
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformationally flexible heterohelicenes as stimuli-controlled soft molecular springs†
Pirudhan
Karak
and
Joyanta
Choudhury
 *
*
Organometallics & Smart Materials Laboratory, Department of Chemistry, Indian Institute of Science Education and Research (IISER) Bhopal, Bhopal 462 066, India. E-mail: joyanta@iiserb.ac.in
First published on 1st September 2022
Abstract
Structurally engineered molecules which can behave as stimuli-controlled mechanical nanomachines such as molecular shuttles, rotors, ratchets, and springs are important in several research areas, including molecular robotics, actuation, sensing, cargo transportation, etc. Helicenes, by virtue of their unique screw-type structures, were proposed as functional models for molecular springs; however, experimental realization has remained an elusive and unmet task until now, because of the lack of appropriate helicene molecules consisting of backbone-decorated dynamic architectures. Aiming to explore this unearthed direction, we present herein a novel class of modular flexible heterohelicenes with a stimuli (acid/base and light)-responsive core and peripheral modules. By applying pH (at core-embedded free imidazole sites) and light (at backbone-tethered dithienylethene units) stimuli, we demonstrate that these flexible heterohelicenes exhibit spring-like movement, with the reversible contraction/extension of the helical pitch. The uniquely functionalized structure of these molecules played a critical role in bestowing such capability, as revealed by crystallographic, spectroscopic and computational data. Careful assessment disclosed that the protonation/deprotonation-induced reversible generation and delocalization of positive charge throughout the π-conjugated helical rim switch the operative interactions between the π clouds of the terminal overlapping arene rings of the helicenes between repulsive and attractive, leading to extension/contraction of the helical pitch. On the other hand, in the case of the light stimulus, it was analyzed that the light-induced ring-closure of the photoactive dithienylethene units created a geometric distortion causing the helicenic wings to bend outward from the helicene rim, which resulted in extension of the helical pitch. The photo-assisted (or thermal) reverse ring-opening reaction converted the system to its original conformation, thus enabling the helicene molecule to display spring-like reversible extension/contraction motion. The new insights on the reversible dynamic features of this class of heterohelicenes under the influence of external stress would guide crucial design principles of helicene-based molecular springs for potential applications.
Introduction
Helicenes and heterohelicenes are an intriguing class of helically twisted, angularly fused polycyclic arenes and heteroarenes respectively, with a unique combination of structural, optical and electronic attributes.1 Owing to the remarkable properties, these architecturally elegant and aesthetically appealing molecules find rapidly widening applications in both fundamental and applied research domains, ranging from crystal engineering2 and supramolecular and host–guest recognition3 to electronic/optoelectronic/non-linear optical materials and devices,4 polarized light emission,5 and chirality-driven biological functions.6 In the field of stimuli-responsive artificial molecular machines, a plethora of non-helicene molecules which respond to various stimuli through the embedded functional units have been designed and applied over the years.7 Among several examples, a light-driven molecular piston cylinder and a chemically driven molecular elevator as illustrated by Stoddart,8 and an electrically-driven molecular vehicle as illustrated by Feringa,9 using structurally sophisticated molecules, are elegant representations which serve as an inspiration for several follow-up developments. Alongside molecule-based systems, several supramolecular and polymer-based stimuli-responsive materials have been fabricated and demonstrated as smart devices,10 shape-changing actuators,11etc. For example, a spring-like liquid-crystalline polymer embedded with azo-benzene photoswitch units, reported by Fletcher and Katsonis, exhibited macroscopic helical motion in the presence of light stimuli, thus working as photoactuators.12 However, for applications in the domain of the abovementioned dynamically functional structures including various other types such as molecular shuttles, rotors, ratchets, and springs,13 which rely on stimuli-induced controlled mechanical movements, suitable (hetero)helicene molecules are yet to be designed. Interestingly, by virtue of their unique helical screw-type structures, these molecules should, in principle, qualify to act as molecular springs mimicking macroscopic spring action (Fig. 1A). However actual realization of their spring-like elastic function by applying external stimuli is extremely challenging. The key challenge relates to the subtle configuration of the helicene structure in order to execute stimuli-induced nanoscale conformational switching, which would result in a reversible extension–contraction process at the molecular level. Helicene molecules with such capability are rarely reported in the literature.14 Tanaka demonstrated solvent-dependent contracted and extended structures in the self-assembled crystalline states of a helicene diol.14a Wang, Müllen and Petrukhina observed gradual contraction of a double [7]helicene upon successive reduction with alkali metals.14b However, the dynamic reversibility of the processes was not demonstrated with these helicene systems. Recently, a new type of helicene, viz., ‘expanded helicene’, in which the entire helical rim is composed of alternate angular and linear ring fusions (Fig. 1B), thus providing flexible helicity, was synthesized by Hirose and Matsuda.15 Due to the special flexible helical rim, this expanded helicene was shown, by computational simulations, to behave as a soft molecular spring. The system exhibited a small spring constant value which was further found to be a function of the inverse of third power of the helical dimeter, in accordance with the property of macroscopic springs.16 However, validating the stimuli-controlled experimental demonstration of this phenomenon has remained an elusive challenge until now, due to the lack of suitable helicene molecules consisting of stimuli-responsive modules within the structural backbone which would induce reversible contraction–expansion of the helical pitch, as well as a suitable monitoring property which is simultaneously correlated with such structural changes. | ||
| Fig. 1 (A) Action of a representative macroscopic spring. (B) The reported expanded helicene proposed as a soft molecular spring. (C) Stimuli responsive sub-expanded heterohelicene core studied in this work. Hypothesis of the (D) acid/base-controlled and (E) light-controlled spring-like behavior of sub-expanded helicenes reported in this work. | ||
We report herein a novel class of heteroatom-doped helicenes consisting of partial linear ring fusion along with the classical angular ring fusions across the helical rim, named as ‘sub-expanded heterohelicenes’ (Fig. 1C), configured with dynamic stimuli-responsive modules to enable stimuli-triggered soft molecular spring-like behavior (Fig. 1C–E), accompanied by simultaneous optical readout to monitor the stimuli-induced structural changes. Our design strategy was based on two hypotheses. In the first design, we expected that the free basic nitrogen atoms of the imidazole-heterocyclic rings embedded in the core of the heterohelicenes (Fig. 1D) would be responsive to protons (acid), making them positively charged imidazolium cations. The positive charge generated thereby would be expected to get delocalized throughout the π-conjugated helical rim, albeit with a gradient. In such a situation, the operative repulsive interactions between the π clouds of the terminal overlapping arene rings of the helical rim are expected to reduce or even to become attractive, which in turn, may decrease the ring-to-ring distance, thus contracting the helical pitch. Upon base treatment, the original helical pitch would be retained. This hypothesis can be endorsed by previous reports showing that the delocalization of oxidatively-generated positive charge on benzene rings in conformationally helical oligomeric ortho-phenylenes17a as well as in π-stacked multilayered para- and meta-cyclophanes17b led to a decrease of the distance between the planes of the overlapping benzene rings, plausibly via inter-ring attractive interactions. The second design strategy relies upon the photochromic dithienylethene (DTE) moieties, decorated at the peripheral positions of the heterohelicenes. Light-induced ring-closure of the DTE units is supposed to create a geometric distortion causing the helicenic wings to bend outward from the helicene rim, which would result in extension of the helical pitch. The photo-assisted (or thermal) reverse ring-opening reaction would convert the system to its original conformation (Fig. 1E). Both the hypothesized processes have been probed with the help of combined experimental–computational analysis, thus providing new insights into the dynamic features of such molecules under the influence of external stress, which would guide crucial design principles of helicene-based molecular springs for potential applications.
Results and discussion
To start our investigation, two series – a cationic (c-H1, c-H2, and c-H3) and a neutral (n-H1 and n-H2) – of flexible sub-expanded polyazahelicenes were synthesized exploiting a unique Cp*Rh(III)-catalyzed rollover π-expansion (RoPE) reaction (Scheme 1).18 The RoPE strategy19 involved concurrent linear and angular ring fusions on readily accessible imidazolium/imidazole-containing pyridines as templates by annulating with alkyne staples, generating a 6-6-5-6-6-6-5-6-6-ring-based hetero[9]-helicene skeleton. The novel dirhoda-hetero[9]-helicene intermediate (Rh-H1) was also synthesized by quadruple C–H activation-rollover metallation of a starting imidazolium-containing pyridine substrate (Scheme S8†), to confirm the origin of the flexible helicity in the derived products. All of the above helicenes were obtained as a racemic mixture of both P and M enantiomers in equal quantity, which could not be separated by methods like fractional crystallization (in the absence and presence of a chiral compound), resolution with chiral counter-anion or chiral chromatography. | ||
| Scheme 1 Synthesis of the sub-expanded heterohelicenes via a catalytic rollover protocol. | ||
Flexible helicity can lead to soft molecular spring-like behavior. To assess whether the present sub-expanded cationic and neutral heterohelicene molecules, consisting of a penta-aza[9]-helicenic core with fully conjugated 6-6-5-6-6-6-5-6-6-ring arrangement, actually qualify to possess flexible helicity along the helicene rim in a quantitative sense toward behaving as soft molecular springs, crystal structures and crystalline-state packing of c-H1, c-H2, n-H1, and n-H2 were analyzed first. It was found that they featured a large helical diameter of approx. 7 Å, due to the laterally expanded helix backbone (Fig. 2A–D and S7–S10†).
 | ||
| Fig. 2 Crystal structure and crystal-state packing of (A) n-H1 (CCDC 2165994), (B) c-H1 (CCDC 2158816), (C) n-H2 (CCDC 2165995), and (D) c-H2 (CCDC 2158813). (E) Racemization barrier (kcal mol−1) for c-ExHHel2 calculated at the 6-31g(d,p) level of theory. (F) Crystal structure and crystal-state packing of the Rh-H1 intermediate (CCDC 2158814). [The helical pitch pAI is defined as the centroid-to-centroid distance between the terminal rings A and I of the helical rim of the helicene molecule. The helical diameter dCH is the longest centroid-to-centroid distance between the two rings C and H of the helical rim. The intersecting angle θAI is defined as the angle between the two planes passing through the terminal rings A & I.] (G) (I) Computational calculation of the spring constant value: the distance between the two terminal carbon atoms (red) was allowed hypothetically to expand by a difference up to 2.0 Å, and the respective relative energies were calculated. (II) The variation of relative energy of the neutral and cationic helicenes n-H1 and c-H1 as a function of the increment of the distance between two terminal carbons. (III) The plot of the spring constant k as a function of the inverse of third power of the helical diameter (dh) of the corresponding helicene molecules considering an elliptical cross-section. Computations were performed at the B3LYP/6-311g(2d,p) level of theory. | ||
Most importantly, they contained (i) a small helical pitch, pAI (centroid–centroid distance between two terminal overlapping rings, i.e., rings A & I) values within the range of 3.48–3.77 Å, and (ii) a small intersecting angle, θAI, within 3.2°–17.4°, between the two planes passing through the terminal rings A & I. These values are smaller in comparison to those of the parent angularly fused rigid carbo[7]helicene (p = 3.87 Å; θ = 32°, CCDC 852537).20 Moreover, the molecules showed an increased helical diameter (approx. 7.0 Å) also in comparison to that of the parent [7]helicene (5.1 Å, CCDC 852537),20b suggesting an expanded and relaxed helical rim. The crystal packing of all of these molecules displayed alternately stacked P and M enantiomers in racemic column structures (Fig. 2A–D and S7–S10†).
As the enantiopure P or M isomers of these compounds could not be obtained by any of the attempted protocols, we decided to investigate the P/M racemization kinetics via a computational method. Thus, the P/M racemization barrier values calculated for both the cationic and neutral molecules lied within the range of 16–18 kcal mol−1 at 25 °C (Fig. 2E, Table S3†) which were comparable with those of previously reported flexible [7]heliphene 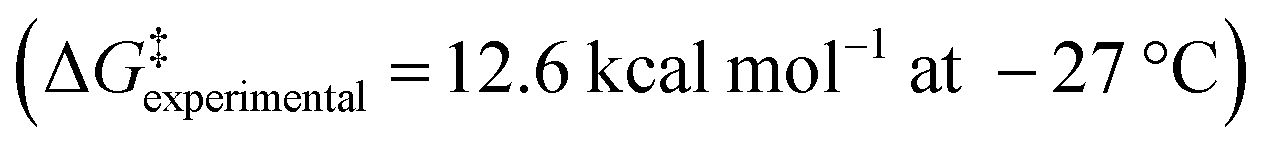 ,21 [13]helicene-based helical kekulene analogues
,21 [13]helicene-based helical kekulene analogues  ,15 and the aza-bora[9]helicene
,15 and the aza-bora[9]helicene 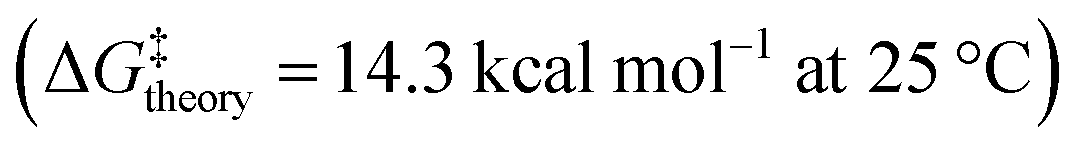 .22 Further, the half-life values (τ1/2) of the helical inversion process were τ1/2 = 0.4, 2.6, 1.7 and 0.1 s at 25 °C for c-H1, c-H2, n-H1 and n-H2 respectively. The low P/M configurational barrier and hence the fast kinetics of helical inversion via easy up-and-down flapping of the two helicenic wings are in accordance with the characteristics of the expanded/flexible helicene rim in these molecules.23
.22 Further, the half-life values (τ1/2) of the helical inversion process were τ1/2 = 0.4, 2.6, 1.7 and 0.1 s at 25 °C for c-H1, c-H2, n-H1 and n-H2 respectively. The low P/M configurational barrier and hence the fast kinetics of helical inversion via easy up-and-down flapping of the two helicenic wings are in accordance with the characteristics of the expanded/flexible helicene rim in these molecules.23
Intriguingly, the crystallographic features of the intermediate dirhoda-hetero[9]-helicene Rh-H1 clearly revealed the genesis of the expanded/flexible helicity at this intermediate stage only, followed by its translation to the corresponding organic helicene products. For example, Rh-H1 possessed a helical structure with a helical diameter of 7.4 Å, thus representing a rare bimetallic metallahelicene with two metal-containing rings being part of the contiguous helical network of mixed ‘carbocycle–heterocycle–metallacycle’ arrangement containing a 6-6-5-5-6-5-5-6-6-ring-based [9]-helicene skeleton (Fig. 2F).24 The crystal packing showed the presence of alternately stacked P and M enantiomers along the vertical direction to form a racemic column structure, reminiscent of the molecular arrangement in the product crystals (Fig. 2B–D). Due to the influence of the two contiguous metallacycle rings having the piano-stool geometry at the metal centre with its ligands within the helical rim, it exhibited a helical pitch (dAI) of 3.84 Å and an intersecting angle (θAI) between the two planes passing through the terminal rings of only 5.5°. Thus, after structural indication of the presence of the typical flexible helicity features in this new set of sub-expanded heterohelicenes, their soft molecular spring-like behavior, in line with the previous expanded helicene compound reported by Hirose and Matsuda,15 was accordingly supported by computational simulations as depicted in Fig. 2G(I and II). The results showed not only the expected small spring constant values of the systems but also a very good correlation between the spring constant and the inverse of third power of the helical dimeter of the molecules (Fig. 2G(III)), in accordance with the equation of the spring constant for macroscopic springs with an elliptical cross-section,25 considering that the present molecules would ideally contain an elliptical cross-section.
To execute the stimuli-controlled spring-like reversible contraction–extension of the helical pitch in the prospective helicenes, we first focused on the pairs of the neutral and positively charged helicenes n-H1/c-H1 and n-H2/c-H2, which are chemically inter-related by quaternization of the embedded imidazole N atoms (free versus alkylated imidazole N). Interestingly, scrutiny of their structural inter-relationship showed that there is a decrease in the helical pitch (by 0.21–0.18 Å) and intersecting angle θAI between the two terminal rings (by 7.8°–14.2°) on going from the neutral to the positively charged analogues (from 3.77 Å and 16.7° in n-H1 to 3.56 Å and 8.9° in c-H1, and from 3.66 Å and 17.4° in n-H2 to 3.48 Å and 3.2° in c-H2). Not only these two parameters, there was also lowering of the distance between the other corresponding pairs of overlapping rings on the two wings of the helical rim, certifying an overall geometrical shrinkage in the positively charged helicene molecules (Fig. S11†). Although not in helicene molecules, similar geometrical shrinkage and contraction of the helical pitch (or the distance between the planes of the overlapping arene rings) were previously observed in conformationally helical oligomeric ortho-phenylenes (by 0.02–0.07 Å)17a as well as in π-stacked multi-layered para- and meta-cyclophanes (by 0.06–0.40 Å),17b upon (oxidative) generation of positive charge in the molecules from the neutral analogues. In the case of the cyclophanes, the angle between the overlapping rings was also reduced in the positively charged molecules, similar to the above observation for the helicenes. The resulting optical readouts were characteristics of the above structural and electronic change. Thus, the absorption and emission maxima were found to be red-shifted from the neutral helicenes n-H1 (418 nm and 506 nm respectively) and n-H2 (440 nm and 523 nm respectively) to the cationic analogues c-H1 (437 nm and 576 nm respectively) and c-H2 (482 nm and 646 nm respectively) (Fig. 3A–D).
 | ||
| Fig. 3 (A) Absorption and (B) emission spectra of n-H1 and c-H1. (C) Absorption and (D) emission spectra of n-H2 and c-H2. All the absorption and emission spectra were recorded in CHCl3 (4 μM) at 25 °C; excitation wavelength = 440 nm, 455 nm, 430 nm, and 432 nm for c-H1, c-H2, n-H1, and n-H2 respectively. (E) Computed change in the helical pitch values in n-H1 and n-H2 upon protonation, calculated at the B3LYP/6-31g(d,p) level of theory. (F) Acid–base responsive changes in the absorption properties of n-H1. (G) Acid–base responsive changes in the emission properties of n-H1. (H) Acid–base responsive changes in the absorption properties of n-H2. (I) Acid–base responsive changes in the emission properties of n-H2. All the absorption spectra were recorded in CHCl3 (20 μM) at 25 °C. All the emission spectra were recorded in CHCl3 (10 μM) at 25 °C; excitation wavelength = 420 nm (for n-H1), 440 nm (n-H2). Molecular electrostatic potential (MEP) maps of the (J) neutral (n-H1) and the positively charged (alkylated c-H1, and protonated n-H1 + 2H+) helicenes, and (K) neutral (n-H2) and the positively charged (alkylated c-H2, and protonated n-H2 + 2H+) helicenes. (calculated by DFT at the B3LYP/6-31g(d,p) level of theory). | ||
The quaternization process (free versus protonated imidazole N) has an advantage of easy reversible deprotonation. Hence, in principle, the same contraction/extension of the helical pitch can be mimicked via an easily feasible reversible protonation–deprotonation strategy, which can analogously convert the system into cationic and neutral respectively. This was executed and monitored experimentally in solution via the optical readout as an output of the protonation–deprotonation, as well as computationally via measuring the change in the helical pitch. Thus, the calculated helical pitch decreased by 0.07 Å and 0.08 Å upon protonation of the neutral helicene n-H1 and n-H2 (Fig. 3E), along with similar (like the alkylated congeners) overall geometrical shrinkage (Fig. S22†). This was accompanied by a simultaneous red-shift of the absorption maximum from 418 nm in n-H1 and 440 nm in n-H2 to 440 nm and 495 nm in the corresponding positively charged protonated species (Fig. 3F and H). The emission maximum followed a similar trend (Fig. 3G and I). The subsequent treatment with a base deprotonated the system, regenerating the original absorption spectral profile. These promising combined experimental/computational results indeed supported the feasibility of executing the acid/base stimuli-induced spring-like motion with the judiciously configured neutral flexible helicene systems in solution. To verify the hypothesized underlying driving force for the spring-like contraction of the helical pitch upon generation of the positive charge in the helicene molecules, we analyzed the molecular electrostatic potential (MEP) maps of the neutral (n-H1 and n-H2) and the positively charged (both alkylated c-H1 and c-H2, and protonated n-H1 + 2H+ and n-H2 + 2H+) helicene molecules. Indeed, the development and delocalization of the positive charge throughout the π-cloud of the heteroarene/arene rings of the helical rim of the cationic molecules, with of course a gradient, was clearly evident (Fig. 3J and K). In contrast, the arene rings at both wings of the helical rim of the neutral molecules contained the negatively charged π-cloud. Therefore, it was highly likely, in accordance with the previous arguments, that the reduction of repulsive interactions or even generation of attractive interactions between the π clouds of the (terminal) overlapping rings in the case of the positively charged helicenes led to the observed contraction of the helical pitch. The similar values of the computationally obtained (0.15–0.16 Å) and crystallographically found (0.21–0.18 Å) decrement of helical pitch in the case of the positively charged N-alkylated c-H1 and c-H2 from the corresponding neutral analogues (n-H1 and n-H2) further emphasized that the geometrical shrinkage was not attributed to the effect of the counter-ions or the crystal-packing.
However, in the above system, the stimuli-responsive spring-like motion was operated in solution with acid/base stimuli which are chemical and invasive in nature.13f Although solution-based spring-like applications are relevant, for applications pertinent to materials chemistry, a physical and non-invasive stimulus is preferred and the system should be capable of displaying solid-state operation, preferably on a solid surface. One of the practical and convenient physical non-invasive stimuli is light. Previously, macroscopic reversible structural changes induced by the light stimulus were demonstrated on a solid state such as single crystals26 and polymer films,27 containing photoswitchable molecules. To execute a light-operated system, we utilized the photo-responsive dithienylethene (DTE) unit-containing analogous cationic helicene c-H3 (Fig. 4A). The system was investigated in solution first to evaluate its efficacy toward photo-switchable conversion, via monitoring through the optical readout. The yellow-colored solution of c-H3 in acetonitrile showed a significant absorption band at 446 nm (ϵ = 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 020 mol−1 cm−1). Irradiation with blue light blue LED, 2.25 mW cm−2 triggered cyclization reaction at the peripheral DTE sites, as suggested by a color change from yellow to green (Fig. 4B). Notably, as c-H3 consisted of two DTE units in one molecule, in principle, irradiation with blue light could form both the mono ring-closed (c-H3′) and double ring-closed (c-H3′′) conformers, depending on the arrangements (parallel/antiparallel) of the methyl groups in the DTE units. The cyclization process was accompanied by an appearance of a new absorption band at 654 nm (ϵ = 9860 mol−1 cm−1) and an increase of the absorbance at 446 nm (ϵ = 44400 mol−1 cm−1) (Fig. 4B). The appearance of the lower-energy absorption band at 654 nm indicates the formation of a more conjugated skeleton (in c-H3′ and c-H3′′) upon photoirradiation via the ring-closing reaction (Fig. 4A). The photoswitchable dynamic behavior of this flexible heterohelicene was confirmed by achieving the reverse reaction, i.e., ring-opening from c-H3′/c-H3′′ to c-H3, using a higher wavelength red light red LED, 1.55 mW cm−2 (Fig. 4C). The system showed excellent reversibility in acetonitrile solution (Fig. 4D). Notably, the ring-closing response was quite fast, whereas a prolonged ring-opening process suggests a relatively stable ring-closed isomer in the photostationary state, similar to hindered diarylethenes.28 However, the ring-opening process could be accelerated by applying heat.
020 mol−1 cm−1). Irradiation with blue light blue LED, 2.25 mW cm−2 triggered cyclization reaction at the peripheral DTE sites, as suggested by a color change from yellow to green (Fig. 4B). Notably, as c-H3 consisted of two DTE units in one molecule, in principle, irradiation with blue light could form both the mono ring-closed (c-H3′) and double ring-closed (c-H3′′) conformers, depending on the arrangements (parallel/antiparallel) of the methyl groups in the DTE units. The cyclization process was accompanied by an appearance of a new absorption band at 654 nm (ϵ = 9860 mol−1 cm−1) and an increase of the absorbance at 446 nm (ϵ = 44400 mol−1 cm−1) (Fig. 4B). The appearance of the lower-energy absorption band at 654 nm indicates the formation of a more conjugated skeleton (in c-H3′ and c-H3′′) upon photoirradiation via the ring-closing reaction (Fig. 4A). The photoswitchable dynamic behavior of this flexible heterohelicene was confirmed by achieving the reverse reaction, i.e., ring-opening from c-H3′/c-H3′′ to c-H3, using a higher wavelength red light red LED, 1.55 mW cm−2 (Fig. 4C). The system showed excellent reversibility in acetonitrile solution (Fig. 4D). Notably, the ring-closing response was quite fast, whereas a prolonged ring-opening process suggests a relatively stable ring-closed isomer in the photostationary state, similar to hindered diarylethenes.28 However, the ring-opening process could be accelerated by applying heat.
 | ||
| Fig. 4 (A) Representation of the reversible photochromism of c-H3. (B) Absorption spectral profile of 30 μM c-H3 (in CH3CN) upon irradiation with blue light (blue LED, 2.25 mW cm−2). (C) Absorption spectral profile of 30 μM c-H3′ + c-H3′′ (in CH3CN) upon irradiation with red light (red LED, 1.55 mW cm−2). (D) Reversibility of the photochromism of c-H3 (10 μM in CH3CN). The absorbance value at λmax = 654 nm was monitored for switching study upon consecutive irradiation with blue light for 3 min (green dots) and red light for 45 min (red dots). (E) Schematic diagram of photochromic film fabrication of c-H3 in the PMMA gel matrix (2.3 wt%) on a PET substrate. (F) Reversibility of the photochromism of c-H3 in the PMMA gel matrix coated on a PET substrate. The absorbance value at λmax = 654 nm was monitored for switching study upon consecutive irradiation with blue light (blue LED, 5.10 mW cm−2) for 3 min (green dots) and heating at 100 °C for 2 min (red dots). (G) Optimized structures along with the helical pitch and inter-planar angle between the two wings of the helical rim values of the ring-opened and ring-closed isomers of c-H3 (calculated by DFT at the B3LYP/6-31g(d,p) level of theory). | ||
After the validation in solution, the photoswitchable process of c-H3 was also examined on a solid surface, in the form of a PMMA (poly(methyl methacrylate)) polymer film matrix (2.3 wt%) on a flexible PET (polyethylene terephthalate) substrate (Fig. 4E). Delightedly, the system was found to be highly efficient and reversible in its operation in the film as well (Fig. 4F). Irradiation with blue light (blue LED, 5.10 mW cm−2) for 3 min led to a rapid color change of the film from yellow to green, as monitored by the change of absorbance at λmax = 654 nm. On the other hand, the reversion of the color from green to yellow was rapidly achieved by heating the film at 100 °C by exposing the film to hot air for just 2 min. Notably, the reverse reaction was also possible by irradiating red light, but the process was found to be expectedly slow in the thin film. In this regard, light-responsive monolayers and assembled thin-films fabricated on solid surfaces were previously explored for various applications such as organic field-effect transistor memories, permeability-controlled charge transport, and reversible trapping/release.29
Interestingly, when the computationally calculated helical pitch of c-H3 was compared between the ring-opened and both mono and double ring-closed conformers, we found an expansion of 0.17–0.26 Å at the molecular level, upon photoinduced ring-closing processes (Fig. 4G), which was promising in the current context. This was plausibly associated with the conformational (mechanical) distortion caused by the corresponding ring-closing stress at the peripheral sites, which eventually transmitted an opposite perturbation (such as an outward bending) at the two wings of the helical rim of the molecule. In fact, the increase in the inter-planar angle between the two wings of the helical rim by 5.6° and 10.6° in the mono and double ring-closed conformers of c-H3 respectively, on changing from their corresponding ring-opened conformations (Fig. 4G), supported the above plausible reverse perturbation (outward bending) of the two helicenic wings. The analysis prompted us to experimentally demonstrate and validate the photo-controlled spring-like expansion–contraction motion on a solid surface through a reversible ring-closing and ring-opening process. We hypothesized that monitoring the change in the thickness of a thin film of c-H3 (in the form of a PMMA polymer matrix) on the surface of a silicon wafer upon light irradiation would be an appropriate and valid experiment which can be examined by spectroscopic ellipsometry (SE) (Fig. 5A).
 | ||
| Fig. 5 (A) Schematic representation of spectroscopic ellipsometry (SE) measurement of the thickness of a thin film of c-H3 in the PMMA matrix on Si wafer. (B) Thickness changes of a thin film of c-H3 in the PMMA matrix upon blue light irradiation (blue LED, 5.10 mW cm−2) and heating (80 °C); MSE = mean squared error of the fitting model applied to fit the data. See ESI† for details. (C) δѰ and (D) δΔ spectroscopic ellipsometry spectral difference of the film before and after the blue light irradiation, where δѰ/Δ = Ѱ/Δ before irradiation, – Ѱ/Δ after irradiation, and Ѱ and Δ are the ellipsometric parameters. (E) Emission spectral changes of a thin film of c-H3 (in PMMA) on Si wafer upon blue light (5.10 mW cm−2) irradiation (for 2 min). | ||
Adsorption of helicene molecules on a solid surface was studied previously, and it was reported that they adsorb on the surface via dispersive interactions of the planar arene part of the backbone with the surface, resulting in parallel (horizontal) alignment.30 Moreover, in solvent-cast films of various polymers, the backbones were found to align preferentially to the plane of the surface.31 In line with these previous studies, it is reasonable to expect that the molecules of c-H3 by virtue of their extended π-surface would adsorb parallel to the surface of the solid silicon substrate, and then would supramolecularly assemble on top of the surface via intermolecular π–π stacking interactions, akin to the crystal packing arrangement present in the congener helicenes (n-H1, n-H2, c-H1 and c-H2). The PMMA polymer would allow the spatial separation of the helicene molecules required for any structural/conformational change. Therefore, for the thin film of c-H3/PMMA on a silicon surface, any change (increase or decrease) of the helical pitch of c-H3 at the molecular level can be correlated with the change in the overall thickness of the assembled film. In situ variable angle spectroscopic ellipsometry (VASE) represents a sensitive technique to probe such thickness changes, which are expected to be in the order of sub-nanometer to nanometer-levels in the case of the assembled film of c-H3 molecules. In fact, the SE technique was previously applied successfully to monitor the light-induced changes in the thickness of thin films of this type of DTE-containing photochromic organic compound dispersed in the PMMA polymer matrix or of DTE-containing co-polymers.31b,32 In our experiment, a suitable thin film of the photochromic helicene c-H3 on a silicon wafer was fabricated by spin coating (angular speed between 400 and 700 rpm) on a silicon substrate using a solution of 12 wt% c-H3 in PMMA in acetonitrile (see ESI† for details). The initial thickness was measured to be 125.08 nm under dark conditions (Fig. 5B). After irradiating blue light (λ = 400–450 nm, blue LED, 5.10 mW cm−2) for 6 min on the thin film in situ without disturbing the position of the wafer sample from the stage of the instrument, the thickness was found to be increased to 127.25 nm. An increment of the thickness of the thin film by about 2.17 nm or 21.7 Å was substantial and commensurate considering the multilayer assembly of the molecules on the surface, and can be correlated with the light-stimulated ring-closure-induced expansion of the helical pitch at the molecular level by approx. 0.17–0.26 Å per molecular layer, as supported by the computational analysis described above. The reverse reaction was promoted by heating the sample in situ at approx. 80 °C for 3.0 min, and the film thickness was measured again. We found that it decreased to 124.83 nm, which was close to the original thickness value, suggesting ring-opening-induced re-contraction of the helical pitch at the molecular level, thus regaining the initial thickness. It is noteworthy to emphasize here that although the magnitude of ≈1.7% for the change of thickness observed in the present film apparently seems to be small, it is comparable to the change one would expect for similar polymer-dispersed films, as per previous literature reports.31b,33 For example, Canepa and co-workers observed ≈1.5% change in the thickness of a thin film prepared on a silicon wafer with a DTE-based cross-linked polymer, upon similar photo-induced ring closure.31b Similarly, for a thin film of an azobenzene-based photoresponsive polymer, the extent of relative change in film thickness was found to be in the range of 0.6–1.6% to a maximum of 1.5–4%, as reported by Barrett and co-worker.33 In contrast to the above examples with thin films (which are in fact relatively less ordered materials), photomechanically induced large geometrical changes were reported in a few cases but only when the photoactive molecules were self-assembled in a highly ordered fashion in macroscopic objects such as crystals26 or cross-linked liquid crystalline polymers27 or nematic elastomers.34 For instance, a class of azobenzene-containing nematic elastomers was reported to contract by around 20% on UV light irradiation.34 Similarly, ≈5–7% geometrical change (contraction/expansion) was observed for large-sized (10–100 μm) single crystals, containing a photoswitchable DTE-containing molecule such as 1,2-bis(2-ethyl-5-phenyl-3-thienyl)perfluorocyclopentene.26 However, on the other hand, even in the case of DTE-type systems in ordered crystalline form, dimension changes ranging from as high as 5.6% to as low as 0.14%, resulting from photo-induced ring closure, were also reported.35
In the ellipsometric measurements of our thin film, the difference in Ψ and Δ spectral profiles of the system before and after the light irradiation indicated evident changes between the two states of the molecules (Fig. 5C and D). Notably, the change of the refractive index (Δn = 0.00635) and extinction coefficient (Δk = 0.00267) was small for the film before and after the light irradiation. Importantly, to minimize the possible aggregation, aggregation-related light scattering, and also interference from light absorption by the thin film containing compound c-H3, the VASE experiments were conducted with films having an optimum thickness value (preferably 100–300 nm in the form of an optimum ≤10–12% dispersion in the host polymer). Moreover, to take care of still any absorption, the ellipsometry data were fitted with a suitable model (B-Spline) for the absorbing film, and validated by low mean squared error (MSE) values, for reliable accuracy. We validated the above phenomenon with additional two samples as well (see ESI† for details). A bare PMMA film assembled on a silicon wafer without containing the photochromic helicene compound c-H3 did not show any increase in thickness upon similar blue light irradiation for the same time. This control experiment confirmed that the reversible thickness change in the case of the photochromic helicene/PMMA film on the silicon wafer was actually associated with the photo-induced ring-closing/ring-opening of the DTE units of the photochromic helicene molecules and the resulting spring-like motion via expansion/re-contraction of the helical pitch, in line with the computational analysis.
Because silicon wafer is non-transparent in nature, the photochromic helicene/PMMA film on this surface was further examined simultaneously through fluorescence spectroscopy to probe the light-induced ring-cyclization process via monitoring the emission readout (Fig. 5E). Evidently, a decrease of the fluorescence intensity upon irradiating blue light on the thin film was observed suggesting the successful ring-closure process due to which efficient energy transfer from the excited helicene core to the ring-closed DTE unit of the molecule could occur as a consequence of significant spectral overlap between the emission band of c-H3 and absorption band of c-H3′/c-H3′′ (Fig. S23j†) (secondary inner-filter effect), thus leading to inhibition of the fluorescence process.36
Conclusions
In summary, we developed a convenient and simple synthetic protocol, termed as rollover π-expansion (RoPE), to access a novel class of flexible expanded heterohelicenes with a stimuli-responsive core and peripheral functional modules. Through careful investigation with two series of dynamically configured pH and light-responsive unique sub-expanded heterohelicenes, the capability of flexible helicene molecules to behave as stimuli-triggered soft spring-like nanoscopic objects was demonstrated experimentally both in solution and on surfaces, supported by crystallographic, spectroscopic and computational data. The critical role of reversible protonation/deprotonation-induced generation and delocalization of positive charge throughout the π-clouds of the helicene rings in enabling the reversible contraction/extension of the helical pitch was scrutinized. On the other hand, for the light-switchable helicene molecules, the reversible geometrical perturbation due to ring-closing/ring-opening at the photo-active peripheral sites was found to be pivotal to induce reversible outward/inward bending of the helicene wings leading to the observed extension/contraction of the molecules. The new insights on helicene-based molecular design principles proposed in this work would stimulate focused research and pave the way toward future development of stimuli-controlled molecular and supramolecular nanoscopic helicene springs for various task-specific actuation and sensing applications.Data availability
All data of this research work are available in the manuscript and in the ESI.†Author contributions
P. K. performed all the experimental as well as computational studies with inputs from J. C. Both P. K. and J. C. designed and wrote the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. C. thanks DST-SERB (grant no. CRG/2019/006038 and EMR/2016/003002) and IISER Bhopal for generous financial support. P. K. thanks IISER Bhopal for an Integrated PhD fellowship. We thank Dr. Madhusudan K. Pandey for help in crystallography. The authors acknowledge the NMR, Mass, SCXRD and Spectroscopic Ellipsometry facilities of CIF, and HPC facility of IISER Bhopal. The authors sincerely thank the anonymous reviewers for their noteworthy and insightful comments.References
- (a) M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC; (b) K. Xu, Y. Fu, Y. Zhou, F. Hennersdorf, P. Machata, I. Vincon, J. J. Weigand, A. A. Popov, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 15876–15881 CrossRef CAS PubMed; (c) Y. Nakakuki, T. Hirose, H. Sotome, H. Miyasaka and K. Matsuda, J. Am. Chem. Soc., 2018, 140, 4317–4326 CrossRef CAS PubMed; (d) K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed; (e) X. Guo, Z. Yuan, Y. Zhu, Z. Li, R. Huang, Z. Xia, W. Zhang, Y. Li and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 16966–16972 CrossRef CAS PubMed; (f) S. K. Pedersen, K. Eriksen and M. Pittelkow, Angew. Chem., Int. Ed., 2019, 58, 18419–18423 CrossRef CAS; (g) T. Otani, T. Sasayama, C. Iwashimizu, K. S. Kanyiva, H. Kawai and T. Shibata, Chem. Commun., 2020, 56, 4484–4487 RSC.
- A. E. Samkian, G. R. Kiel, C. G. Jones, H. M. Bergman, J. Oktawiec, H. M. Nelson and T. D. Tilley, Angew. Chem., Int. Ed., 2021, 60, 2493–2499 CrossRef CAS.
- L. Zhang, H.-X. Wang, S. Li and M. Liu, Chem. Soc. Rev., 2020, 49, 9095–9120 RSC.
- (a) T. Verbiest, V. Elshocht Sven, M. Kauranen, L. Hellemans, J. Snauwaert, C. Nuckolls, J. Katz Thomas and A. Persoons, Science, 1998, 282, 913–915 CrossRef CAS PubMed; (b) D.-W. Zhang, M. Li and C.-F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- (a) T. Otani, A. Tsuyuki, T. Iwachi, S. Someya, K. Tateno, H. Kawai, T. Saito, K. S. Kanyiva and T. Shibata, Angew. Chem., Int. Ed., 2017, 56, 3906–3910 CrossRef CAS PubMed; (b) W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Chem. Commun., 2019, 55, 13793–13803 RSC; (c) T. Mori, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed; (d) X. Xiao, S. K. Pedersen, D. Aranda, J. Yang, R. A. Wiscons, M. Pittelkow, M. L. Steigerwald, F. Santoro, N. J. Schuster and C. Nuckolls, J. Am. Chem. Soc., 2021, 143, 983–991 CrossRef CAS PubMed.
- W. Ma, L. Xu, L. Wang, C. Xu and H. Kuang, Adv. Funct. Mater., 2019, 29, 1805512 CrossRef.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- (a) P. R. Ashton, V. Balzani, O. Kocian, L. Prodi, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1998, 120, 11190–11191 CrossRef CAS; (b) J. D. Badjić, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef PubMed.
- T. Kudernac, N. Ruangsupapichat, M. Parschau, B. Maciá, N. Katsonis, S. R. Harutyunyan, K.-H. Ernst and B. L. Feringa, Nature, 2011, 479, 208–211 CrossRef CAS PubMed.
- R. Yerushalmi, A. Scherz, M. E. van der Boom and H.-B. Kraatz, J. Mater. Chem., 2005, 15, 4480–4487 RSC.
- G. Stoychev, A. Kirillova and L. Ionov, Adv. Opt. Mater., 2019, 7, 1900067 CrossRef.
- S. Iamsaard, S. J. Aßhoff, B. Matt, T. Kudernac, J. J. L. M. Cornelissen, S. P. Fletcher and N. Katsonis, Nat. Chem., 2014, 6, 229–235 CrossRef CAS PubMed.
- (a) H. Murakami, A. Kawabuchi, K. Kotoo, M. Kunitake and N. Nakashima, J. Am. Chem. Soc., 1997, 119, 7605–7606 CrossRef CAS; (b) T. R. Kelly, I. Tellitu and J. P. Sestelo, Angew. Chem., Int. Ed. Engl., 1997, 36, 1866–1868 CrossRef CAS; (c) O.-S. Jung, Y. J. Kim, Y.-A. Lee, J. K. Park and H. K. Chae, J. Am. Chem. Soc., 2000, 122, 9921–9925 CrossRef CAS; (d) B. L. Feringa, Acc. Chem. Res., 2001, 34, 504–513 CrossRef CAS PubMed; (e) V. Serreli, C.-F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523–527 CrossRef CAS PubMed; (f) C.-B. Huang, A. Ciesielski and P. Samorì, Angew. Chem., Int. Ed., 2020, 59, 7319–7330 CrossRef CAS.
- (a) K. Tanaka, H. Osuga and Y. Kitahara, J. Chem. Soc., Perkin Trans., 2000, 2, 2492–2497 RSC; (b) Z. Zhou, L. Fu, Y. Hu, X.-Y. Wang, Z. Wei, A. Narita, K. Müllen and M. A. Petrukhina, Angew. Chem., Int. Ed., 2020, 59, 15923–15927 CrossRef CAS PubMed.
- Y. Nakakuki, T. Hirose and K. Matsuda, J. Am. Chem. Soc., 2018, 140, 15461–15469 CrossRef CAS PubMed.
- X. Chen, S. Zhang, D. A. Dikin, W. Ding, R. S. Ruoff, L. Pan and Y. Nakayama, Nano Lett., 2003, 3, 1299–1304 CrossRef CAS.
- (a) E. Ohta, H. Sato, S. Ando, A. Kosaka, T. Fukushima, D. Hashizume, M. Yamasaki, K. Hasegawa, A. Muraoka, H. Ushiyama, K. Yamashita and T. Aida, Nat. Chem., 2011, 3, 68–73 CrossRef CAS PubMed; (b) M. Fujitsuka, S. Tojo, M. Shibahara, M. Watanabe, T. Shinmyozu and T. Majima, J. Phys. Chem. A, 2011, 115, 741–746 CrossRef CAS PubMed.
- P. Karak, C. Dutta, T. Dutta, A. L. Koner and J. Choudhury, Chem. Commun., 2019, 55, 6791–6794 RSC.
- (a) D. Ghorai, C. Dutta and J. Choudhury, ACS Catal., 2016, 6, 709–713 CrossRef CAS; (b) P. Karak, S. S. Rana and J. Choudhury, Chem. Commun., 2022, 58, 133–154 RSC.
- (a) P. T. Beurskens, G. Beurskens and T. E. M. van den Hark, Cryst. Struct. Commun., 1976, 5, 241–246 CAS; (b) Z. Qiu, C.-W. Ju, L. Frédéric, Y. Hu, D. Schollmeyer, G. Pieters, K. Müllen and A. Narita, J. Am. Chem. Soc., 2021, 143, 4661–4667 CrossRef CAS PubMed.
- S. Han, A. D. Bond, R. L. Disch, D. Holmes, J. M. Schulman, S. J. Teat, K. P. C. Vollhardt and G. D. Whitener, Angew. Chem., Int. Ed., 2002, 41, 3223–3227 CrossRef CAS PubMed.
- J. Full, S. P. Panchal, J. Götz, A.-M. Krause and A. Nowak-Król, Angew. Chem., Int. Ed., 2021, 60, 4350–4357 CrossRef CAS PubMed.
- (a) G. R. Kiel, S. C. Patel, P. W. Smith, D. S. Levine and T. D. Tilley, J. Am. Chem. Soc., 2017, 139, 18456–18459 CrossRef CAS; (b) G. R. Kiel, K. L. Bay, A. E. Samkian, N. J. Schuster, J. B. Lin, R. C. Handford, C. Nuckolls, K. N. Houk and T. D. Tilley, J. Am. Chem. Soc., 2020, 142, 11084–11091 CrossRef CAS; (c) K. Fujise, E. Tsurumaki, K. Wakamatsu and S. Toyota, Chem.–Eur. J., 2021, 27, 4548–4552 CrossRef CAS PubMed.
- E. S. Gauthier, R. Rodríguez and J. Crassous, Angew. Chem., Int. Ed., 2020, 59, 22840–22856 CrossRef CAS.
- K. Nagaya, Y. Hirata, T. Tsurumi, S. Takeda, K.-i. Nagai and K. Tanifuji, J. Mater. Des., 1992, 114, 667–669 Search PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS.
- J.-i. Mamiya, Polym. J., 2013, 45, 239–246 CrossRef CAS.
- W. Li, C. Jiao, X. Li, Y. Xie, K. Nakatani, H. Tian and W. Zhu, Angew. Chem., Int. Ed., 2014, 53, 4603–4607 CrossRef CAS.
- (a) A. Gulino, F. Lupo, G. G. Condorelli, M. E. Fragalà, M. E. Amato and G. Scarlata, J. Mater. Chem., 2008, 18, 5011–5018 RSC; (b) Y. J. Jeong, E. J. Yoo, L. H. Kim, S. Park, J. Jang, S. H. Kim, S. W. Lee and C. E. Park, J. Mater. Chem. C, 2016, 4, 5398–5406 RSC; (c) R. Balgley, G. de Ruiter, G. Evmenenko, T. Bendikov, M. Lahav and M. E. van der Boom, J. Am. Chem. Soc., 2016, 138, 16398–16406 CrossRef CAS PubMed; (d) R. Balgley, Y. M. Algavi, N. E. Dov, M. Lahav and M. E. van der Boom, Angew. Chem., Int. Ed., 2018, 57, 13459–13464 CrossRef CAS PubMed.
- (a) R. Fasel, A. Cossy, K. H. Ernst, F. Baumberger, T. Greber and J. Osterwalder, Chem. Phys., 2001, 115, 1020–1027 CAS; (b) R. Fasel, M. Parschau and K.-H. Ernst, Angew. Chem., Int. Ed., 2003, 42, 5178–5181 CrossRef CAS PubMed; (c) E. Tuca and I. Paci, Phys. Chem. Chem. Phys., 2019, 21, 9189–9199 RSC.
- (a) W. M. Prest and D. J. Luca, J. Appl. Phys., 1979, 50, 6067–6071 CrossRef CAS; (b) C. Toccafondi, L. Occhi, O. Cavalleri, A. Penco, R. Castagna, A. Bianco, C. Bertarelli, D. Comoretto and M. Canepa, J. Mater. Chem. C, 2014, 2, 4692–4698 RSC.
- C. Bertarelli, A. Bianco, F. D'Amore, M. C. Gallazzi and G. Zerbi, Adv. Funct. Mater., 2004, 14, 357–363 CrossRef CAS.
- O. M. Tanchak and C. J. Barrett, Macromolecules, 2005, 38, 10566–10570 CrossRef CAS.
- H. Finkelmann, E. Nishikawa, G. G. Pereira and M. Warner, Phys. Rev. Lett., 2001, 87, 015501 CrossRef CAS PubMed.
- (a) M. Irie, T. Lifka, S. Kobatake and N. Kato, J. Am. Chem. Soc., 2000, 122, 4871–4876 CrossRef CAS; (b) S. Kobatake, K. Shibata, K. Uchida and M. Irie, J. Am. Chem. Soc., 2000, 122, 12135–12141 CrossRef CAS; (c) T. Kodani, K. Matsuda, T. Yamada, S. Kobatake and M. Irie, J. Am. Chem. Soc., 2000, 122, 9631–9637 CrossRef CAS; (d) M. Irie, S. Kobatake and M. Horichi, Science, 2001, 291, 1769–1772 CrossRef CAS PubMed; (e) K. Uchida, N. Izumi, S. Sukata, Y. Kojima, S. Nakamura and M. Irie, Angew. Chem., Int. Ed., 2006, 45, 6470–6473 CrossRef CAS PubMed.
- C. Li, H. Yan, L.-X. Zhao, G.-F. Zhang, Z. Hu, Z.-L. Huang and M.-Q. Zhu, Nat. Commun., 2014, 5, 5709 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2158813, 2158814, 2158816, 2165994 and 2165995. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc04006a |
| This journal is © The Royal Society of Chemistry 2022 |