
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkoxysulfonyl radical species: acquisition and transformation towards sulfonate esters through electrochemistry†
Chun
Zhang
a,
Man
Yang
a,
Yanjie
Qiu
a,
Meijun
Song
a,
Hongyan
Wang
a,
Min
Yang
 a,
Wenlin
Xie
d,
Jie
Wu
*abc and
Shengqing
Ye
*a
a,
Wenlin
Xie
d,
Jie
Wu
*abc and
Shengqing
Ye
*a
aSchool of Pharmaceutical and Materials Engineering & Institute for Advanced Studies, Taizhou University, 1139 Shifu Avenue, Taizhou 318000, China. E-mail: jie_wu@fudan.edu.cn; shengqing.ye@tzc.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
cSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
dSchool of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan 411201, China
First published on 30th September 2022
Abstract
Sulfonyl radical mediated processes have been considered as a powerful strategy for the construction of sulfonyl compounds. However, an efficient and high atom-economical radical approach to the synthesis of sulfonate esters is still rare, owing to the limited tactics to achieve alkoxysulfonyl radicals. Herein, an electrochemical anodic oxidation of inorganic sulfites with alcohols is developed to afford alkoxysulfonyl radical species, which are utilized in subsequent alkene difunctionalization to provide various sulfonate esters. This transformation features excellent chemoselectivity and broad functional group tolerance. This new discovery presents the potential prospect for the construction of sulfonate esters, and enriches the electrochemical reaction type.
Introduction
Owing to their great importance in organic synthesis, pharmaceuticals, and materials science, studies on sulfonyl derivatives have gained remarkable attention in organic synthesis.1–10 The construction of sulfonyl compounds through a sulfonyl radical involving process has developed rapidly over the past decades.11–21 However, compared to the well explored sulfone radicals or sulfamoyl radicals in the construction of sulfones and sulfonamides, the study of alkoxysulfonyl radicals in the synthesis of sulfonate esters is still limited, since the pioneering work on the generation of alkoxysulfonyl radicals from chlorosulfate derivatives in 1989 by Chatgilialoglu and co-workers22 (Scheme 1a). So far, strategies reported for the construction of alkoxysulfonyl radicals have been limited to radical exchange processes, in which radical species would react with chlorosulfonate, arylsulfonate or allylsulfonate to generate alkoxysulfonyl radicals.23–26 With this strategy, direct synthesis of sulfonate esters could be achieved. However, prefunctionalized sulfonate esters were utilized as the alkoxysulfonyl radical sources, which restricted the applicability of these methods. The development of more direct and efficient routes to afford alkoxysulfonyl radicals is highly desirable (Scheme 1b).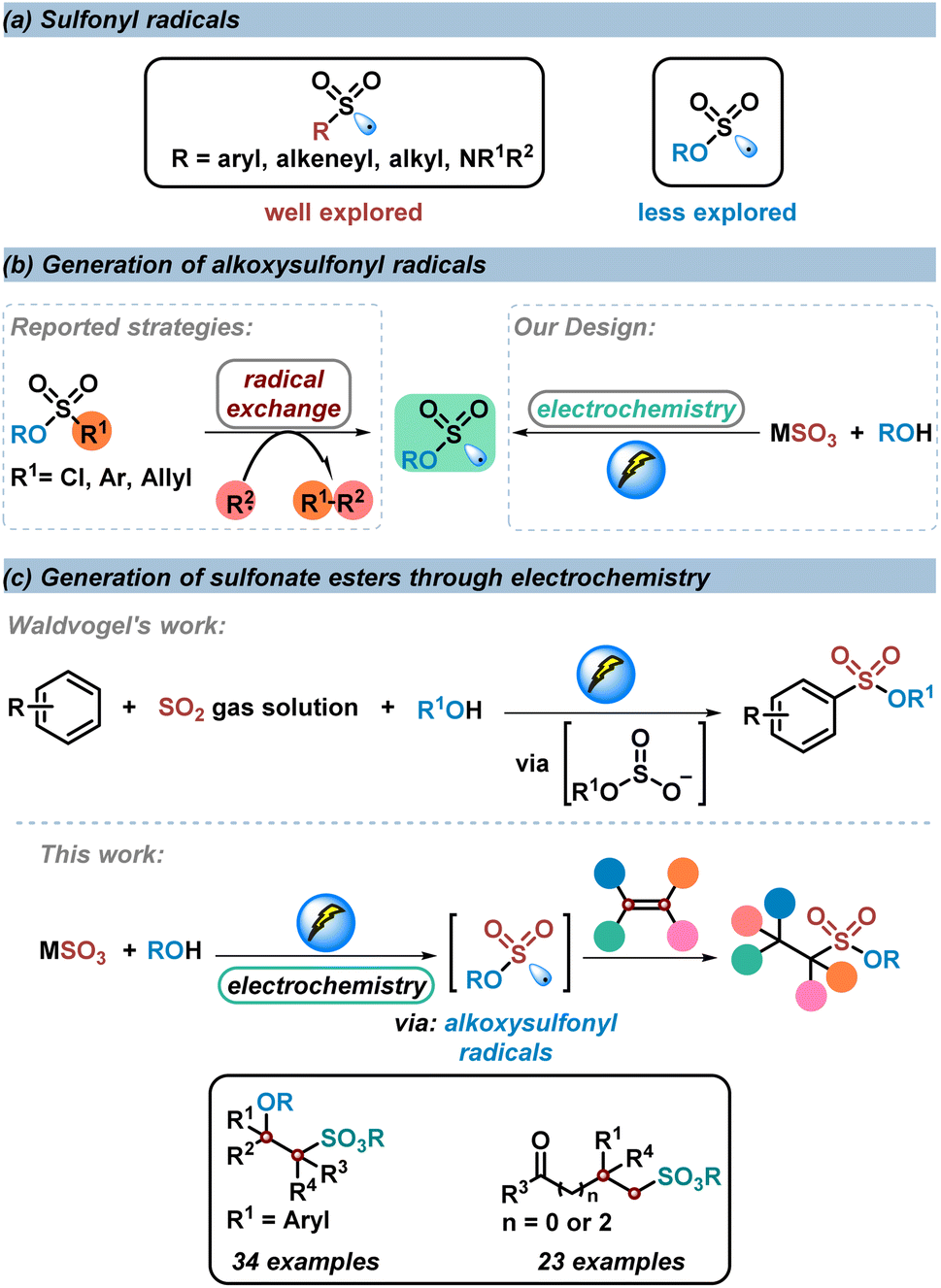 | ||
| Scheme 1 Generation of sulfonate esters through electrochemistry. | ||
In recent years, electrochemical anodic oxidation, as an alternative to chemical oxidation, represents an efficient and environmental strategy in organic synthesis.27–40 Considering that inorganic sulfites (SO32−, HSO3−, S2O52−) are cheap, easy-to-handle, and easy-to-obtain sulfur dioxide surrogates in the synthesis of sulfonyl compounds,41,42 and based on our continuous work on the construction of sulfonyl compounds by using inorganic sulfites as sulfur dioxide surrogates, we design a direct strategy to construct alkoxysulfonyl radicals through electrochemical anodic oxidation of inorganic sulfites with alcohols. The S(IV) of inorganic sulfites could be activated by electrochemical anodic oxidation to afford corresponding radicals or radical anions (the voltammograms show that the oxidation peak of Na2SO3 aqueous solution is near 0.6 V).43–45 Compared with the reported pathway to the synthesis of alkoxysulfonyl radicals, the new strategy features a more efficient and diversified route to construct alkoxysulfonyl radicals (Scheme 1b).
Given that alkenes are general radical acceptors, we conceive that this alkoxysulfonyl radical would be trapped by subsequent alkene difunctionalization to provide sulfonate esters. In 2020, Waldvogel's research group described an electrochemical reaction of electro-rich arenes with sulfur dioxide gas solution to produce alkyl arylsulfonate esters.46,47 In this transformation, a nucleophilic addition process of alkyl sulfite with an arene radical cation is involved, rather than an alkoxysulfonyl radical process. There have been no reports on the electrochemical anodic oxidation of sulfur dioxide until now.48 Herein, we develop an electrochemical three-component reaction of alkenes, inorganic sulfites, and alcohols to produce various sulfonate esters in good yields with excellent chemoselectivity and broad functional group tolerance. During this transformation, the key alkoxysulfonyl radical species was generated through an electrochemical anodic oxidation of inorganic sulfites and subsequent alcohol trapping (Scheme 1c).
Results and discussion
We commenced our design with 4-phenylstyrene 1a, methanol 2a, and K2S2O5 as model substrates. Initially, this model reaction was carried out under a constant current of 4 mA for 4 h at room temperature in an undivided electrolytic cell with a graphite anode and a platinum cathode, by using nBu4NBF4 as the electrolyte and MeCN as the solvent. Encouragingly, the desired sulfonate ester 3aa was isolated in 33% yield (Table 1, entry 1). The yield could be improved by extending the reaction time to 12 h, giving rise to 3aa in 81% yield (Table 1, entries 2–3). To our delight, the yield of 3aa could be increased to 90% (in 86% isolated yield) when inorganic sulfite was changed from K2S2O5 to NaHSO3 (Table 1, entries 4–6). No better results could be obtained when other electrolytes (LiBF4 or LiPF6) or solvents (THF or DMAc) were tested (Table 1, entries 7–10). Changing the electric current or replacing the cathode to graphite or nickel could not give a high yield of 3aa (Table 1, entries 11–14). Only 5% of 3aa was obtained when the amount of methanol was reduced to 1 mL (Table 1, entry 15).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | “SO2” | Solvent | Electrolyte | I cell (mA) | t (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), MeOH (2 mL), “SO2” (4.0 equiv.), solvent (8 mL), graphite anode, Pt cathode, undivided cell, constant current, room temperature. b Yield determined by 1H NMR analysis based on 1a (using 1,3,5-trimethoxybenzene as the internal standard), isolated yield in brackets. c Graphite anode, Ni cathode. d Graphite anode, graphite cathode. e MeOH (1 mL), MeCN (9 mL). | ||||||
| 1 | K2S2O5 | MeCN | n Bu4NBF4 | 4 | 4 | 35 (33) |
| 2 | K2S2O5 | MeCN | n Bu4NBF4 | 4 | 8 | 73 |
| 3 | K2S2O5 | MeCN | n Bu4NBF4 | 4 | 12 | 81 |
| 4 | Na2S2O5 | MeCN | n Bu4NBF4 | 4 | 12 | 84 |
| 5 | Na2SO3 | MeCN | n Bu4NBF4 | 4 | 12 | 10 |
| 6 | NaHSO3 | MeCN | n Bu4NBF4 | 4 | 12 | 90 (86) |
| 7 | NaHSO3 | MeCN | LiBF4 | 4 | 12 | 44 |
| 8 | NaHSO3 | MeCN | LiPF6 | 4 | 12 | 33 |
| 9 | NaHSO3 | THF | n Bu4NBF4 | 4 | 12 | 42 |
| 10 | NaHSO3 | DMAc | n Bu4NBF4 | 4 | 12 | <5 |
| 11 | NaHSO3 | MeCN | n Bu4NBF4 | 2 | 24 | 64 |
| 12 | NaHSO3 | MeCN | n Bu4NBF4 | 6 | 8 | 68 |
| 13c | NaHSO3 | MeCN | n Bu4NBF4 | 4 | 12 | 62 |
| 14d | NaHSO3 | MeCN | n Bu4NBF4 | 4 | 12 | 36 |
| 15e | NaHSO3 | MeCN | n Bu4NBF4 | 4 | 12 | 5 |
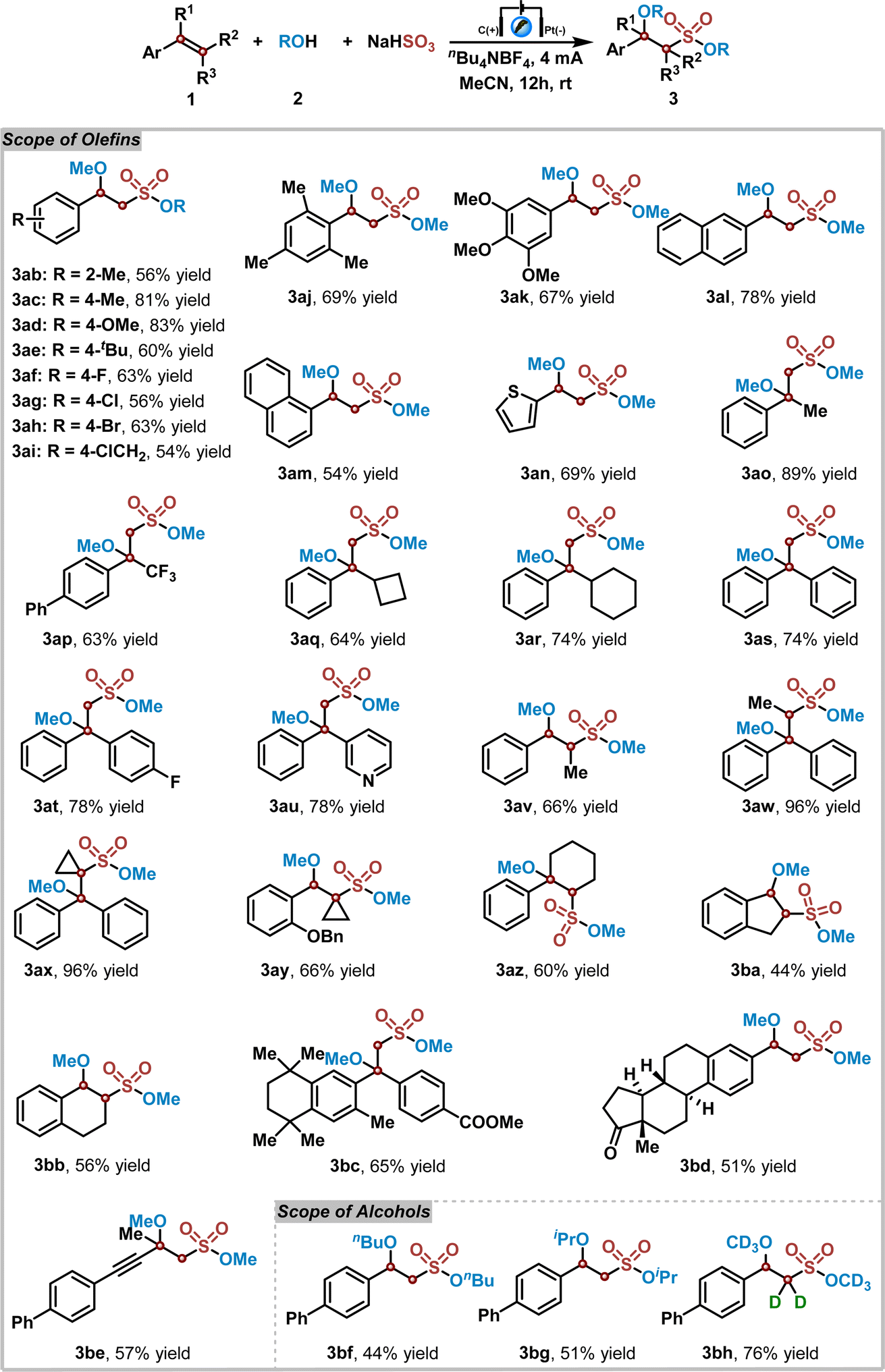
Subsequently, the substrate scope of this electrochemical reaction of alkenes 1, alcohols 2, and NaHSO3 was explored under the optimized reaction conditions. The results are shown in Table 2. At the outset, a series of substituted styrenes were examined in the reaction of methanol 2a with NaHSO3. Various electron-donating or electron-withdrawing functional groups on the styrene benzene ring were tolerated well in this reaction, yielding the corresponding products 3ab–3ak in 54% to 83% yields. Notably, large sterically hindered styrenes, such as 2,4,6-trimethylstyrene, also participated well in this transformation, giving rise to the corresponding sulfonate ester 3aj in 69% yield. Additionally, naphthyl and thienyl substituted vinyls were suitable for this reaction as well (3al–3an). Furthermore, the desired products 3ao–3av could be obtained in good to excellent yields, when disubstituted vinylic substrates were utilized in this electrochemical reaction. The chemical structure of product 3as was confirmed by X-ray diffraction analysis (see ESI†).49 In contrast, tri- and even tetra-substituted vinylic substrates were compatible in this reaction, affording the corresponding products 3aw–3ay in excellent yields. As we expected, this reaction was also compatible with cyclic styrenic substrates, leading to sulfonate esters 3az–3bb in 44–60% yields. Moreover, bioactive molecules, such as bexarotene- and estrone-derived substrates, could provide the corresponding sulfonate ester derivatives 3bc and 3bd in 65% and 51% yields. An alkynyl-containing substrate was also investigated, which afforded the desired product 3be in 57% yield with excellent chemoselectivity. Next, several alcohols were utilized in the reaction of 4-phenylstyrene 1a with NaHSO3. It was found that n-butanol and isopropyl alcohol all worked well in this reaction, delivering the sulfonate ester products 3bf and 3bg in 44% and 51% yields. Interestingly, when this electrochemical reaction was carried out with deuterated methanol, the α-hydrogen deuterated sulfonate ester 3bh was obtained in 76% yield. Further investigation indicated that the α-hydrogen of corresponding sulfonate ester could undergo a hydrogen–deuterium exchange process with deuterated methanol under the standard reaction conditions (see ESI† for details).
| a Isolated yield based on alkenes 1. |
|---|
|
Considering that the intramolecular radical functional group migration process features a powerful approach for the difunctionalization of unactivated alkenes.50–52 We proposed that the alkoxysulfonyl radical species produced by the electrochemical anodic oxidation process could provide the sulfonated products of unactivated alkenes through a radical functional group migration strategy. To our delight, the sulfonated product 5a could be achieved in 90% yield, when this electrochemical oxidative sulfonation reaction was performed by using 2-methyl-1,1-diphenylprop-2-en-1-ol 4a, methanol 2a, and K2S2O5 as substrates under slightly modified reaction conditions (see ESI† for details). The reaction scope with other allyl alcohols is summarized in Table 3. It was found that 1,1-diaryl allyl alcohols with various functional groups on the aromatic ring reacted smoothly under the standard reaction conditions, giving rise to the corresponding sulfonate esters 5a–5i in excellent yields. Additionally, 1-alkyl-1-aryl allyl alcohols were also workable in this transformation, and aryl migrated products could be produced in good yields with excellent chemoselectivity (5j–5p). The chemical structure of product 5m was confirmed by X-ray diffraction analysis (see ESI†).53 However, when this reaction was carried out with 1-cyclopropyl-2-methyl-1-phenylprop-2-en-1-ol 4q or 1-cyclobutyl-2-methyl-1-phenylprop-2-en-1-ol 4r, the mixture of aryl group and cycloalkyl group migrated products could be obtained (5qa–5ra and 5qb–5rb). Notably, 1,1-dialkyl allylic alcohols were compatible in this reaction as well, affording corresponding products 5s and 5t in 81% and 74% yields. Furthermore, ring expanding product 5u could be obtained in 80% yield through this migration process. Not only 1,2-migration, but also remote heteroaryl ipso-migration was realized as well, leading to the corresponding 1,4-migration products 5v and 5w in 60% and 74% yields.

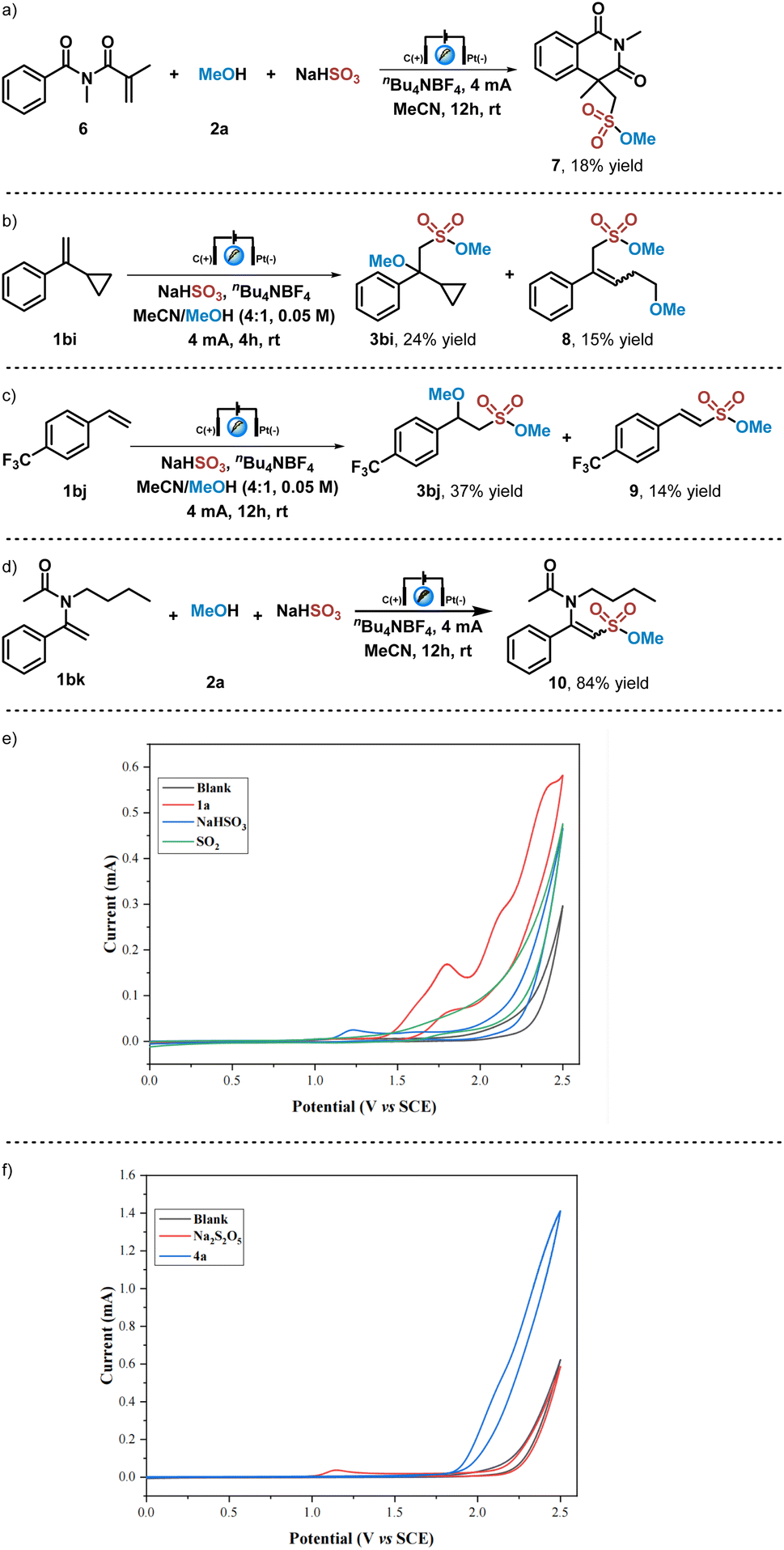
Several control experiments were then carried out to further understand the reaction mechanism. As shown in Scheme 2a, when this electrochemical reaction was performed with N-methacryloyl-N-methylbenzamide 6, which has been utilized as a radical trapper in radical processing reactions,54 the cyclized sulfonate ester 7 could be obtained in 18% yield. When cyclopropyl substituted styrene 1bi was applied in this electrochemical reaction with methanol and NaHSO3, the desired sulfonated product 3bi and the radical ring-opening product 8 were obtained in 24% and 15% yields (Scheme 2b). These results indicated that alkoxysulfonyl radical species are generated in this electrochemical reaction, and an alkoxysulfonyl radical addition process is involved in this transformation. When this electrochemical reaction was carried out with a strong electron-withdrawing functional group substituted styrene, such as trifluoromethyl substituted styrene 1bj, not only the difunctionalization product 3bi could be obtained in 37% yield, but also alkene 9 could be isolated in 14% yield. Then enamide 1bk was employed in this electrochemical reaction, giving rise to the coupling sulfonate 10 in 84% yield. These results showed that cationic species were formed by anode oxidation of radical species after the alkoxysulfonyl radical addition process, which could undergo a proton elimination process to afford corresponding coupling products. Subsequently, several CV (Cyclic Voltammetry) experiments were performed. No oxidation peak was observed when the CV experiment on the solution of sulfur dioxide gas in a mixture solvent of MeCN/MeOH (V/V = 4/1) was carried out (Scheme 2c). This result excluded the possibility of building alkoxysulfonyl radical species from the oxidation of sulfur dioxide nor alkyl sulfite (generated in situ from sulfur dioxide and methanol), which has been demonstrated in a previous report.47 In contrast, inorganic sulfite anions could be easily oxidized that an oxidation peak of NaHSO3 solution in MeCN/MeOH (V/V = 4/1) was found at 1.23 V (vs. SCE). On the other hand, the oxidation peak of olefin 1a solution in MeCN/MeOH (V/V = 4/1) was observed at 1.80 V (vs. SCE), which indicated that anodic oxidation of inorganic sulfite NaHSO3 is more preferential (Scheme 2c). In addition, CV experiments of inorganic sulfite Na2S2O5 and alkene 4a also gave similar results, showing that inorganic sulfite Na2S2O5 could be oxidized at the anode (oxidation peak) at 1.15 V (vs. SCE), while no obvious oxidation peak of 4a was found (Scheme 2d). Moreover, the CV experiment of alkene 6 showed no oxidation peak as well (see ESI† for details). All these results suggested that the alkoxysulfonyl radical species involved in this reaction are generated by electrochemical anodic oxidation of inorganic sulfites and subsequent alcohol capture.
 | ||
| Scheme 2 Control experiments. | ||
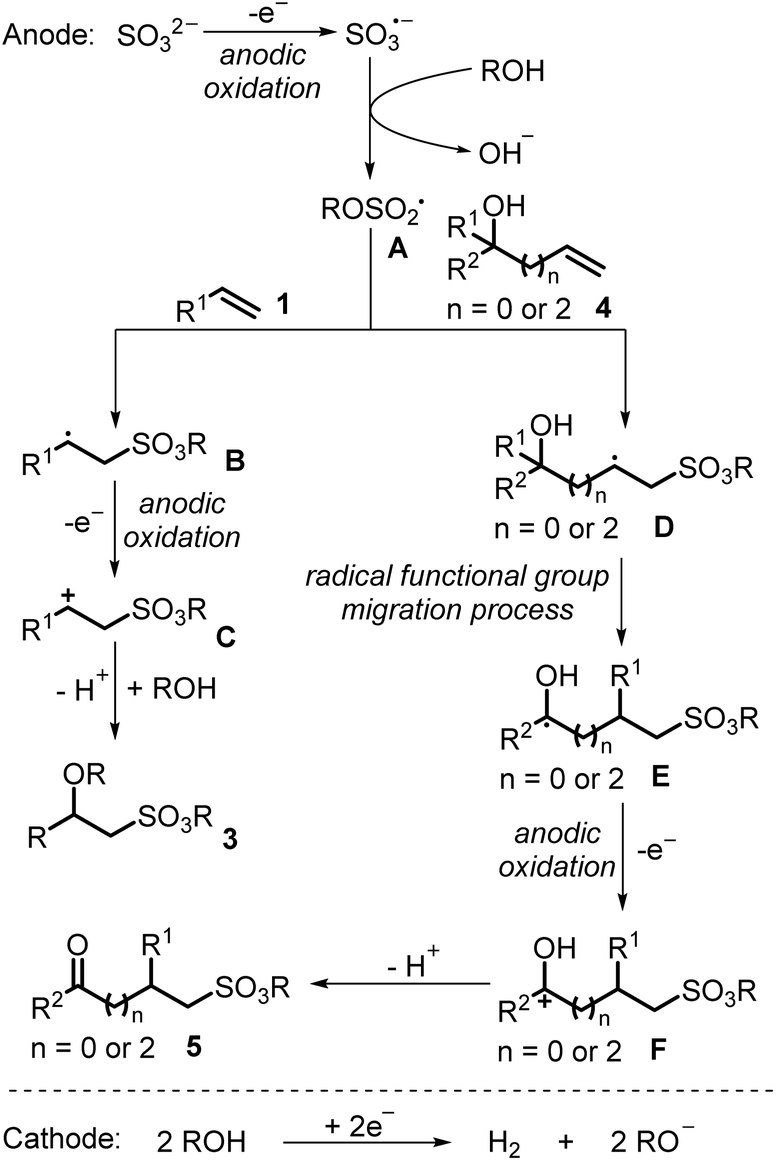
Based on the above results and previous reports, a possible reaction pathway for this electrochemical alkene sulfonation process is proposed in Scheme 3. The inorganic sulfite could be oxidized to the sulfite radical anion at the anode through a single-electron transfer oxidation, which would be subsequently trapped by alcohols to afford alkoxysulfonyl radical species A. Then, alkoxysulfonyl radical species A would undergo radical addition with alkene 1 to provide radical intermediate B. Further anodic single-electron transfer oxidation of radical intermediate B would provide carbocation C, which would subsequently react with alcohols to generate sulfonation product 3. On the other hand, the radical addition of alkoxysulfonyl radical species A with alkene 4 would provide radical intermediate D. Followed by radical functional group migration, anodic single-electron transfer oxidation and deprotonation, the sulfonation product 5 could be obtained.
 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In summary, we have developed an efficient and applicable route to provide alkoxysulfonyl radical species via the electrochemical anodic oxidation of inorganic sulfites with alcohols. Various sulfonate esters could be obtained by subsequent alkene difunctionalization with these alkoxysulfonyl radical species. This method exhibits broad functional group tolerance and excellent chemoselectivity. With this new discovery, potential prospects for the construction of sulfonate esters are revealed.Data availability
The data supporting this study are available within the article and the ESI.† The X-ray crystallography coordinates for structures of 3as and 5m have been deposited at the Cambridge Crystallographic Data Center (CCDC: 2131889 and 2131892).Author contributions
S. Y. conceived the study. C. Z. and M. Y. conducted the experiments and analysed the data. Y. Q., M. S., H. W. and M. Y. conducted preparation of starting materials. S. Y. and J. W. directed the project. C. Z. and W. X. conducted the experiments during manuscript revision. S. Y. prepared the manuscript. C. Z. prepared the supplemental information. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21901178), the Natural Science Foundation of Zhejiang Province (LY21B020002), the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (No. 2019R01005), and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (2020ZD04) is gratefully acknowledged.Notes and references
- W. R. Roush, S. L. Gwaltney, J. Cheng, K. A. Scheidt, J. H. McKerrow and E. Hansell, J. Am. Chem. Soc., 1998, 120, 10994–10995 CrossRef CAS.
- J. W. Choi, S. J. Shin, H. J. Kim, J.-H. Park, H. J. Kin, E. H. Lee, A. N. Pea, Y. S. Bahn and K. D. Park, ACS Med. Chem. Lett., 2019, 10, 1061–1067 CrossRef CAS.
- S. M. Pauff and S. C. Miller, Org. Lett., 2011, 13, 6196–6199 CrossRef CAS PubMed.
- D. C. Meadows, T. B. Mathews, T. W. North, M. J. Hadd, C. L. Kuo, N. Neamati and J. Gervay-Hague, J. Med. Chem., 2005, 48, 4526–4534 CrossRef CAS.
- D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 26, 793–814 CrossRef CAS PubMed.
- A.-N. R. Alba, X. Companyo and R. Rios, Chem. Soc. Rev., 2010, 39, 2018–2033 RSC.
- M. Kakimoto, S. J. Grunzinger and T. Hayakawa, Polym. J., 2010, 42, 697–705 CrossRef CAS.
- H. Sasabe, Y. Seino, M. Kimura and J. Kido, Chem. Mater., 2012, 24, 1404–1406 CrossRef CAS.
- N. S. Simpkins, Sulfones in Organic Synthesis, Pergamon Press, Oxford, 1993 Search PubMed.
- P. Vogel, M. R. Turks, L. Bouchez, D. Marković, A. Varela-Álvarez and J. Á. Sordo, Acc. Chem. Res., 2007, 40, 931–942 CrossRef CAS PubMed.
- C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS.
- Q. Lu, J. Zhang, G. Zhao, Y. Qi, H. Wang and A. Lei, J. Am. Chem. Soc., 2013, 135, 11481–11484 CrossRef CAS PubMed.
- X.-J. Tang and W. R. Dolbier Jr, Angew. Chem., Int. Ed., 2015, 54, 4246–4249 CrossRef CAS PubMed.
- A. García-Domínguez, S. Müller and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 9949–9952 CrossRef.
- Y. Ning, Q. Ji, P. Liao, E. A. Anderson and X. Bi, Angew. Chem., Int. Ed., 2017, 56, 13805–13808 CrossRef CAS PubMed.
- D. Zheng, Y. An, Z. Li and J. Wu, Angew. Chem., Int. Ed., 2014, 53, 2451–2454 CrossRef CAS PubMed.
- D. Zheng, J. Yu and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 11925–11929 CrossRef CAS PubMed.
- F. Liu, J.-Y. Wang, P. Zhou, G. Li, W.-J. Hao, S.-J. Tu and B. Jiang, Angew. Chem., Int. Ed., 2017, 56, 15570–15574 CrossRef CAS PubMed.
- Y. Meng, M. Wang and X. Jiang, Angew. Chem., Int. Ed., 2020, 59, 1346–1353 CrossRef CAS PubMed.
- K. Liu and A. Studer, J. Am. Chem. Soc., 2021, 143, 4903–4909 CrossRef CAS PubMed.
- S. M. Hell, C. F. Meyer, G. Laudadio, A. Misale, M. C. Willis, T. Noël, A. A. Trabanco and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 720–725 CrossRef CAS PubMed.
- C. Chatgilialoglu, D. Griller and S. Rossini, J. Org. Chem., 1989, 54, 2734–2737 CrossRef CAS.
- L. Cala, O. García-Pedrero, R. Rubio-Presa, F. J. Fañanás and F. Rodríguez, Chem. Commun., 2020, 56, 13425–13428 RSC.
- J. J. Douglas, H. Albright, M. J. Sevrin, K. P. Cole and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2015, 54, 14898–14902 CrossRef CAS PubMed.
- M. Bossart, R. Fässler, J. Schoenberger and A. Studer, Eur. J. Org. Chem., 2002, 2742–2757 CrossRef CAS.
- X. Dong, W. Jiang, D. Hua, X. Wang, L. Xu and X. Wu, Chem. Sci., 2021, 12, 11762–11768 RSC.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605–621 RSC.
- J. Yoshida, K. Kataoka, R. Horacjada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed.
- P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed.
- S. D. Minteer and P. S. Baran, Acc. Chem. Res., 2020, 53, 545–546 CrossRef CAS PubMed.
- L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed.
- K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T. S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS PubMed.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed.
- Y. Yuan, Y. Cao, Y. Lin, Y. Li, Z. Huang and A. Lei, ACS Catal., 2018, 8, 10871–10875 CrossRef CAS.
- S. Ye, G. Qiu and J. Wu, Chem. Commun., 2019, 55, 1013–1019 RSC.
- S. Ye, M. Yang and J. Wu, Chem. Commun., 2020, 56, 4145–4155 RSC.
- A. G. Zelinsky and B. Y. Pirogov, Electrochim. Acta, 2017, 231, 371–378 CrossRef CAS.
- T. Luo, Y. Peng, L. Chen, J. Li, F. Wu and D. Zhou, Environ. Sci. Technol., 2020, 54, 10261–10269 CrossRef PubMed.
- A. G. Zelinsky, Electrochim. Acta, 2016, 188, 727–733 CrossRef CAS.
- S. P. Blum, D. Schollmeyer, M. Turks and S. R. Waldvogel, Chem.–Eur. J., 2020, 26, 8358–8362 CrossRef CAS PubMed.
- S. P. Blum, T. Karakaya, S. Scheollmeyer, A. Klapars and S. R. Waldvogel, Angew. Chem., Int. Ed., 2021, 60, 5056–5062 CrossRef CAS PubMed.
- S. P. Blum, K. Hofman, G. Manolikakes and S. R. Waldvogel, Chem. Commun., 2021, 57, 8236–8249 RSC.
- CCDC 2131889 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
- W. Li, W. Xu, J. Xie, S. Yu and C. Zhu, Chem. Soc. Rev., 2018, 47, 654–667 RSC.
- P. Sivaguru, Z. Wang, G. Zanoni and X. Bi, Chem. Soc. Rev., 2019, 48, 2615–2656 RSC.
- X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS PubMed.
- CCDC 2131892 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
- Q. Liu, Y. Lu, H. Sheng, C.-S. Zhang, X.-D. Su, Z.-X. Wang and X.-Y. Chen, Angew. Chem., Int. Ed., 2021, 60, 25477–25484 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data, copies of 1H and 13C NMR spectra. CCDC 2131889 and 2131892. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc04027a |
| This journal is © The Royal Society of Chemistry 2022 |