. S24†). As we can see in Fig. S16a (see ESI Section S7†), coronene is a strong stabilizer, with −87.3 kcal per mol per ligand average adsorption energy, compared to −118.2 kcal mol−1 for 1 and −115.9 kcal mol−1 for 2. The COHP and DOS profiles of Ru57H37(coronene)3, plotted in Fig. S17,† are similar to those of the Ru@1 and Ru@2 models, with almost identical d-band center values (surface: 2.89 eV, core: 3.41 eV) and the same metal–ligand interaction characterized both by low-lying MLCT states and by a mixing between some d states of the metal moiety and some π MOs of coronene (see also the selection of MOs plotted in Fig. S17†). The electron withdrawing effect of coronene is however weaker (1.32|e|). Coronene remains almost planar, in sharp contrast to the ligands in the Ru@1 and Ru@2 models. One can speculate that it involves less flexibility and adaptability during the growth process. Since a polycyclic aromatic hydrocarbon like coronene is not able to stabilize RuNPs and the nanographene 2 is, we can consider that the curvature is essential to stabilize RuNPs. In fact, to the best of our knowledge, Ru@2 is the first example of MNPs stabilized by a purely aromatic ligand, where π-interactions between the aromatic rings and the metal surface are more than enough to stabilize metal nanoparticles. Once observed the importance of the curvature, we decided to investigate the role of the carbonyl group present in nanographene 1 in the stabilization of RuNPs. The ability of oxygen donor ligands such as alcohols,38 cyclodextrines39 and carboxylic acids16,40 to stabilize RuNPs is very well known. However, there is no example about ketones-stabilized MNPs prepared by the organometallic approach. We first decomposed Ru(COD)(COT) in THF under 3 bar H2 in the presence of benzophenone (Fig. 8b), obtaining a black dispersion. Once purified, TEM analysis revealed the presence of two populations of nanoparticles with mean sizes of 1.4 ± 0.3 and 2.7 ± 0.7 nm, respectively, besides a large number of agglomerates (see ESI Section S9, Fig. S25†). Although benzophenone presents a carbonyl group together with two phenyl groups, we observed that this molecule is not an effective ligand to stabilize RuNPs. Going further with these coordination studies, we also employed dibenzosuberone (Fig. 8c) as MNP stabilizer. Apart from the carbonyl and two phenyl groups, this organic molecule also bears a heptacycle ring. Interestingly, the curvature caused by the presence of the seven membered ring enhances the stabilization of RuNPs. After the decomposition of Ru(COD)(COT) under usual conditions (3 bar H2, THF, r.t.) using dibenzosuberone as stabilizer, small RuNPs were obtained. TEM micrographs of these RuNPs after purification by washing with pentane showed disperse and well distributed nanoparticles with a mean diameter of 1.3 ± 0.6 nm (see ESI Section S9, Fig. S26†). Regarding the coordination of benzophenone, as can be seen in Fig. S16c (see ESI Section S7†), the ligands are mainly coordinated via the CO moiety or at least by this group and by the ipso and ortho carbons of the phenyl groups, which explains the rather low coordination energy around −46.9 kcal mol−1 for this ligand. For dibenzosuberone (see ESI Section S7, Fig. S16b†), the presence of the heptacycle ring allows, in addition to CO coordination, the formation of a strongly stabilizing μ3-η2:η2:η2 face-capping interaction between one of the phenyl groups and the RuNP surface. In this case, the coordination energy per ligand is 1.5 times higher than that of benzophenone (−70.6 vs. −46.9 kcal mol−1). Therefore, we can assume that although the lone pair electrons of the CO group may help to stabilize RuNPs, the most important stabilizing feature in nanographene 1 is its curvature, which favors an optimal coordination on the surface.
 | ||
| Fig. 8 2D and 3D structures of the polyaromatic hydrocarbons used in control experiments: (a) coronene, (b) benzophenone and (c) dibenzosuberone. Hydrogen atoms are not shown for the sake of simplicity. | ||
Catalytic studies
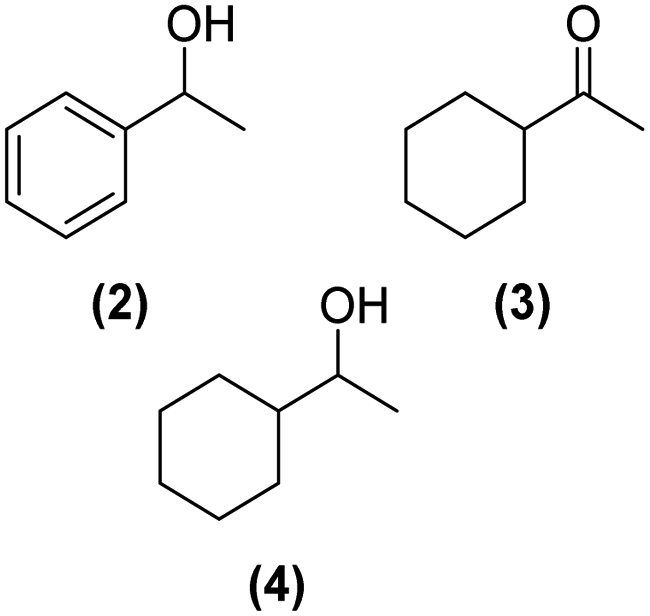
Once observed that non-planar hept-HBC nanographenes are capable of stabilizing small RuNPs, the effect of this distorted polycyclic aromatic hydrocarbon on their catalytic activity has been investigated. Ligand-stabilized RuNPs have been previously reported as efficient nanocatalysts for mild hydrogenation reactions.1,2,3a Thus, in a first approach, acetophenone hydrogenation was used as a model reaction to probe the surface reactivity of Ru@1 and Ru@2 under mild conditions (50 °C, 10 bar H2, 20 h), since it presents the possibility of evaluating the selectivity between aromatic and carbonyl hydrogenation. In fact, acetophenone can be hydrogenated to 1-cyclohexylethanol through two different pathways (routes A and B of Fig. 9a). An interesting impact of the molecular structure of hept-HBCs on the activity and selectivity of the RuNPs was observed. More specifically, it was found that although both catalysts prefer to hydrogenate the aromatic ring rather than the ketone (preferably go through route B), Ru@2 is clearly more active than Ru@1 (Fig. 9b and c). Comparing their initial conversion rates, Ru@2 hydrogenates the phenyl group faster than Ru@1. After 1 hour reaction, Ru@2 exhibits an estimated conversion of around 60% (estimated TOF = 12.5 h−1),41 with the 1-cyclohexylethanone/1-phenylethanol ratio being 2.7, whereas the conversion with Ru@1 at the same time is only 35% (estimated TOF = 7.3 h−1) with a much lower ratio (i.e. 1.6) (see kinetic data in Fig. 9b and c). The higher the 1-cyclohexylethanone/1-phenylethanol ratio during the first stages of the reaction, the higher the capability of the catalyst to hydrogenate the aromatic ring. However, at higher reaction time (i.e. 20 hours), complete conversion is reached with both catalysts with slight differences in terms of selectivity (Table 1, entries 1 and 2). When working with Ru@2, 1-cyclohexylethanol is the main product (i.e. 85% of selectivity), with the only side product being 1-cyclohexylethanone. On the other hand, Ru@1 appears to be less selective, producing a mixture of 1-phenylethanol, 1-cyclohexylethanone and 1-cyclohexylethanol. The latter catalytic results illustrate the high activity of Ru@1 and Ru@2 in the mild hydrogenation of aromatic rings. | ||
| Fig. 9 (a) Different catalytic pathways in the hydrogenation of acetophenone. Hydrogenation of acetophenone using (b) Ru@1 and (c) Ru@2 as catalysts. Reaction conditions: acetophenone (0.15 mmol), 2 mg RuNPs (7.2 × 10−3 mmol Ru assuming ∼60% Ru from TGA/ICP and ∼60% Ru(s)), THF (2 mL), hydrogen (10 bar), 50 °C. | ||
| Entry | Catalyst | Substrates | Products | Conversionb | Selectivityb |
|---|---|---|---|---|---|
| a Reactions conditions: substrate (0.15 mmol), 2 mg RuNPs (7.2 × 10−3 mmol Ru assuming ∼60% Ru from TGA/ICP and ∼60% Ru(s)), THF (2 mL), hydrogen (10 bar), 50 °C, 20 h. b Conversions and selectivities were determined by GC using dodecane as the internal standard, and confirmed by GC-MS. c Cyclohexanol was isolated as a pure product with a yield of 88.8%. d Reaction temperature (70 °C). | |||||
| 1 | Ru@1 |

|
|
>99% | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:4 = 4:22:74 :3:4 = 4:22:74 |
| 2 | Ru@2 | >99% | 2:3:4 = 0:15:85 |
||
| 3 | Ru@1 |
|
|
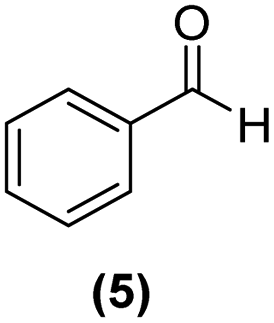
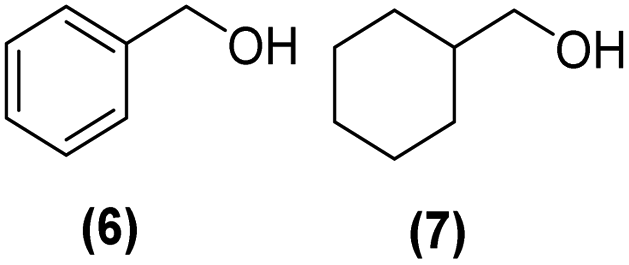
>99% | 6:7 = 67:33 |
| 4 | Ru@2 | >99% | 6:7 = 56:44 |
||
| 5 | Ru@1 |

|
|
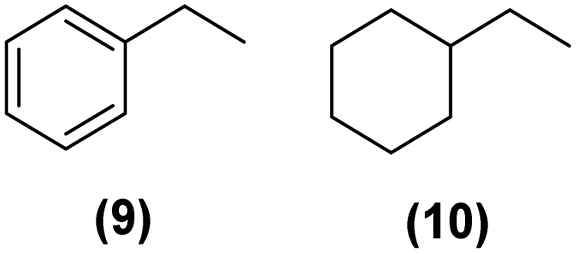
>99% | 9:10 = 6:94 |
| 6 | Ru@2 | >99% | 9:10 = 0:100 |
||
| 7 | Ru@1 |

|
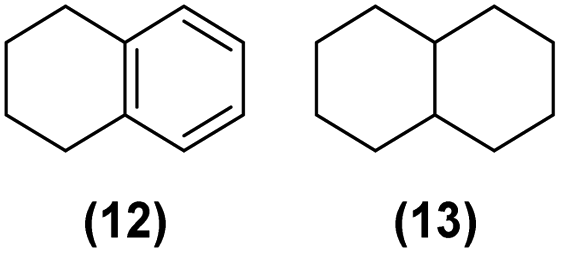
|
>99% | 12:13 = 45:55 |
| 8 | Ru@2 | >99% | 12:13 = 16:84 |
||
| 9 | Ru@1 |

|

|
>99% | 15:16 = 59:41 |
| 10 | Ru@2 | >99% | 15:16 = 21:79 |
||
| 11 | Ru@1 |

|

|
>99% | — |
| 12 | Ru@2 | >99% | — | ||
| 13 | Ru@1 |

|

|
>99% | — |
| 14 | Ru@2 | >99% | — | ||
| 15 | Ru@1 |

|

|
94% | — |
| 16 | Ru@2c | >99% | — | ||
| 17 | Ru@1 |
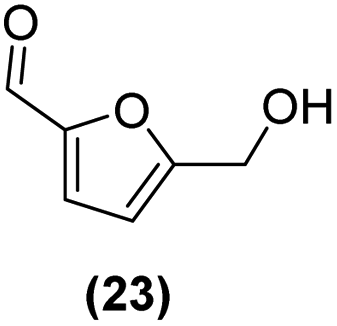
|

|
98% | 24:25 = 77:23 |
| 18 | Ru@2 | 97% | 24:25 = 50:50 |
||
| 19 | Ru@1 |
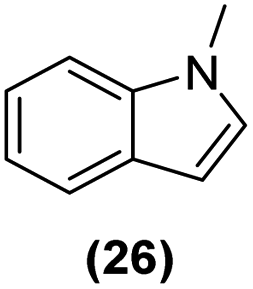
|

|
49% | 27:28:29 = 13:6:81 |
| >99%d | 27:28:29 = 5:0:95 |
||||
| 20 | Ru@2 | 69% | 27:28:29 = 12:3:85 |
||
| >99%d | 27:28:29 = 1:0:99 |
||||
Motivated by the high activity of these canopy RuNPs in the hydrogenation of aromatic rings, we decided to further investigate the catalytic differences of Ru@1 and Ru@2 in the hydrogenation of a series of arenes under the previously used conditions (50 °C, 10 bar H2, 20 h). As a rule, Ru@2 is a more active catalytic system than Ru@1 (similar tendency as in the case of acetophenone). For example, in the hydrogenation of benzaldehyde (Table 1, entries 3 and 4), the conversion with both catalysts is >99% but Ru@2 produces a higher percentage of the over-reduced product. Similar results were found in the hydrogenation of styrene, naphthalene and biphenyl (Table 1 entries 5–10), where, in all cases Ru@2 yields a higher amount of the totally hydrogenated product. On the other hand, toluene and anisole were successfully hydrogenated resulting in a complete reduction of the aromatic ring with both catalysts (Table 1 entries 11–14). Phenol and hydroxymethylfurfural (HMF) were also effectively hydrogenated, highlighting the potential of these canopy RuNPs for the transformation of biomass derivatives. Again, Ru@2 showed a higher reactivity in the hydrogenation of these biomass platform molecules (Table 1, entries 15–18). Finally, the activity of Ru@1 and Ru@2 was also evaluated in the hydrogenation of 1-methyl indole, which is a promising LOHC due to its high hydrogen content (5.76 wt%) and low melting point (−20 °C). Under these moderate conditions, Ru@2 shows a conversion of 69%, with 85% selectivity towards the octahydro-1-methylindole, while Ru@1 presents a conversion of only 49% with a similar selectivity (Table 1, entries 19 and 20). On the other hand, when the reaction was performed at 70 °C instead of 50 °C, the conversion with both catalysts is >99% with high selectivities towards the product 29 (99 and 95%), incorporating up to four hydrogen molecules into the initial substrate. Normally, the hydrogenation of N-indole, or others LOHCs, requires harsher conditions,42 demonstrating again the great potential of these NPs in the hydrogenation of aromatic compounds.
Given the similar DFT adsorption energies (Fig. 5a), d band centers and DOS and COHP profiles (see ESI Section S7, Fig. S13†) found between Ru@1 models and their Ru@2 counterparts, the origin of the higher activity of Ru@2 in comparison with Ru@1 is not obvious. However, two different effects could be involved. On one hand, the slightly weaker interaction between 2 and the metal surface together with a slightly enhanced drift effect could facilitate the approach of the aromatic substrates to the ruthenium (Fig. 6). On the other hand, Fig. 9 shows a slower conversion of 1-phenylethanol into 1-cyclohexylethanol by Ru@1. It could be related to the formation of hydrogen bonds between 1-phenylethanol and the CO group of 1 that could compete with the π coordination of the phenyl group, and even favor an exchange of 1-phenylethanol with the second coordination sphere, thus delaying the hydrogenation of the phenyl group. In any case, an interesting ligand effect was observed, since a minor modification on the hept-HBC structure (CCH2 instead of CO) was reflected in a substantial increase in the aromatic hydrogenation activity of the herein presented RuNPs.
To investigate the stability of Ru@1 and Ru@2 NPs a set of experiments were carried out. TEM and HRTEM analyses after the hydrogenation of acetophenone, (Table 1, entries 1 and 2) showed MNPs with the same morphology, size and crystallinity as the corresponding as-synthesized ones (see ESI Section S9, Fig. S27 and S28†), demonstrating their robustness under reaction conditions (50 °C, 10 bar H2, 20 h). Going further with stability studies, a multi-addition experiment was performed. Here, the hydrogenation of anisole was done in a multiple addition way. More specifically, each 12 hours new starting material was added to the reaction medium, analysing the anisole conversion just before each addition. The reactivity of Ru@1 and Ru@2 NPs remains practically unaltered for at least 4 addition cycles (conversions between 98 and 94%). After that, the conversion slightly decreases to 90–80%, remaining constant until the 8th cycle (see ESI Section S10, Fig. S31a and b†). TEM micrographs of Ru@1 and Ru@2 after the multi-addition experiments revealed a small increase in size and distribution, from ca. 1.6 nm to ca. 2.0 nm (see ESI Section S9, Fig. S29 and S30†), which could explain the slight decrease in the activity after the 4th cycle. In any case, these results point to a good stability of these canopied nanoparticles under reaction conditions during long reaction times (up to 96 h). Finally, the hydrogenation of toluene was performed under neat conditions by using Ru@2 as a catalyst, observing a remarkable increase in the turnover number (TON) of the catalyst. While under standard conditions (2 mL THF, 10 bar H2, 50 °C and 20 h) Ru@2 showed a TON of 20.8 (Table 1, entry 12), in neat toluene the TON increases to a value of 1420 (see ESI Section S11 and Fig. S32†).43 This notable increase probably is due to a concentration effect, and a substrate-THF competition for their coordination to the MNP surface. The high number of catalytic cycles performed per surface Ru atom (maximum TON = 1420) together with the activity of the catalysts with time (up to 96 h), highlights the great stability of the nanocatalyst and its high potential as a hydrogenation catalyst.
Conclusions
We have successfully stabilized RuNPs with non-planar hept-HBC nanographenes by following an organometallic approach (Ru@1 and Ru@2). Normally polycyclic aromatic hydrocarbons are not effective stabilizing ligands, but interestingly the curvature of hept-HBCs makes them efficient MNP stabilizers. A combined theoretical/experimental study allowed us to investigate the coordination modes and dynamics of the heptagon-containing saddle-shaped nanographenes used as stabilizers. We have shown that hept-HBC 1, functionalized with a carbonyl group, coordinates to the ruthenium surface through a double interaction: (i) through the carbonyl group by σ/π-donation and (ii) via interactions of the σ or π MOs of the aromatic rings with the metal surface, which also results in a significant metal-to-ligand charge transfer. In a similar way, hept-HBC 2 that bears the vinyl group coordinates to the ruthenium surface atoms via both the methylene carbon and the HBC moiety. In any case, the drift of these ligands on the NP surface is a thermodynamically facile process. Non-planar hept-HBC-stabilized RuNPs showed an interesting application in catalysis, since they were highly active in the hydrogenation of aromatic substrates under mild conditions. In addition to a wide number of aromatic substrates, Ru@1 and Ru@2 efficiently hydrogenated platform molecules derived from biomass (i.e. HMF) or LOHCs (i.e. N-indole). Moreover, an interesting ligand effect was observed; Ru@2 stabilized with the vinylic hept-HBC was clearly more active than Ru@1. Subtle modification of the nanographenes used as stabilizers had a notable effect on the catalytic performance of the Ru NPs in the aromatic hydrogenation activity. Finally, the stability of the obtained RuNPs was investigated by multiple addition experiments, proving to be stable catalysts for at least 96 h. Therefore, we have effectively stabilized for the first time colloidal RuNPs with polycyclic aromatic hydrocarbons, finding interesting insights about their coordination and dynamics. The similarity with carbon supports in general, and with graphenes in particular, makes these hept-HBCs very attractive compounds to stabilize MNPs and further investigate graphene–metal interactions. In addition, the remarkably activity (maximum TON = 1420) and stability (up to 96 h) of these nanographene-stabilized RuNPs make them promising catalysts for reactions of industrial interest.Author contributions
C. C.-N. and A. G.-Z. performed the synthesis and structural/spectroscopic characterization of the nanocatalysts and the catalytic experiments and participated in writing – review and editing. A. H. G. D. and M. D. C. performed the synthesis and characterization of the heptagon-containing saddle-shaped nanographenes. P. O.-B. performed the liquid NMR study. I. d. R. and R. P. carried out the DFT calculations and participated in writing – review and editing. A. G. C. and L. M. M.-P. coordinated the tasks between researchers, conceived the idea of the project and wrote the original draft with input from other authors. All authors contributed to the design of the experiments, analysis of the results and preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Instituto de Tecnología Química (ITQ), Instituto de investigaciones Químicas (IIQ), Consejo Superior de Investigaciones Científicas (CSIC), Universitat Politècnica de València (UPV), University of Granada (UGR) and University of Seville (US) for the facilities. The authors also acknowledge the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (GA 677023), FEDER(EDRF) Junta de Andalucía-Consejería de Transformación Económica, Industria, Conocimiento y Universidades (P18-FR-2877), the Agencia Estatal de Investigación (AEI) and “Ramón y Cajal” programme (RYC2020-030031-I) for financial support. We also acknowledge the Electron Microscopy Service of the UPV for TEM facilities. C. Cerezo-Navarrete gratefully acknowledges the Generalitat Valenciana Predoctoral Fellowship (GVA: ACIF/2019/076). I. d. R. and R. P. thank the HPC CALcul en MIdi-Pyrénées (CALMIP, OLYMPE machine, grant P0611) for a very generous allocation of computer time on this project.References
-
(a)
U. Heiz and U. Landman, in Nanocatalysis, Springer, Berlin, 2007 CrossRef
; (b) D. Astruc, in Nanoparticles and Catalysis, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed
-
(a) A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS PubMed
-
(a) C. A. Schoenbaum, D. K. Schwartz and J. W. Medlin, Acc. Chem. Res., 2014, 47, 1438–1445 CrossRef CAS PubMed
-
(a) I. Schrader, J. Warneke, J. Backenköhler and S. Kunz, J. Am. Chem. Soc., 2015, 137, 905 CrossRef CAS PubMed
- L. M. Martínez-Prieto, C. Urbaneja, P. Palma, J. Campora, K. Philippot and B. Chaudret, Chem. Commun., 2015, 51, 4647 RSC
-
(a) L. M. Martínez-Prieto, I. Cano, A. Márquez, E. A. Baquero, S. Tricard, L. Cusinato, I. del Rosal, R. Poteau, Y. Coppel, K. Philippot, B. Chaudret, J. Campora and P. W. N. M. van Leeuwen, Chem. Sci., 2017, 8, 2931 RSC
-
(a) A. Hashimoto, K. Suenaga, A. Gloxer, K. Urita and S. Iijima, Nature, 2004, 430, 867 CrossRef PubMed
-
(a) P. Y. Huang, C. S. Ruiz-Vargas, A. M. van der Zande, W. S. Whitney, M. P. Levendorf, J. W. Kevek, S. Garg, J. S. Alden, C. J. Hustedt, Y. Zhu, J. Park, P. L. McEuen and D. A. Muller, Nature, 2011, 469, 389 CrossRef CAS PubMed
-
(a) E. D. Grayfer, L. S. Kibis, A. I. Stadnichenko, O. Y. Vilkov, A. I. Boronin, E. M. Slavinskaya, O. A. Stonkus and V. E. Fedorov, Carbon, 2015, 89, 290 CrossRef CAS
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Coord. Chem. Rev., 2016, 312, 99 CrossRef CAS
-
(a) M. A. Petrukhina, Y. Sevryugina, A. Y. Rogachev, E. A. Jackson and L. T. Scott, Organometallics, 2006, 25, 5492–5495 CrossRef CAS
- For recent reviews, see:
(a) S. H. Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630 CrossRef CAS PubMed
- A. H. G. David, S. Míguez-Lago, C. M. Cruz, J. M. Cuerva, V. Blanco and A. G. Campaña, Org. Mat., 2021, 3, 51 CAS
- Examples for solid-state NMR:
(a) T. Gutmann, A. Grünberg, N. Rothermel, M. Werner, M. Srour, S. Abdulhussain, S. Tan, Y. Xu, H. Breitzke and G. Buntkowsky, Solid State Nucl. Magn. Reson., 2013, 55–56, 1–11 CrossRef CAS PubMed
-
(a) C. Taglang, L. M. Martínez-Prieto, I. del Rosal, L. Maron, R. Poteau, K. Philippot, B. Chaudret, S. Perato, A. Sam Lone, C. Puente, C. Dugave, B. Rousseau and G. Pieters, Angew. Chem., Int. Ed., 2015, 54, 10474 CrossRef CAS PubMed
- R. González-Gómez, L. Cusinato, C. Bijani, Y. Coppel, P. Lecante, C. Amiens, I. del Rosal, K. Philippot and R. Poteau, Nanoscale, 2019, 11, 9392 RSC
-
(a) A. Gual, C. Godard, S. Castillón and C. Claver, Dalton Trans., 2010, 39, 11499 RSC
-
(a)
K. Weissermel and H.-J. Arpe, in Industrial Organic Chemistry, Wiley-VCH, Weinheim, 2008 Search PubMed
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023 CrossRef CAS PubMed
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74 CrossRef CAS PubMed
-
J. March, in Advanced Organic Chemistry: Reaction, Mechanism and Structure, Wiley-Interscience, New York, 1992 Search PubMed
-
R. L. Augustine, in Heterogeneous Catalysis for the Synthetic Chemistry, Marcel Dekker Inc., New York, 1996 Search PubMed
- See for example:
(a) X. Cui, A.-E. Surkus, K. Junge, C. Topf, J. Radnik, C. Kreyenschulte and M. Beller, Nat. Commun., 2016, 7, 11326 CrossRef PubMed
-
(a) D. González-Gálvez, P. Lara, O. Rivada-Wheelaghan, S. Conejero, B. Chaudret, K. Philippot and P. W. N. M. van Leeuwen, Catal.: Sci. Technol., 2013, 3, 99 RSC
-
(a) I. R. Márquez, N. Fuentes, C. M. Cruz, V. Puente-Muñoz, L. Sotorrios, M. L. Marcos, D. Choquesillo-Lazarte, B. Biel, L. Crovetto, E. Gómez-Bengoa, M. T. González, R. Martin, J. M. Cuerva and A. G. Campaña, Chem. Sci., 2017, 8, 1068 RSC
- Metal contents are in good agreement with the ruthenium values (wt%) obtained for Ru@1 and Ru@2 by ICP (see Experimental part†).
- J. M. Asensio, S. Tricard, Y. Coppel, R. Andrés, B. Chaudret and E. de Jesús, Angew. Chem., Int. Ed., 2017, 56, 865 CrossRef CAS PubMed
- F. Novio, K. Philippot and B. Chaudret, Catal. Lett., 2010, 140, 1 CrossRef CAS
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS
-
(a) S. Maintz, V. L. Deringer, A. L. Tchougreeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030 CrossRef CAS PubMed
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211 CrossRef CAS
- See for example:
(a) Z. Song, T. Cai, J. C. Hanson, J. A. Rodriguez and J. Hrbek, J. Am. Chem. Soc., 2004, 126, 8576 CrossRef CAS PubMed
- See for example:
(a) Y. Zhang, F. Lu, H.-Y. Zhang and J. Zhao, Catal. Lett., 2017, 147, 20 CrossRef CAS
- See for example:
(a) J. Kang, S. Zhang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2009, 48, 2565 CrossRef CAS PubMed
- See for example:
(a) M. Lashdaf, A. Hase, E. Kauppinen and A. O. I. Krause, Catal. Lett., 1998, 52, 199 CrossRef CAS
- See for example:
(a) A. B. Ayusheev, O. P. Taran, I. A. Seryak, O. Y. Podyacheva, C. Descorme, M. Besson, L. S. Kibis, A. I. Boronin, A. I. Romanenko, Z. R. Ismagilov and V. Parmon, Appl. Catal., B, 2014, 146, 177 CrossRef CAS
- See for example:
(a) M. Gopiraman, S. Ganesh Babu, Z. Khatri, W. Kai, Y. A. Kim, M. Endo, R. Karvembu and I. S. Kim, J. Phys. Chem. C, 2013, 117, 23582 CrossRef CAS
-
(a) O. Vidoni, K. Philippot, C. Amiens, B. Chaudret, O. Balmes, J.-O. Malm, J.-O. Bovin, F. Senoq and M.-J. Casanove, Angew. Chem., Int. Ed., 1999, 38, 3736 CrossRef CAS
-
(a) A. Nowicki, Y. Zhang, B. Léger, J.-P. Rolland, H. Bricout, E. Monflier and A. Roucoux, Chem. Commun., 2006, 296 RSC
- F. Leng, I. C. Gerber, P. Lecante, A. Bentaleb, A. Muñoz, B. M. Illescas, N. Martín, G. Melinte, O. Ersen, H. Martinez, M. R. Axet and P. Serp, Chem.–Eur. J., 2017, 23, 13379–13386 CrossRef CAS PubMed
- Turn over frequencies (TOFs) were calculated considering the number of moles of acetophenone consumed and the number of surface Ru atoms, which in turn were estimated by using the NP size and the magic number approach (see ESI Section S2 and Table S1†), as previously reported: A. P. Umpierre, E. de Jesús and J. Dupont, ChemCatChem, 2011, 3, 1413–1418 CrossRef CAS
-
(a) M. Yang, G. Cheng, D. Xie, T. Zhu, Y. Dong, H. Ke and H. Cheng, Int. J. Hydrogen Energy, 2018, 43, 8868 CrossRef CAS
- TONs were calculated considering mol of toluene formed per mol of surface Ru atoms, which were estimated by using the NP size and the magic number approach (see ESI Section S2 and Table S1†), as previously reported: A. P. Umpierre, E. de Jesús and J. Dupont, ChemCatChem, 2011, 3, 1413–1418 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04228b |
| This journal is © The Royal Society of Chemistry 2022 |