
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot chemical pyro- and tri-phosphorylation of peptides by using diamidophosphate in water†
Huacan
Lin
 *,
Luke J.
Leman
* and
Ramanarayanan
Krishnamurthy
*
*,
Luke J.
Leman
* and
Ramanarayanan
Krishnamurthy
*
Department of Chemistry, The Scripps Research Institute, La Jolla, California 92037, USA. E-mail: hulin@scripps.edu; lleman@scripps.edu; rkrishna@scripps.edu
First published on 31st October 2022
Abstract
Protein (pyro)phosphorylation is emerging as a post-translational modification (PTM) in signalling pathways involved in many cellular processes. However, access to synthetic pyrophosphopeptides that can serve as tools for understanding protein pyrophosphorylation is quite limited. Herein, we report a chemical phosphorylation method that enables the synthesis of pyrophosphopeptides in aqueous medium without the need for protecting groups. The strategy employs diamidophosphate (DAP) in a one-pot sequential phosphorylation-hydrolysis of mono-phosphorylated peptide precursors. This operationally simple method exploits the intrinsic nucleophilicity of a phosphate moiety installed on serine, threonine or tyrosine residues in complex peptides with excellent chemoselectivity and good yields under mild conditions. We demonstrate the installation of the pyrophosphate group within a wide range of model peptides and showcase the potential of this methodology by selectively pyrophosphorylating the highly functionalized Nopp140 peptide fragment. The potential to produce higher (poly)phosphorylated peptides was demonstrated as a proof-of-principle experiment where we synthesized the triphosphorylated peptides using this one-pot strategy.
Introduction
Post-translational modifications (PTMs) play significant roles in modulating protein function and activity.1 Among a broad range of PTMs,2–6 protein phosphorylation arguably is the most widely studied PTM and plays a crucial role in regulating many other cellular processes. Since its discovery in 1959,7 phosphorylation has been closely linked with many cellular functions in the cell cycle,8 apoptosis,9 differentiation10 and others. Various techniques for enrichment of phosphorylated proteins as well as their identification and quantitation are accessible today for the analysis of phosphorylation.11 In contrast to protein phosphorylation, protein pyrophosphorylation is a poorly characterized PTM. It is known to be mediated by a group of second messengers termed inositol pyrophosphates (PP-InsPs)12 which are capable of transferring the beta-phosphoryl group from 5PP-InsP5 in the presence of Mg2+ to a phosphorylated serine residue in protein substrates to generate a pyrophosphorylated protein (Fig. 1a).13,14 Some significant advancements have been made in understanding the important roles that inositol pyrophosphates play in various pathways.15–18 Even so, the biological functions of protein pyrophosphorylation remain largely unknown due to the difficulty in identifying pyrophosphorylated proteins in complex cellular contexts and the few efficient methodologies for chemically synthesizing pyrophosphorylated peptides and proteins in vitro. Thus, there is a need to develop new methods and expand the toolbox for accessible peptide and protein pyrophosphorylation.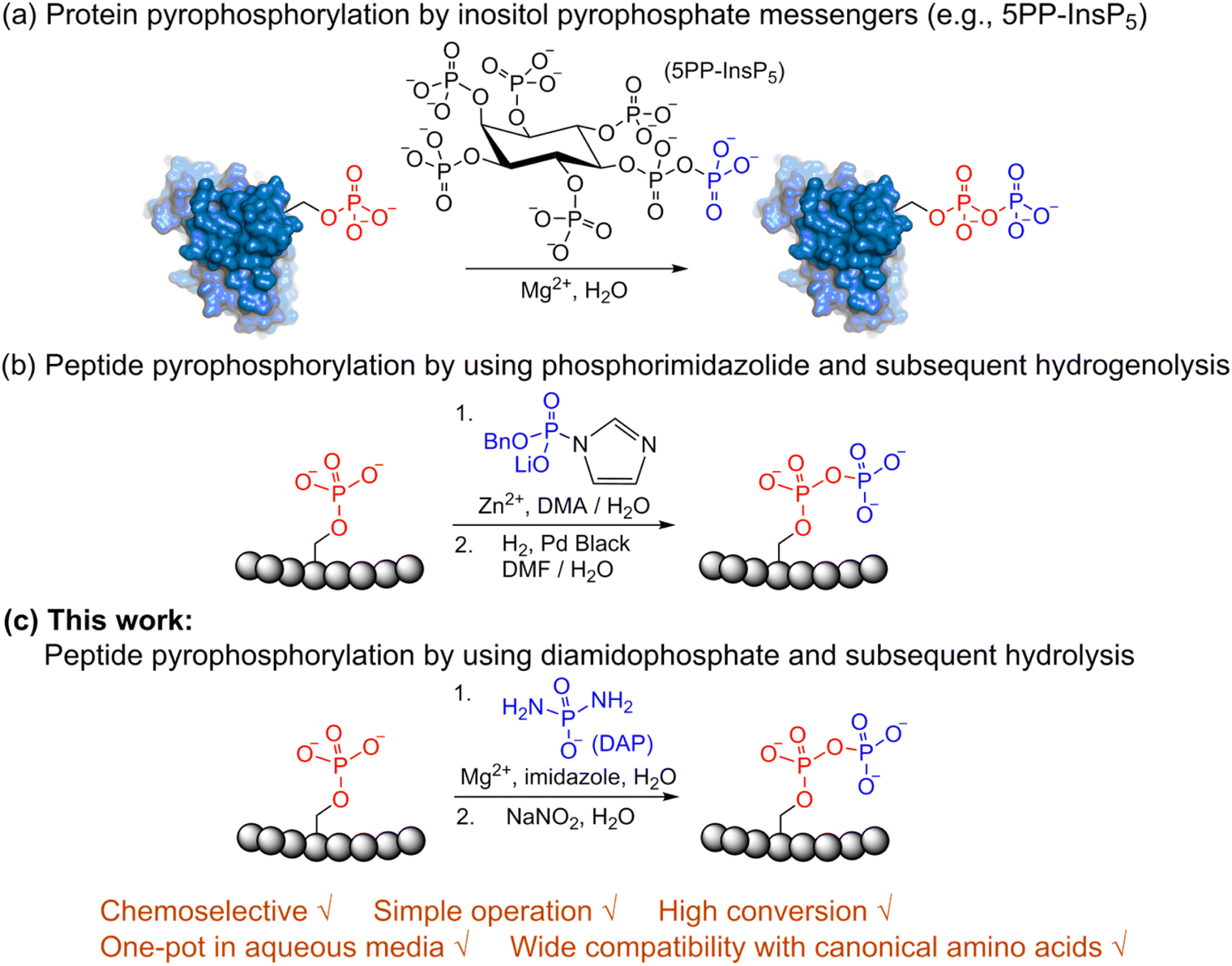 | ||
| Fig. 1 Site-selective peptide pyrophosphorylation via a one-pot sequential amidophosphorylation-hydrolysis methodology. (a) Inositol pyrophosphate messenger-mediated protein pyrophosphorylation in the presence of magnesium. (b) Chemical pyrophosphorylation of peptides using phosphorimidazolide reagents.20 (c) Diamidophosphate-mediated site-selective pyrophosphorylation of peptides. The use of diamidophosphate in aqueous medium, presented in this work, offers several advantages. Circles represent amino acids; DMA, dimethylacetamide; DMF, dimethylformamide. | ||
In 2014, Fiedler and co-workers reported a synthetic methodology to access pyrophosphorylated peptides and proteins from phosphorylated precursors (Fig. 1b).19,20 This protocol involves pyrophosphorylation of a phosphoserine residue by using a benzyl phosphorimidazolide reagent in water/dimethylacetamide co-solvent as the first step, followed by hydrogenolysis of the isolated benzyl-protected intermediates in water/dimethylformamide co-solvent to provide the pyrophosphopeptides. This protocol also proceeded in water but with slower reaction kinetics. Surprisingly, there are no other methods available that allow easy access to pyrophosphorylated peptides, despite the considerable number of synthetic approaches for polyphosphorylation of small molecules such as nucleosides.21–24 Here, we present a simple one-pot chemical strategy for the facile synthesis of pyrophosphopeptides. Our approach starts from a peptide precursor bearing a phosphorylated Ser, Thr or Tyr residue, which site-selectively reacts with diamidophosphate (DAP) in water to generate an amidopyrophosphorylated intermediate, followed by nitrous acid-induced hydrolysis to afford the expected pyrophosphopeptide (Fig. 1c). Distinct from the existing method, our approach does not require protecting/deprotecting group chemistry, or isolation of peptide intermediates. Furthermore, this higher yielding one-pot transformation displays excellent selectivity for phosphate moieties over the other nucleophilic peptide side chains, and wide applicability for accessing various synthetic and native pyrophosphopeptides.
Results and discussion
Previous work from our group showed that DAP, which was prepared via saponification of phenyl phosphorodiamidate, is a versatile phosphorylating agent.25 Particularly, DAP reacted with 5′-nucleoside monophosphates (5′-NMPs) and 5′-nucleoside diphosphates (5′-NDPs) to generate the corresponding 5′-amidodiphosphate and 5′-amidotriphosphate derivatives, respectively, and converted 2′,3′-ribonucleotides to the corresponding 2′,3′-cyclic-phosphates.26 Recently, we showed that DAP enables the conversion of 5′-NMPs and 5′-oligonucleotide monophosphates into the corresponding nucleoside triphosphates (5′-NTPs) and 5′-oligonucleotide triphosphates via amidophosphates in a one-pot amidophosphorylation-hydrolysis setting in water.27 Inspired by these observations, we reasoned that phosphopeptides might also undergo a similar amidophosphorylation-hydrolysis to afford the corresponding pyrophosphopeptides in one-pot,28,29 considering the plausible reactive groups in the side chains (e.g., lysine, aspartate, glutamate, cysteine, etc.).To test this hypothesis, 1 mM peptide 1 ( , phosphoserine indicated in bold and red, prepared by the incorporation of commercially available Fmoc-O-benzyl-phosphoserine, ESI Fig. S2†),30 was treated with an excess of DAP (30 equiv.), MgCl2 (10 equiv.) and imidazole (10 equiv.) at pH 5.5 in water at room temperature, 45 °C or −20 °C (in a freezer). The formation of the amidopyrophosphopeptide 19 in the crude reaction mixture was monitored by reverse-phase high-performance liquid chromatography (HPLC) and mass spectrometry (MS). Imidazole acts as a catalyst to increase the efficiency of the amidophosphorylation reaction of the phosphopeptide by forming the amidophosphorimidazolide intermediate in situ.26 Encouragingly, we observed almost full consumption of the starting peptide 1 after 48 hours to produce amidopyrophosphopeptide 19 with 91% conversion at −20 °C as indicated by HPLC, while significantly lower conversions of 22% and 20% were obtained at room temperature and 45 °C, respectively (ESI Fig. S23–S25†). The excellent conversion at −20 °C was consistent with our previous results indicating that the eutectic concentrating environment of water-ice enabled efficient formation of ribonucleoside 2′,3′-cyclophosphate from the corresponding ribonucleoside 3′-monophosphates (3′-NMPs) in the presence of DAP, MgCl2 and imidazole.31 We attribute this significantly higher yield observed at −20 °C to (a) the freeze concentration effect32 in co-existing water-ice phases, an effect seen in other contexts27,33 and (b) a much slower hydrolytic degradation of DAP (when compared to room temperature and 45 °C), which allows the DAP to be available for the phosphorylation reaction over a longer period of time (see ESI Fig. S26–S31† for details). Through further investigations, we found that decreasing the amounts of DAP (5 equiv.), MgCl2 (2 equiv.) and imidazole (2 equiv.) still produced 19 with 86% conversion (Table 1, entry 1, and ESI Fig. S34†). Thus, we adopted these conditions for future amidophosphorylation reactions.
, phosphoserine indicated in bold and red, prepared by the incorporation of commercially available Fmoc-O-benzyl-phosphoserine, ESI Fig. S2†),30 was treated with an excess of DAP (30 equiv.), MgCl2 (10 equiv.) and imidazole (10 equiv.) at pH 5.5 in water at room temperature, 45 °C or −20 °C (in a freezer). The formation of the amidopyrophosphopeptide 19 in the crude reaction mixture was monitored by reverse-phase high-performance liquid chromatography (HPLC) and mass spectrometry (MS). Imidazole acts as a catalyst to increase the efficiency of the amidophosphorylation reaction of the phosphopeptide by forming the amidophosphorimidazolide intermediate in situ.26 Encouragingly, we observed almost full consumption of the starting peptide 1 after 48 hours to produce amidopyrophosphopeptide 19 with 91% conversion at −20 °C as indicated by HPLC, while significantly lower conversions of 22% and 20% were obtained at room temperature and 45 °C, respectively (ESI Fig. S23–S25†). The excellent conversion at −20 °C was consistent with our previous results indicating that the eutectic concentrating environment of water-ice enabled efficient formation of ribonucleoside 2′,3′-cyclophosphate from the corresponding ribonucleoside 3′-monophosphates (3′-NMPs) in the presence of DAP, MgCl2 and imidazole.31 We attribute this significantly higher yield observed at −20 °C to (a) the freeze concentration effect32 in co-existing water-ice phases, an effect seen in other contexts27,33 and (b) a much slower hydrolytic degradation of DAP (when compared to room temperature and 45 °C), which allows the DAP to be available for the phosphorylation reaction over a longer period of time (see ESI Fig. S26–S31† for details). Through further investigations, we found that decreasing the amounts of DAP (5 equiv.), MgCl2 (2 equiv.) and imidazole (2 equiv.) still produced 19 with 86% conversion (Table 1, entry 1, and ESI Fig. S34†). Thus, we adopted these conditions for future amidophosphorylation reactions.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate peptide sequencea | Sequence origin | Amidopyrophosphorylated productc (% conversion) | Pyrophosphorylated productc (% conversion over two steps) |
| a Amino acids highlighted in bold and red bear a phosphate group. Substrate numbers are given in bold. b Peptides that do not contain a phosphate group. c % conversions were estimated by reverse phase HPLC analysis based on the area-under-the-curve of the amidopyrophosphopeptide intermediates 19–34 and pyrophosphopeptides 35–50vs. sum area of all peptidic-peaks. d The isolated yield (after two steps) of the purified pyrophosphopeptide by reverse phase HPLC purification. e The yield (after two steps) determined using standard curves of the purified pyrophosphopeptides. f NA = not applicable. g A nitrothioite peptide was observed by LC-MS as the product from oxidation of the thiol group by HNO2. Pyrophosphopeptide 38 (containing the thiol) was formed from reduction of the nitrothioite-intermediate by the addition of excess 4-mercaptophenylacetic acid (MPAA). Reaction conditions: (1) peptide (1 mM, 1.0 equiv.), DAP (5 mM, 5.0 equiv.), MgCl2 (2 mM, 2.0 equiv.), imidazole (2 mM, 2.0 equiv.), H2O, pH = 5.5, −20 °C, 48–72 h; (2) NaNO2 (5 mM, 5.0 equiv.), H2O, pH = 3.0, −20 °C, 20 h. | ||||
| 1 |
1

|
Random | 19 (86) | 35 (85) |
| 2 |
2

|
Random | 20 (96) | 36 (95) |
| 3 |
3

|
Random | 21 (97) | 37 (96) (65)d |
| 4 |
4

|
Random | NR | NAf |
| 5 |
5

|
Random | 22 (95) | 38 (66)g |
| 6 |
6

|
Random | NR | NAf |
| 7 |
7

|
Random | 23 (94) | 39 (92) |
| 8 |
8
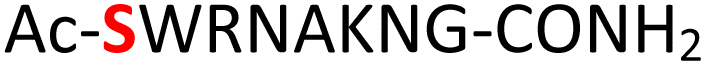
|
Random | 24 (93) | 40 (86) |
| 9 |
9
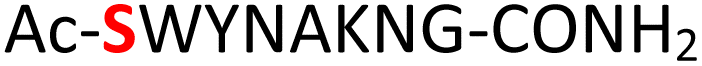
|
Random | 25 (96) | 41 (90) (63)d (83)e |
| 10 |
10

|
Random | 26 (94) | 42 (86) |
| 11 |
11
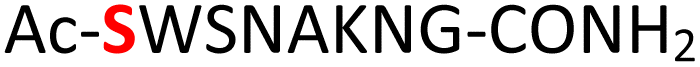
|
Random | 27 (96) | 43 (90) (71)d (87)e |
| 12 |
12

|
Random | 28 (94) | 44 (93) (68)d (85)e |
| 13 |
13

|
Random | 29 (90) | 45 (83) |
| 14 |
14
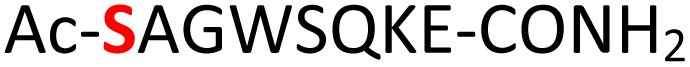
|
Random | 30 (95) | 46 (94) |
| 15 |
15

|
RPA190![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 34 |
31 (82) | 47 (79) |
| 16 |
16

|
IC2C35 | 32 (63) | 48 (60) |
| 17 |
17

|
Gcr136,37 |
33 (88) | 49 (86) |
| 18 |
18

|
EIF2S237,38 |
34 (70) | 50 (68) |
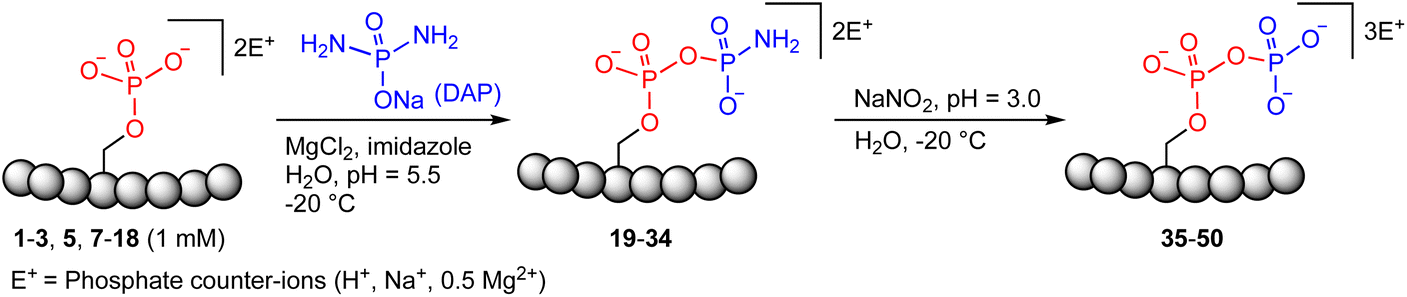
For the hydrolysis step, 5 equiv. of sodium nitrite were added to the crude 19 in the same pot and the pH was adjusted to 3.0 and left at −20 °C. Monitoring by HPLC showed complete conversion of amidopyrophosphopeptide 19 into pyrophosphopeptide 35 after an additional 20 hours at −20 °C. This resulted in overall 85% conversion over two steps starting from 1, as confirmed by LC-MS (Table 1, entry 1 and ESI Fig. S35†), suggesting that the nitrous acid–induced hydrolysis of the amidopyrophosphate group in 19 was almost quantitative. Having established appropriate conditions, we sought to evaluate the functional group tolerance by incorporating other amino acids with nucleophiles in their side chains. Peptide 2 ( , and ESI Fig. S3†) bearing a reactive lysine proceeded efficiently and produced the expected amidopyrophosphopeptide 20 with near quantitative conversion at 96% as confirmed by LC-MS (Table 1, entry 2 and ESI Fig. S36†). Treatment of crude 20 with NaNO2 at pH 3.0 produced pyrophosphopeptide 36 as the desired product with 95% conversion over two steps (Table 1, entry 2 and ESI Fig. S37†).
, and ESI Fig. S3†) bearing a reactive lysine proceeded efficiently and produced the expected amidopyrophosphopeptide 20 with near quantitative conversion at 96% as confirmed by LC-MS (Table 1, entry 2 and ESI Fig. S36†). Treatment of crude 20 with NaNO2 at pH 3.0 produced pyrophosphopeptide 36 as the desired product with 95% conversion over two steps (Table 1, entry 2 and ESI Fig. S37†).
As a representative example for investigating the effect of the position of the phosphate group on the efficiency and selectivity, we chose peptide 3 ( , and ESI Fig. S4†), which has a phosphoserine at the N-terminal residue with a neighboring histidine unit (Table 1, entry 3). Almost full conversion to the amidopyrophosphopeptide 21 (97%) and pyrophosphopeptide 37 (96% over two steps from the starting peptide 3) were observed under the conditions as indicated by HPLC analysis (Fig. 2). No other by-products were detectable during the formation of 21 and 37. Although the retention times for 21 and 37 were close, the 1-unit difference in [M − H]− and 0.5-unit difference in [M − 2H]2− between the amidopyrophosphopeptide ion and pyrophosphopeptide ion in the MS spectra clearly showed the transformation from –NH2 in the phosphate group to –OH (Fig. 2).
, and ESI Fig. S4†), which has a phosphoserine at the N-terminal residue with a neighboring histidine unit (Table 1, entry 3). Almost full conversion to the amidopyrophosphopeptide 21 (97%) and pyrophosphopeptide 37 (96% over two steps from the starting peptide 3) were observed under the conditions as indicated by HPLC analysis (Fig. 2). No other by-products were detectable during the formation of 21 and 37. Although the retention times for 21 and 37 were close, the 1-unit difference in [M − H]− and 0.5-unit difference in [M − 2H]2− between the amidopyrophosphopeptide ion and pyrophosphopeptide ion in the MS spectra clearly showed the transformation from –NH2 in the phosphate group to –OH (Fig. 2).
 | ||
| Fig. 2 Crude LC-MS chromatograms of reaction mixtures confirming the formation of pyrophosphorylated peptide 37 by a one-pot sequential amidophosphorylation-hydrolysis scenario. (i) The starting material 3 and the production of (ii) amidopyrophosphopeptide intermediate 21 and (iii) desired pyrophosphopeptide 37. The conversion in each step, as judged by using the HPLC traces, was nearly quantitative. | ||
Furthermore, a series of model peptides with a sequence similar to  where X = cysteine (5), aspartic acid (7), arginine (8), tyrosine (9), proline (10) or serine (11) were synthesized and investigated for the scope of this chemistry (ESI Fig. S6, S8–S12†). As outlined by the results in Table 1 (entries 7–11), the reactions from phosphopeptides 7–11 proceeded smoothly to afford the amidopyrophosphopeptide intermediates 23–27 with 93–96% and the corresponding pyrophosphopeptides 39–43 with 86–92% conversion over two steps (ESI Fig. S46–S55†). Despite the presence of other nucleophilic residues, the clean conversion indicated exclusive amidophosphorylation in the phosphoserine residue without any interferences from the existing tryptophan, lysine, histidine, aspartic acid, arginine, tyrosine, proline, or (non-phosphorylated) serine. In contrast, reactions of peptides lacking a phosphorylated serine gave no amidopyrophosphopeptide products (entries 4 and 6, and ESI Fig. S40 and S45†) even in the presence of a reactive hydroxyl and thiol group, respectively. It is noteworthy that no significant side reactions were observed during the nitrous acid–induced hydrolysis with a nucleophilic NH2 group in lysine, tryptophan, histidine, or arginine residues, thus suggesting the high reactivity and chemoselectivity of the amino moiety in the amidophosphate group under our conditions.
where X = cysteine (5), aspartic acid (7), arginine (8), tyrosine (9), proline (10) or serine (11) were synthesized and investigated for the scope of this chemistry (ESI Fig. S6, S8–S12†). As outlined by the results in Table 1 (entries 7–11), the reactions from phosphopeptides 7–11 proceeded smoothly to afford the amidopyrophosphopeptide intermediates 23–27 with 93–96% and the corresponding pyrophosphopeptides 39–43 with 86–92% conversion over two steps (ESI Fig. S46–S55†). Despite the presence of other nucleophilic residues, the clean conversion indicated exclusive amidophosphorylation in the phosphoserine residue without any interferences from the existing tryptophan, lysine, histidine, aspartic acid, arginine, tyrosine, proline, or (non-phosphorylated) serine. In contrast, reactions of peptides lacking a phosphorylated serine gave no amidopyrophosphopeptide products (entries 4 and 6, and ESI Fig. S40 and S45†) even in the presence of a reactive hydroxyl and thiol group, respectively. It is noteworthy that no significant side reactions were observed during the nitrous acid–induced hydrolysis with a nucleophilic NH2 group in lysine, tryptophan, histidine, or arginine residues, thus suggesting the high reactivity and chemoselectivity of the amino moiety in the amidophosphate group under our conditions.
Previous studies on peptide ligation39 and isocyanate formation40 showed that the optimal pH value for NaNO2–mediated oxidation of hydrazides was 3.0–4.0 where all amines from standard amino acid residues were protonated and thus rendered unreactive as nucleophiles. In our case, the amidopyrophosphate group remained active due to its low value of pKa ≈ 1.225,26 as well as the limited NaNO2 added (equivalent amount to DAP), probably explaining the chemoselective HNO2-induced hydrolysis. In this context, the reaction of peptide 5 containing a cysteine is notable. As expected peptide 5 underwent, in the first step of amidophosphorylation, almost quantitative conversion (95%) to amidopyrophosphopeptide 22 (Table 1, entry 5, and ESI Fig. S41†). However, in the hydrolysis step with NaNO2, we observed the formation of the peptide nitrothioite (RSNO) as the major product – as indicated by the new peak in the LC-MS being +29 Da heavier than that of the free thiol peptide. It is known that the thiol group (RSH) could be oxidized using HNO2 to generate nitrothioite (RSNO).41 Nevertheless, the nitrothioite was cleanly reduced back to the original thiol by adding excess 4-mercaptophenylacetic acid (MPAA). By utilizing this protocol, the desired pyrophosphopeptide 38 with native cysteine was obtained with 66% conversion (ESI Fig. S42–S44†). Methionine, the other sulfur-containing residue, was reported to be stable with nitrous acid treatment,39 and is, therefore, not expected to interfere in this protocol as well.
To expand the potential application of this methodology further, we subjected phosphothreonine- and phosphotyrosine-containing peptides (12 and 13, and ESI Fig. S13 and S14†) to the one-pot reaction conditions (Table 1, entries 12 and 13). Gratifyingly, both reactions furnished the expected pyrophosphopeptides 44 and 45 with 93% and 83% conversions via amidopyrophosphopeptides 28 and 29, respectively (ESI Fig. S56–S59†). The reaction of phosphopeptide 14 (ESI Fig. S15†) containing multiple nucleophilic amino acid residues proceeded well to afford the expected pyrophosphopeptide 46 with 94% conversion (Table 1, entry 14, and ESI Fig. S60 and S61†). To demonstrate the practicality of the reactions, we scaled-up the reactions of 3, 9, 11 and 12 and the progress was monitored by LC-MS (ESI Fig. S62–S69†). All reactions proceeded to near complete conversion over the two steps to afford the desired pyrophosphopeptides, each of which were purified and isolated by preparative HPLC in 65%, 63%, 71% and 68% yields, respectively (Table 1, entries 3, 9, 11 and 12). The identities of the pyrophosphopeptide products were also established by 31P-NMR (ESI Fig. S73–S78†). Four additional known sequences (15, 16, 17 and 18, and ESI Fig. S16–S19†) containing free N- and C-termini, derived from the DNA-directed RNA polymerase I subunit RPA19034 (RPA190, Table 1, entry 15), the cytoplasmic dynein 1 intermediate chain 235 (IC2C, Table 1, entry 16), the glycolytic gene transcriptional activator36,37 (Gcr1, Table 1, entry 17) and the mammalian protein eukaryotic translation initiation factor 237,38 (EIF2S2, Table 1, entry 18) were selected as substrates. The one-pot reactions of all these fully unprotected peptides 15, 16, 17 and 18 proceeded to give desired pyrophosphopeptides 47, 48, 49 and 50 with 79%, 60%, 86% and 68% conversion, respectively (ESI Fig. S79–S86†). The slight drop in yield for 16 and 18 may be a sequence effect and not due to the unprotected N- or C-terminus since the unprotected amino and carboxylic side chains (e.g., in sequences 2 and 7) have not impacted the yields. The reaction displayed excellent chemoselectivity considering the presence of multiple nucleophilic amino acids such as glutamic acid, aspartic acid, serine, and lysine in these peptides.
Tandem mass spectrometry (MS/MS) was reported to be used as a fragmentation technique for the identification of serine and threonine pyrophosphorylation.42 To further confirm the constitutional integrity of the pyrophosphopeptide product, peptide 43 was characterized by collision-induced dissociation (CID) MS/MS spectrometry (Fig. 3). The observed fragment ion (M-178), generated from 43 by loss of the pyrophosphate motif, is a clear indicator for the pyrophosphorylated peptide and supports the assignment of phosphorylated serine as the site of pyrophosphorylation.42 The b-ions and y-ions with varying lengths of amino acids retained by the amino and carboxyl-terminal part of M-178 were identified, precisely confirming the sequence of 43 as  (ESI Fig. S87 and S88†). The MS/MS analysis revealed no modification of tryptophan, serine, or lysine side chains in 43. Combined with the exclusively single peak observed in HPLC for 43 (ESI Fig. S67†) and 31P NMR (ESI Fig. S75†), this analysis again demonstrated the efficiency and chemoselectivity of the pyrophosphorylation reaction.
(ESI Fig. S87 and S88†). The MS/MS analysis revealed no modification of tryptophan, serine, or lysine side chains in 43. Combined with the exclusively single peak observed in HPLC for 43 (ESI Fig. S67†) and 31P NMR (ESI Fig. S75†), this analysis again demonstrated the efficiency and chemoselectivity of the pyrophosphorylation reaction.
 | ||
Fig. 3 CID MS/MS analysis of the pyrophosphopeptide 43 in positive ion mode, confirming the sequence as  . The sequence was identified by matching b-ion (red) and y-ion (blue) fragments. M-178 was generated from 43 losing the pyrophosphate motif. The b-ions were retained by the amino-terminal part of M-178 and the y-ions were retained by the carboxyl-terminal part of M-178. . The sequence was identified by matching b-ion (red) and y-ion (blue) fragments. M-178 was generated from 43 losing the pyrophosphate motif. The b-ions were retained by the amino-terminal part of M-178 and the y-ions were retained by the carboxyl-terminal part of M-178. | ||
Next, we assessed the generality of the one-pot aqueous pyrophosphorylation strategy with a highly functionalized substrate, the Nopp140 peptide fragment containing amino acids 76–100, which is known to be pyrophosphorylated by inositol pyrophosphate in vitro.14 The Nopp140 peptide fragment, a 25-residue peptide bearing a single phosphoserine, seven serines/threonines, five lysines, and eight glutamic/aspartic acids, seemed a challenging substrate to explore (Fig. 4a). The substrate 52 was synthesized by post-assembly phosphitylation and oxidation in the solid phase30,43 in high efficiency and characterized by LC-MS (Fig. 4b, and ESI Fig. S1, S20 and S21†). Peptide 52 was reacted with DAP for 20 hours and was efficiently amidophosphorylated to form amidopyrophosphopeptide 53 in 75% conversion (Fig. 4a, and ESI Fig. S89†). Remarkably, subsequent treatment of 53 with NaNO2 in the same pot successfully afforded pyrophosphopeptide 54 with overall 70% conversion over two steps (Fig. 4a, and ESI Fig. S90†). The identities of 53 and 54 were confirmed by high-resolution mass spectrometry (HRMS) (Fig. 4c and d). The efficient one-pot pyrophosphorylation of the highly functionalized 52 leading to the formation of 54 once again highlighted the high intrinsic nucleophilicity and selectivity of the phosphoserine residue towards DAP and the superior reactivity of the peptide-amidophosphate towards HNO2 even in a complex sequence. And yields of this one-pot strategy were much better than the previous results (ca. 11%) of the benzyl protected diphosphate derivative as reported in the literature.20
 | ||
| Fig. 4 Formation of the pyrophosphopeptide 54 from the highly functionalized Nopp140 peptide fragment. (a) General reaction scheme showing the pyrophosphorylation of the Nopp140 peptide fragment 52 to generate 54via a one-pot sequential amidophosphorylation-hydrolysis scenario. (b)–(d) High resolution ESI-MS (m/z) spectra in positive ion mode of the starting peptide 52, amidopyrophosphopeptide intermediate 53, and desired pyrophosphopeptide 54, respectively. Conversions were calculated by analytical HPLC based on the area-under-the-curve of 53 and 54vs. sum area of all peptidic peaks. | ||
To further validate our methodology on producing valuable polyphosphorylated peptides, as a proof-of-principle experiment, we selected phosphoserine- and phosphothreonine-containing peptides (3 and 12, respectively) to generate triphosphopeptides via a one-pot sequential amidophosphorylation-hydrolysis scenario in water (Fig. 5). As observed previously, reactions of phosphopeptides 3 and 12 reached completion to quantitatively afford the pyrophosphopeptides 37 and 44via amidopyrophosphopeptides 21 and 28, respectively (ESI Fig. S92, S93, S97 and S98†). By simply repeating the second amidophosphorylation and subsequent hydrolysis steps in the same pot, satisfactory conversions to final triphosphopeptides 56 and 58 were achieved in 61% and 70% yield, via amidotriphosphopeptides 55 and 57, respectively, as analysed by HPLC and anion exchange chromatography (ESI Fig. S94–S96 and S99–S101†). Successful triphosphorylation of phosphopeptides 3 and 12 could potentially broaden the applicability of this method in generating polyphosphorylated peptides.
 | ||
| Fig. 5 Synthesis of triphosphopeptides 56 and 58 from respective phosphopeptides 3 and 12via a one-pot four step sequential amidophosphorylation-hydrolysis scenario in water. | ||
Conclusions
In summary, we have developed a one-pot strategy that enables efficient peptide pyrophosphorylation in water via a sequential amidophosphorylation-hydrolysis scenario. This method is operationally simple and highly chemoselective, obviating the need for orthogonal protecting groups for the canonical amino acid residues within the peptide. The efficacy of the pyrophosphorylation reaction was demonstrated by the broad scope of phosphopeptide substrates with different amino acid compositions. Notably, this method allowed the site-selective pyrophosphorylation of complex phosphopeptide targets. Furthermore, triphosphorylation of phosphopeptides was achieved by simply repeating the two steps of amidophosphorylation and hydrolysis in the same pot. Given the high yields and lack of byproducts, it is plausible that the thus-generated crude pyrophosphopeptides could be used directly in subsequent exploratory studies. For example, a repeat of this DAP-mediated phosphorylation has the potential to produce peptide- and protein-polyphosphates44,45 in the same pot. Thus, we anticipate that this method will serve as a practical tool to quickly generate biologically relevant pyrophosphopeptides and explore their biochemistry in the emerging area of protein pyrophosphorylation.Data availability
The data supporting this article are available in the ESI.†Author contributions
H. Lin, L. J. Leman and R. Krishnamurthy conceived the project. H. Lin performed the experiments and analysed the data. H. Lin, L. J. Leman and R. Krishnamurthy wrote the manuscript and approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSF and the NASA Astrobiology Program under the Centre for Chemical Evolution (CHE-150421) and a grant from the Simons Foundation to R. K. (327124FY19).References
- Y. L. Deribe, T. Pawson and I. Dikic, Nat. Struct. Mol. Biol., 2010, 17, 666–672 CrossRef CAS PubMed
.
- K. K. Biggar and S. S. Li, Nat. Rev. Mol. Cell Biol., 2015, 16, 5–17 CrossRef CAS PubMed
- C. Choudhary and M. Mann, Nat. Rev. Mol. Cell Biol., 2010, 11, 427–439 CrossRef CAS PubMed
- S. J. Humphrey, D. E. James and M. Mann, Trends Endocrinol. Metab., 2015, 26, 676–687 CrossRef CAS PubMed
- K. W. Moremen, M. Tiemeyer and A. V. Nairn, Nat. Rev. Mol. Cell Biol., 2012, 13, 448–462 CrossRef CAS PubMed
- E. Verdin and M. Ott, Nat. Rev. Mol. Cell Biol., 2015, 16, 258–264 CrossRef CAS PubMed
- E. H. Fischer, D. J. Graves, E. R. S. Crittenden and E. G. Krebs, J. Biol. Chem., 1959, 234, 1698–1704 CrossRef CAS PubMed
- T. Garcia-Garcia, S. Poncet, A. Derouiche, L. Shi, I. Mijakovic and M. F. Noirot-Gros, Front. Microbiol., 2016, 7, 184 Search PubMed
- I. Kitazumi and M. Tsukahara, FEBS J., 2011, 278, 427–441 CrossRef CAS PubMed
- D. V. Hoof, J. Munoz, S. R. Braam, M. W. H. Pinkse, R. Linding, A. J. R. Heck, C. L. Mummery and J. Krijgsveld, Cell Stem Cell, 2009, 5, 214–226 CrossRef PubMed
- M. Mann, S. E. Ong, M. Gronborg, H. Steen, O. N. Jensen and A. Pandey, Trends Biotechnol., 2002, 20, 261–268 CrossRef CAS PubMed
- A. Chakraborty, S. Kim and S. H. Snyder, Sci. Signaling, 2011, 4, re1 CrossRef PubMed
- A. Saiardi, R. Bhandari, A. C. Resnick, A. M. Snowman and S. H. Snyder, Science, 2004, 306, 2101–2105 CrossRef CAS PubMed
- R. Bhandari, A. Saiardi, Y. Ahmadibeni, A. M. Snowman, A. C. Resnick, T. Z. Kristiansen, H. Molina, A. Pandey, J. K. Werner, K. R. Juluri, Y. Xu, G. D. Prestwich, K. Parang and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15305–15310 CrossRef CAS PubMed
- A. Burton, C. Azevedo, C. Andreassi, A. Riccio and A. Saiardi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18970–18975 CrossRef CAS PubMed
- A. Chakraborty, M. A. Koldobskiy, N. T. Bello, M. Maxwell, J. J. Potter, K. R. Juluri, D. Maag, S. Kim, A. S. Huang, M. J. Dailey, M. Saleh, A. M. Snowman, T. H. Moran, E. Mezey and S. H. Snyder, Cell, 2010, 143, 897–910 CrossRef CAS PubMed
- T. S. Lee, J. Y. Lee, J. W. Kyung, Y. Yang, S. J. Park, S. Lee, I. Pavlovic, B. Kong, Y. S. Jho, H. J. Jessen, D. H. Kweon, Y. K. Shin, S. H. Kim, T. Y. Yoon and S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8314–8319 CrossRef CAS PubMed
- J. Worley, X. Luo and A. P. Capaldi, Cell Rep., 2013, 3, 1476–1482 CrossRef CAS PubMed
- A. M. Marmelstein, J. A. M. Morgan, M. Penkert, D. T. Rogerson, J. W. Chin, E. Krause and D. Fiedler, Chem. Sci., 2018, 9, 5929–5936 RSC
- A. M. Marmelstein, L. M. Yates, J. H. Conway and D. Fiedler, J. Am. Chem. Soc., 2014, 136, 108–111 CrossRef CAS PubMed
- T. Durr-Mayer, D. Qiu, V. B. Eisenbeis, N. Steck, M. Haner, A. Hofer, A. Mayer, J. S. Siegel, K. K. Baldridge and H. J. Jessen, Nat. Commun., 2021, 12, 5368 CrossRef PubMed
- T. M. Haas, S. Wiesler, T. Durr-Mayer, A. Ripp, P. Fouka, D. Qiu and H. J. Jessen, Angew. Chem., Int. Ed., 2022, 61, e202113231 CAS
- B. Roy, A. Depaix, C. Perigaud and S. Peyrottes, Chem. Rev., 2016, 116, 7854–7897 CrossRef CAS PubMed
- S. M. Shepard, H. J. Jessen and C. C. Cummins, J. Am. Chem. Soc., 2022, 144, 7517–7530 CrossRef CAS PubMed
- A. Osumah and R. Krishnamurthy, ChemBioChem, 2021, 22, 3001–3009 CrossRef CAS PubMed
- C. Gibard, S. Bhowmik, M. Karki, E. K. Kim and R. Krishnamurthy, Nat. Chem., 2018, 10, 212–217 CrossRef CAS PubMed
- H. Lin, E. I. Jimenez, J. T. Arriola, U. F. Muller and R. Krishnamurthy, Angew. Chem., Int. Ed., 2022, 61, e202113625 CAS
- M. Ikehara, S. Uesugi and T. Fukui, Chem. Pharm. Bull., 1967, 15, 440–447 CrossRef CAS PubMed
- W. P. Jencks and M. Gilchrist, J. Am. Chem. Soc., 1964, 86, 1410–1417 CrossRef CAS
- T. J. Attard, N. O'Brien-Simpson and E. C. Reynolds, Int. J. Pept. Res. Ther., 2007, 13, 447–468 CrossRef CAS
- E. Y. Song, E. I. Jimenez, H. Lin, K. L. Vay, R. Krishnamurthy and H. Mutschler, Angew. Chem., Int. Ed., 2021, 60, 2952–2957 CrossRef CAS PubMed
- L. Y. An, Z. Dai, B. Di and L. L. Xu, Molecules, 2021, 26, 750 CrossRef CAS PubMed
- A. Kanavarioti, P. Monnard and D. W. Deamer, Astrobiology, 2001, 1, 271–281 CrossRef CAS PubMed
- S. G. Thota, C. P. Unnikannan, S. R. Thampatty, R. Manorama and R. Bhandari, Biochem. J., 2015, 466, 105–114 CrossRef CAS PubMed
- M. Chanduri, A. Rai, A. B. Malla, M. Wu, D. Fiedler, R. Mallik and R. Bhandari, Biochem. J., 2016, 473, 3031–3047 CrossRef CAS PubMed
- Z. Szijgyarto, A. Garedew, C. Azevedo and A. Saiardi, Science, 2011, 334, 802–805 CrossRef CAS PubMed
- L. M. Yates and D. Fiedler, ChemBioChem, 2015, 16, 415–423 CrossRef CAS PubMed
- V. K. Pathak, P. J. Nielsen, H. Trachsel and J. W. B. Hershey, Cell, 1988, 54, 633–639 CrossRef CAS
- G. M. Fang, Y. M. Li, F. Shen, Y. C. Huang, J. B. Li, Y. Lin, H. K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS PubMed
- A. A. Vinogradov, M. D. Simon and B. L. Pentelute, Org. Lett., 2016, 18, 1222–1225 CrossRef CAS PubMed
- L. Grossi and P. C. Montevecchi, J.
Org. Chem., 2002, 67, 8625–8630 CrossRef CAS PubMed
- M. Penkert, L. M. Yates, M. Schumann, D. Perlman, D. Fiedler and E. Krause, Anal. Chem., 2017, 89, 3672–3680 CrossRef CAS PubMed
- D. M. Andrews, J. Kitchin and P. W. Seale, Int. J. Pept. Protein Res., 1991, 38, 469–475 CrossRef CAS PubMed
- C. Azevedo, T. Livermore and A. Saiardi, Mol. Cell, 2015, 58, 71–82 CrossRef CAS PubMed
- C. Azevedo, J. Singh, N. Steck, A. Hofer, F. A. Ruiz, T. Singh, H. J. Jessen and A. Saiardi, ACS Chem. Biol., 2018, 13, 1958–1963 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthetic schemes, HPLC traces and MS spectra. See DOI: https://doi.org/10.1039/d2sc04160j |
| This journal is © The Royal Society of Chemistry 2022 |