
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral diboranes as catalysts for the stereoselective organopolymerization of epoxides†
Ayla
Sirin-Sariaslan
and
Stefan
Naumann
 *
*
University of Stuttgart, Institute of Polymer Chemistry, 70569 Stuttgart, Germany. E-mail: stefan.naumann@ipoc.uni-stuttgart.de
First published on 31st August 2022
Abstract
It is demonstrated that stereoselective polymerization of epoxides, long a domain of metal-based compounds, can also be achieved via the application of organocatalysts. A simple two-step synthesis starting from widely available 1,1′-bi-2-naphthol (BINOL) backbones yields diboranes which, in tandem with organobases, deliver isotactic-enriched (it) polyethers from the homopolymerization of racemic propylene oxide (PO) and other epoxides. Thereby, isotactic diad contents of up to 88% can be achieved, resulting in well-defined (1.1 < ĐM < 1.3) polyethers with high molar masses (Mn > 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1). Notably, it is also possible to grow it-enriched sequences of PPO on aliphatic polyester-type initiators, thus enabling the incorporation of stereocontrolled polyether blocks in more complex polymer architectures. It is expected that this ability will greatly benefit the preparation of polyether-containing additives. The BINOL-type diboranes can be readily modified, suggesting further potential as a platform from which optimized catalysts can be developed.
000 g mol−1). Notably, it is also possible to grow it-enriched sequences of PPO on aliphatic polyester-type initiators, thus enabling the incorporation of stereocontrolled polyether blocks in more complex polymer architectures. It is expected that this ability will greatly benefit the preparation of polyether-containing additives. The BINOL-type diboranes can be readily modified, suggesting further potential as a platform from which optimized catalysts can be developed.
Introduction
To date, the stereoselective polymerization of substituted epoxides such as propylene oxide (PO) has remained an exclusive domain of relatively few metal-based polymerization systems.1 First described in the early 1950s in the form of a heterogeneous FeCl3 setup (Pruitt–Baggett catalyst) to deliver fractions of isotactic (it-)PPO,2 later examples also include Al- and Zn-based compounds3 (i.e., the Vandenberg catalyst4,5).A further significant breakthrough occurred in 2005–2008 when Coates and co-workers published their findings on (bimetallic) Co(III) catalysts.6,7 Conversion of rac-PO and many other epoxides was observed, resulting in excellent selectivity and high molar masses. The obtained it-polyethers were found to be semi-crystalline with sometimes surprisingly high melting points.8 Bimetallic ring-opening and an optimized Co–Co distance, embedded in a chiral environment, were reported to be essential.9 The Coates system is strictly enantioselective.
Despite these advances, to the best knowledge of the authors, there is currently no commercial epoxide polymerization process dedicated to deliver it- or it-enriched PPO or other aliphatic polyethers, with the exception of perhaps a single example.10 In view of the substantial amounts manufactured annually – PO is the second most important epoxide monomer after ethylene oxide – and the broad applicability of aliphatic polyethers,11 there is certainly no lack of interest. Especially the introduction of isotactic (enriched) polyether segments in more complex polymer architectures (i.e., multiblocks containing also polyester sequences) seems promising in this regard, potentially opening up a new frontier to tailor the properties of polyether-based additives, where fine-tuning for applications in drug-delivery, tissue-engineering, rheology control or hydrogel formation can be conceived.12
Such requirements suggest potential benefits in the use of organocatalytic polymerization catalysts,13–16 where the controlled conversion of substituted epoxides has experienced a step change in the last few years.17,18 Especially the rise of a simple borane co-catalyst (triethylborane, Et3B)19,20 has enabled the preparation of very well-defined polyether-sequences with short reaction times under mild conditions.21,22 This borane can be employed together with a range of organobases, including very simple nitrogen bases.23 Intriguingly, elegant work by Zhao and Ling has demonstrated that with such a setup even poly(ester-ether) multiblock motifs can be constructed – excessive transesterification is avoided during growth of the polyether blocks.24 What deserves particular attention is the proposed mechanism, which necessitates two equivalents of Et3B per organobase, a stoichiometry inherently linked to the simultaneous monomer activation and chain-end deactivation/coordination, which is also well-known from Lewis-pair polymerization (Scheme 1).25 The viability of using multi-centered, onium-bridged (achiral) boranes for polymerization catalysis has been impressively demonstrated by Wu and others,26–28 while Du's work highlighted the potential of in situ-formed chiral diboranes for, i.e., asymmetric imine hydrogenation.29–31 Herein it will be shown that these characteristics can be exploited further to engender stereoselective polymerization of rac-PO and other epoxides by using simple, chiral diboranes as co-catalysts, notably retaining the benefits of a well-controlled polymerization and a useful tolerance towards polyester-type initiators.
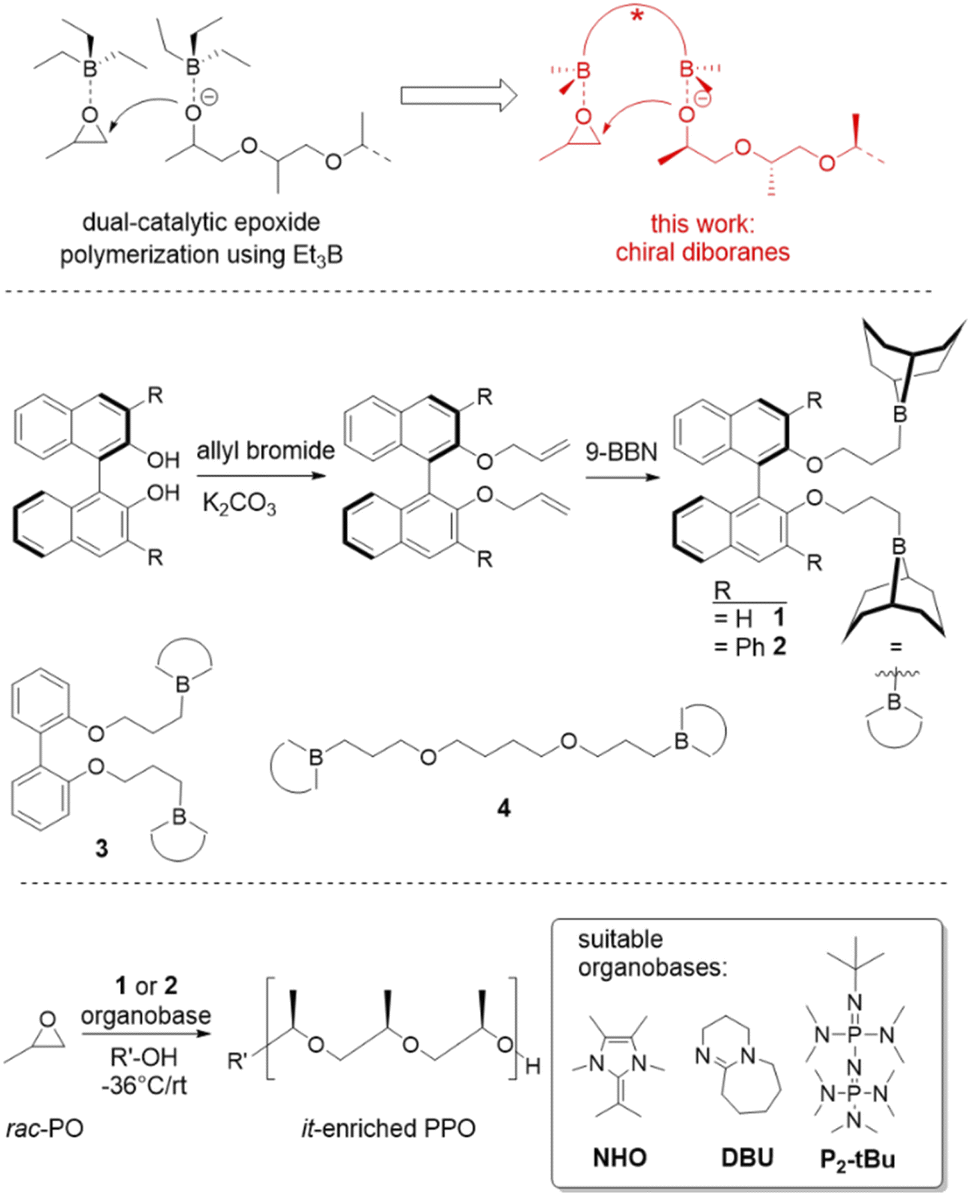 | ||
| Scheme 1 Research rationale (top), diborane synthesis (middle) and polymerization setup (bottom) for the preparation of it-enriched PPO. | ||
Results and discussion
In a first step, a strategy was developed to connect two borane functionalities to a chiral and well-available backbone. To this end, enantiopure (R)-BINOL (1,1′-bi-2-naphthol) and derivatives were first subjected to a Williamson etherification followed by hydroboration with 9-BBN (Scheme 1). This user-friendly two-step route delivered catalysts 1 and 2; crystal structure analysis confirmed expectations (Fig. 1). Analogously, control compounds 3 (biphenol-based) and 4 (linear aliphatic) were received.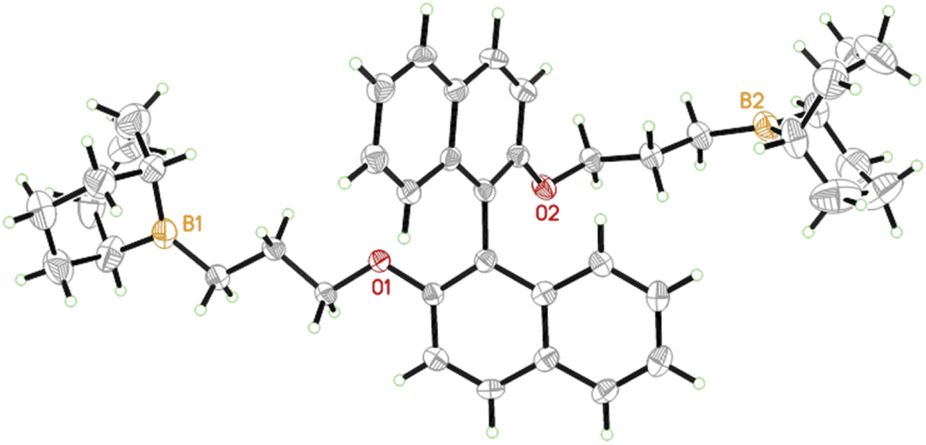 | ||
| Fig. 1 Single crystal X-ray structure of chiral diborane 1. Naphthyl–naphthyl dihedral angle = 114°. See ESI and CCDC file 2168081.† | ||
A series of experiments was conducted, using 1 in combination with a range of organobases (including a specific N-heterocyclic olefin (NHO),32,33 a phosphazene and simple DBU, Scheme 1) and benzyl alcohol as initiator (Table 1). As a catalyst (Cat)/base (B) molar ratio a stoichiometry of at least 2:1 was chosen to ensure an excess of borane functionalities over propagating chain-ends. This was thought to enable monomer activation in spatial proximity to the growing polymeric species (Scheme 1, top) in a potentially stereoselective environment.
| # | Cat | B | B/Cat/BnOH/POa | Solvent [PO] | T [°C] | t [h] | M n (calc.) [g mol−1] | Đ M | m [%] | Conversionb [%] |
|---|---|---|---|---|---|---|---|---|---|---|
| a Molar ratio. b Determined via1H NMR. c Determined via GPC (CHCl3). d Determined via13C NMR. | ||||||||||
| 1 | 1 | NHO | 1:2:5:2000 |
Bulk | 50 | 3.0 | 9000 | 1.11 | 68 | 40 |
| 2 | 1 | NHO | 1:2:5:2000 |
Bulk | 25 | 1.5 | 13000 |
1.18 | 62 | 53 |
| 3 | 1 | NHO | 1:2:5:2000 |
Bulk | −36 | 2.0 | 16000 |
1.18 | 65 | 70 |
| 4 | 1 | NHO | 1:2:5:2000 |
Bulk | −78 | 3.0 | 15000 |
1.17 | 66 | 59 |
| 5 | 1 | P2-tBu | 1:2:5:2000 |
Bulk | 25 | 2.0 | 12000 |
1.24 | 69 | 49 |
| 6 | 1 | DBU | 1:2:5:2000 |
Bulk | 25 | 24 | 6000 | 1.18 | 62 | 27 |
| 7 | 1 | NHO | 1:2:2.5:1000 |
Toluene [2 M] | −36 | 24 | 25000 |
1.14 | 82 | 80 |
| 8 | 1 | NHO | 1:2:2.5:10000 |
Toluene [2 M] | −36 | 24 | 123000 |
1.18 | 79 | 63 |
| 9 | 2 | NHO | 1:4:5:1000 |
Bulk | −36 | 24 | 5000 | 1.17 | 74 | 22 |
| 10 | 2 | NHO | 1:4:5:10000 |
Bulk | −36 | 24 | 8000 | 1.11 | 80 | 7 |
| 11 | 2 | NHO | 1:2:2.5:1000 |
THF [4 M] | 25 | 24 | 16000 |
1.25 | 78 | 70 |
| 12 | 2 | NHO | 1:2:2.5:1000 |
THF [2 M] | 25 | 8 d | 4000 | 1.14 | 88 | 16 |
| 13 | 3 | DBU | 1:4:5:2000 |
Bulk | 25 | 0.5 | 15000 |
1.06 | 56 | 65 |
| 14 | 4 | DBU | 1:4:5:2000 |
Bulk | 25 | 24 | 12000 |
1.18 | 57 | 52 |
These initial experiments, conducted in the bulk (entries 1–6), indicated high conversion and a rapid increase in viscosity alongside a unimodal, narrow molar mass distribution. Accordingly, MALDI-ToF MS analyses underlined full control over the BnOH-derived end groups (Fig. 2) and thus excellent end group fidelity. Allyl-type termination, undesired and frequently observed in conventional PO polymerization as a result of transfer reactions,11 was absent according to mass spectrometry and 1H NMR spectroscopy studies, rendering this approach also suitable for constructing more complex polymer architectures (see below). Conversion and determined molar masses correlate linearly, as expected (Fig. S13†).
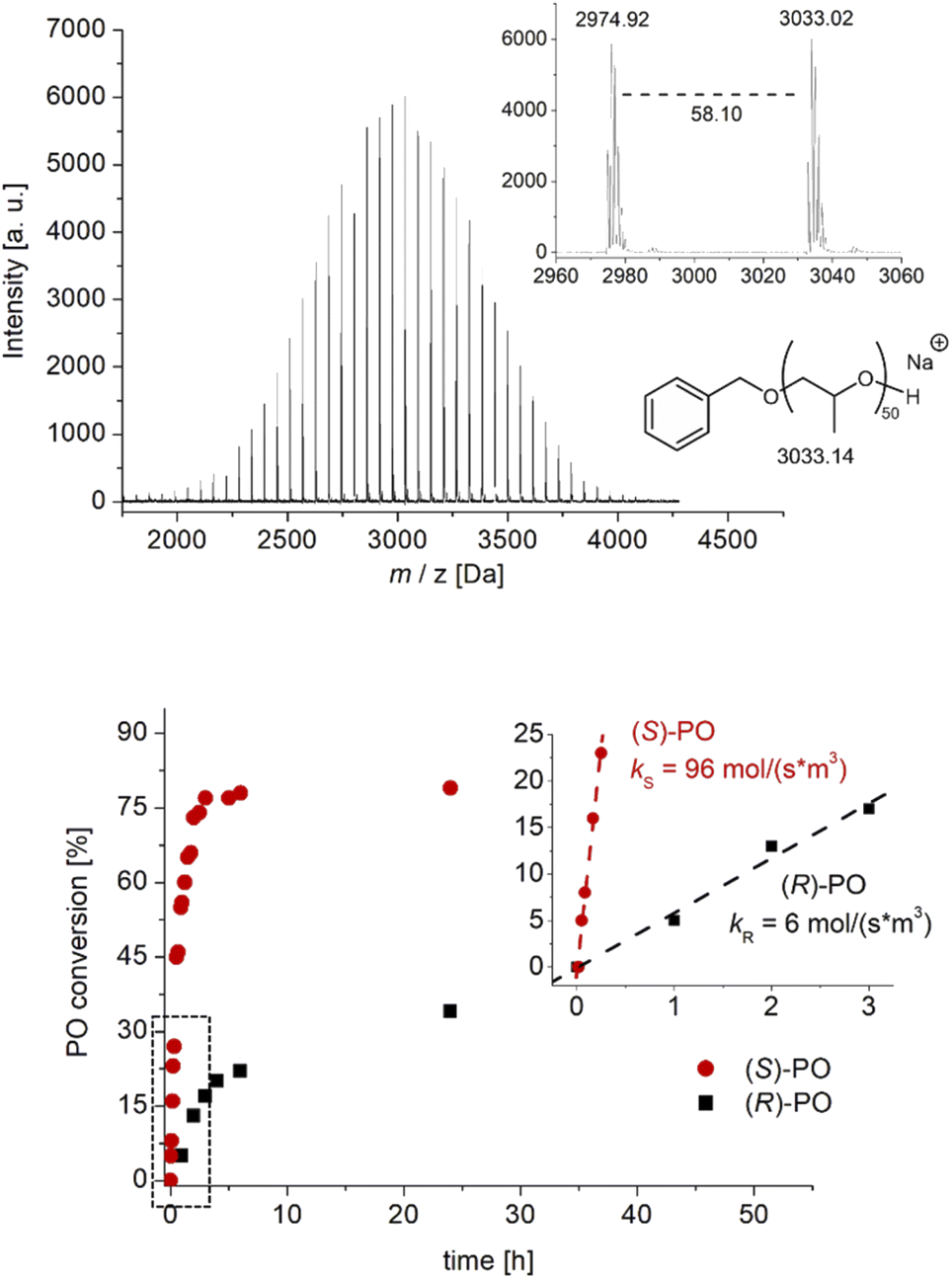 | ||
| Fig. 2 Top: MALDI-ToF MS analysis of PPO prepared by the action of DBU/1 (bulk, −36 °C) with BnOH as initiator. Bottom: Polymerization kinetics using enantiopure monomer (2 M in THF, ambient, NHO/1/BnOH/PO = 1:2:5:2000 molar ratio). | ||
The observed degree of isotacticity in these first experiments, however, was only moderate. As determined via13C NMR analyses, the proportion of isotactic diad placement (m) was found to be 62–69%, almost independent of the reaction temperature (−78 °C to 50 °C) and type of organobase. However, a reduction of [Cat]0 by application of solvent pushed m up to > 80% (entry 7), indicating that bimolecular interaction between diboranes may be detrimental.29 Even at very high PO loading (10000 eq.) the polymerization runs to > 50% conversion and >120000 g mol−1 (Mn) within 24 h (entry 8).
Kinetic studies using enantiopure PO revealed the (S)-PO to be the clearly preferred enantiomer for catalyst 1 (Fig. 2 and S14†). Notably this preference cleanly reverses when the (S)-enantiomer of 1 is employed, where a faster consumption of (R)-PO is observed (Fig. S16†). Zero order in monomer conversion was found, especially in the early stages of polymerization, which is in accordance with the particular characteristics of Lewis pair polymerizations using achiral diboranes (constant concentration of activated (borane-coordinated) monomer).25,26 Deviations from linear behavior can possibly be attributed to increasing viscosity (or even solidification) of the reaction mixture at higher conversions. Nonetheless, from these data the selectivity factor s can be approximated as the ks/kr ratio. For example, s = 8 for bulk polymerization at room temperature (Fig. S14†) improves to s = 16 under dilution (2 M in THF, Fig. 2). These findings nicely mirror the selectivity determined by 13C NMR under these conditions (Table 1) and also underline the positive impact of dilution on selectivity. To put this into perspective, a perfectly isoselective setup can be expected to deliver s > 100.8 1H NMR experiments, comparing the position of the PO monomer signal to that found in the presence of diborane, revealed no observable shift of the monomer or catalyst signals (Fig. S22†). This is in agreement with previous observations targeting Et3B and indicates a relatively weak monomer activation, potentially also supporting alternative polymerization models.24 In any case, the monomer activation/chain-end deactivation mechanism (Schemes 1 and 2) must be considered as simplified and certainly requires further investigation.
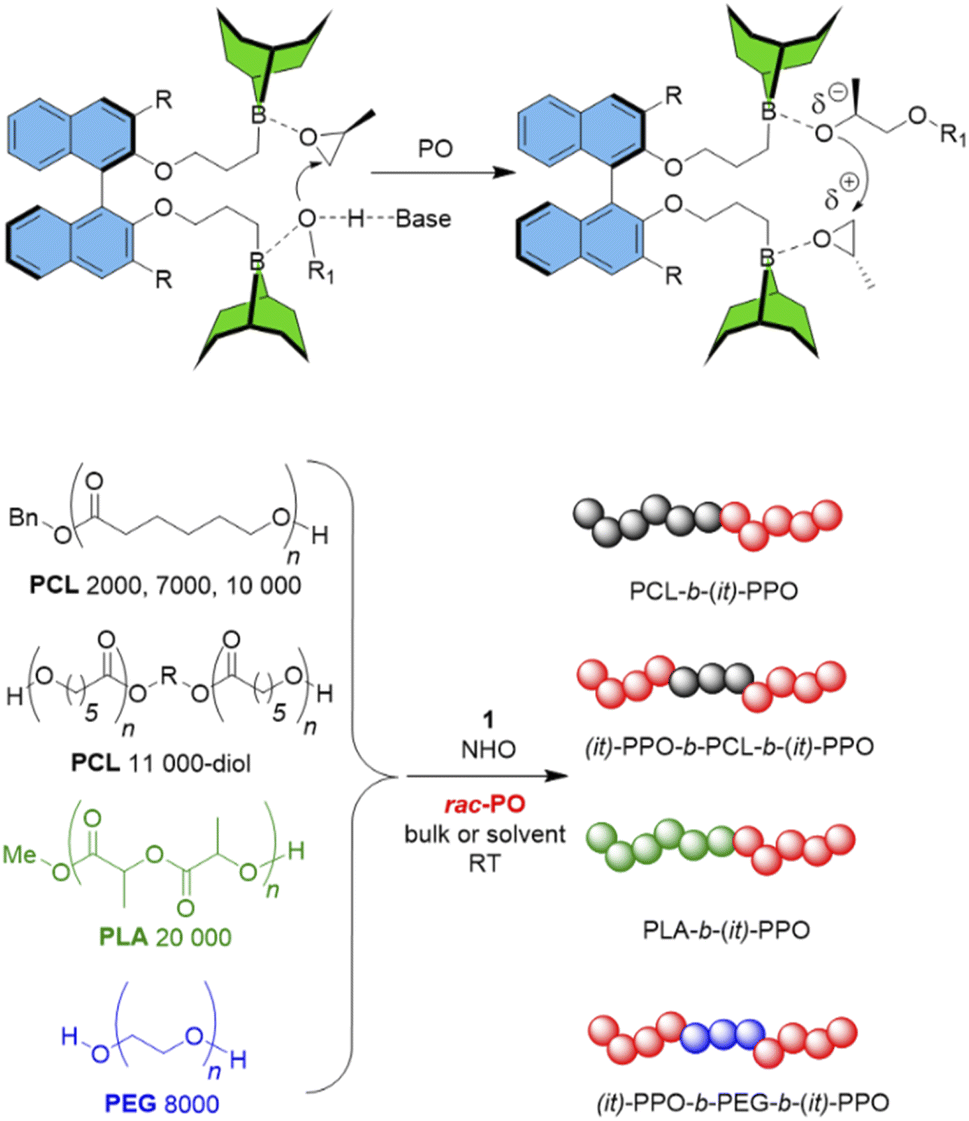 | ||
| Scheme 2 Top: Simplified cooperative mechanism in the presence of diborane, organobase, PO and initiator/propagating chain end (R1-OH). Bottom: Block copolymers with isotactic-enriched (it) polyether moieties. | ||
Next, a (R)-BINOL-backbone carrying phenyl groups ortho to the hydroxyl functionalities was employed, aiming for a more efficient transfer of stereoinformation. Gratifyingly, application of diborane 2 entailed a further increase in selectivity. In the bulk, m was found to be 74–80%, while under dilute conditions an isotactic diad placement of m = 88% was observed. Molar mass distributions remained well-controlled (1.1 < ĐM <1.3), even up to relatively high molar masses (Mn > 40000 g mol−1). Compared to 1, this higher selectivity is accompanied by notably slower polymerization kinetics (Table 1, entries 9–12). To put this degree of selectivity into context, the observed bias for isotactic placement of PO by Al-porphyrin complexes is m = 69% at 20 °C.3 In contrast, the bimetallic catalysts reported by Coates are far superior in this regard (mm > 99%, 0 °C).8
Control experiments employing the racemic mixture of 1 (rac-1) as well as 3 and 4 provided further insight. While de facto atactic PPO was observed by employing both the biphenyl derivative 3 and its linear congener 4 (Table 1), m remained unchanged by the switch from 1 to rac-1 (m = 82% and 80%, respectively at −36 °C, see Table S1† for further examples). This is intriguing from the perspective of practical application since using racemic mixtures may simplify future synthetic schemes for diborane preparation. Moreover, the overall monomer consumption is faster and expectedly identical for both PO enantiomers (Fig. S15†). The underlying reason for this behavior is most likely the strong association between the propagating polyether chain end (which is a secondary alkoxide) and borane functionality; very frequent chain exchange between diboranes seems unlikely in view of these results. The loss in selectivity observed when using biphenyl-based 3 could be attributed to its lower rotational barrier along the chirality axis and the higher flexibility regarding the phenyl–phenyl dihedral angle. Nonetheless, this compound may be a powerful co-catalyst if atactic polyether architectures are required (65% conversion after 30 minutes at 0.2 mol% diborane loading, entry 13).
In a next step, further epoxide monomers were screened, including 1-butylene oxide (BO), allyl glycidyl ether (AGE) and styrene oxide (SO). While SO could not be polymerized successfully, BO and AGE were polymerized to produce well-controlled polyethers with molar masses of 3000–31000 g mol−1. Stereoselectivity in general was comparable to that observed for PO (m = 85% (BO) and 78% (AGE), respectively). Polymerizations using enantiopure BO revealed the same tendency as described above for PO, namely a preference of 1 for the (S)-enantiomer of the monomer (Fig. S17†). A summary of all polymerizations is provided (Tables S1–S4†).
The ensuing polyether homopolymers (PPO, PBO, PAGE) are predominantly amorphous (Fig. S24–S26†); only those with the highest m values (>85%) display moderately defined melting points (PPO with Tm up to 45 °C, Fig. S23,† as compared to 67 °C for perfectly isotactic material1). TGA analyses of atactic and it-enriched PPO revealed comparable behavior (Fig. S27†).
However, even if the degree of isotacticity is not sufficient to provide semi-crystalline behavior, polymeric additives can profit from isotactic enrichment. For example, it-enriched block copolyether additives have a systematic effect on the viscosity and storage/loss moduli if they are employed in aqueous environment to form hydrogels. A perfectly isotactic, semi-crystalline congener, on the other hand, failed under identical conditions on account of solubility issues.12c
A further aspect of particular relevance for additives technology is the incorporation of it-enriched polyether moieties in more complex architectures, especially in conjunction with polyester blocks. The latter can so far only be included in a polyether-first approach, which means the polyester block is grown on a (typically atactic) polyether macroinitiator. The reverse (polyester-first) is not readily possible since the polyester is susceptible to degradation under the reaction conditions used for epoxide conversion, including the conventional KOH-mediated polymerization but also organocatalytic- and metal-catalyzed polymerizations. To the best knowledge of the authors, this is also true for the previously reported iso- or enantioselective metal-based approaches for epoxide polymerization.1 In sharp contrast, pronounced functional group tolerance regarding ester moieties should be inherent to borane co-catalysis, as was impressively demonstrated using Et3B,24 raising the possibility that polyester-first strategies and related polymer architectures could also be achievable for the chiral diborane catalysts presented here.
To investigate this, commercial and custom-made polyester macroinitiators (polycaprolactone (PCL) and polylactide (PLA), Scheme 2) were subjected to the same polymerization procedure as described above. This resulted in the corresponding diblock copolymers PCL-b-(it)-PPO and PLA-b-(it)-PPO as well as triblock copolymers ((it)-PPO-b-PCL-b-(it)-PPO). Block formation was substantiated by 1H DOSY NMR (Fig. S28–S32†) and GPC analysis (Fig. S34–S36†). Notably, the narrow molar mass distribution confirms that no substantial transesterification occurs, and indeed it is possible to stir commercial PCL for 24 h (THF) in the presence of NHO/1 without any observable change in the molar mass of the macroinitiator (Fig. S33†). Once PO is added, the catalyst becomes active and leads to growth of the it-enriched polyether block. Likewise, it was possible to use α,ω-dihydroxy PEG 8000 as a bifunctional macroinitiator, resulting in isotactic-enriched variants of the well-known Reverse Pluronic-type triblock copolyethers, (it)-PPO-b-PEG-b-(it)-PPO. The data for these block copolymers are summarized in Table S4.†
Conclusions
In summary, an organocatalytic, cooperative catalyst based on readily available chiral diboranes enables the preparation of isotactic enriched aliphatic polyethers. Notably, the resulting polyether moieties are well-defined up to high molar masses and can be incorporated in block copolymers, including “polyester-first”-based architectures. It is believed that these abilities will be especially valuable for polymer additives manufacturing. The BINOL-type diborane platform is amenable to a plethora of structural modifications, which are currently studied in our group regarding their impact on stereoselectivity.Data availability
Experimental setups, procedures and additional data (kinetics, 13C NMR, thermal analysis, GPC data) are available in the ESI.†Author contributions
Experiments were planned and conducted by Ayla Sirin-Sariaslan under the supervision of Stefan Naumann. Writing was done by Ayla Sirin-Sariaslan with review by Stefan Naumann.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Wolfgang Frey and Dr Dongren Wang (both University of Stuttgart) are gratefully acknowledged for single crystal X-ray structure analysis and MALDI-ToF MS measurements, respectively. S. N. gratefully acknowledges funding by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG, NA 1206/3-1).Notes and references
- M. I. Childers, J. M. Longo, N. J. van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- M. E. Pruitt and J. M. Baggett, US Pat., 2706181, 1955 Search PubMed.
- See for example: N. Takeda and S. Inoue, Makromol. Chem., 1978, 179, 1377–1381 CrossRef CAS.
- E. J. Vandenberg, J. Polym. Sci., 1960, 47, 486–489 CrossRef CAS.
- R. C. Ferrier, S. Pakhira, S. E. Palmon, C. G. Rodriguez, D. J. Goldfeld, O. O. Iyiola, M. Chwatko, J. L. Mendoza-Cortes and N. A. Lynd, Macromolecules, 2018, 51, 1777–1786 CrossRef CAS.
- K. L. Peretti, H. Ajiro, C. T. Cohen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 11566–11567 CrossRef CAS.
- W. Hirahata, R. M. Thomas, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2008, 130, 17658–17659 CrossRef CAS PubMed.
- R. M. Thomas, P. C. B. Widger, S. M. Ahmed, R. C. Jeske, W. Hirahata, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 16520–16525 CrossRef CAS PubMed.
- S. M. Ahmed, A. Poater, M. I. Childers, P. C. B. Widger, A. M. LaPointe, E. B. Lobkovsky, G. W. Coates and L. Cavallo, J. Am. Chem. Soc., 2013, 135, 18901–18911 CrossRef CAS.
- As a possible exception, a reviewer has pointed out that commercially available Hydrin©-elastomers are most likely prepared under application of the Vandenberg catalyst which allows for high-molar mass copolyethers and a moderate isotacticity (albeit it is not clear to what degree the latter is decisive for product properties).
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS.
- See for example: (a) W. Shi, A. J. McGrath, Y. Li, N. A. Lynd, C. J. Hawker, G. H. Fredrickson and E. J. Kramer, Macromolecules, 2015, 48, 3069 CrossRef CAS; (b) A. J. McGrath, W. Shi, C. G. Rodriguez, E. J. Kramer, C. J. Hawker and N. A. Lynd, Polym. Chem., 2015, 6, 1465–1473 RSC; (c) F. Markus, J. R. Bruckner and S. Naumann, Macromol. Chem. Phys., 2020, 221, 1900437 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813 CrossRef CAS.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409 CrossRef CAS PubMed.
- M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142 RSC.
- Y. Xia and J. Zhao, Polymer, 2018, 143, 343 CrossRef CAS.
- J. Raynaud, W. N. Ottou, Y. Gnanou and D. Taton, Chem. Commun., 2010, 46, 3203 RSC.
- S. Naumann, A. W. Thomas and A. P. Dove, Angew. Chem., Int. Ed., 2015, 54, 9550 CrossRef CAS PubMed.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117 CrossRef CAS PubMed.
- S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Nat. Commun., 2019, 10, 293 CrossRef PubMed.
- Y. Chen, J. Shen, S. Liu, J. Zhao, Y. Wang and G. Zhang, Macromolecules, 2018, 51, 8286 CrossRef CAS.
- C.-J. Zhang, H.-Y. Duan, L.-F. Hu, C.-H. Zhang and X.-H. Zhang, ChemSusChem, 2018, 11, 4209 CrossRef CAS.
- C. Vogler and S. Naumann, RSC Adv., 2020, 10, 43389 RSC.
- S. Liu, T. Bai, K. Ni, Y. Chen, J. Zhao, J. Ling, X. Ye and G. Zhang, Angew. Chem., Int. Ed., 2019, 58, 15478 CrossRef CAS PubMed.
- M. L. McGraw and E. Y.-X. Chen, Macromolecules, 2020, 53, 6102 CrossRef CAS.
- G.-W. Yang, Y.-Y. Zhang, R. Xie and G.-P. Wu, Angew. Chem., Int. Ed., 2020, 59, 16910 CrossRef CAS PubMed.
- G.-W. Yang, Y.-Y. Zhang and G.-P. Wu, Acc. Chem. Res., 2021, 54, 4434 CrossRef CAS PubMed.
- X. Wang, J. Hui, M. Shi, X. Kou, X. Li, R. Zhong and Z. Li, ACS Catal., 2022, 12, 8434 CrossRef CAS.
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 6810 CrossRef CAS PubMed.
- S. Wei and H. Du, J. Am. Chem. Soc., 2014, 136, 12261 CrossRef CAS PubMed.
- Z. Zhang and H. Du, Angew. Chem., Int. Ed., 2015, 54, 623 CAS.
- S. Kronig, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2013, 2013, 2301 CrossRef CAS.
- S. Naumann, Chem. Commun., 2019, 55, 11658 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2168081. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03977j |
| This journal is © The Royal Society of Chemistry 2022 |