
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cooperative adsorbent for the switch-like capture of carbon dioxide from crude natural gas†
Rebecca L.
Siegelman
 ab,
Joshua A.
Thompson‡
c,
Jarad A.
Mason§
a,
Thomas M.
McDonald
a and
Jeffrey R.
Long
*abd
ab,
Joshua A.
Thompson‡
c,
Jarad A.
Mason§
a,
Thomas M.
McDonald
a and
Jeffrey R.
Long
*abd
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: jrlong@berkeley.edu
bMaterials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cChevron Energy Technology Company, Richmond, CA 94801, USA
dDepartment of Chemical and Biomolecular Engineering, University of California, Berkeley, CA 94720, USA
First published on 13th September 2022
Abstract
Natural gas constitutes a growing share of global primary energy due to its abundant supply and lower CO2 emission intensity compared to coal. For many natural gas reserves, CO2 contamination must be removed at the wellhead to meet pipeline specifications. Here, we demonstrate the potential of the diamine-appended metal–organic framework ee-2–Mg2(dobpdc) (ee-2 = N,N-diethylethylenediamine; dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate) as a next-generation CO2 capture material for high-pressure natural gas purification. Owing to a cooperative adsorption mechanism involving formation of ammonium carbamate chains, ee-2–Mg2(dobpdc) can be readily regenerated with a minimal change in temperature or pressure and maintains its CO2 capacity in the presence of water. Moreover, breakthrough experiments reveal that water enhances the CO2 capture performance of ee-2–Mg2(dobpdc) by eliminating “slip” of CO2 before full breakthrough. Spectroscopic characterization and multicomponent adsorption isobars suggest that the enhanced performance under humid conditions arises from preferential stabilization of the CO2-inserted phase in the presence of water. The favorable performance of ee-2–Mg2(dobpdc) is further demonstrated through comparison with a benchmark material for this separation, zeolite 13X, as well as extended pressure cycling. Overall, these results support continued development of ee-2–Mg2(dobpdc) as a promising adsorbent for natural gas purification.
Introduction
Natural gas currently supplies over 23% of global primary energy,1–4 and growing reliance on natural gas drove a 25% increase in worldwide gas production between 2010 and 2020.3 Both economic and environmental incentives have stimulated this growth: gas reserves are abundant and largely untapped, and natural gas burns with a lower carbon intensity compared to coal, while also emitting less NOx, mercury, and particulate matter.3,5Despite the abundance of crude gas reserves, approximately 25% of U.S. reserves5,6 and up to 50% of worldwide reserves7 are estimated to be sub-quality due to CO2 contamination in excess of 2%. This CO2 must be removed from the crude gas prior to transport in a pipeline to avoid dilution of the heating value of the gas and to prevent corrosion in the presence of moisture. Aqueous amine solutions, which utilize acid–base chemistry to absorb and separate CO2 by forming ammonium carbamate or bicarbonate, remain the industry standard for natural gas purification.8 Despite the maturity of aqueous amine technology, these solutions suffer from a number of disadvantages, including high regeneration energies, low CO2 capacities, and issues with corrosivity, oxidation, and volatilization.8 The limitations of amine solutions and the burgeoning market for natural gas purification have led to exploration of a number of alternative technologies in various stages of development. Physical absorption,9 low-temperature (<0 °C) separations,10 membrane-based processes,11 separations using clathrate hydrates,12 and adsorptive separations have all received growing interest in recent years.
Pressure-swing adsorption (PSA)13 processes have generated particular interest as modular systems with the potential to reduce the energy intensity and environmental hazards of natural gas purification relative to amine solutions.14,15 In PSA, a porous solid selectively binds CO2 from a crude natural gas feed at elevated pressure, generating a methane-enriched product stream. Following saturation of the adsorbent bed with CO2, desorption is achieved by depressurization and purge steps, after which the bed is repressurized to begin a new cycle. Two or more adsorption beds are used in parallel to allow continuous operation through concurrent adsorption and desorption steps.
PSA for CO2 removal has seen early commercial-scale success, notably in Guild Associates' Molecular Gate™ process16 and Xebec's Rotary-Valve, Fast-Cycle PSA process,17 which incorporate proprietary titanosilicate and metal-based adsorbents, respectively. Further advances in material design are attractive to achieve improved performance in PSA processes. New adsorbents for natural gas conditioning must (1) possess high capacity and selectivity for CO2, (2) co-adsorb minimal CH4 to maximize recovery, (3) release the bound CO2 with minimal energetic input, and (4) demonstrate rapid adsorption–desorption kinetics. Additionally, adsorbents with the ability to function in the presence of moisture are highly desirable, as natural gas feeds are typically saturated with water.5 Because water tends to displace CO2 on adsorbents that utilize exposed metal ions as binding sites,18–21 the impact of water must be carefully managed through upstream dehydration, use of a sacrificial adsorbent layer, or periodic high-temperature regeneration. Adsorbents with segregated binding sites for CO2 and H2O could therefore offer avenues for more economical process configurations. At minimum, such adsorbents would add robustness in case of process deviations during upstream dehydration.
Amine-functionalized adsorbents are promising candidates to meet each of the above criteria. As with amine solutions, these materials selectively bind CO2 through acid–base chemistry to form ammonium carbamate or bicarbonate species. This strategy has already been applied in the design of amine-grafted mesoporous silicas for natural gas purification.22,23 Amine-functionalized metal–organic frameworks have likewise received growing attention for CO2 removal from humid gas streams.24 These materials feature inorganic ions or clusters bridged by organic ligands to form highly porous and crystalline coordination solids. Amine functionality can be incorporated within the ligand25,26 or post-synthetically introduced by grafting to open metal coordination sites within the pores.27,28
The latter approach led to the recent discovery of a new class of frameworks of the form M2(dobpdc)(diamine)2 (M = Mg, Mn, Fe, Co, Ni, Zn; dobpdc4− = 4,4′- dioxidobiphenyl-3,3′-dicarboxylate) that feature one-dimensional hexagonal channels densely lined with metal(II) cations, each bearing an appended diamine (Fig. 1a).29,30 The ideal spacing between metal sites facilitates a unique adsorption mechanism in which CO2 inserts into the metal–amine bonds (Fig. 1b); the resulting metal-bound carbamate and dangling ammonium groups interact with neighboring sites to form chains of ammonium carbamate running along the framework channel.30 This switch-like mechanism manifests as sharp, high-capacity steps in the CO2 adsorption isotherms, and the full capacity of bound CO2 can accordingly be desorbed with minimal changes in temperature or pressure (Fig. 1c). The threshold pressure for CO2 insertion can be precisely tailored to a given CO2 separation process through judicious choice of the constituent metal and diamine.31
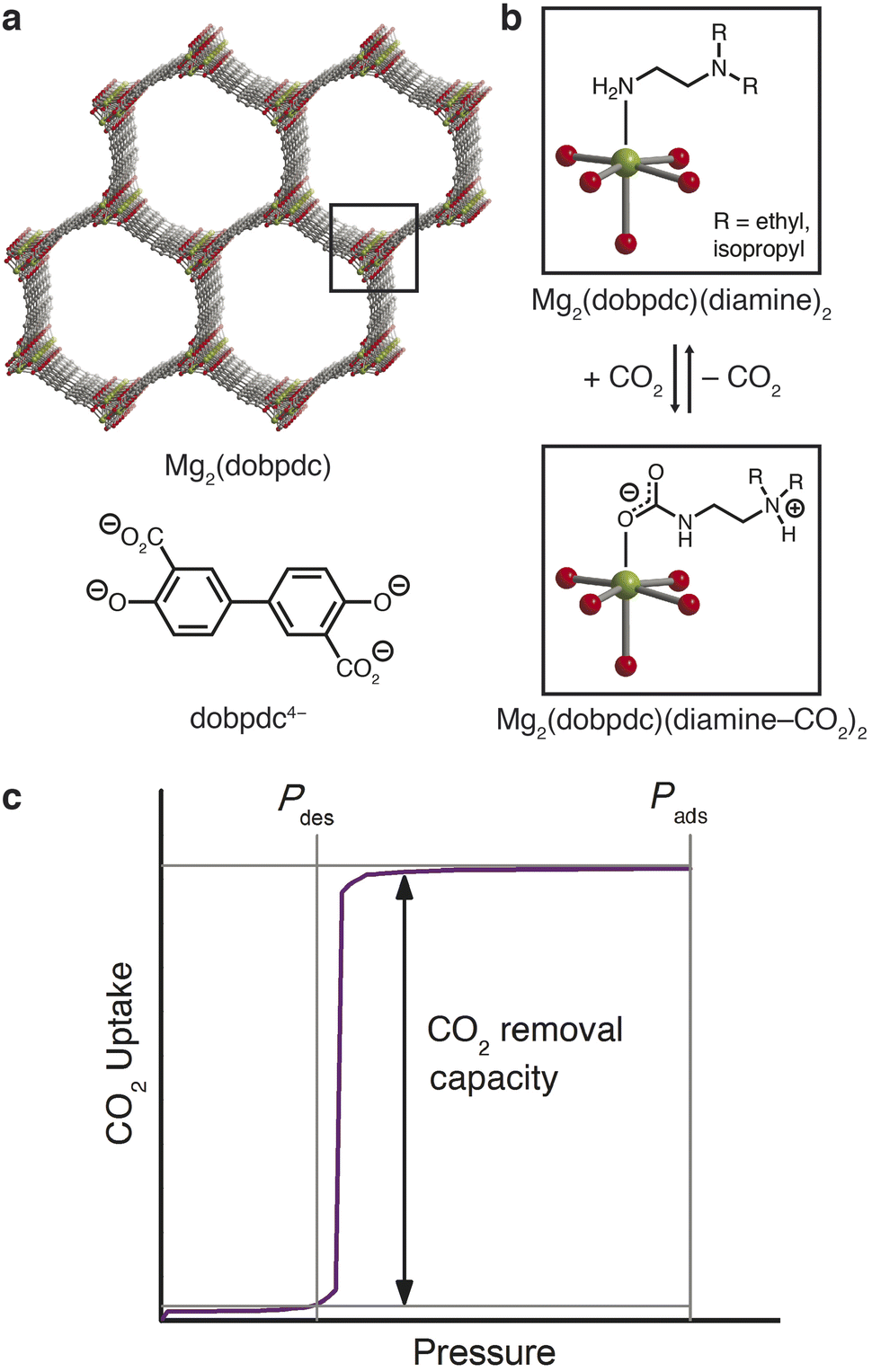 | ||
| Fig. 1 (a) Structure of the metal–organic framework Mg2(dobpdc) (top) and the ligand dobpdc4− (bottom). Green, gray, and red spheres correspond to Mg, C, and O atoms, respectively; H atoms have been omitted for clarity. (b) Post-synthetic functionalization of the open Mg(II) coordination sites with diamines to yield Mg2(dobpdc)(diamine)2 (top), and chemisorption of CO2 through insertion into the metal–amine bond (bottom), resulting in cooperative formation of ammonium carbamate chains along the pore axis. (c) Hypothetical isotherm and schematic of a pressure-swing adsorption process with a cooperative adsorbent, for which the full adsorption capacity of the material can be accessed as working capacity. | ||
Previous exploration of diamine-appended M2(dobpdc) has been limited to CO2 capture from low-pressure air, flue gas, or process streams in temperature-swing systems.29,30,32–37 Here, we demonstrate the design of a diamine-appended framework as a next-generation adsorbent for pressure-swing conditioning of CO2-rich natural gas. We further demonstrate that humidity enhances the CO2 capture performance of these materials and discuss the mechanistic effects underlying this advantageous behavior.
Experimental
General materials and methods
The ligand 4,4′-dihydroxy-(1,1′-biphenyl)-3,3′-dicarboxylic acid (H4dobpdc) was obtained from Hangzhou Trylead Chemical Technology Co. and used as received; or was obtained from Combi-Blocks and purified by recrystallization from a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v:v) acetone:water mixture; or was synthesized as previously reported.38 All other reagents and solvents were obtained from commercial suppliers at reagent grade purity or higher and were used without further purification. Synthetic procedures were conducted under air unless otherwise noted. Zeolite 13X was obtained from Sigma Aldrich as 60–80 mesh pellets (product code 20305, Supelco). Full characterization of this material is included in ESI Section 7.† The metal–organic framework Mg2(dobpdc) was prepared as a gram-scale powder and characterized following a previously reported procedure (ESI Section 1†).31 Ultrahigh purity (99.999%) He, N2, CH4, and H2 and research grade (99.998%) CO2 were used for all adsorption experiments. Elemental analyses were conducted at the Microanalytical Laboratory of the University of California, Berkeley, using a PerkinElmer 2400 Series II combustion analyzer. 1H NMR spectra were collected on a Bruker AMX 300 MHz NMR spectrometer. Details regarding use of dynamic scanning calorimetry for heat capacity measurements and kinetics experiments are given in ESI Sections 8 and 9,† respectively. Powder X-ray diffraction (PXRD) patterns were collected with a laboratory Bruker AXS D8 Advance diffractometer with CuKα radiation (λ = 1.5418 Å) or at the Advanced Photon Source with synchrotron radiation (λ = 0.45399 Å), as specified in the figure captions. Details for synchrotron PXRD experiments under controlled atmospheres are given in ESI Section 17.† Single-crystal synthesis and X-ray diffraction details are included in ESI Section 18.†
:1 (v:v) acetone:water mixture; or was synthesized as previously reported.38 All other reagents and solvents were obtained from commercial suppliers at reagent grade purity or higher and were used without further purification. Synthetic procedures were conducted under air unless otherwise noted. Zeolite 13X was obtained from Sigma Aldrich as 60–80 mesh pellets (product code 20305, Supelco). Full characterization of this material is included in ESI Section 7.† The metal–organic framework Mg2(dobpdc) was prepared as a gram-scale powder and characterized following a previously reported procedure (ESI Section 1†).31 Ultrahigh purity (99.999%) He, N2, CH4, and H2 and research grade (99.998%) CO2 were used for all adsorption experiments. Elemental analyses were conducted at the Microanalytical Laboratory of the University of California, Berkeley, using a PerkinElmer 2400 Series II combustion analyzer. 1H NMR spectra were collected on a Bruker AMX 300 MHz NMR spectrometer. Details regarding use of dynamic scanning calorimetry for heat capacity measurements and kinetics experiments are given in ESI Sections 8 and 9,† respectively. Powder X-ray diffraction (PXRD) patterns were collected with a laboratory Bruker AXS D8 Advance diffractometer with CuKα radiation (λ = 1.5418 Å) or at the Advanced Photon Source with synchrotron radiation (λ = 0.45399 Å), as specified in the figure captions. Details for synchrotron PXRD experiments under controlled atmospheres are given in ESI Section 17.† Single-crystal synthesis and X-ray diffraction details are included in ESI Section 18.†
Gram-scale synthesis of diamine-appended M2(dobpdc)
Diamine-grafting conditions for preparing gram-scale diamine-appended Mg2(dobpdc) powders were adapted from a previous report.30 A 20% (v/v) solution of diamine in toluene was dried by stirring over CaH2 for 3 h at 60 °C. Separately, several grams of washed, methanol-solvated Mg2(dobpdc) was dried under flowing N2 at 250 °C for 12 h. The desolvated parent framework was then cooled to room temperature under N2, and a quantity of diamine solution corresponding to 10 equivalents of diamine per MgII site was added by cannula transfer. (We note that at the milligram scale, direct diamine grafting has been demonstrated without activation of the methanol-solvated parent framework or diamine drying agents such as CaH2 (ref. 31)). The reaction vessel was sonicated for 15 min under N2 and then left undisturbed under N2 for a minimum of 12 h at room temperature. The solid was isolated by filtration and washed three times with 100 mL portions of toluene and three times with 100 mL portions of hexanes in air at room temperature. Prior to adsorption measurements, the sample was desolvated (activated) by heating in vacuo or under flowing N2 at 100 °C for 12 h. Diamine loadings were determined by collecting NMR spectra of ∼5 mg of activated, diamine-appended framework samples digested with a solution of ∼20 μL of 35 wt% DCl in ∼0.75 mL of DMSO-d6. The ratio of the diamine to ligand peak integrals indicated typical diamine loadings of 100% within error (±5%). Combustion elemental analysis calculated for Mg2(dobpdc)(ee-2)2 (C26H38Mg2N4O6; ee-2 = N,N-diethylethylenediamine): C, 56.65; H, 6.95; N, 10.16. Found: C, 56.42; H, 6.77; N, 10.15. Combustion elemental analysis calculated for Mg2(dobpdc)(ii-2)2.1 (C30.8H48Mg2N4.2O6; ii-2 = N,N-diisopropylethylenediamine): C, 59.50; H, 7.78; N, 9.46. Found: C, 59.50, H, 7.62; N, 9.46.Volumetric gas adsorption experiments
Prior to sample measurement, an empty sample holder was used to collect background CO2 adsorption isotherms at the analysis temperatures and target pressure points. A small negative background was observed at high pressures and can likely be attributed to volume or temperature calibration errors or errors in the equation of state used to correct for non-ideality. The background adsorption was found to be consistent over several measurements, and polynomial fits of replicate data sets at each temperature were used to perform background subtraction on the experimental data sets (Fig. S13†). A negligible background was observed for high-pressure CH4 measurements, and these datasets were therefore not corrected. For each material, total adsorption (ntot) was calculated from the experimentally measured, background-corrected excess adsorption data (nex) using the Gurvich rule:42
| ntot = nex + Vpρbulk(T,P) | (1) |
The pore volume (Vp) was determined from the 77 K N2 adsorption experiments (Fig. S3, S8 and S33†), and the bulk gas density was determined from the NIST REFPROP database41 at each experimental temperature and pressure.
| s = (qCO2/qCH4)/(pCO2/pCH4) | (2) |
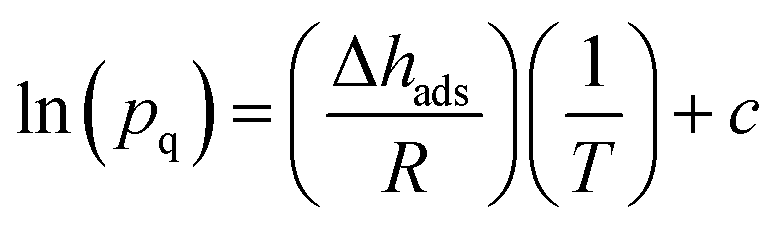 | (3) |
Thermogravimetric analysis
Dry thermogravimetric analysis (TGA) experiments were conducted at atmospheric pressure using a TA Instruments TGA Q5000. Humid TGA experiments were conducted using a TA Instruments TGA Q50 by passing the incident gas stream through two water bubblers in series, leading to an estimated water content of 1.3% at 25 °C (∼30% R.H.). All adsorption and desorption isobars were collected at a ramp rate of 1 °C min−1. Pre-mixed CO2/N2 blends (5%, 10%, 15%, 20%, 30%, and 50% CO2 in N2) were purchased from Praxair. Initial sample activation was performed at 120 °C under flowing N2 for 20–30 min until stabilization of the mass was observed. Decomposition experiments were conducted at a ramp rate of 1.5 °C min−1 under dry N2.Breakthrough experiments and pressure cycling
For all dynamic breakthrough measurements, pellets of adsorbent were prepared by mechanical compression. Prior to activation from toluene or hexanes, the powdered diamine-appended material was placed in a stainless-steel cylinder between highly polished faces of a stainless-steel platform and corresponding plunger. A mechanical press was used to compress the powder between the platform and plunger to form a tablet. This tablet was then broken to the desired particle size between copper mesh sieves (Fig. S50†) to produce pellets of either 25–45 mesh (350–700 μm) or 60–80 mesh (180–250 μm), as specified below. Pellets were activated by heating in vacuo or under flowing N2 at 100 °C for 12 h.Quantitative, dry breakthrough measurements at 1, 35, and 70 bar were conducted on a custom-built breakthrough apparatus at the Chevron Energy Technology Company in Richmond, CA (Fig. S51†). A 5 cm3 stainless steel column (30.5 cm length, 0.46 cm diameter) was packed with 25–45 mesh pellets of activated ii-2–Mg2(dobpdc) (2.50 g) or ee-2–Mg2(dobpdc) (3.31 g). Glass wool was placed before the end fittings to secure the adsorbent in place. Capacities for CO2 are reported with an error of ±0.3 mmol g−1, as calculated by assuming a 30 s error in integrated breakthrough time and a flow rate deviation of ±1%.
Qualitative dry and humid breakthrough experiments at 7 and 50 bar and extended pressure cycling were performed on a customized PSA-1000 instrument built by L&C Science and operated at the Chevron Energy Technology Company. A 3.2 cm3 stainless steel bed (16.5 cm length, 0.5 cm diameter) was packed with 0.90 g of activated ee-2–Mg2(dobpdc) or 1.98 g of activated zeolite 13X as 60–80 mesh pellets. Additional experimental details and breakthrough experiments are included in ESI Sections 13–15.†
Results and discussion
Material design considerations
Effective use of diamine-appended M2(dobpdc) for PSA requires thoughtful manipulation of several design features unique to this class of materials. Critically, for adsorbents with step-shaped isotherms, CO2 binding is only possible at partial pressures exceeding the step pressure. Once the adsorbate partial pressure in the column drops below this threshold pressure, the switch-like mechanism is disabled, and any remaining CO2 will “slip” through the bed with the CH4 product (Fig. 2). More specifically, the breakthrough profile for adsorption can be predicted from the CO2 isotherm using a graphical method known as Golden's rule, or the “rubber band” or “string” rule.32,44–46 In short, an operating curve is constructed by stretching an imaginary rubber band (green curve, Fig. 2) beneath the adsorption isotherm between the initial state (0% CO2) and the feed state (10% CO2). In regions bounded by points of tangency with the isotherm, a compressive “shock” is anticipated in the breakthrough profile. In regions where the rubber band runs along the isotherm, a dispersive wave is generated. With step-shaped isotherms, a multi-moment “shock–wave–shock” breakthrough profile is therefore frequently observed, with the initial “shock” corresponding to CO2 slip at concentrations beneath the step. (See ESI† Section 12 for additional details). As a result, the adsorbent must be designed such that the adsorption step occurs not simply below the CO2 partial pressure in the feed, but instead at a minimum partial pressure dictated by the desired product purity and/or CO2 capture rate. For natural gas purification, this variable is fixed by the maximum allowable CO2 content of 2 mol% in the product CH4 stream.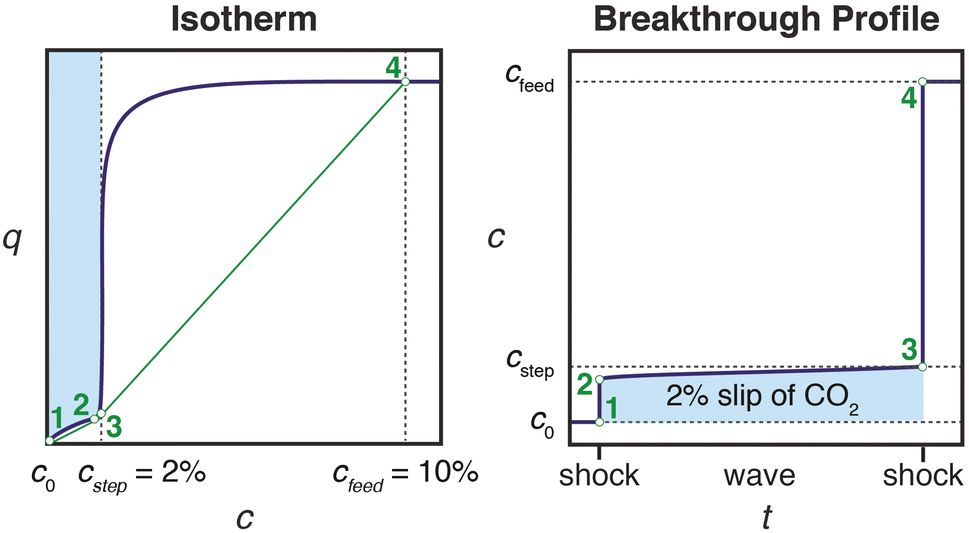 | ||
| Fig. 2 Schematic of a “shock–wave–shock” breakthrough profile (right, concentration, c, vs. time, t) anticipated for an adsorbent with a step-shaped isotherm (left, quantity adsorbed, q, vs. c). The operating curve used to predict the breakthrough profile from the isotherm is shown in green, and the numerical labels indicate corresponding points in the isotherm and breakthrough profile. At concentrations beneath the step, CO2 “slips” through the bed (shaded blue area). To achieve a CH4 product purity of ≥98%, an adsorbent with <2% slip is desirable. Figure adapted from ref. 32. | ||
Crude natural gas may be recovered from reserves coupled with crude oil (associated gas) or in independent reserves (non-associated or wellhead gas) and can therefore exhibit a broad range of total pressures (up to ∼70 bar), compositions (up to ∼80% CO2), and temperatures.5,7 For this study, we chose two representative cases: (1) simulated non-associated gas, 70 bar, 10% CO2, and (2) simulated associated gas, 7 bar, 10% CO2, both in the range of 25–50 °C. To reduce the CO2 concentration below 2%, a diamine-appended framework must operate with a step pressure of ≤1.4 bar for the non-associated gas in case (1) and ≤140 mbar for the associated gas in case (2) (Table 1).
| Wellhead gas | Associated gas | |
|---|---|---|
| Target total pressure | 70 bar | 7 bar |
| Target CO2 partial pressure | 7 bar | 0.7 bar |
| Target CO2 adsorption step pressure | ≤1.4 bar | ≤0.14 bar |
Previous exploration of diamine-appended frameworks for cooperative CO2 capture has generated a library of adsorbents with step pressures ranging from 10−4 bar to 1 bar at 40 °C.31 For the high-pressure separations in this work, the previously reported adsorbents ee-2–Mg2(dobpdc) (ee-2 = N,N-diethylethylenediamine) and ii-2–Mg2(dobpdc) (ii-2 = N,N-diisopropylethylenediamine)31,35 were identified as promising candidate materials for low-energy CO2 capture. The primary/tertiary (1°/3°) diamines in these materials promote high thermal stability due to the strong interaction between the magnesium centers and the primary amines. The sterically bulky tertiary amines further supress amine volatilization (see Fig. S12† for a two-year stability test) and destabilize the CO2-adsorbed phase, leading to high CO2 step pressures and low differential enthalpies of CO2 adsorption (−Δhads < 70 kJ mol−1). These favorable characteristics supported further investigation of 1°/3° diamine-appended frameworks for PSA.
Pressure-swing adsorption with a cooperative adsorption threshold at atmospheric pressure
The target CO2 insertion step pressure of 1.4 bar for non-associated gas purification suggests an attractive possibility for energy-efficient PSA: following saturation of the adsorbent at high pressure, the bound CO2 could be completely desorbed at atmospheric pressure without need for vacuum or heat. The adsorbent ii-2–Mg2(dobpdc) was previously found to have a step pressure of 1 bar at 40 °C (ref. 31) and was therefore initially investigated for CO2 capture from non-associated gas.High-pressure, single component CO2 and CH4 isotherms were first collected for ii-2–Mg2(dobpdc) (Fig. 3). Step-like CO2 isotherms were observed at all temperatures, with the step capacity corresponding to the anticipated binding of 1 CO2 molecule per diamine (3.3 mmol g−1). Application of the Clausius–Clapeyron relationship indicated differential adsorption enthalpies of −45 ± 4 kJ mol−1 for CO2 and –13.4 ± 0.6 kJ mol−1 for CH4 at a loading of 1 mmol g−1 (See Fig. S16 and S20† for plots of −Δhads as a function of loading for CO2 and CH4, respectively.). Compared to other amine-based adsorbents, which frequently exhibit low-coverage adsorption enthalpies with magnitudes of >70 kJ mol−1 CO2,27,47,48 the moderate adsorption enthalpies of 1°/3° diamine-appended frameworks may reduce both heat management requirements upon adsorption and the energetic input required for desorption. For a target stream of 10% CO2 at 70 bar, non-competitive CO2/CH4 selectivities of 17, 19, and 20 were calculated from the single-component isotherms at 25, 40, and 50 °C under the target conditions for wellhead gas purification. These values are anticipated to represent lower bounds for selectivity, as CO2 and CH4 have separate primary binding sites in diamine-appended frameworks.
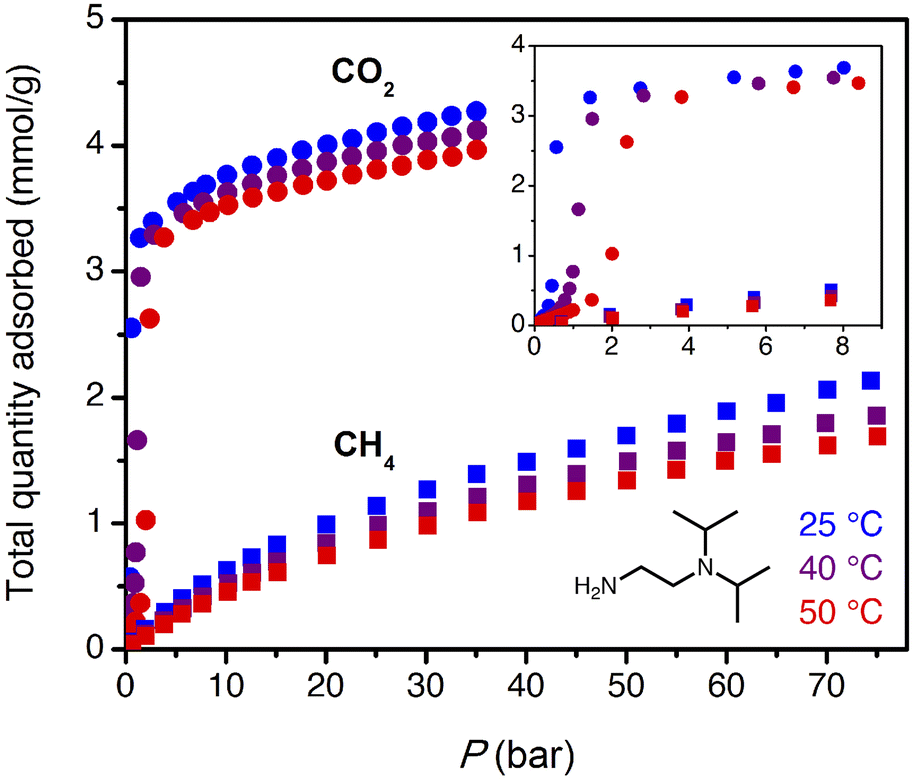 | ||
| Fig. 3 High-pressure CO2 (circles) and CH4 (squares) adsorption isotherms for ii-2–Mg2(dobpdc) at 25 °C (blue), 40 °C (purple), and 50 °C (red). The inset shows an enlarged view of the low-pressure region. | ||
High-pressure, dry breakthrough experiments were conducted to analyze the CO2 capacity and mixed-gas selectivity of ii-2–Mg2(dobpdc) from dry, simulated wellhead gas. With 300 sccm of 10% CO2 in CH4 at 70 bar and 30 °C, a shock–wave–shock breakthrough profile was observed (Fig. 4), consistent with the general schematic shown in Fig. 2. (See ESI† Section 14 for a duplicate measurement and additional experiments with different temperatures, pressures, and gas compositions.). Integrating over the full breakthrough profile yielded a high CO2 exhaustion capacity of 3.3 mmol g−1 (equivalent to 1 CO2 per diamine) and a moderate CO2/CH4 selectivity of 7.7. Despite these promising performance metrics, several limitations were noted that would inhibit the realistic application of this material for PSA. Importantly, the breakthrough experiments show greater CO2 “slip” than would be anticipated based on the single-component, equilibrium isotherms. Specifically, while application of Golden's rule predicted a CO2 slip of <1 mol% (Table S9†), the initial shock in the CO2 breakthrough curve instead yielded a slip concentration of 3 mol% CO2 (2 bar of CO2). As a result, the CH4 product purity was limited to ∼97%, and the material displayed negligible useable CO2 capacity for production of pipeline-quality CH4 (≥98%).
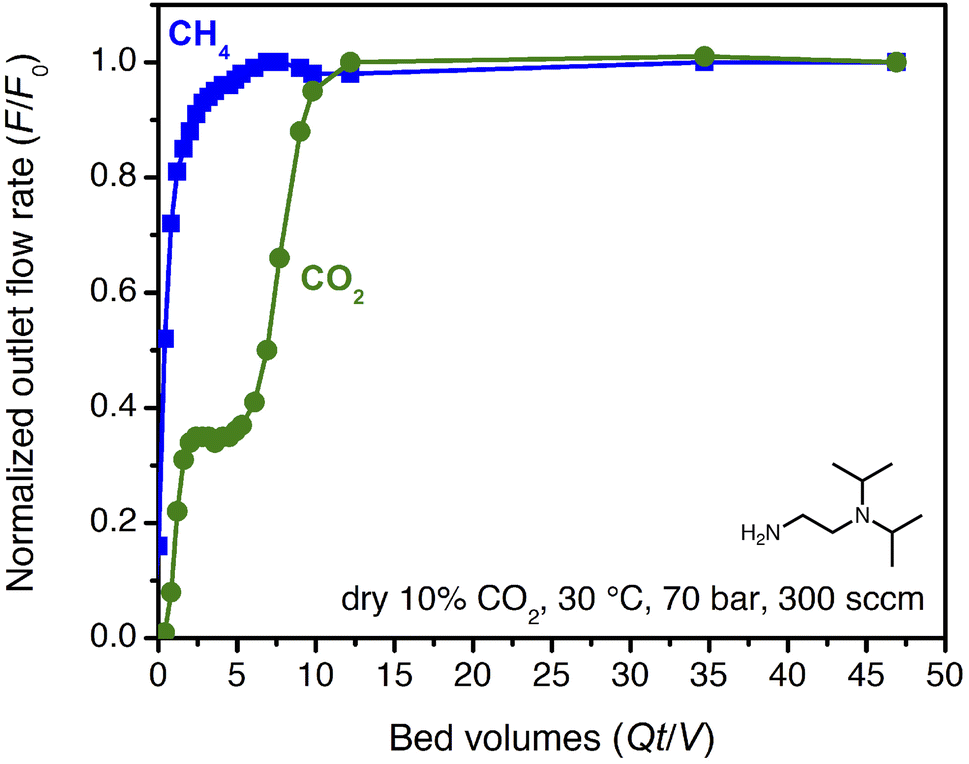 | ||
| Fig. 4 Breakthrough profile for ii-2–Mg2(dobpdc) under 300 sccm of dry 10% CO2 in CH4 at 70 bar and 30 °C. The CO2 in the initial shock corresponds to a slip concentration of 3 mol%. | ||
The partial pressure of CO2 in the initial shock is consistent with an effective step temperature of 53 °C, as calculated from the adsorption isotherms and the differential enthalpy of adsorption (ESI Section 5†). In part, this can be attributed to temperature rise in the bed during adsorption. Thermocouples at 3′′ and 9′′ from the inlet along the 12′′ bed indicated a small temperature rise during the experiment, to a maximum of 38 °C (Fig. S52†). Kinetics limitations may also contribute to the high level of CO2 slip observed in breakthrough experiments. Examination of the temperature dependence of the rate of CO2 adsorption by dynamic scanning calorimetry revealed slow CO2 adsorption kinetics at conditions approaching the step (ESI Section 9†). Furthermore, a 25 °C adsorption isotherm collected for the pelletized adsorbent revealed a shift in the step pressure to >0.6 bar, vs. ∼0.4 bar for the powder under the same equilibration criteria (Fig. S21†). Extending the time allotted for equilibration shifted the pellet isotherm step position back to that of the powder, indicating that kinetics limitations likely contribute to the complex CO2 breakthrough profiles observed for this material and that the adsorption properties may be sensitive to external stimuli, including compaction. Similar kinetics limitations were recently discussed for this class of adsorbents in the context of direct air capture.36 Note that this behavior is anticipated for such materials, because the driving force for reactive CO2 adsorption is small at conditions near the cooperative adsorption step but increases at lower temperature or with an increasing partial pressure of CO2 beyond the step (Fig. S38 and S39†). Regardless, these results suggest that predictions made from single-component adsorption isotherms alone may not provide an accurate depiction of adsorbent performance under flow conditions (additional discussion below).
In addition, the equilibrium adsorption isotherms show hysteresis, with the desorption branch of the isotherm closing at 0.25 bar at 25 °C and 0.5 bar at 50 °C (Fig. S14†). As a result, vacuum or heat would be required to desorb CO2 between cycles. In sum, ii-2–Mg2(dobpdc) may be valuable as an adsorbent for bulk separation of CO2 from high-pressure streams, but to produce pipeline-quality natural gas, the material would either need to be paired with a polishing adsorbent or replaced with an adsorbent with a slightly lower step pressure. The latter strategy motivated us to explore ee-2–Mg2(dobpdc) for both associated and non-associated gas separations.
Evaluation of ee-2–Mg2(dobpdc) for natural gas purification
The smaller steric profile of the tertiary amine in N,N-diethylethylenediamine (ee-2) results in lower CO2 insertion pressures for ee-2–Mg2(dobpdc) as compared to ii-2–Mg2(dobpdc), as a result of stronger ion pairing in the ammonium carbamate chains formed upon CO2 adsorption.31 As shown in Fig. 5, the inflection point of the first CO2 adsorption step for this material shifts from ∼0.11 bar at 40 °C to 0.24 bar at 50 °C and exceeds 1 bar slightly below 75 °C, suggesting that regeneration could be achieved with a moderate concentration swing or combined pressure–vacuum or pressure–temperature swing process. The two-step CO2 adsorption profile has been attributed to steric crowding within the pore, which necessitates a rearrangement at half capacity to allow full occupancy of ammonium carbamate chains.¶35 The full capacity of both adsorption steps (3.6 mmol g−1) is consistent with the binding of 1 CO2 molecule per diamine, with a slight increase in the gravimetric capacity from that of ii-2–Mg2(dobpdc) due to the smaller appended diamine. In addition, the high-pressure isotherms show greater uptake of CO2 beyond the step as compared to the material grafted with ii-2, consistent with a larger accessible surface area (911 ± 4 m2 g−1, vs. 490 ± 4 m2 g−1 for ii-2–Mg2(dobpdc); Fig. S3 and S8†) that likely facilitates faster diffusion through the pores. High-pressure methane isotherms likewise show increased uptake compared to that of ii-2–Mg2(dobpdc), leading to a non-competitive selectivity of 15–16 over a temperature range of 25–50 °C for wellhead gas containing 10% CO2 at 70 bar. Desorption isotherms for CO2 show closure of the hysteresis loop at 0.01, 0.04, and 0.1 bar at 25, 40, and 50 °C, respectively (Fig. 5a). Differential enthalpies of adsorption of −65 ± 1 and −13.1 ± 0.2 kJ mol−1 were calculated for CO2 and CH4, respectively, at a loading of 1 mmol g−1 (Fig. S23 and S28†). Note, however, that the CH4 adsorption enthalpy within the diamine-appended framework may deviate from that in the CO2-inserted framework, in which the pores are lined with ion-paired chains of ammonium carbamate.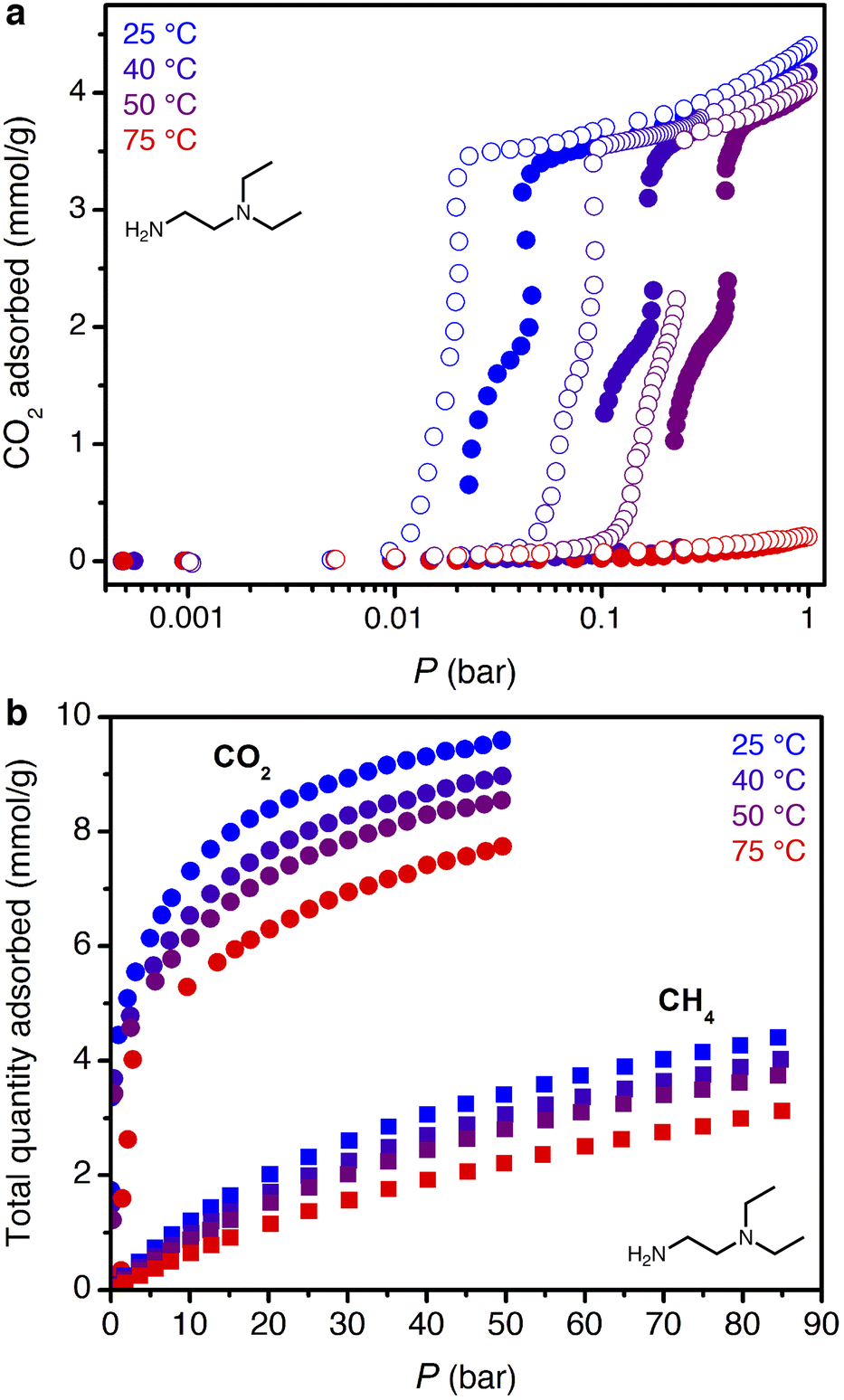 | ||
| Fig. 5 (a) Low-pressure CO2 adsorption (filled circles) and desorption (open circles) isotherms for ee-2–Mg2(dobpdc). (b) High-pressure CO2 (circles) and CH4 (squares) adsorption isotherms for ee-2–Mg2(dobpdc). | ||
The multicomponent performance of ee-2–Mg2(dobpdc) was first evaluated in dry breakthrough experiments. The powdered adsorbent was compressed and sieved to form binderless pellets, which were found to perform identically to the powder (see Fig. S32†). For a simulated wellhead gas stream containing 10% CO2 at 70 bar, 30 °C, and 300 sccm, a CH4 purity of 99.6% was achieved (Fig. 6a), easily satisfying the target pipeline specification of 98% (see Fig. S58† for a duplicate experiment). Exothermic CO2 adsorption produced a moderate ΔT of ∼8 °C (Fig. 6b). Notably, heat loss through the column walls in this experiment likely dampened the temperature rise, which is expected to be much greater (ΔT ∼ 40 °C) under adiabatic conditions in a full-scale system. A small amount of “roll-up” (normalized outlet flow rate, F/F0, >1) is visible in the CH4 breakthrough curve as strong binding of CO2 displaces adsorbed CH4. Co-adsorption of CH4 was higher than anticipated from the single-component adsorption isotherms, reaching 7.8 mmol g−1 and resulting in a mixture selectivity of 4.8 for CO2. However, we note that the CH4 capacity and associated CO2 selectivity calculations are susceptible to large errors due to the short breakthrough times for CH4. For example, a 5% error for a typical integrated CH4 breakthrough time of 160 s would result in an error of ±0.5 mmol g−1 in calculated CH4 capacity. In a subsequent breakthrough experiment at a lower total pressure of 35 bar, a product purity of 99.2% and CO2/CH4 selectivity of 5.8 were achieved (Fig. S59†). For varying total pressures with 10% CO2 at 30 °C, consistent CO2 slip of ∼0.3 bar was observed, signifying an effective step temperature of 52 °C, as calculated from the adsorption enthalpy and adsorption isotherms (see ESI Section 6, Table S10, and Fig. S60†). These results indicate that, like ii-2–Mg2(dobpdc), ee-2–Mg2(dobpdc) shows greater slip than anticipated from the equilibrium CO2 isotherms alone. However, breakthrough results suggest that ee-2–Mg2(dobpdc) possesses sufficiently favorable thermodynamics and kinetics of CO2 adsorption to produce pipeline quality methane from dry wellhead gas feeds.
 | ||
| Fig. 6 (a) Breakthrough profile for ee-2–Mg2(dobpdc) under 300 sccm of dry 10% CO2 in CH4 at 70 bar and 30 °C. (b) Corresponding temperature profile, as measured with two internal thermocouples. | ||
Performance under humid conditions
The promising CO2 capture performance of ee-2–Mg2(dobpdc) from dry, high-pressure feeds prompted our further investigation of this material for CO2 capture under humid conditions. Water isotherms (Fig. S29†) were first collected and indicated minimal adsorption below condensation at pressures of ∼15, 20, and 40 mbar at 25, 30, and 40 °C, respectively. A differential enthalpy of H2O adsorption of −49 ± 1 kJ mol−1 was calculated from the adsorption isotherms at a loading of 3.6 mmol g−1 (1 H2O per diamine, Fig. S30†); however, this value does not account for potential differences in the binding site and interaction energy of water with the framework after CO2 insertion.The mixture selectivity of ee-2–Mg2(dobpdc) was then evaluated through breakthrough experiments under humidified gas feeds. With simulated associated gas (7 bar, 10% CO2 in CH4, 30 °C, 300 sccm), a five-fold increase in the CO2 breakthrough volume and elimination of the CO2 slip were observed in the second cycle of a feed at 55% relative humidity (RH) as compared to a dry feed (Fig. 7a; activation performed at 30 °C with a vacuum pressure of 0.02 mbar between cycles). The relative humidity level of 55% corresponds to the inflection point in the single-component H2O isotherms of ee-2–Mg2(dobpdc) (Fig. S29†), suggesting that the material shows strong CO2 capture performance even at conditions approaching H2O saturation. Moving forward, additional experiments are needed to understand the effect of H2O on the breakthrough performance of ee-2–Mg2(dobpdc) as a function of humidity.
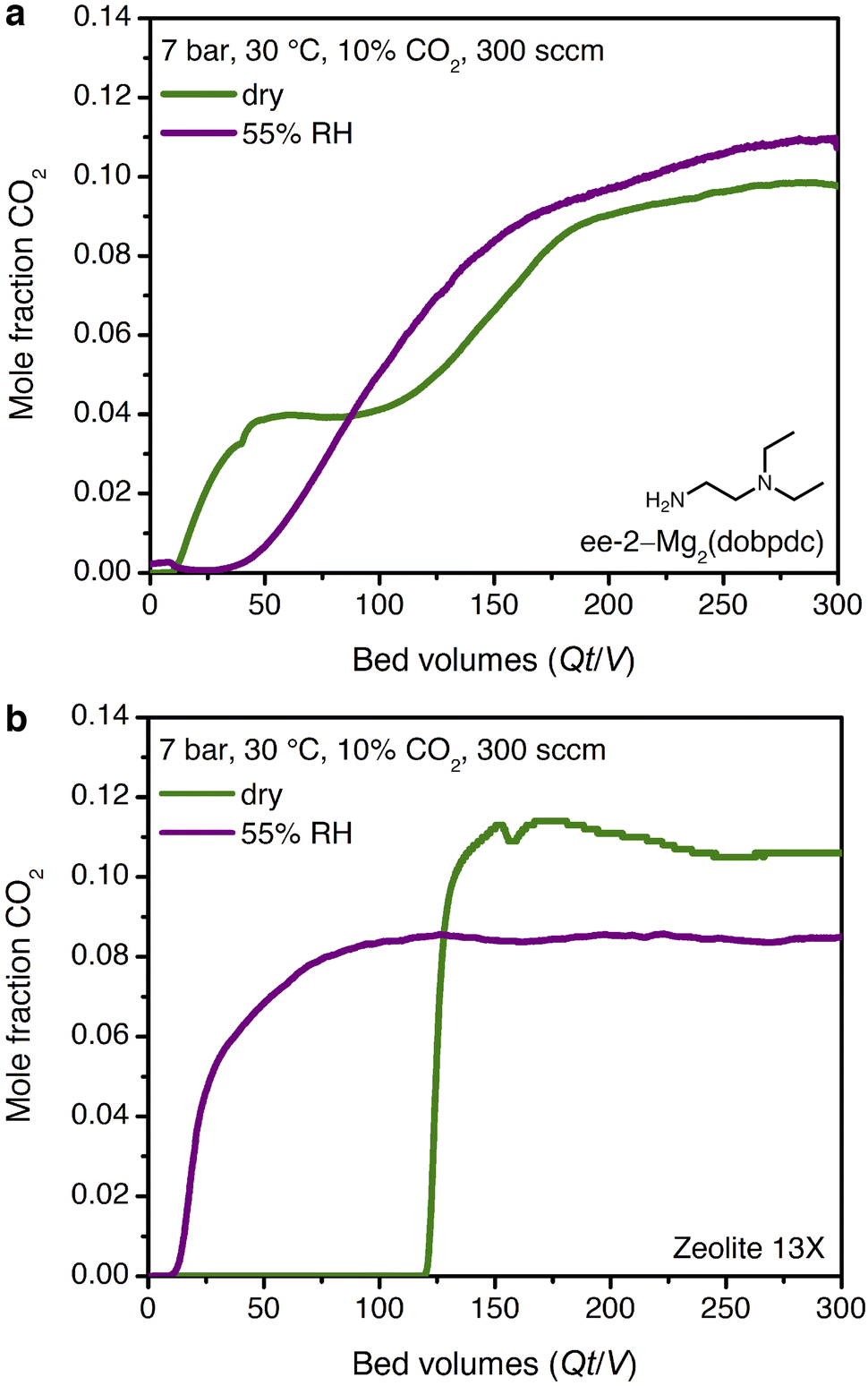 | ||
| Fig. 7 Dry (green) and humid (purple, 55% RH, second cycle) qualitative breakthrough curves for (a) ee-2–Mg2(dobpdc) and (b) zeolite 13X. Humidity eliminates CO2 slip for ee-2–Mg2(dobpdc) and generates a five-fold increase in breakthrough volume, while zeolite 13X shows an eight-fold decrease in breakthrough volume under humid conditions. Materials were evacuated at 30 °C with a vacuum pressure of 0.02 mbar for 12 h between cycles. | ||
Similarly improved performance was observed under humid conditions for a breakthrough experiment simulating non-associated gas. Following pre-saturation of the bed with water at 30 °C, the bed was evacuated at 30 °C and fed a dry mixture of 10% CO2 in CH4 at 50 bar, 30 °C, and 300 sccm. Negligible slip and an increased breakthrough time were observed compared to an equivalent experiment with a dry bed and dry feed (Fig. S61†). To corroborate these results, equilibrium CO2 isotherms were collected for the powdered and pelletized materials before and after saturation with water (P/P0 = ∼0.7) and after saturation and subsequent evacuation with a turbomolecular pump at 30 °C (Fig. S32†). The step-shaped CO2 isotherms of the water-exposed materials matched those of the pristine material, although a 5% decrease in post-step CO2 capacity was observed at 1 bar (Fig. S32†).
With confirmation in hand of the stability and improved performance of ee-2–Mg2(dobpdc) under humid conditions, a key next step in evaluating ee-2–Mg2(dobpdc) for practical application will be to develop an understanding of thermal gradients in the bed under realistic conditions. Unfortunately, the temperature rise in the bed could not be tracked directly for the humid breakthrough experiments in this work. Considering a previous study of a related diamine-appended framework, water was found to increase the binding energy of CO2 by stabilizing the ammonium carbamate chains formed upon CO2 adsorption.49 Consistent results were observed in this study for ee-2–Mg2(dobpdc) (see mechanistic section below), suggesting that humidity may exacerbate the thermal excursion observed under dry conditions (Fig. 6). Larger-scale experiments with internal temperature monitoring will be needed to understand the true magnitude of the reinforcing temperature wave coupled with the switch-like adsorption front. To advance toward commercialization, innovative contactor and/or process designs will be critical to manage this sorption heat economically and maintain the bed temperature beneath the cooperative CO2 adsorption threshold.
Despite these challenges, the performance of ee-2–Mg2(dobpdc) under humid conditions is compelling when compared to an industry benchmark adsorbent, zeolite 13X. In a second breakthrough cycle with simulated associated gas at 55% RH, zeolite 13X showed an eight-fold decrease in CO2 breakthrough volume due to passivation of the CO2 binding sites by water (Fig. 7b). As a result, if the H2O front were to advance through the bed in a PSA process, utilizing zeolite 13X as the adsorbent would require either costly pre-drying of the stream or periodic full regeneration at temperatures of 250–300 °C. Employing amine-functionalized adsorbents for direct removal of CO2 from humid streams may therefore provide significant energy savings in a PSA process, although post-purification drying of CH4 would still be required prior to pipeline transport. Several previous reports have noted equivalent or improved CO2 capture performance in the presence of water for amine-functionalized silicas and diamine-appended frameworks.21,23,33,50–55 However, to our knowledge, the dramatic humidity-induced improvement in capture rate in the breakthrough profile of ee-2–Mg2(dobpdc) has not been observed previously for other classes of amine-functionalized adsorbents. We therefore sought to determine the mechanism underlying the improved performance of ee-2–Mg2(dobpdc) under humid conditions.
Mechanism of CO2 adsorption and proposed role of water
X-ray diffraction, infrared spectroscopy, and thermogravimetric analysis were used to probe the structural nuances underlying differences in the observed CO2 adsorption behavior under dry and humid conditions. Previous characterization of ee-2–Mg2(dobpdc) by 1H, 13C, and 15N NMR spectroscopy, combined with DFT calculations, confirmed that ee-2–Mg2(dobpdc) forms ammonium carbamate chains upon CO2 adsorption;56 however, crystallographic characterization of the chain structure has not been reported to date. While diffraction-quality crystals could not be obtained for the magnesium framework, single-crystals of the corresponding ee-2–Zn2(dobpdc) framework were synthesized and could be analyzed by single-crystal X-ray diffraction. Here, we report the first CO2-inserted single-crystal structure for a 1°/3° diamine-appended framework (Fig. 8; see Fig. S69† for a thermal ellipsoid plot). Insertion of CO2 into the Zn–N bonds generates planar, O-bound carbamate species, with corresponding proton transfer to each unbound amine to yield ion-paired ammonium groups. | ||
| Fig. 8 Structure of ee-2–Zn2(dobpdc) before (top) and after (bottom) CO2 insertion to form ammonium carbamate chains, as observed by single-crystal X-ray diffraction. Light blue, blue, red, gray, and white spheres represent Zn, N, O, C, and H atoms, respectively. The structure of ee-2–Zn2(dobpdc) prior to CO2 adsorption was obtained from ref. 31. | ||
Attempts at crystallographic observation of water binding within the CO2-inserted structure are ongoing but have not yet proven successful. As an alternative, we probed the interaction of water with the framework through collection of in situ infrared spectra of ee-2–Mg2(dobpdc) under N2, dry CO2, and humid CO2 (Fig. S40†). Upon exposure to CO2, characteristic ammonium carbamate C–N and C–O vibrations are observed at 1320 and 1629 cm−1, respectively. When the CO2-inserted sample is exposed to humidity, the C–N vibration persists but shifts to slightly higher energy (∼1326 cm−1; note that the C–O vibration is obscured by the H–O–H bend at 1630 cm−1). These observations are consistent with preservation of the CO2 insertion mechanism in the presence of water and suggest hydrogen bonding of water with the carbamate, which would stabilize the CO2-inserted phase.
Multicomponent isobars were collected to study the effect of water on the CO2 insertion threshold. Due to safety considerations preventing the use of flammable gases in the TGA furnace, pre-mixed cylinders of CO2 in N2 were used to approximate the behavior of ee-2–Mg2(dobpdc) under CO2/CH4 mixtures. Dry isobaric adsorption and desorption profiles were compared to humid isobars obtained by bubbling the incident CO2/N2 mixtures through two room-temperature water bubblers prior to the furnace inlet (Fig. 9; see Fig. S41–S48† for other mixture compositions, as well as dry and humid N2 isobars). Under humid streams (∼1.3% H2O), the step temperature for each mixture increases relative to the step temperature in the corresponding dry isobar, equivalent to a lower CO2 step pressure in an adsorption isotherm. This shift is again consistent with preferential stabilization of the CO2-bound phase under humid conditions, which can likely be attributed to hydrogen bonding of water with the metal-bound carbamate species. Similar results have been reported previously for related diamine-appended frameworks.35,49 Importantly, step-like adsorption and desorption behavior is still observed in the humid isobars. Under humid CO2 at atmospheric pressure, the hysteresis loop closes by 75 °C, supporting a small temperature swing as one potential process configuration. Vacuum-swing or concentration-swing processes may also be viable for this material, and optimization of process design is currently in progress.
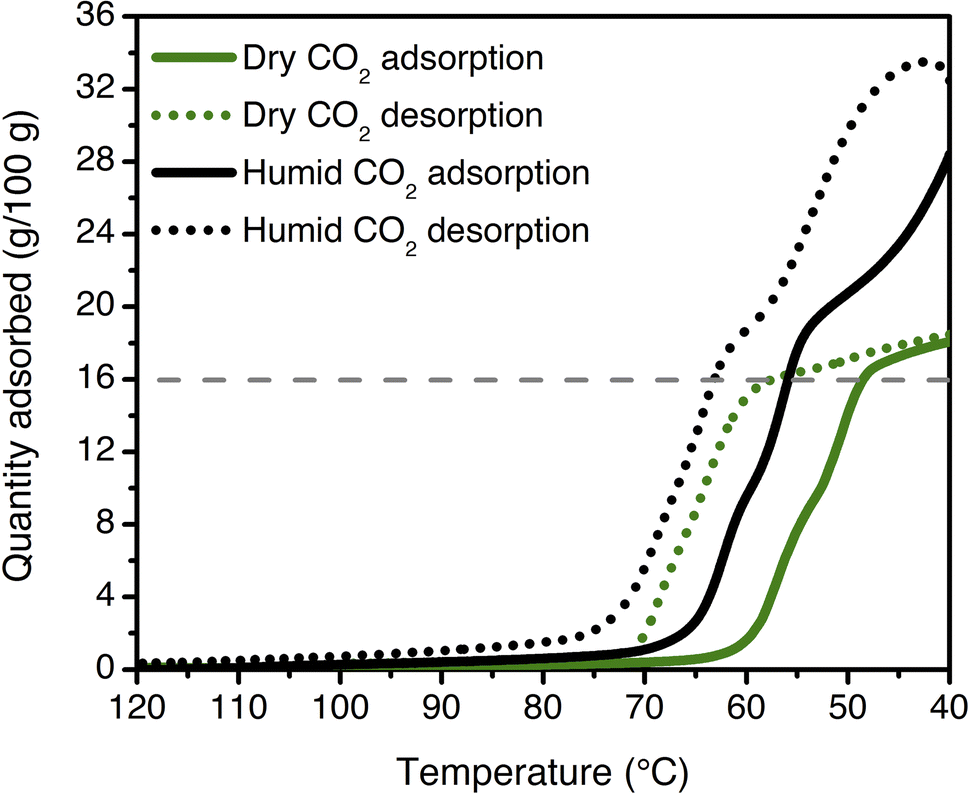 | ||
| Fig. 9 Dry (green) and humid (black) CO2 adsorption (solid lines) and desorption (dotted lines) isobars for ee-2–Mg2(dobpdc) at atmospheric pressure (ramp rate: 1 °C min−1). The cooperative adsorption threshold shifts to higher temperatures under humid conditions, indicating more favorable adsorption in the presence of H2O. The grey dashed line indicates the capacity anticipated for binding 1 CO2 per diamine (16.0 g/100 g). The isobars shown here are consistent with those measured for a previously synthesized and tested batch of ee-2–Mg2(dobpdc).49 | ||
In light of the humid infrared spectra and isobaric data, the elimination of CO2 slip in humid breakthrough experiments can be explained using Golden's rule, as introduced in Fig. 2. By reducing the effective step pressure for cooperative CO2 adsorption, water removes the intersection between the operating curve and the adsorption isotherm, resulting in a favorable single-shock breakthrough profile (Fig. 10). In ongoing work, we are investigating the correlation between relative humidity and capture rate in greater detail, as well as studying the influence of humidity on the kinetics of CO2 adsorption.
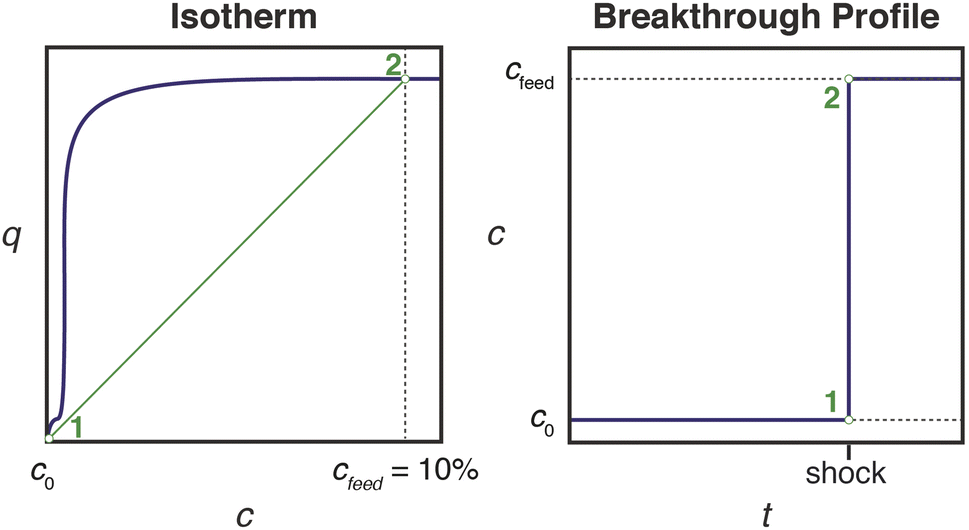 | ||
| Fig. 10 Schematic showing generation of a single-shock breakthrough profile (right, concentration vs. time) for an adsorbent with a step-shaped isotherm (left, quantity adsorbed vs. concentration). The operating curve used to predict the breakthrough profile from the isotherm is shown in green, and the numerical labels indicate corresponding points in the isotherm and breakthrough profile. When the operating curve does not intersect the isotherm, CO2 slip is not observed. | ||
Conclusions
As natural gas demand continues to grow, new strategies are needed to remove CO2 contamination from currently unusable reserves. Here, we have shown that the framework ee-2–Mg2(dobpdc) is a promising candidate for natural gas purification in a PSA process, the first-such demonstration with a diamine-appended metal–organic framework. Using dry breakthrough experiments, we have also reiterated the importance of considering CO2 “slip” for adsorbents with step-shaped adsorption isotherms.32,36,49 Humid breakthrough experiments subsequently revealed that water significantly improves the CO2 capture performance of ee-2–Mg2(dobpdc) by eliminating pre-breakthrough slip. In contrast, an equivalent experiment with the benchmark material zeolite 13X revealed a rapid loss of CO2 capacity following saturation with water, an observation consistent with previous results for adsorbents relying on exposed metal cations as binding sites. Through spectroscopic characterization and multicomponent isobars, we have traced the origin of improved performance under humid conditions to stabilizing H2O–carbamate interactions. Finally, we have shown that ee-2–Mg2(dobpdc) retains its cooperative adsorption properties following extended mechanical pressure cycling under both dry and humid simulated natural gas feeds. These results support further development of ee-2–Mg2(dobpdc) for this important separation.In future work, we aim to model the influence of both the relative humidity and temperature on the CO2 capture performance of ee-2–Mg2(dobpdc). Additionally, we will evaluate the potential for co-removal of H2S, which can be present in crude natural gas streams at concentrations as high as 16 mol% and must be reduced to a level of ∼5–15 ppm.58 A number of recent studies have explored the interaction of H2S with metal–organic frameworks,58–65 but the effect of H2S on diamine-appended Mg2(dobpdc) frameworks remains untested. Furthermore, shaped particles or other structured forms are needed to evaluate the material in a full PSA process and to determine the CH4 recovery and purity achievable in an optimized cycle configuration. Co-adsorption of heavier hydrocarbons and the influence of heat effects should also be evaluated at that stage. Finally, as these materials advance toward commercialization, innovative contactor and/or process design strategies will be critical to leverage the cooperative adsorption mechanism while managing the corresponding thermal front.
Data availability
Crystallographic data for CO2-inserted ee-2–Zn2(dobpdc) have been deposited as CCDC 1912757. Additional experimental details and data are provided in the ESI,† including isotherms, dynamic scanning calorimetry, thermogravimetric analysis, breakthrough data, infrared spectra, and powder and single-crystal X-ray diffraction details.Author contributions
R. L. S., J. A. T., T. M. M., and J. R. L. formulated the project. R. L. S. synthesized the materials and measured and analyzed the gas adsorption data, with contributions from T. M. M. and J. A. M. J. A. T. collected and analyzed the breakthrough and pressure cycling data. R. L. S. collected and analyzed the single-crystal X-ray diffraction data. R. L. S. and J. R. L. wrote the manuscript, and all authors contributed to revising the manuscript.Conflicts of interest
The authors declare the following competing financial interests: The University of California, Berkeley and Chevron Energy Technology Company have applied for a joint patent for the materials discussed herein, on which J. R. L, R. L. S., J. A. T., and T. M. M. are listed as inventors.Acknowledgements
We gratefully acknowledge Chevron Energy Technology Company for financial support of this work, and for providing partial graduate fellowship support of R. L. S. Structural and spectroscopic studies were supported through the Center for Gas Separations, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0001015. The latest stages of this research were supported by the U.S. Department of Energy Office of Basic Energy Sciences under award DE-SC0019992. Single-crystal X-ray diffraction data were collected on Beamline 12.2.1 at the Advanced Light Source at Lawrence Berkeley National Laboratory, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Synchrotron powder X-ray diffraction data were collected at the Advanced Photon Source, a U.S. Department of Energy Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank Levi Merrell (Chevron Energy Technology Company), Dr Eugene Kim (UC Berkeley), and Dr Kristen Colwell (UC Berkeley) for experimental assistance; Dr Stephen DeWitt (Georgia Institute of Technology) and Dr Miguel I. Gonzalez, Dr Jeffrey Martell, Dr Phillip Milner, Dr Jonathan Bachman, and Ever Velasquez (all of UC Berkeley) for helpful discussions; and Dr Katie Meihaus (UC Berkeley) for editorial assistance.Notes and references
- IEA, Key World Energy Statistics 2021, OECD Publishing, Paris, 2021, DOI:10.1787/2ef8cebc-en.
- BP Statistical Review of World Energy 2021, BP, 2021 Search PubMed.
- International Energy Outlook 2021, U.S. Energy Information Administration Search PubMed.
- Outlook for Energy, ExxonMobil, 2021 Search PubMed.
- W. R. Parrish and A. J. Kidnay, Fundamentals of Natural Gas Processing, CRC Press, 2006 Search PubMed.
- D. Leppin and H. S. Meyer, in SPE-21505-MS, Society of Petroleum Engineers, SPE, 1991 Search PubMed.
- W. F. J. Burgers, P. S. Northrop, H. S. Kheshgi and J. A. Valencia, Energy Procedia, 2011, 4, 2178–2184 CrossRef.
- T. E. Rufford, S. Smart, G. C. Y. Watson, B. F. Graham, J. Boxall, J. C. Diniz da Costa and E. F. May, J. Pet. Sci. Eng., 2012, 94, 123–154 CrossRef.
- B. Burr and L. Lyddon, A Comparison of Physical Solvents for Acid Gas Removal, Bryan Research & Engineering, Inc, 2008 Search PubMed.
- K. Maqsood, A. Mullick, A. Ali, K. Kargupta and S. Ganguly, Rev. Chem. Eng., 2014, 30, 453–477 CAS.
- Y. Alcheikhhamdon and M. Hoorfar, Chem. Eng. Process., 2017, 120, 105–113 CrossRef CAS.
- M. van Denderen, E. Ineke and M. Golombok, Ind. Eng. Chem. Res., 2009, 48, 5802–5807 CrossRef CAS.
- D. M. Ruthven, S. Farooq and K. S. Knaebel, Pressure Swing Adsorption, VCH Publishers, New York, N.Y., 1994 Search PubMed.
- M. Tagliabue, D. Farrusseng, S. Valencia, S. Aguado, U. Ravon, C. Rizzo, A. Corma and C. Mirodatos, Chem. Eng. J., 2009, 155, 553–566 CrossRef CAS.
- S. Sircar, Sep. Sci. Technol., 1988, 23, 519–529 CrossRef.
- Guild Associates, Inc., Molecular Gate® Carbon Dioxide Removal Systems,https://www.moleculargate.com/carbon-dioxide-removal-adsorption-system.html, accessed October 1, 2017.
- J. Toreja, N. Chan, B. VanNostrand and J. P. Dickinson, in Laurance Reid Gas Conditioning Conference, University of Oklahoma Norman, OK, 2011 Search PubMed.
- J. Liu, Y. Wang, A. I. Benin, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2010, 26, 14301–14307 CrossRef CAS PubMed.
- Y. Wang and M. D. LeVan, J. Chem. Eng. Data, 2010, 55, 3189–3195 CrossRef CAS.
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS.
- J. A. Mason, T. M. McDonald, T.-H. Bae, J. E. Bachman, K. Sumida, J. J. Dutton, S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2015, 137, 4787–4803 CrossRef CAS PubMed.
- Y. Belmabkhout, G. De Weireld and A. Sayari, Langmuir, 2009, 25, 13275–13278 CrossRef CAS PubMed.
- H. Y. Huang, R. T. Yang, D. Chinn and C. L. Munson, Ind. Eng. Chem. Res., 2003, 42, 2427–2433 CrossRef CAS.
- I. A. Ibarra and E. González-Zamora, Mater. Chem. Front., 2017, 1, 1471–1484 RSC.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed.
- R. W. Flaig, T. M. Osborn Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef CAS.
- C. S. Hong, M. Kang and D. W. Kang, Dalton Trans., 2019, 48, 2263–2270 RSC.
- T. M. McDonald, D. M. D'Alessandro, R. Krishna and J. R. Long, Chem. Sci., 2011, 2, 2022–2028 RSC.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- R. L. Siegelman, T. M. McDonald, M. I. Gonzalez, J. D. Martell, P. J. Milner, J. A. Mason, A. H. Berger, A. S. Bhown and J. R. Long, J. Am. Chem. Soc., 2017, 139, 10526–10538 CrossRef CAS PubMed.
- M. Hefti, L. Joss, Z. Bjelobrk and M. Mazzotti, Faraday Discuss., 2016, 192, 153–179 RSC.
- P. J. Milner, R. L. Siegelman, A. C. Forse, M. I. Gonzalez, T. Runčevski, J. D. Martell, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2017, 139, 13541–13553 CrossRef CAS PubMed.
- H. Jo, W. R. Lee, N. W. Kim, H. Jung, K. S. Lim, J. E. Kim, D. W. Kang, H. Lee, V. Hiremath, J. G. Seo, H. Jin, D. Moon, S. S. Han and C. S. Hong, ChemSusChem, 2017, 10, 541–550 CrossRef CAS PubMed.
- P. J. Milner, J. D. Martell, R. L. Siegelman, D. Gygi, S. C. Weston and J. R. Long, Chem. Sci., 2018, 9, 160–174 RSC.
- L. Darunte, T. Sen, C. Bhawanani, K. S. Walton, D. S. Sholl, M. J. Realff and C. W. Jones, Ind. Eng. Chem. Res., 2019, 58, 366–377 CrossRef CAS.
- D. S. Choi, D. W. Kim, D. W. Kang, M. Kang, Y. S. Chae and C. S. Hong, J. Mater. Chem. A, 2021, 9, 21424–21428 RSC.
- A. S. Lindsey and H. Jeskey, Chem. Rev., 1957, 57, 583–620 CrossRef CAS.
- K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552–8556 CrossRef CAS PubMed.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- E. W. Lemmon, M. L. Huber and M. O. McLinden, REFPROP: Reference fluid thermodynamic and transport properties, version 9, National Institute of Standards and Technology, 2010 Search PubMed.
- L. Gurvich, J. Phys. Chem. Soc. Russ., 1915, 47, 805 CAS.
- NIST WebBook, https://webbook.nist.gov/.
- F. M. Golden, Ph.D thesis, University of California, Berkeley, 1973.
- F. G. Helfferich and P. W. Carr, J. Chromatogr. A, 1993, 629, 97–122 CrossRef CAS.
- W. Zhang, Y. Shan and A. Seidel-Morgenstern, J. Chromatogr. A, 2006, 1107, 216–225 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS.
- M. A. Alkhabbaz, P. Bollini, G. S. Foo, C. Sievers and C. W. Jones, J. Am. Chem. Soc., 2014, 136, 13170–13173 CrossRef CAS PubMed.
- R. L. Siegelman, P. J. Milner, A. C. Forse, J.-H. Lee, K. A. Colwell, J. B. Neaton, J. A. Reimer, S. C. Weston and J. R. Long, J. Am. Chem. Soc., 2019, 141, 13171–13186 CrossRef CAS PubMed.
- X. Xu, C. Song, B. G. Miller and A. W. Scaroni, Ind. Eng. Chem. Res., 2005, 44, 8113–8119 CrossRef CAS.
- R. Serna-Guerrero, Y. Belmabkhout and A. Sayari, Adsorption, 2010, 16, 567–575 CrossRef CAS.
- P. Bollini, S. A. Didas and C. W. Jones, J. Mater. Chem., 2011, 21, 15100–15120 RSC.
- S. A. Didas, M. A. Sakwa-Novak, G. S. Foo, C. Sievers and C. W. Jones, J. Phys. Chem. Lett., 2014, 5, 4194–4200 CrossRef CAS PubMed.
- H. Zhang, A. Goeppert, G. A. Olah and G. K. S. Prakash, J. CO2 Util., 2017, 19, 91–99 CrossRef CAS.
- J. Yu, Y. Zhai and S. S. C. Chuang, Ind. Eng. Chem. Res., 2018, 57, 4052–4062 CrossRef CAS.
- A. C. Forse, P. J. Milner, J.-H. Lee, H. N. Redfearn, J. Oktawiec, R. L. Siegelman, J. D. Martell, B. Dinakar, L. B. Porter-Zasada, M. I. Gonzalez, J. B. Neaton, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2018, 140, 18016–18031 CrossRef CAS PubMed.
- C. W. Skarstrom, US Pat., US2944627A, 1960.
- M. S. Shah, M. Tsapatsis and J. I. Siepmann, Chem. Rev., 2017, 117, 9755–9803 CrossRef CAS PubMed.
- J. Ethiraj, F. Bonino, C. Lamberti and S. Bordiga, Microporous Mesoporous Mater., 2015, 207, 90–94 CrossRef CAS.
- J. Liu, Y. Wei, P. Li, Y. Zhao and R. Zou, J. Phys. Chem. C, 2017, 121, 13249–13255 CrossRef CAS.
- P. M. Bhatt, Y. Belmabkhout, A. H. Assen, Ł. J. Weseliński, H. Jiang, A. Cadiau, D.-X. Xue and M. Eddaoudi, Chem. Eng. J., 2017, 324, 392–396 CrossRef CAS.
- K. C. Kim, J. Organomet. Chem., 2018, 854, 94–105 CrossRef CAS.
- Y. Belmabkhout, P. M. Bhatt, K. Adil, R. S. Pillai, A. Cadiau, A. Shkurenko, G. Maurin, L. Gongping, W. J. Koros and M. Eddaoudi, Nat. Energy, 2018, 3, 1059–1066 CrossRef CAS.
- J. N. Joshi, G. Zhu, J. J. Lee, E. A. Carter, C. W. Jones, R. P. Lively and K. S. Walton, Langmuir, 2018, 34, 8443–8450 CrossRef CAS.
- Y. Belmabkhout, R. S. Pillai, D. Alezi, O. Shekhah, P. M. Bhatt, Z. Chen, K. Adil, S. Vaesen, G. D. Weireld, M. Pang, M. Suetin, A. J. Cairns, V. Solovyeva, A. Shkurenko, O. E. Tall, G. Maurin and M. Eddaoudi, J. Mater. Chem. A, 2017, 5, 3293–3303 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures, adsorption isotherms and fits, crystallographic information. CCDC 1912757. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03570g |
| ‡ Current address: Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA. |
| § Current address: Harvard University, Cambridge, MA 02138, USA. |
| ¶ In light of the double-stepped adsorption profiles observed here for ee-2–Mg2(dobpdc) and observed previously for Mg2(dobpdc) functionalized with bulky 1°/2° diamines,35 the single-stepped adsorption profile of ii-2–Mg2(dobpdc), which bears an even larger diisopropyl-substituted tertiary amine, is surprising. This result suggests that the 50% CO2-inserted structure in ii-2–Mg2(dobpdc) is energetically unfavorable, and further work is needed to elucidate the structure of the CO2-adsorbed phase for this material. |
| This journal is © The Royal Society of Chemistry 2022 |