
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical oxidative rearrangement of tetrahydro-β-carbolines in a zero-gap flow cell†
Yiting
Zheng
ac,
Yuen Tsz
Cheung
b,
Lixin
Liang
 b,
Huiying
Qiu
b,
Lei
Zhang
c,
Anson
Tsang
a,
Qing
Chen
*ab and
Rongbiao
Tong
*b
b,
Huiying
Qiu
b,
Lei
Zhang
c,
Anson
Tsang
a,
Qing
Chen
*ab and
Rongbiao
Tong
*b
aDepartment of Mechanical and Aerospace Engineering, and Energy Institute, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, P. R. China. E-mail: chenqing@ust.hk
bDepartment of Chemistry, The Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong SAR, China. E-mail: rtong@ust.hk; Fax: +852 23581594; Tel: +852 23587357
cState Key Laboratory of Electrical Insulation and Power Equipment, Xi'an Jiaotong University, Xi'an, 710049, China
First published on 25th August 2022
Abstract
Oxidative rearrangement of tetrahydro-β-carbolines (THβCs) is one of the most efficient methods for the synthesis of biologically active spirooxindoles, including natural products and drug molecules. Here, we report the first electrochemical approach to achieve this important organic transformation in a flow cell. The key to the high efficiency was the use of a multifunctional LiBr electrolyte, where the bromide (Br−) ion acts as a mediator and catalyst and lithium ion (Li+) acts as a likely hydrophilic spectator, which might considerably reduce diffusion of THβCs into the double layer and thus prevent possible nonselective electrode oxidation of indoles. Additionally, we build a zero-gap flow cell to speed up mass transport and minimize concentration polarization, simultaneously achieving a high faradaic efficiency (FE) of 96% and an outstanding productivity of 0.144 mmol (h−1 cm−2). This electrochemical method is demonstrated with twenty substrates, offering a general, green path towards bioactive spirooxindoles without using hazardous oxidants.
Introduction
Oxidative rearrangement of tetrahydro-β-carbolines (THβCs)1 (Fig. 1) was proposed as an enzyme-mediated reaction2–4 responsible for biosynthesis of many biologically active spiro[indolizidine-1,3-oxindole] (i.e., spirooxindoles) natural products such as mitraphylline, rhynchophylline, (iso)corynoxeine, corynoxine, corynoxine B, spirotryprostatins A and B, and alstonisine (Fig. 1). With chemical oxidants [Pb(OAc)4, OsO4, t-BuOCl, N-bromosuccinimide (NBS), etc.] identified for such oxidative rearrangement, it has become a widely employed method for the construction of spiro[indolizidine-1,3-oxindole],5–11 which is associated with significant biological activities (antiviral,12 anticancer,13 and antibacterial14) and regarded as a pharmaceutically privileged structural motif.15–18 However, these chemical oxidants are hazardous and toxic and would generate stoichiometric amounts of harmful chemical waste. Recently, two green approaches using Oxone-halide19 and Fenton-halide20 for the oxidative rearrangement of THβCs were reported to reduce the impact on the environment and human health. While Oxone as an inorganic triple salt (KHSO5·0.5KHSO4·0.5K2SO4) has admirable bench stability, unique reactivity towards halides, and safe/easy operation, it generates two equivalents of potassium sulfate as a byproduct with an often-criticized high E-factor. On the other hand, hydrogen peroxide is widely considered to be an ideal terminal oxidant in organic synthesis since the only byproduct is water. However, hydrogen peroxide (typically 30 wt% in water was used in laboratory) is unstable at room temperature with an inconsistent concentration and quality and might decompose explosively. Herein, we report the first electrochemical method for the oxidative rearrangement of THβCs without using any chemical oxidant.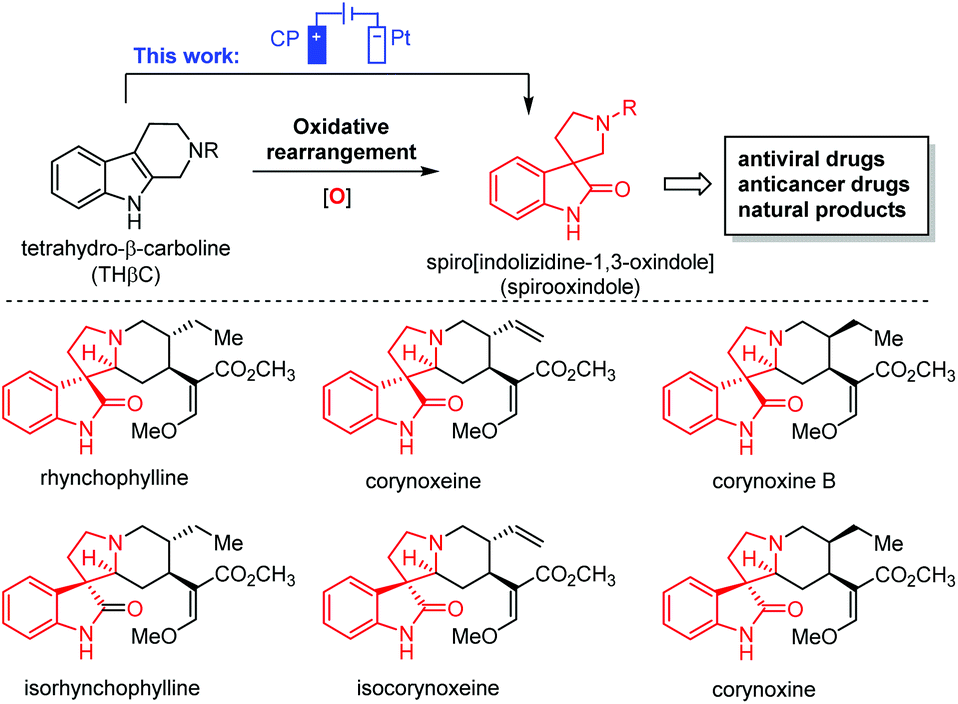 | ||
| Fig. 1 Oxidative rearrangement of tetrahydro-β-carboline and spirooxindole natural products. | ||
Electrochemical synthesis, a potentially sustainable and atom-economic means towards organic compounds, has received increasing attention in the past decade.21–24 The use of mediators for oxidation/reduction is one of the most effective strategies to achieve high regio- and chemo-selectivity through minimizing the direct non-selective oxidation/reduction of substrates on electrodes.25,26 Halides have emerged as one of the most successful mediators for selective electrochemical organic synthesis.27 Mechanistically, the halide mediators NaX, KX, NH4X or n-Bu4NX (X− is Cl−, Br− or I−) are oxidized at the anode into reactive halogenating species (RHS), which then react with substrates to generate an intermediate that can undergo further transformation(s) to yield a product. Inspired by this general mechanism, we envisioned that the electrochemically generated RHS could be used as an oxidant for the oxidative rearrangement of THβCs (Fig. 2(A)) because RHS generated in situ from chemical oxidation of halide (Oxone/halide or Fenton-halide) was successfully used for this reaction.19,20 The challenge is to minimize the competing nonselective anodic oxidation of THβCs over a halide because related direct electro-oxidation of indoles was reported by Oliveira-Brett,28 Mount,29 Vincent,30,31 Lei,32 Fang,33etc. Therefore, the selectivity between a halide and indole (i.e., THβCs) is critical to the success of the oxidative rearrangement of THβCs.
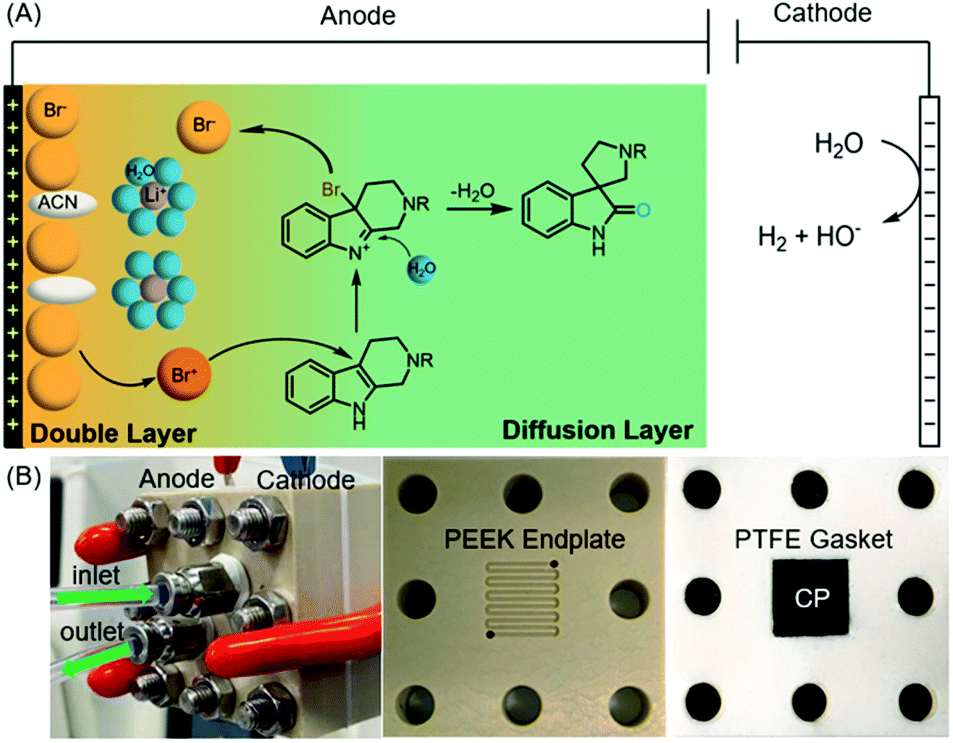 | ||
| Fig. 2 Reaction mechanism and cell design. (A) The proposed bromide-mediated electrochemical oxidative rearrangement of tetrahydro-β-carboline to spirooxindole; (B) photos of the screw assembled flow cell (top), the PEEK endplate (bottom left), and the cathode (10 mm × 10 mm × 0.6 mm) fitted into the PTFE gasket (bottom right). | ||
We planned to address this selectivity challenge by (1) evaluating different electrodes and supporting electrolytes and (2) devising a flow cell. As we illustrate in Fig. 2(A), the oxidative rearrangement of THβCs should occur preferably at the diffusion layer but not at the double layer, where the choices of the anode and even the supporting electrolyte can determine the selectivity: the generation of RHS or the undesired nonselective oxidation. Secondly, a high-rate divided flow cell would considerably reduce the substrate diffusion into the double layer for possible direct oxidation on the electrode, while it could achieve a desirable rapid transport of the halide towards the electrode for an adequate local concentration of RHS necessary for the rearrangement and at the same time avoiding concentration polarization that may result in side reactions.34,35 In this article, we disclose the results of our research efforts to achieve the first electrochemical oxidative rearrangement.
Results and discussion
With THβC 1a as the model compound, we designed the electrochemical synthesis as follows. The electrolyte contained two common solvents: water for high salt solubility and high conductivity, and acetonitrile (MeCN) for dissolving organic molecules. We added acetic acid (AcOH), which was shown to facilitate the generation of RHS in aqueous media.36 The volume ratio of MeCN, AcOH, and H2O was optimized to be 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.4:2 (Fig. S1†). The anode was a commercial carbon paper (CP, SGL-39AA) and baked in air at 400 °C for 24 hours to improve the wetting with the electrolyte and further suppress the oxygen evolution reaction (OER).37 The cathode was Pt due to its catalytic activity towards the hydrogen evolution reaction (HER). The electrodes (1 cm2 in area) were then assembled in a zero-gap flow cell as shown in Fig. 2(B) with two PEEK endplates and sealed with PTFE gaskets (0.5 mm thick) as shown in Fig. S3 and S4.† The term zero-gap refers to the design wherein the anode, the membrane, and the cathode were pressed using a torque wrench together with no gap among them, so the cell resistance can be minimized, and the flow can be accelerated with a flow field. Through a serpentine flow channel engraved in the endplate, as presented in Fig. S3†, a peristaltic pump circulated the electrolyte at a relatively fast flow rate (>1 mL min−1). The membrane was Nafion® 117, whose effectiveness in preventing the reduction of product 2a at the counter electrode was obvious as compared with an undivided beaker cell under otherwise the same conditions.
:2.4:2 (Fig. S1†). The anode was a commercial carbon paper (CP, SGL-39AA) and baked in air at 400 °C for 24 hours to improve the wetting with the electrolyte and further suppress the oxygen evolution reaction (OER).37 The cathode was Pt due to its catalytic activity towards the hydrogen evolution reaction (HER). The electrodes (1 cm2 in area) were then assembled in a zero-gap flow cell as shown in Fig. 2(B) with two PEEK endplates and sealed with PTFE gaskets (0.5 mm thick) as shown in Fig. S3 and S4.† The term zero-gap refers to the design wherein the anode, the membrane, and the cathode were pressed using a torque wrench together with no gap among them, so the cell resistance can be minimized, and the flow can be accelerated with a flow field. Through a serpentine flow channel engraved in the endplate, as presented in Fig. S3†, a peristaltic pump circulated the electrolyte at a relatively fast flow rate (>1 mL min−1). The membrane was Nafion® 117, whose effectiveness in preventing the reduction of product 2a at the counter electrode was obvious as compared with an undivided beaker cell under otherwise the same conditions.
Under the optimal conditions (Table 1), the oxidative rearrangement of 1a achieved 97% yield of 2a and 96% faradaic efficiency (FE). Under a fixed applied cell potential, the yield was attained with the full conversion as monitored by thin layer chromatography (TLC). The amount of the product was also converted to charge via Faraday's law, the ratio of which over the charge input during the synthesis was the FE (calculated from eqn (1) in the ESI†). As shown in Table 1, the amount of bromide ion was determined to be two equivalents: 1.2 equivalents of LiBr led to significant OER, while 3.0 equivalents resulted in bromination of spirooxindoles (∼10%) (entries 2 and 3). The use of a Pt mesh anode in place of carbon paper (entry 6) drastically diminished the yield and FE probably due to Pt-catalyzed side reactions (i.e., C1 oxidation/ring-opening and/or the OER).34 Chloride and iodide (Cl− and I−) (entries 7 and 8) were also examined but were not effective. This was not unexpected given that the oxidation potential of the chloride ion might be higher than that of nonselective indole oxidation, while the easily generated iodonium ion [I+] at the low oxidation potential promoted sluggish oxidative rearrangement.38
|
|
|||
|---|---|---|---|
| Entry | Deviation from optimized conditions | Yieldb (%) | FE (%) |
| a The optimal conditions were room temperature with an anolyte of THβC (0.2 mmol), and LiBr (0.4 mmol, 2.0 eq.). The reaction was performed in a zero-gap (Nafion 117) flow cell as per General Procedure A in the ESI. b NMR yield was obtained. c The potentials were calibrated vs. RHE from Ag/AgBr in an undivided cell with IR compensation. | |||
| 1 | Nonea | 97 | 96 |
| 2 | LiBr: 1.2 eq. | 67 | 67 |
| 3 | LiBr: 3.0 eq. | 88 | 88 |
| 4 | Undivided cell (1.7 V)c | 80 | 68 |
| 5 | Undivided cell (1.2 V)c | 25 | 11 |
| 6 | Pt anode | 24 | 9 |
| 7 | No halide | 0 | 0 |
| 8 | LiCl instead of LiBr | 7 | 5 |
| 9 | LiI instead of LiBr | 32 | 7.5 |
| 10 | E: 0.85 V | 55 | 55 |
| 11 | E: 1.0 V | 71 | 71 |
| 12 | E: 1.6 V | 50 | 50 |
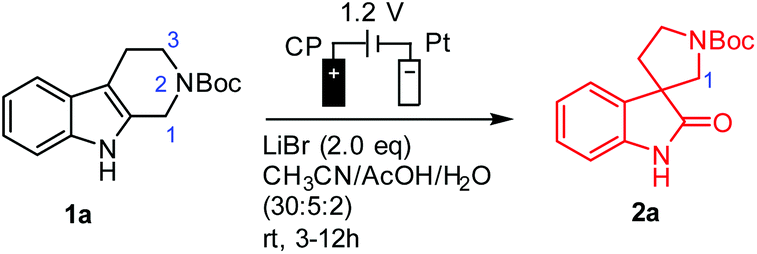
Table 1 clearly shows the impact of key variables on the reaction: the cell type (H-cell vs. flow cell), electrode, electrolyte, working voltage, and mediator (LiBr), some of which are further elucidated below. A variable unique to the electrochemical synthesis is the metal cation in the electrolyte. Despite its lack of direct involvement in the reactions, we observed substantial differences in the FE in the order of Li+ > Na+ > K+ > Bu4N+ (as Fig. 3(A) shows). We speculated that the cations acted as spectators, as broadly discussed in the literature on the HER and OER where the catalytic activities were correlated to the hydration energies of the cations, an indicator of the degree of blockage of active sites at the electrode by the hydrated cations.39–41 A similar correlation was found between FE and the hydration energy in our case. The cation was less likely to affect the bromide oxidation since the current density increased along with the hydration energy, as shown in Fig. 3(A). Instead, it was more likely to reduce the side reactions (direct electrode oxidation of indoles) through minimizing the diffusion of the substrate into the double layer. In the case of the lithium ion (Li+), the high hydration energy resulted in water aggregation around the double layer39,41 and thus prevented the hydrophobic THβC from diffusing into the double layer near the electrode (illustrated in Fig. 2(A)).
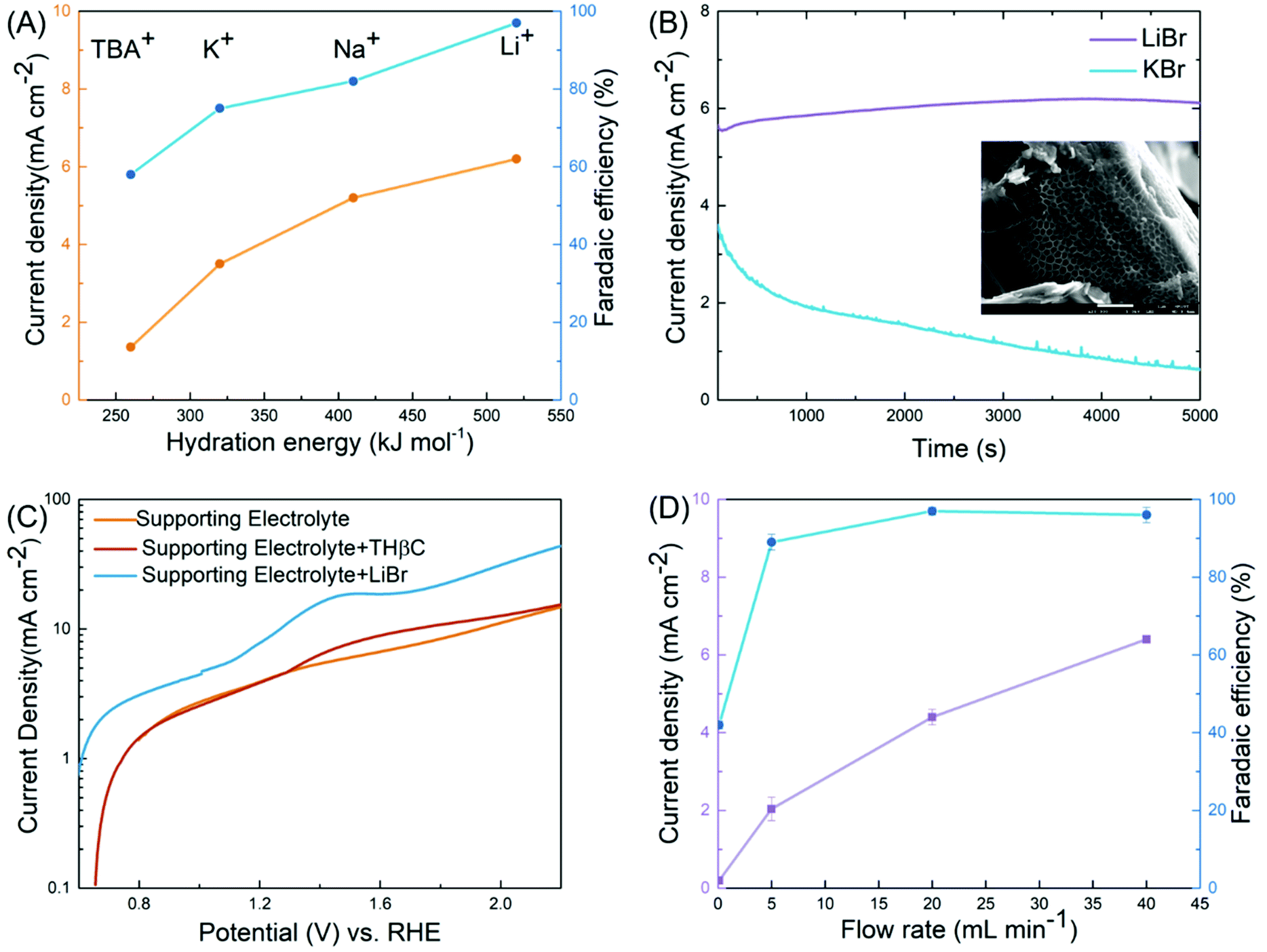 | ||
| Fig. 3 Key variables of the electro-oxidative rearrangement of THβC. (A) The effect of cations on the current density (orange) and the FE (blue). (B) Current–time profiles with LiBr (purple) and KBr (blue) as the salts. The inset is the SEM image of the carbon fiber in the carbon paper used in the synthesis reaction with KBr, on which a porous organic layer is likely the polymer product of the side reaction. (C) Linear sweep voltammetry (LSV) in a three-electrode cell (Fig. S6†) at 50 mV s−1 of a supporting electrolyte (0.8 M LiClO4, orange), the supporting electrolyte with THβC (red), and the supporting electrolyte with LiBr (blue). (D) The impact of flow rates on the current density (purple) and the FE (blue). | ||
This rationalization was further supported by the current–time profile (Fig. 3(B)). The oxidation current was stable with LiBr at the beginning stage of the reaction but decayed quickly with KBr, which suggested passivation. When examining the carbon paperby scanning electron microscopy (SEM) (the inset of Fig. 3(B)), we observed porous organic matter derived from THβC when KBr was used as the mediator. In contrast, we did not detect similar organic matter in the carbon paper electrode for the LiBr-mediated reaction. In a control experiment, we also observed a much larger oxidation peak in LSV with KBr than that with LiBr in the supporting electrolyte (see Fig. S6(A)†). Although the complex CP did not permit full elucidation of the spectator effect, our experimental results reveal the great impact of the spectating cation on selectivity of halide versus THβC oxidation, which facilitates future investigations.
The optimal potential was determined to be 1.2 V across the two electrodes. At a lower potential (0.85 and 1.0 V, Table 1, entries 9 and 10), insufficient RHS [Br+] was produced for the rearrangement; at a higher potential (1.6 V, entry 12), the OER and C1 oxidation/ring-opening were observed. This is consistent with linear sweeping voltammetry (LSV, Fig. 3(C)) performed in a three-electrode cell. The potential of LSV was converted to a scale vs. the relative hydrogen electrode (RHE) so that the value was comparable with the cell potential of the flow cell given the hydrogen evolution reaction in the positive side of the cell. We characterized three electrolytes, a blank supporting electrolyte of 0.8 M LiClO4, the supporting electrolyte with THβCs, and the electrolyte with LiBr. Although addition of the supporting salt, necessary for its ionic conductivity in the characterization, potentially complicated the reaction mechanism, we could estimate the onset potentials of the reaction, in good agreement with the optimal potential seen in the flow cell. The oxidation of the bromide ion was about 0.2 V earlier than a small oxidation peak, which was likely associated with the C1 oxidation/ring-opening reaction of THβC and responsible for the low yield when no mediator was used in the flow cell (entry 7). The OER took place at a higher potential. Therefore, the highest efficiency was achieved in a ∼0.2 V window.
Another key variable is the rate of electrolyte flow. The zero-gap design permits a high flow rate for rapid oxidation of the bromide ion despite its relatively low concentration, up to ∼6 mA cm−2 at 40 mL min−1, translating to ∼10% of bromide oxidation per pass of the flow (Fig. 3(D)). A high current density led to an increase of productivity (calculated from eqn (2) in the ESI†). The higher the productivity, the shorter the time, the smaller the cell required, and the more the cost-effectiveness. In addition, the FE was increased slightly to 97% at the highest flow rate, which displayed the other key advantage of rapid flow. As the bromide oxidation was limited by mass transport (Fig. S6(B)†), the higher the flow rate, the more vigorous the convective transport, the higher the interface concentration of RHS [Br+], and the more selective the reaction towards the rearrangement. This advantage allowed the flow cell to deliver superior yields over both the undivided cell (Table 1, entry 4 and 5) and flow cells that operate at slow flow rates (<1 mL min−1 in Fig. 3(D)).
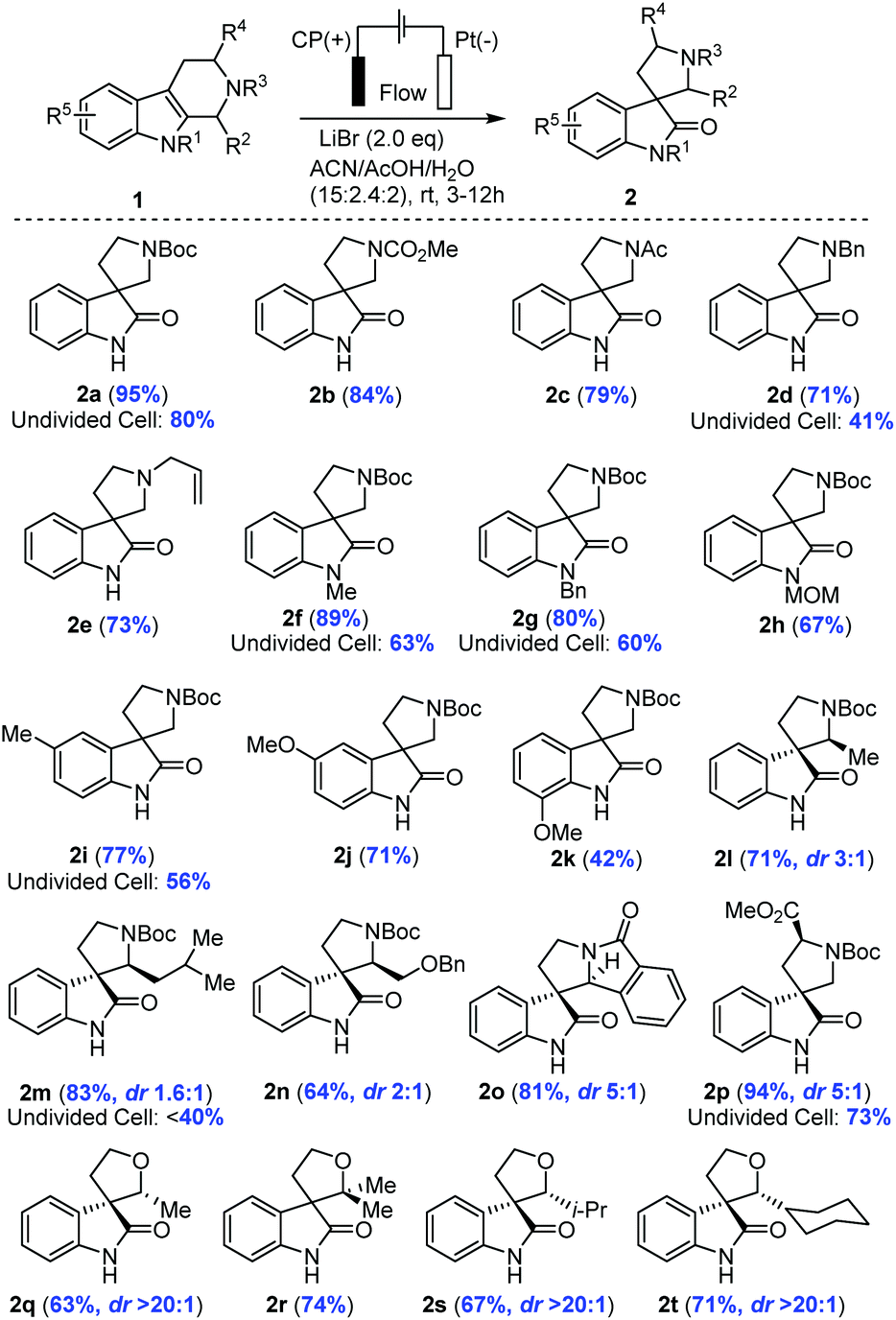
The electrochemical method was applicable to a broad scope of substrates as summarized in Table 2. Various N2 protecting groups (NR3) including N-CO2Me, N-Ac, N-Bn and N-allyl (2b–2e) were tolerated under the electrochemical conditions with good to excellent yields (71–95%). Notably, N-Bn and N-allyl of THβCs were suitable substrates for this electrochemical oxidation, which was interesting in light of the well-established Shono oxidation42,43 that was the major pathway for tertiary amines or amides under conventional electrochemical oxidation conditions. Our studies (Table 2 and Fig. 3(C)) suggested that Li+ might impede the direct Shono oxidation of THβCs at the anode. This finding was important for mediator-promoted electrochemical oxidation/reduction in organic synthesis and for application of our electrochemical oxidative rearrangement of THβCs in synthesis of bioactive spirooxindole molecules containing an N-alkyl group, particularly tertiary amines.
| a The reaction was carried out at room temperature as per General Procedure A using a zero-gap flow cell. Isolated yield was obtained. |
|---|
|
Next, we examined the protecting group on indole nitrogen (NR1) and found that only electron-donating groups such N-Me (2f), N-Bn (2g), and N-MOM (2h) were suitable for the oxidative rearrangement with good yields (67–89%), not a surprise as an electron-withdrawing group (N-Ac, N-Ts, or N-Boc) would increase the oxidation potential to overlap with the direct indole oxidation and water oxidation. Electron-rich THβCs with a methyl or methoxy group on the benzene ring were good substrates for the electrochemical oxidative rearrangement to provide spirooxindoles with good yields (2i–2k: 42–77%). Different C1 substituents (R2) of THβCs did not reduce the yields (64–83%), but a diastereomeric mixture (2l–2o) was often obtained with a ratio ranging from 1.6:1–5:1. Fortunately, these diastereomers could be separated by flash column chromatography on silica gel and assigned reliably by comparison of the NMR spectra with reported ones.19 When tryptophan-derived THβC was used, the spirooxindole (2p) was isolated in excellent yield (94%) under the optimal conditions, although diastereoselectivity was only moderate (dr. 5:1). Lastly, we further extended our electrochemical oxidative rearrangement to tetrahydropyrano[3,4-b]indoles (THPIs).19 All THPIs with monosubstituted or disubstituted alkyl groups at the C2 position underwent the expected oxidative rearrangement to provide 2q–2t in good yields. Interestingly, the diastereoselectivity (dr. >20:1) was excellent as compared to THβC substrates.
The advantage of the zero-gap flow cell over an undivided beaker cell (2a, 2d, 2f, 2g, 2i, 2m, and 2p, Table 2) was significant in the extended substrate scope. In the undivided cell, we carried out the synthesis similarly under a constant potential but with a reference electrode (AgBr/Ag, calibrated to be stable at −0.26 V vs. RHE) to avoid inconsistency caused by the large cell resistance (∼120 ohms). To attain a current at the same order of magnitude as that of the flow cell, the potential approached 1.7 V vs. RHE. Under these conditions, the undivided cell could merely deliver 40 to 60% of yield, inferior to the generally high yields (>70%) in the flow cell. In addition to the use of the ion-selective membrane in the flow cell to separate the product from the cathode as discussed earlier, another key factor was mass transport. Take the transformation of 1m to 2m as an example (details in ESI, Section 6†). The main byproduct in the undivided cell was from arene bromination, likely because this large molecule cannot diffuse through the porous electrode. Concentration polarization thus led to a large overpotential and a low selectivity (<40% in the undivided cell). The polarization was minimized by the rapid electrolyte flow, resolving the trade-off between a high productivity and a high selectivity encountered in many previous studies on similar transformations as we summarize in Fig. S7.†
To shed light on the mechanism of this electrochemical oxidative rearrangement, we performed some mechanistic studies as shown in Scheme 1. First, an oxygen labelling experiment was performed to identify the source of oxygen on spirooxindoles (Scheme 1a). When H2O was replaced with H218O as the reaction medium, more than 87% 18O (determination by mass spectrum analysis) was incorporated in the spirooxindoles (1a), which suggested that water was involved in the reaction and was the main oxygen source of spirooxindole. Secondly, we attempted to capture the reactive brominating species (RBS, Scheme 1b), which was believed to be responsible for the oxidative rearrangement. Anisole (3) was easily brominated in excellent yield (91%) in the absence of THβC. When a 1:1 ratio of anisole and THβC was used, spirooxindole was isolated as the sole product without detection of bromoanisole. These results suggest that RBS was generated in the reaction, and interestingly, oxidative rearrangement occurred faster than bromination. This also explained why bromination of spirooxindoles was rarely observed. Thirdly, we attempted to elucidate the possible molecular structure of RBS using UV absorption and fluorescence spectra of the HOBr probe44 (Scheme 1c, also see Fig. S8 in the ESI†), which proved the generation of HOBr as the RBS for the rearrangement and bromination reactions. The obvious color change of the reaction mixture (brown-orange-clear) suggested the presence of a small amount of bromine, which could be converted into HOBr under acidic aqueous reaction conditions. Based on the above results and the well-established bromide-mediated electrooxidation, we proposed a plausible mechanism of electrochemical oxidative rearrangement of indole (Scheme 1d). Electrooxidation of LiBr generated HOBr, which oxidized the THβC into bromo indoline intermediate I. Water addition followed by semipinacol rearrangement furnished spirooxindole.
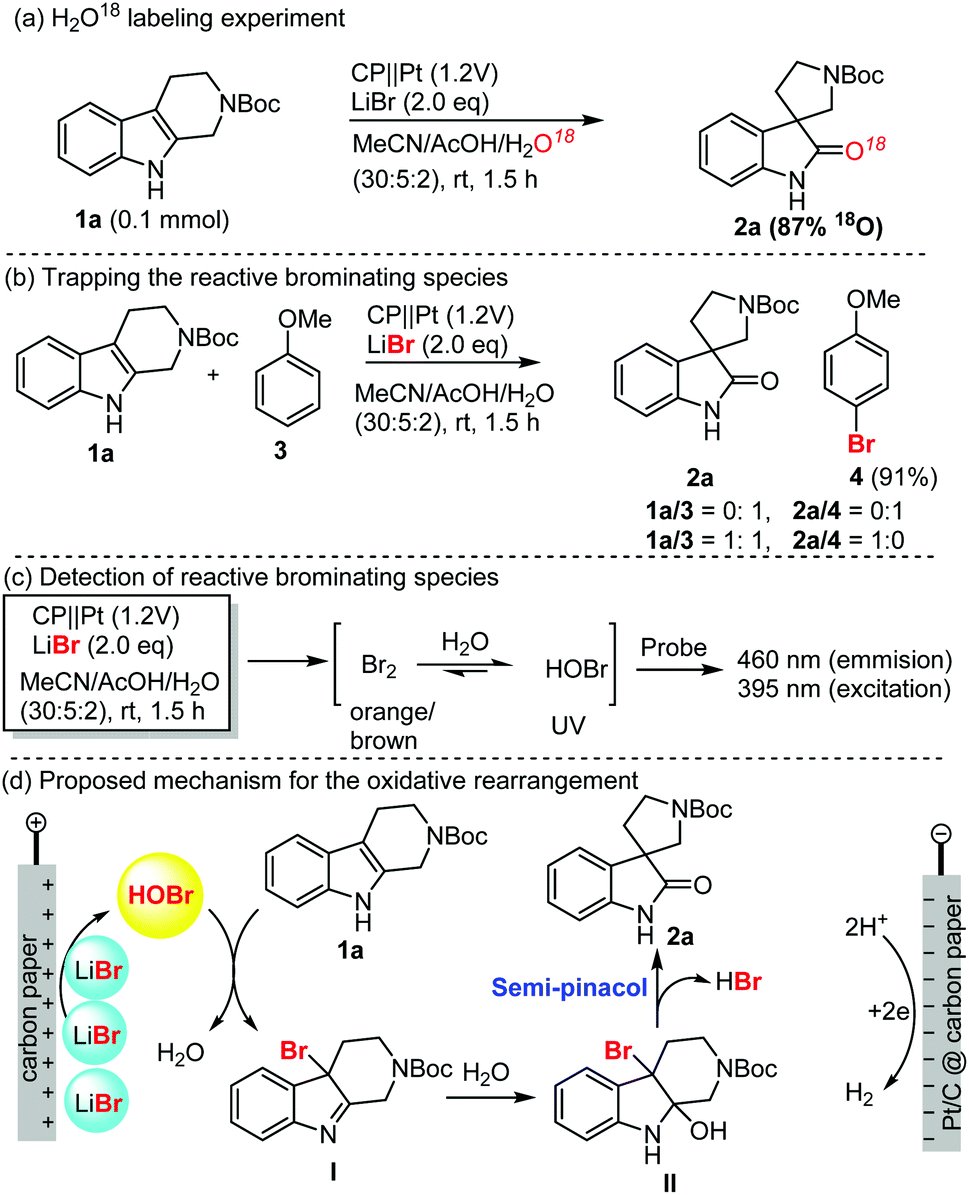 | ||
| Scheme 1 Mechanistic studies and the proposed mechanism for electrochemical oxidative rearrangement of tetrahydro-β-carbolines. (A) Isotopic labelling experiment; (B) RBS trapping experiments; (C) detection of RBS; the (D) proposed mechanism for electrochemical oxidative rearrangement of THβC. | ||
Conclusion
We have designed a zero-gap flow cell and demonstrated efficient, high-rate LiBr-mediated electro-oxidative rearrangement of tetrahydro-β-carbolines to spirooxindoles using commercial electrodes. The generality of this electrochemical oxidative rearrangement was demonstrated with 20 examples with good to excellent yields. Importantly, we discovered that the lithium ion played a key role as a spectator in the double layer to suppress the direct oxidation of the substrate and improve the yield. Compared to recent literature reports, our designed zero-gap flow cell delivers the highest FE and productivity. We expect that our catalytic system and cell design could be further applied to other electro-organic reactions.Data availability
Experimental procedures and characterization data are available within this article and its ESI.† Data are also available from the corresponding author on request.Author contributions
Y. Zheng and Y. T. Cheung contributed equally to this work.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research was financially supported by the Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (SMSEGL20Sc01-B), Research Grants Council of Hong Kong (C6026-19G, 16307219, 16304618, and 16306920), the National Foundation of Natural Science, China (52022002), and the Electrical Insulation and Power Equipment in Xi'an Jiaotong University and State Key Laboratory (Grant No. EIPE21205). The authors are thankful for the technical assistance from Fanny and Alex from the Materials Characterization and Preparation Facilities (MCPF) at HKUST.Notes and references
- N. Finch and W. I. Taylor, J. Am. Chem. Soc., 1962, 84, 1318–1320 CrossRef CAS.
- Z. Liu, F. Zhao, B. Zhao, J. Yang, J. Ferrara, B. Sankaran, B. V. Venkataram Prasad, B. B. Kundu, G. N. Phillips, Y. Gao, L. Hu, T. Zhu and X. Gao, Nat. Commun., 2021, 12, 4158 CrossRef CAS PubMed.
- L. Szabó, Molecules, 2008, 13, 1875–1896 CrossRef PubMed.
- S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532 RSC.
- S. D. Edmondson and S. J. Danishefsky, Angew. Chem., Int. Ed., 1998, 37, 1138–1140 CrossRef CAS PubMed.
- S. Edmondson, S. J. Danishefsky, L. Sepp-Lorenzino and N. Rosen, J. Am. Chem. Soc., 1999, 121, 2147–2155 CrossRef CAS.
- F. von Nussbaum and S. J. Danishefsky, Angew. Chem., Int. Ed., 2000, 39, 2175–2178 CrossRef CAS PubMed.
- M. Ito, C. W. Clark, M. Mortimore, J. B. Goh and S. F. Martin, J. Am. Chem. Soc., 2001, 123, 8003–8010 CrossRef CAS PubMed.
- H. Takayama, R. Fujiwara, Y. Kasai, M. Kitajima and N. Aimi, Org. Lett., 2003, 5, 2967–2970 CrossRef CAS PubMed.
- S. F. Martin, J. E. Hunter, B. Benage, L. S. Geraci and M. Mortimore, J. Am. Chem. Soc., 1991, 113, 6161–6171 CrossRef CAS.
- C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2003, 2209–2219 CrossRef.
- N. Ye, H. Chen, E. A. Wold, P.-Y. Shi and J. Zhou, ACS Infect. Dis., 2016, 2, 382–392 CrossRef CAS PubMed.
- Y. Zhao, S. Yu, W. Sun, L. Liu, J. Lu, D. McEachern, S. Shargary, D. Bernard, X. Li, T. Zhao, P. Zou, D. Sun and S. Wang, J. Med. Chem., 2013, 56, 5553–5561 CrossRef CAS PubMed.
- G. Bhaskar, Y. Arun, C. Balachandran, C. Saikumar and P. T. Perumal, Eur. J. Med. Chem., 2012, 51, 79–91 CrossRef CAS PubMed.
- S. S. Panda, R. A. Jones, P. Bachawala and P. P. Mohapatra, Mini-Rev. Med. Chem., 2017, 1515–1536 CAS.
- L.-M. Zhou, R.-Y. Qu and G.-F. Yang, Expert Opin. Drug Discov., 2020, 15, 603–625 CrossRef CAS PubMed.
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed.
- B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed.
- J. Xu, L. Liang, H. Zheng, Y. R. Chi and R. Tong, Nat. Commun., 2019, 10, 4754 CrossRef PubMed.
- G. Zhao, L. Liang, E. Wang, S. Lou, R. Qi and R. Tong, Green Chem., 2021, 23, 2300–2307 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed.
- S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed.
- C. A. Malapit, M. B. Prater, J. R. Cabrera-Pardo, M. Li, T. D. Pham, T. P. McFadden, S. Blank and S. D. Minteer, Chem. Rev., 2022, 122, 3180–3218 CrossRef CAS PubMed.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- H.-T. Tang, J.-S. Jia and Y.-M. Pan, Org. Biomol. Chem., 2020, 18, 5315–5333 RSC.
- T. A. Enache and A. M. Oliveira-Brett, Electroanalysis, 2011, 23, 1337–1344 CrossRef CAS.
- P. Jennings, A. C. Jones, A. R. Mount and A. D. Thomson, Faraday Trans., 1997, 93, 3791–3797 RSC.
- J. Wu, Y. Dou, R. Guillot, C. Kouklovsky and G. Vincent, J. Am. Chem. Soc., 2019, 141, 2832–2837 CrossRef CAS PubMed.
- H. Abou-Hamdan, C. Kouklovsky and G. Vincent, Synlett, 2020, 31, 1775–1788 CrossRef CAS.
- K. Liu, W. Song, Y. Deng, H. Yang, C. Song, T. Abdelilah, S. Wang, H. Cong, S. Tang and A. Lei, Nat. Commun., 2020, 11, 3 CrossRef PubMed.
- H. Qin, Z. Yang, Z. Zhang, C. Liu, W. He, Z. Fang and K. Guo, Chem.–Eur. J., 2021, 27, 13024–13028 CrossRef CAS PubMed.
- A. J. J. Jebaraj, N. S. Georgescu and D. A. Scherson, J. Phys. Chem. C, 2016, 120, 16090–16099 CrossRef CAS.
- A. Zolfaghari, Electrochim. Acta, 2002, 47, 1173–1187 CrossRef CAS.
- A. Butler, Curr. Opin. Chem. Biol., 1998, 2, 279–285 CrossRef CAS PubMed.
- Q. Chen, M. R. Gerhardt, L. Hartle and M. J. Aziz, J. Electrochem. Soc., 2016, 163, A5010–A5013 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- D. Strmcnik, K. Kodama, D. van der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, Nat. Chem., 2009, 1, 466–472 CrossRef CAS PubMed.
- C. Stoffelsma, P. Rodriguez, G. Garcia, N. Garcia-Araez, D. Strmcnik, N. M. Marković and M. T. M. Koper, J. Am. Chem. Soc., 2010, 132, 16127–16133 CrossRef CAS PubMed.
- C. Qian, P. Li and J. Sun, Angew. Chem., Int. Ed., 2021, 60, 5871–5875 CrossRef CAS PubMed.
- K. Yamamoto, M. Kuriyama and O. Onomura, Chem. Rec., 2021, 21, 2239–2253 CrossRef CAS PubMed.
- K. Yamamoto, M. Kuriyama and O. Onomura, Acc. Chem. Res., 2020, 53, 105–120 CrossRef CAS PubMed.
- Z. Hou, X. Wang, R. Yang, Z. Li, Y. Sun, L. Qu and H. Zeng, Sens. Actuators, B, 2020, 315, 128125 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03951f |
| This journal is © The Royal Society of Chemistry 2022 |