
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insight into electrocatalytic glyoxal reduction on copper and its relation to CO2 reduction†
Andreas M.
Reichert‡
 a,
Oriol
Piqué‡
d,
Walter A.
Parada
a,
Ioannis
Katsounaros
*a and
Federico
Calle-Vallejo
*bcd
a,
Oriol
Piqué‡
d,
Walter A.
Parada
a,
Ioannis
Katsounaros
*a and
Federico
Calle-Vallejo
*bcd
aHelmholtz Institute Erlangen-Nürnberg for Renewable Energy (IEK-11), Forschungszentrum Jülich GmbH, Cauerstr. 1, 91058 Erlangen, Germany. E-mail: i.katsounaros@fz-juelich.de
bNano-Bio Spectroscopy Group and European Theoretical Spectroscopy Facility (ETSF), Department of Polymers and Advanced Materials: Physics, Chemistry and Technology, University of the Basque Country UPV/EHU, Avenida Tolosa 72, 20018 San Sebastián, Spain. E-mail: federico.calle@ehu.es
cIKERBASQUE, Basque Foundation for Science, Plaza de Euskadi 5, 48009 Bilbao, Spain
dDepartment of Materials Science and Chemical Physics & Institute of Theoretical and Computational Chemistry (IQTC), University of Barcelona, Martí i Franquès 1, 08028 Barcelona, Spain
First published on 6th September 2022
Abstract
Copper electrodes produce several industrially relevant chemicals and fuels during the electrochemical CO2 reduction reaction (CO2RR). Knowledge about the reaction pathways can help tune the reaction selectivity toward higher-value products. To probe the uncertain role of the C2 molecule glyoxal, we electrochemically reduced it on polycrystalline Cu and quantified its liquid-phase products, namely, ethanol, ethylene glycol, and acetaldehyde. The gas phase contained hydrogen and traces of ethylene. In contrast with previous hypothesis, a one-to-one comparison with CO2RR on Cu indicates that glyoxal is neither a major intermediate in the pathway toward ethylene nor in the pathway toward ethanol. In addition, great possibilities for the selective, low-temperature production of ethylene glycol are open, as computational modelling shows that ethylene glycol and ethanol are produced on different active sites. Thus, apart from the mechanistic insight into CO2RR, this study gives new directions to facilitate the electrification of chemical processes at refineries.
Introduction
The electrochemical CO2 reduction reaction (CO2RR) offers a route for the synthesis of fuels and chemicals (e.g. ethanol, carbon monoxide or ethylene) which are widely used in the current industrial infrastructure.1–4 If the electrolysis is powered by electrical energy from renewable sources (e.g. wind or solar), then useful commodity chemicals and synthetic, storable fuels can be generated in a carbon-neutral way that alternates from the traditional energy-intensive thermal catalysis processes. Besides the use of selective electrocatalysts toward high-value products, this route can become economically feasible with the continuous decrease in the price of renewable electricity and the progress in the design of appropriate electrode and/or electrolyzer configurations that will enable CO2 conversion in large scales.5–9Among the active metals for the CO2RR, gold, silver and zinc reduce CO2 to CO with high selectivity, while post-transition metals (tin, bismuth, lead) produce formate.10 Copper is so far the only known catalyst able to convert CO2 to C2+ products, e.g. hydrocarbons and alcohols.11–14 The fact that numerous (more than ten)14 compounds are simultaneously formed from the CO2RR on copper highlights the complexity of the reaction mechanism for the formation of the various intermediate and final C1, C2 and C3 products.13–16 In view of these characteristics, copper electrodes have been the focus of extensive research efforts,17 and numerous copper-based materials and morphology modifications have been explored to explain and take advantage of the unique electrocatalytic properties of copper, understand the reaction mechanism,18–20 and improve the selectivity toward C2+ products.21–24
The chemistry underlying the peculiar electrocatalytic properties of Cu apparently stems from a combination of intermediate adsorption energies of hydrogen and CO.25 Besides, the ability to form ethylene and ethanol is due to an anomalous stabilization of key C2 intermediates,26 and schemes have been proposed to understand the selectivity of Cu facets.19,27–29 Some experimentally hypothesized or computationally predicted intermediates have been electrochemically reduced13,28,30 in order to confirm or discard their participation in the CO2RR reaction pathways. In addition, several reaction intermediates have been experimentally detected.31–33 In particular, recent reports proposed adsorbed glyoxal as an intermediate in the pathway from CO2 to ethanol.34,35 Here we present a combined experimental/computational study of glyoxal reduction reaction (GRR) on Cu electrodes. We conclude that glyoxal is not a major CO2RR intermediate on copper, but its electroreduction offers a promising means of producing ethylene glycol at room temperature. Finding suitable electrochemical routes is desirable in the foreseen electrification of refineries,3,36 in particular in this case because (i) most ethylene glycol is currently produced in industry from either petroleum, natural gas or coal,37 (ii) the major gas-phase route involves ethylene oxide hydration, which is impractical under electrochemical conditions as ethylene oxide is selectively electroreduced to ethylene.13,28 Apart from that, the conventional route has known shortcomings, such as the insufficient selectivity of ethylene oxidation, the use of Ag catalysts, and the production of higher ethylene glycol homologues, which imposes the need for downstream distillation units.38
Results and discussion
Experimental results
We performed all GRR experiments by adding glyoxal in a deaerated phosphate buffer, instead of the typical bicarbonate solution, to prevent the electrolyte from acting as a source of CO2 (from the CO2/bicarbonate equilibrium) and hence ensure that glyoxal is the only species reduced to the detected products. The phosphate buffer was adjusted to pH 6.8, that is the same as in CO2RR experiments with the most often used 0.1 mol L−1 KHCO3 electrolyte, to facilitate the comparison of the results obtained for GRR with those for CO2RR.Fig. 1 shows the average current density during electrolysis at the respective cathode potential, as well as the partial current density assigned to each product, jproduct, using the determined formation rates and Faraday's law (see the ESI, Section S2†). Note that the electrolysis time was restricted to 40 min to avoid a substantial decrease in the concentration of glyoxal in solution as a result of its conversion, which would lead to changes in the formation rate during the GRR experiment.
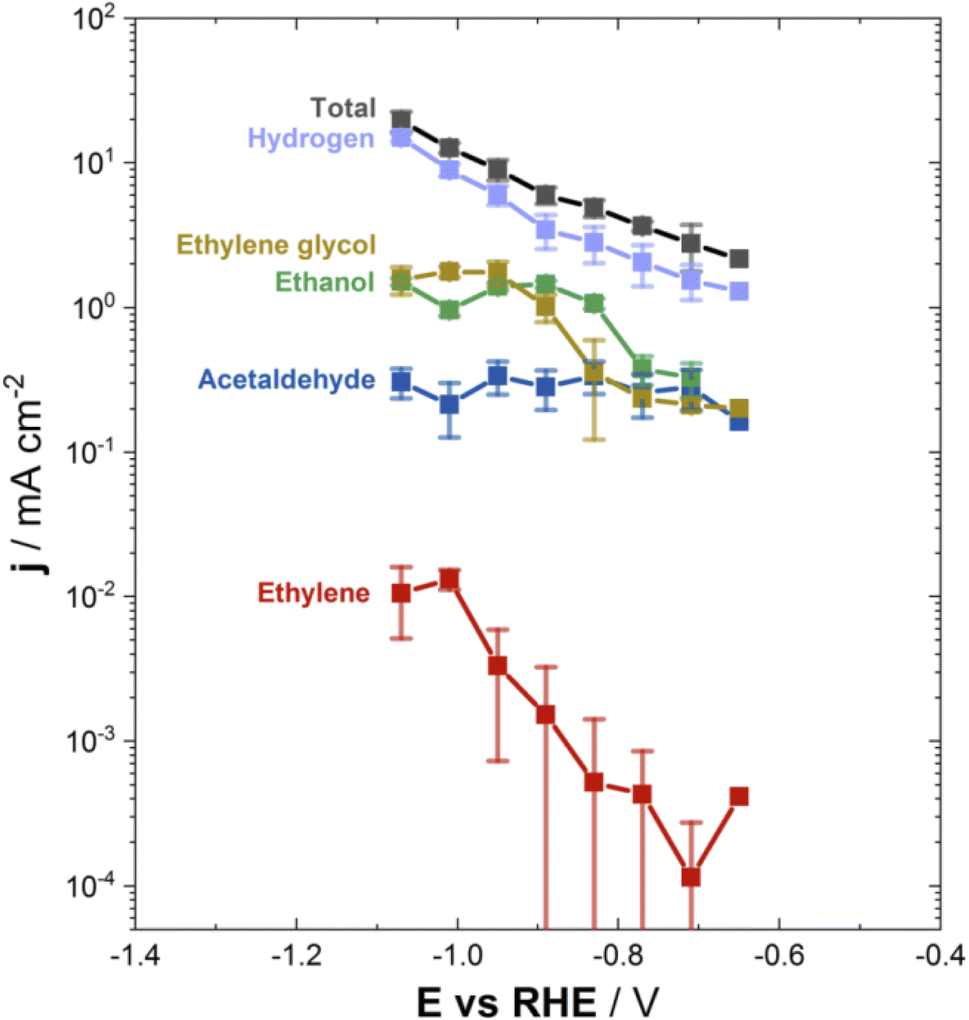 | ||
| Fig. 1 Total (recorded) current density, and partial current densities calculated for each product of glyoxal or water reduction, after their quantification with appropriate analytical methods. Working electrode: copper. Electrolyte: 0.02 mol L−1 glyoxal in a 0.1 mol L−1 phosphate buffer (adjusted to pH 6.8), saturated in Argon. Electrolysis time: 40 min. | ||
The main products of glyoxal reduction were acetaldehyde, ethanol and ethylene glycol, with (negative) current densities ranging roughly from 0.2 to 2.0 mA cm−2 for all products in the potential range investigated. On the contrary, jC2H4was lower by one to three orders of magnitude, compared to the other products. If the GRR and CO2RR14 product distributions are correlated in the same potential range and pH, then the formation of acetaldehyde and ethanol is a shared characteristic of both reactions. However, there are striking differences in the formation of ethylene and ethylene glycol. In particular, ethylene is a major product only for the CO2RR,14 whereas ethylene glycol forms in significant rates only from the GRR (Fig. 1), always when compared in the same potential and pH regions.
This conspicuous discrepancy in the formation of ethylene and ethylene glycol from the two reactions is an indication that glyoxal is not involved as a main intermediate in the pathway from CO2 to ethylene and ethanol during the copper-catalyzed CO2RR. Otherwise, both ethanol and ethylene would be formed in comparable rates from the GRR, and ethylene glycol would be formed in significant rates from the CO2RR. Certainly, as acetaldehyde and ethanol are produced from both CO2RR14 and GRR, one cannot completely exclude that part of the acetaldehyde and ethanol produced during the CO2RR originates from glyoxal as an intermediate. However, if it exists, such a glyoxal-mediated pathway will not be the main route for the formation of C2 products and will be responsible only for a small portion of the ethanol formed during the reduction of CO2.
Looking into the dependence of the potential on the current density for each product, jCH3CHO remained almost constant within the examined potential window, whereas jC2H5OH and j(CH2OH)2 reached a plateau for potentials more negative than ca. −0.9 VRHE, likely related to limitations in the transport of glyoxal, even though experiments at controlled mass-transport conditions would be required to confirm this hypothesis. The constant trend of jCH3CHO is attributed to the sequential reduction of acetaldehyde to ethanol,13,30,34,39 indicating the establishment of an equilibrium between the formation and consumption of acetaldehyde. In an independent experiment under the same conditions, with ethylene glycol in the initial solution instead of glyoxal, hydrogen was the only product while ethylene glycol was not reactive, in accordance with previous studies.13,40 Mechanistically, this indicates that ethylene glycol is not an intermediate of glyoxal reduction to ethanol and, thus, ethylene glycol is formed via an independent pathway compared to the acetaldehyde/ethanol pathway.
If the product distribution is expressed in terms of faradaic efficiencies (Fig. 2a, see also the ESI, Section S2†), the FEC2H5OH and FE(CH2OH)2 reached a maximum of 24% and 20%, respectively, at ca. −0.9 VRHE, while the FECH3CHO was decreasing with overpotential, from 10% (at ca. −0.7 VRHE) to 2% (at ca. −1.1 VRHE). Among the gaseous compounds, the FEC2H4 was negligible (always 0.1% or less) and the FEH2 from water reduction was 50% or higher at any potential investigated.
 | ||
| Fig. 2 (a) Faradaic efficiency and (b) carbon balance for the various electrolysis products. Glycolic acid is not shown in (a) because it is formed homogeneously from base-catalyzed glyoxal decomposition, without the transfer of electrons. Glyoxal included in (b) refers to the amount of glyoxal that did not react and was detected in the electrolyte. Working electrode: copper. Electrolyte: 0.02 mol L−1 glyoxal in a 0.1 mol L−1 phosphate buffer (adjusted to pH 6.8), saturated in argon. Electrolysis time: 40 min. | ||
When the concentration of glyoxal in the electrolyte was doubled (to 0.04 mol L−1) in experiments at −0.87 VRHE, the formation of ethylene glycol was clearly favored, whereas the rates for the formation of ethanol and acetaldehyde decreased. The partial current density for hydrogen was not affected, indicating that the concentration of glyoxal is not sufficiently high to inhibit the hydrogen evolution reaction. A comparison of the current densities for all products is given in the ESI (Fig. S2†), together with the associated faradaic efficiencies. We note that exploring higher concentrations of glyoxal in the starting solution is not possible because the buffer capacity is insufficient to maintain the local pH.
Considering the carbon balance (Fig. 2b, see also the ESI, Section S2†), most of the carbon remained in the form of glyoxal while conversion reached 50% only at ca. −1.0 VRHE. At this potential, carbon is found in the products of the electrochemical GRR in a descending order from ethylene glycol (yield ca. 12%), to ethanol (ca. 6%), to acetaldehyde (ca. 1.5%), to ethylene (<0.05%). Moreover, around 14% of carbon (at −1.0 VRHE) is found in glycolate, the conjugate base of glycolic acid. It is known that glycolate forms from glyoxal via a base-catalyzed homogeneous reaction.41 The detection of glycolate in our experiments (at solution pH 6.8) indicates that the pH at the interface increases under this combination of buffer solution, current density and mass-transport regime,42 thus favoring the interfacial base-catalyzed reaction. This enhancement is more pronounced at more negative potentials where the rate of OH− production (from either glyoxal or water reduction) is higher. The latter also explains why the amount of detected glycolate depends on the potential, even though glycolate is not involved per se in any electrochemical reaction. The relatively low total FE at low overpotentials probably arises from the decreased sensitivity to H2, which is the dominant product in this potential region.
Computational results
We inspected the structure-sensitive reduction of glyoxal to the two most abundant products found in the experimental investigations (Fig. 2), namely, ethylene glycol and ethanol. To do so, we considered five different surface models spanning a wide range of coordination numbers: Cu(111), the most stable surface termination of copper; Cu(100), known to be selective for CO2RR to ethylene and enhance C–C coupling due to its square geometry;15,26,43,44 Cu(211), a surface with step edges active toward acetaldehyde reduction;30 4AD@Cu(100), a square, four-atom island identified in a previous work as the ethanol-producing active site from CO2RR in OD-Cu;45 and 4AD@Cu(111), a four-atom island with hexagonal symmetry. A top view of all five surface models is shown in Fig. S3.†First, we investigated the GRR to ethylene glycol (standard equilibrium potential E0 = 0.36 V vs. RHE). Note that the DFT-calculated E0 matches the experimental one because gas-phase corrections were used,46 see ESI Section S4† for further details. Free-energy diagrams showing the energy variations along the proton-electron transfers for the most favorable pathway to ethylene glycol are shown in Fig. S6.† In addition, schematics of the adsorbed intermediates appear in Fig. 3, and ESI Section S7† shows the trends in glyoxal adsorption energies and its bidentate adsorption configuration on all Cu sites under study.
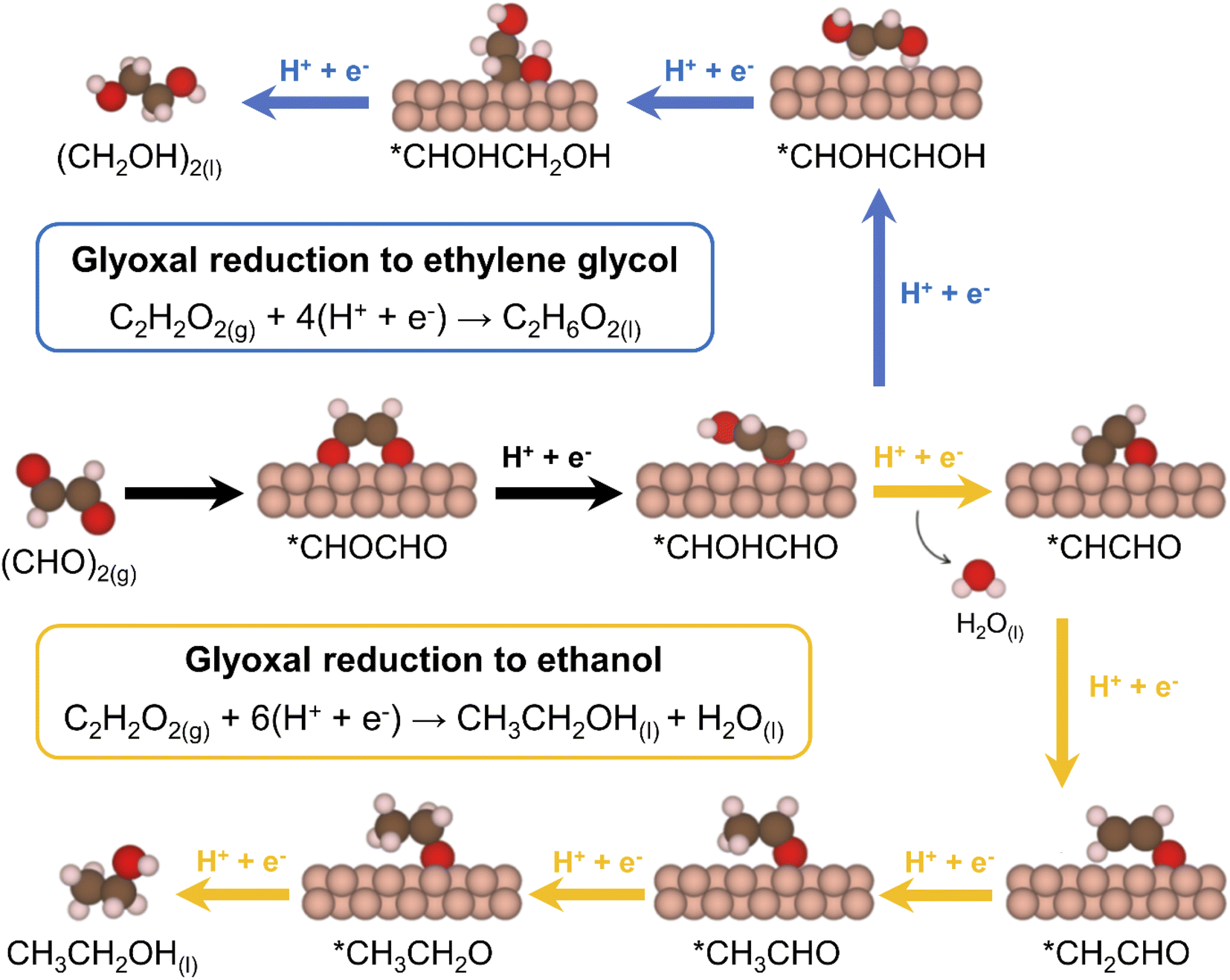 | ||
| Fig. 3 Most favorable electrocatalytic pathway for the GRR to ethylene glycol (blue arrows) and ethanol (orange arrows) on Cu electrodes. The black arrows connect shared reactants and intermediates. | ||
In Fig. 4 (in blue) we provide a coordination–activity plot for the GRR to ethylene glycol calculated on the basis of the energies and generalized coordination numbers59 ( ) in ESI Section S6.† Except for Cu(111), the potential-limiting step (PLS) corresponds to the hydrogenation of *CHOHCHO to form *CHOHCHOH (PLS1 in Fig. 4) on the strong-binding side of the plot, where
) in ESI Section S6.† Except for Cu(111), the potential-limiting step (PLS) corresponds to the hydrogenation of *CHOHCHO to form *CHOHCHOH (PLS1 in Fig. 4) on the strong-binding side of the plot, where  . For Cu(111), the PLS is the subsequent hydrogenation of CHOHCHOH to CHOHCH2OH (PLS2 in Fig. 4). The top of the coordination–activity plot is found at
. For Cu(111), the PLS is the subsequent hydrogenation of CHOHCHOH to CHOHCH2OH (PLS2 in Fig. 4). The top of the coordination–activity plot is found at  , which is close to the coordination of Cu(100) and not far from Cu(111) (
, which is close to the coordination of Cu(100) and not far from Cu(111) ( and 7.50, respectively). These results suggest that terrace sites, in particular the square ones, are more active than undercoordinated defects for GRR to ethylene glycol. However, the competition with *H for active sites might hinder the activity of Cu(111).
and 7.50, respectively). These results suggest that terrace sites, in particular the square ones, are more active than undercoordinated defects for GRR to ethylene glycol. However, the competition with *H for active sites might hinder the activity of Cu(111).
 | ||
Fig. 4 Trends in the activity and selectivity of glyoxal reduction to ethylene glycol (blue) and ethanol (orange). EL is the DFT-calculated limiting potential and  is the generalized coordination number of the active sites (see ESI Section S6†). The dashed lines indicate the equilibrium potentials. PLS: potential-limiting step; the chemical equations corresponding to PLS1 to PLS4 are given in ESI Section S7.† The apices of the blue and orange curves are located at is the generalized coordination number of the active sites (see ESI Section S6†). The dashed lines indicate the equilibrium potentials. PLS: potential-limiting step; the chemical equations corresponding to PLS1 to PLS4 are given in ESI Section S7.† The apices of the blue and orange curves are located at  . Inset: selectivity map for glyoxal reduction based on the trends in the main panel. The orange/blue regions are selective toward ethanol/ethylene glycol. The change of selectivity takes place at . Inset: selectivity map for glyoxal reduction based on the trends in the main panel. The orange/blue regions are selective toward ethanol/ethylene glycol. The change of selectivity takes place at  . The red line is the calculated average onset potential for hydrogen evolution on various copper facets taken from ref. 28. . The red line is the calculated average onset potential for hydrogen evolution on various copper facets taken from ref. 28. | ||
Moreover, we combined our results with those of previous works30,45 to build free–energy diagrams with the energetics of the GRR to ethanol (E0 = 0.38 V vs. RHE), as shown in Fig. S7,† see schematics of the intermediates in Fig. 3. Again, the DFT-calculated and experimental E0 coincide as a result of gas-phase corrections.46 Moreover, in Fig. 4 (in orange) we provide a coordination–activity plot for the GRR to ethanol. For 4AD@Cu(100) and 4AD@Cu(111), the PLS of the reaction is the hydrogenation of *CH2CHO to form acetaldehyde (PLS3 in Fig. 4). On Cu(211), Cu(100) and Cu(111) the PLS is the formation of *CH3CH2O from acetaldehyde (PLS4 in Fig. 4). The top of the coordination–activity plot is found at  , which is close to the coordination of 4AD@Cu(100) (
, which is close to the coordination of 4AD@Cu(100) (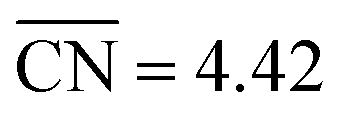 ). These results suggest that square islands of Cu adatoms at Cu(100) are the most active sites for GRR to ethanol, which is in line with their high activity and selectivity for CO reduction to ethanol.45
). These results suggest that square islands of Cu adatoms at Cu(100) are the most active sites for GRR to ethanol, which is in line with their high activity and selectivity for CO reduction to ethanol.45
Discussion
The inset of Fig. 4 is a structure-sensitive selectivity map for GRR to ethylene glycol and ethanol on copper. Step edges at Cu(211) together with (100) and (111) terraces are inclined toward the production of ethylene glycol during GRR, while adatom islands are more selective for ethanol production. In the entire coordination range, there is a competition between GRR and hydrogen evolution (red line in the inset of Fig. 4). At a potential (EL) of −0.7 V vs. RHE, Fig. 4 shows that the two catalytic pathways are open on most active sites, except for Cu(111), such that a polycrystalline electrode will concurrently form ethylene glycol and ethanol together with H2, which agrees with the experimental results in Fig. 1 and 2. Increasing the proportion of (100) terraces in the catalysts seems to be a suitable way of enhancing the production of ethylene glycol, whereas rough electrodes will tend to produce more ethanol by virtue of their undercoordinated defects.Furthermore, our electrochemical measurements show that (i) a negligible amount of ethylene is produced from GRR, and (ii) ethylene glycol is produced from GRR with high faradaic efficiencies. Previous works have indicated that copper catalysts may have product-specific active sites.44,47 Specifically, it has been repeatedly reported that CO2RR on Cu produces ethanol on undercoordinated sites and ethylene on (100) terrace sites.15,28,30,34 Besides, only traces of ethylene glycol are formed during CO2RR,14 and ethylene glycol seems to be a dead-end product.13 Altogether, these facts lead us to conclude that glyoxal cannot be a key intermediate of CO2RR to the C2 products ethanol and ethylene. According to our computational modelling, the ethanol/ethylene glycol pathway bifurcation occurs on the second proton-electron transfer, such that ethylene glycol is not an intermediate in the ethanol pathway, also in agreement with experiments.
A direct mechanistic implication of glyoxal not being a key CO2RR intermediate is that the coupling of two *CHO moieties can be ruled out as a major source of C2+ products on Cu, in spite of its low associated kinetic barriers.35 Furthermore, if *COCHO is the first hydrogenated intermediate in the C2 pathway, as proposed in previous studies,48 the next hydrogenation may not take place on the C atom. More generally, if C2+ products are formed via CO dimerization, as proposed by Hori and coworkers,49 since the first two hydrogenation steps may not both take place on the C atoms, at least one of the O atoms is to be hydrogenated. This condition is fulfilled by the spectroscopically detected *COCOH species.32
Finally, we note that the surface coverage of C2 species could be different during glyoxal reduction compared to CO2 reduction. We anticipate that the C2 coverage is likely smaller in the latter case because the making of C–C bonds is known to be energy-intensive2,15,17,35 and also because glyoxal is chemisorbed on Cu surfaces (see ESI Section S7†). However, the large faradaic efficiencies of H2 in Fig. 1 and 2 suggest that the local environment is similar for the two reduction reactions, in the sense that a given C2-producing site is probably surrounded by several H2-producing sites in both cases.
Conclusions
The intricate mechanism of CO2 electroreduction is yet to be fully elucidated and the most active sites found. Recent works proposed adsorbed glyoxal as an early intermediate in the reduction pathway toward ethanol on Cu electrodes. Here, we put that hypothesis to the test through a combination of experimental and computational tools.We experimentally identified ethanol, acetaldehyde, and ethylene glycol as the major products of glyoxal reduction on Cu. Even though the formation of ethanol and acetaldehyde is characteristic of both glyoxal reduction and CO2 reduction, there are two conspicuous differences: first, ethylene glycol is a minor product during CO2RR on Cu, but forms with a high rate from glyoxal reduction and cannot be further reduced. Therefore, if glyoxal were formed from CO2RR in significant quantities, ethylene glycol would be a major product, which is not the case. Second, glyoxal reduction does not lead to abundant ethylene, which is in contrast a major product of CO2RR on copper-based catalysts. Hence, we conclude that glyoxal is not likely a major intermediate in the electrocatalytic pathway from CO2 to ethanol and ethylene and that the C–C coupling during this reaction does not happen via the coupling of two *CHO moieties.
By means of structure-sensitive modelling we determined that polycrystalline Cu produces a mix of products because its undercoordinated sites favor ethanol production while the more coordinated sites favor ethylene glycol production. Ethylene glycol is conventionally produced via ethylene oxide hydration in gas-phase heterogeneous catalysis. Here we showed a promising room-temperature electrochemical route for its production via glyoxal reduction on copper electrodes with abundant (100) terraces. Alternatively, ethanol can also be produced when the catalysts contain plentiful undercoordinated defects.
Methods
Electrode preparation
Prior to each measurement, a polycrystalline copper foil (2 mm, 99.99+%, GoodFellow) was grinded and polished with a semi-automatic polishing machine (LaboForce-100, Struers) and afterwards electropolished in concentrated H3PO4 (min. 85% ACS, Honeywell Fluka) for 2 min. The metal foil was finally rinsed thoroughly with ultrapure water and used for glyoxal electrolysis.Electrolysis experiments
Steady-state electrolysis was performed in 0.1 mol L−1 phosphate buffer (Na2HPO4/NaH2PO4 concentrations adjusted to pH 6.8) with 0.02 mol L−1 glyoxal, which was continuously purged with 10 sccm Ar. All electrolytes were prepared in ultrapure water (18.2 MΩ cm, total organic level below 4 parts per billion; Milli-Q IQ 7000, Merck) and the gas flow was controlled by a mass flow controller (EL-Flow prestige, Bronkhorst). Electrochemical measurements were conducted in a three-electrode configuration in an in-house-developed, two-compartment PTFE electrolysis cell (ESI, Fig. S1†). The electrolytes in the two half-cells were separated by a bipolar membrane (Fumasep), so glyoxal was present only in the catholyte, whereas the anolyte consisted entirely of the phosphate buffer, free from glyoxal. The electrochemical measurements were controlled by a potentiostat (Reference 600, Gamry). A Ag/AgCl (3 mol L−1 KCl) reference electrode (BASi), an Ir-MMO counter electrode (Metakem) and the respective copper foil as working electrode with an exposed area of 1 cm2 were used as the three electrodes. Only one side of the electropolished copper foil was in contact with the electrolyte. The electrolysis was performed for 40 min and repeated at least two times to assure reproducibility. The measurements were conducted at ambient conditions. To compensate for ohmic losses, the electrolyte resistance was determined with electrochemical impedance spectroscopy prior to the experiments, and the electrode potential was corrected for 85% of the iR drop during the electrolysis using the positive feedback mode of the potentiostat. The remaining 15% of the iR drop was post-corrected after the electrolysis, taking into account the uncompensated resistance and the average current measured during the electrolysis, as described in a previous work.14 The potentials recorded versus the Ag/AgCl reference were converted to the reversible hydrogen electrode (RHE) scale by taking into account the pH of the electrolyte and the potential of the Ag/AgCl electrode versus the standard hydrogen electrode. Hence, all potentials are given here in the RHE scale. The current densities reported in the manuscript were calculated by dividing the recorded current to the geometric area of the copper foil exposed to the electrolyte (1 cm2).Product characterization
The gas outlet of the working electrode compartment was connected to an online gas chromatograph (GC) (Clarus 580 – Arnel Engineered, PerkinElmer) to analyze the gas-phase products that were withdrawn from the cell by the continuous Ar flow. The separation was performed with a Hayesep N (NR021501, PerkinElmer) and a Molecular Sieve 13X (NR022501, PerkinElmer) columns, with Ar as carrier gas (flow rate: 30 mL min−1). The detection was done with a serially connected thermal conductivity detector and a flame ionization detector equipped with a methanizer. Injections were done at 2, 20, 38 min of the electrolysis, and the current recorded during the last 100 s prior to an injection was averaged and used for the calculation of the faradaic efficiency of the product (FEproduct). To analyze dissolved products offline, the catholyte was collected directly after the end of electrolysis. Volatile products were detected with gas chromatography-mass spectrometry (GC-MS) (Clarus 580 coupled to Clarus SQ8T, both PerkinElmer). A thermostated headspace autosampler (PerkinElmer TurboMatrix 40) was used to inject an aliquot from the vapor of the heated sample after the establishment of the gas–liquid equilibrium, to prevent the injection of the salt-containing electrolyte into the gas chromatograph. A fused-silica capillary column (Elite-624Sil MS, PerkinElmer) separated the gas mixture. For the mass spectrometric analysis, electron ionization (70 eV) and a quadrupole mass analyzer were used. The electrolyte was additionally analyzed with high performance liquid chromatography (HPLC) (1260 Infinity II, Agilent), using a HI-Plex H column (8 μm, 7.7 × 300 mm, Agilent) for the separation. A sample aliquot was automatically injected and ultrapure water was used as eluent with a flow rate of 0.7 mL min−1. The column oven temperature was kept at 60 °C while the refractive index detector had a working temperature of 55 °C. Moreover, for the detection of glycolate, an in-house developed combination of HPLC with mass spectrometry (MS) was employed, using direct analysis in real time (DART) ionization and a custom-made enclosed ionization interface (V 2.1S + AZURA CT 2.1, Knauer, coupled to JMS-T100LP AccuTOF LC-plus 4 G + DART, JEOL). As for simple HPLC analysis, the HiPlex-H column (Agilent) was again used for HPLC-DART-MS. Details on the coupling of HPLC with DART-MS can be found in a previous publication.50Computational methods
The DFT calculations were carried out using the Vienna ab initio simulation package (VASP)51 with the Perdew–Burke–Ernzerhof (PBE)52 exchange-correlation functional and the projector augmented-wave (PAW)53 method. In order to capture the heterogeneity of a real electrocatalyst, we modelled five different Cu surface models: Cu(100), Cu(111), Cu(211), 4AD@Cu(100) (a four-atom island on top of a Cu(100) slab), and 4AD@Cu(111) (a four-atom island sitting on top of a Cu(111) slab). All surfaces (except Cu(211)) were modelled with 4 × 4 supercell slabs containing 4 atomic layers and Monkhorst–Pack grids54 were used to sample the k-space. The k-point grids guaranteed convergence of the adsorption energies within ±0.05 eV. The converged PBE lattice constant of bulk Cu (3.64 Å) was used to build the surface models. The adsorbates, the metal adatoms, and the top two layers of the slabs were allowed to relax in every direction, while the two bottommost layers were fixed at the converged bulk equilibrium distances. A plane-wave cutoff of 450 eV was used. The Methfessel-Paxton approach55 was used to smear the Fermi level with an electronic temperature of 0.2 eV and the total energies were extrapolated to 0 K. The periodically repeated images were separated by more than 12 Å of vacuum in the vertical direction and dipole corrections were also applied. The conjugate-gradient algorithm was used to optimize the geometries, carrying out iterations until the maximal force on all atoms was below 0.05 eV Å−1. Asymmetric boxes of 9 × 10 × 11 Å3 were used to calculate the isolated molecules (H2, H2O, O2, glyoxal, acetaldehyde, ethanol, and ethylene glycol), sampling the Γ-point only, using Gaussian smearing and an electronic temperature of 0.001 eV. The total energies of the molecules were semi-empirically corrected to ensure that the calculated equilibrium potentials matched the experimental ones,46 see Table S1.† The reaction free energies were calculated as ΔG ≈ ΔEDFT + ΔZPE − TΔS + ΔEsolvation, where ΔEDFT is the DFT-calculated reaction energy, ΔZPE is the change in the zero-point energy, TΔS is the corresponding entropy change at 298.15 K, and ΔEsolvation contains the solvent-adsorbate stabilization corrections calculated on Cu(111) using the method described in ref. 56. The values of S for the free molecules were obtained from thermodynamic tables, while for adsorbates S only includes the vibrational contributions. Making use of the harmonic-oscillator approximation, vibrational-frequency analyses were performed to obtain the ZPE and Svib values. The computational hydrogen electrode (CHE) was used to model proton-electron pairs.57 We did not calculate kinetic barriers in this study, because Rossmeisl et al. recently noted that there is “not (yet) a method to obtain electrochemical barriers between realistic states at constant electrochemical conditions”.58 The assessment of generalized coordination numbers59 is illustrated in ESI Section S6.† Further computational details appear in the ESI, Sections S4–S8.†Data availability
Electrochemical cell, calculations of partial current density, faradaic efficiency and yield (carbon balance), gas and liquid phase corrections, contributions to the free energies of solvent-adsorbate interactions, free energies and active sites, intermediates of the most favourable pathways and optimized geometries are provided in the ESI.†Author contributions
F. C.-V. and I. K. conceived the idea behind this work and designed the calculations and experiments. A. M. R. and W. A. P. performed the electrochemical measurements and the GC/MS analysis. W. A. P. performed HPLC and HPLC-DART-MS analysis. O. P. carried out the DFT calculations. All authors carried out the data analysis. A. M. R., O. P. and F. C.-V. wrote the first draft of the manuscript based on input from all authors. I. K. and F. C.-V. supervised the research work, edited and finalized the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Peyman Khanipour and Dmitry V. Vasilyev for useful discussions. The grants RYC-2015-18996, MDM-2017-0767 and RTI2018-095460-B-I00 were funded by MCIN/AEI/10.13039/501100011033 and by the European Union. This work was also partly supported by Generalitat de Catalunya via the grant 2017SGR13.References
- D. T. Whipple and P. J. A. Kenis, Prospects of CO2 Utilization via Direct Heterogeneous Electrochemical Reduction, J. Phys. Chem. Lett., 2010, 1(24), 3451–3458, DOI:10.1021/jz1012627.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Advances and Challenges in Understanding the Electrocatalytic Conversion of Carbon Dioxide to Fuels, Nat. Energy, 2019, 4(9), 732–745, DOI:10.1038/s41560-019-0450-y.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What Would It Take for Renewably Powered Electrosynthesis to Displace Petrochemical Processes?, Science, 2019, 364(6438), eaav3506, DOI:10.1126/science.aav3506.
- R. G. Grim, Z. Huang, M. T. Guarnieri, J. R. Ferrell, L. Tao and J. A. Schaidle, Transforming the Carbon Economy: Challenges and Opportunities in the Convergence of Low-Cost Electricity and Reductive CO2 Utilization, Energy Environ. Sci., 2020, 13(2), 472–494, 10.1039/c9ee02410g.
- C. T. Dinh, T. Burdyny, G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. Pelayo García De Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, CO2 Electroreduction to Ethylene via Hydroxide-Mediated Copper Catalysis at an Abrupt Interface, Science, 2018, 360(6390), 783–787, DOI:10.1126/science.aas9100.
- J. M. Spurgeon and B. Kumar, A Comparative Technoeconomic Analysis of Pathways for Commercial Electrochemical CO2 Reduction to Liquid Products, Energy Environ. Sci., 2018, 11(6), 1536–1551, 10.1039/c8ee00097b.
- A. Ozden, Y. Wang, F. Li, M. Luo, J. Sisler, A. Thevenon, A. Rosas-Hernández, T. Burdyny, Y. Lum, H. Yadegari, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, Cascade CO2 Electroreduction Enables Efficient Carbonate-Free Production of Ethylene, Joule, 2021, 5(3), 706–719, DOI:10.1016/j.joule.2021.01.007.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Technical Photosynthesis Involving CO2 Electrolysis and Fermentation, Nat. Catal., 2018, 1(1), 32–39, DOI:10.1038/s41929-017-0005-1.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Towards Membrane-Electrode Assembly Systems for CO 2 Reduction: A Modeling Study, Energy Environ. Sci., 2019, 12(6), 1950–1968, 10.1039/C9EE00909D.
- Y. Hori, Electrochemical CO2 Reduction on Metal Electrodes, Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer, 2008, vol. 42, pp. 89–189, DOI: DOI:10.1007/978-0-387-49489-0_3.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Production of Methane and Ethylene in Electrochemical Reduction of Carbon Dioxide At Copper Electrode in Aqueous Hydrogencarbonate Solution, Chem. Lett., 1986, 15(6), 897–898, DOI:10.1246/cl.1986.897.
- L. Wang, S. Nitopi, A. B. Wong, J. L. Snider, A. C. Nielander, C. G. Morales-Guio, M. Orazov, D. C. Higgins, C. Hahn and T. F. Jaramillo, Electrochemically Converting Carbon Monoxide to Liquid Fuels by Directing Selectivity with Electrode Surface Area, Nat. Catal., 2019, 2(8), 702–708, DOI:10.1038/s41929-019-0301-z.
- K. J. P. Schouten, Y. Kwon, C. J. M. Van Der Ham, Z. Qin and M. T. M. Koper, A New Mechanism for the Selectivity to C1 and C2 Species in the Electrochemical Reduction of Carbon Dioxide on Copper Electrodes, Chem. Sci., 2011, 2(10), 1902–1909, 10.1039/c1sc00277e.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces, Energy Environ. Sci., 2012, 5(5), 7050–7059, 10.1039/c2ee21234j.
- F. Calle-Vallejo and M. T. M. Koper, Theoretical Considerations on the Electroreduction of CO to C2 Species on Cu(100) Electrodes, Angew. Chem., Int. Ed., 2013, 52(28), 7282–7285, DOI:10.1002/anie.201301470.
- A. J. Garza, A. T. Bell and M. Head-Gordon, Mechanism of CO2 Reduction at Copper Surfaces: Pathways to C2 Products, ACS Catal., 2018, 8(2), 1490–1499, DOI:10.1021/acscatal.7b03477.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119(12), 7610–7672, DOI:10.1021/acs.chemrev.8b00705.
- X. Chang, A. Malkani, X. Yang and B. Xu, Mechanistic Insights into Electroreductive C-C Coupling between CO and Acetaldehyde into Multicarbon Products, J. Am. Chem. Soc., 2020, 142(6), 2975–2983, DOI:10.1021/jacs.9b11817.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, Facet Dependence of CO2 Reduction Paths on Cu Electrodes, ACS Catal., 2016, 6(1), 219–229, DOI:10.1021/acscatal.5b01967.
- A. Rendón-Calle, Q. H. Low, S. H. L. Hong, S. Builes, B. S. Yeo and F. Calle-Vallejo, How Symmetry Factors Cause Potential- and Facet-Dependent Pathway Shifts during CO2 Reduction to CH4 on Cu Electrodes, Appl. Catal. B, 2021, 285, DOI:10.1016/j.apcatb.2020.119776.
- P. Iyengar, M. J. Kolb, J. R. Pankhurst, F. Calle-Vallejo and R. Buonsanti, Elucidating the Facet-Dependent Selectivity for CO 2 Electroreduction to Ethanol of Cu–Ag Tandem Catalysts, ACS Catal., 2021, 11(8), 4456–4463, DOI:10.1021/acscatal.1c00420.
- L. Wang, D. C. Higgins, Y. Ji, C. G. Morales-Guio, K. Chan, C. Hahn and T. F. Jaramillo, Selective Reduction of CO to Acetaldehyde with CuAg Electrocatalysts, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(23), 12572–12575, DOI:10.1073/pnas.1821683117.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, Probing the Active Surface Sites for CO Reduction on Oxide-Derived Copper Electrocatalysts, J. Am. Chem. Soc., 2015, 137(31), 9808–9811, DOI:10.1021/jacs.5b06227.
- L. R. L. Ting, R. García-Muelas, A. J. Martín, F. L. P. Veenstra, S. T. J. Chen, Y. Peng, E. Y. X. Per, S. Pablo-García, N. López, J. Pérez-Ramírez and B. S. Yeo, Electrochemical Reduction of Carbon Dioxide to 1-Butanol on Oxide-Derived Copper, Angew. Chem., Int. Ed., 2020, 59(47), 21072–21079, DOI:10.1002/anie.202008289.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Electrochemical CO2 Reduction: A Classification Problem, ChemPhysChem, 2017, 18(22), 3266–3273, DOI:10.1002/cphc.201700736.
- H. Li, Y. Li, M. T. M. Koper and F. Calle-Vallejo, Bond-Making and Breaking between Carbon, Nitrogen, and Oxygen in Electrocatalysis, J. Am. Chem. Soc., 2014, 136(44), 15694–15701, DOI:10.1021/ja508649p.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Electrochemical CO2 Reduction: Classifying Cu Facets, ACS Catal., 2019, 9(9), 7894–7899, DOI:10.1021/acscatal.9b01899.
- O. Piqué, Q. H. Low, A. D. Handoko, B. S. Yeo and F. Calle-Vallejo, Selectivity Map for the Late Stages of CO and CO2 Reduction to C2 Species on Copper Electrodes, Angew. Chem., Int. Ed., 2021, 60(19), 10784–10790, DOI:10.1002/anie.202014060.
- F. L. P. Veenstra, N. Ackerl, A. J. Martín and J. Pérez-Ramírez, Laser-Microstructured Copper Reveals Selectivity Patterns in the Electrocatalytic Reduction of CO2, Chem, 2020, 6(7), 1707–1722, DOI:10.1016/j.chempr.2020.04.001.
- I. Ledezma-Yanez, E. P. Gallent, M. T. M. Koper and F. Calle-Vallejo, Structure-Sensitive Electroreduction of Acetaldehyde to Ethanol on Copper and Its Mechanistic Implications for CO and CO2 Reduction, Catal. Today, 2016, 262, 90–94, DOI:10.1016/j.cattod.2015.09.029.
- S. Zhu, B. Jiang, W.-B. Cai and M. Shao, Direct Observation on Reaction Intermediates and the Role of Bicarbonate Anions in CO 2 Electrochemical Reduction Reaction on Cu Surfaces, J. Am. Chem. Soc., 2017, 139(44), 15664–15667, DOI:10.1021/jacs.7b10462.
- E. Pérez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, Spectroscopic Observation of a Hydrogenated CO Dimer Intermediate During CO Reduction on Cu(100) Electrodes, Angew. Chem., Int. Ed., 2017, 56(13), 3621–3624, DOI:10.1002/anie.201700580.
- A. Wuttig, C. Liu, Q. Peng, M. Yaguchi, C. H. Hendon, K. Motobayashi, S. Ye, M. Osawa and Y. Surendranath, Tracking a Common Surface-Bound Intermediate during CO 2 -to-Fuels Catalysis, ACS Cent. Sci., 2016, 2(8), 522–528, DOI:10.1021/acscentsci.6b00155.
- E. Bertheussen, A. Verdaguer-Casadevall, D. Ravasio, J. H. Montoya, D. B. Trimarco, C. Roy, S. Meier, J. Wendland, J. K. Nørskov, I. E. L. Stephens and I. Chorkendorff, Acetaldehyde as an Intermediate in the Electroreduction of Carbon Monoxide to Ethanol on Oxide-Derived Copper, Angew. Chem., Int. Ed., 2016, 55(4), 1450–1454, DOI:10.1002/anie.201508851.
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, Insights into C-C Coupling in CO2 Electroreduction on Copper Electrodes, ChemCatChem, 2013, 5(3), 737–742, DOI:10.1002/cctc.201200564.
- C. Tang, Y. Zheng, M. Jaroniec and S. Z. Qiao, Electrocatalytic Refinery for Sustainable Production of Fuels and Chemicals, Angew. Chem., Int. Ed., 2021, 60(36), 19572–19590, DOI:10.1002/anie.202101522.
- H. Yue, Y. Zhao, X. Ma and J. Gong, Ethylene Glycol: Properties, Synthesis, and Applications, Chem. Soc. Rev., 2012, 41(11), 4218–4244, 10.1039/c2cs15359a.
- S. Rebsdat and D. Mayer, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000, DOI: DOI:10.1002/14356007.a10_101.Ethylene Glycol.
- M. Löffler, P. Khanipour, N. Kulyk, K. J. J. Mayrhofer and I. Katsounaros, Insights into Liquid Product Formation during Carbon Dioxide Reduction on Copper and Oxide-Derived Copper from Quantitative Real-Time Measurements, ACS Catal., 2020, 10(12), 6735–6740, DOI:10.1021/acscatal.0c01388.
- B. Schmid, C. Reller, S. S. Neubauer, M. Fleischer, R. Dorta and G. Schmid, Reactivity of Copper Electrodes towards Functional Groups and Small Molecules in the Context of CO2 Electro-Reductions, Catalysts, 2017, 7(5), 161, DOI:10.3390/catal7050161.
- A. R. Fratzke and P. J. Reilly, Kinetic Analysis of the Disproportionation of Aqueous Glyoxal, Int. J. Chem. Kinet., 1986, 18(7), 757–773, DOI:10.1002/kin.550180704.
- M. Auinger, I. Katsounaros, J. C. Meier, S. O. Klemm, P. U. Biedermann, A. A. Topalov, M. Rohwerder and K. J. J. Mayrhofer, Near-Surface Ion Distribution and Buffer Effects during Electrochemical Reactions, Phys. Chem. Chem. Phys., 2011, 13(36), 16384–16394, 10.1039/c1cp21717h.
- K. J. P. Schouten, Z. Qin, E. P. Gallent and M. T. M. Koper, Two Pathways for the Formation of Ethylene in CO Reduction on Single-Crystal Copper Electrodes, J. Am. Chem. Soc., 2012, 134(24), 9864–9867, DOI:10.1021/ja302668n.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, Electrochemical Reduction of Carbon Dioxide at Various Series of Copper Single Crystal Electrodes, J. Mol. Catal. A, 2003, 199(1–2), 39–47, DOI:10.1016/S1381-1169(03)00016-5.
- O. Pique, F. Vines, F. Illas and F. Calle-Vallejo, Elucidating the Structure of Ethanol-Producing Active Sites at Oxide-Derived Cu Electrocatalysts, ACS Catal., 2020, 10(18), 10488–10494, DOI:10.1021/acscatal.0c01880.
- L. P. Granda-Marulanda, A. Rendón-Calle, S. Builes, F. Illas, M. T. M. Koper and F. Calle-Vallejo, A Semiempirical Method to Detect and Correct DFT-Based Gas-Phase Errors and Its Application in Electrocatalysis, ACS Catal., 2020, 10(12), 6900–6907, DOI:10.1021/acscatal.0c01075.
- Y. Lum and J. W. Ager, Evidence for Product-Specific Active Sites on Oxide-Derived Cu Catalysts for Electrochemical CO2 Reduction, Nat. Catal., 2019, 2(1), 86–93, DOI:10.1038/s41929-018-0201-7.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Metal Ion Cycling of Cu Foil for Selective C–C Coupling in Electrochemical CO2 Reduction, Nat. Catal., 2018, 1(2), 111–119, DOI:10.1038/s41929-017-0009-x.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, Electrochemical Reduction of CO at a Copper Electrode, J. Phys. Chem. B, 1997, 101(36), 7075–7081, DOI:10.1021/jp970284i.
- R. Kloth, P. Khanipour, K. J. J. Mayrhofer and I. Katsounaros, Implementation of an Enclosed Ionization Interface for the Analysis of Liquid Sample Streams with Direct Analysis in Real Time Mass Spectrometry, Rapid Commun. Mass Spectrom., 2021, 35, e9091, DOI:10.1002/rcm.9091.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775, DOI:10.1103/PhysRevB.59.1758.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13(12), 5188–5192, DOI:10.1103/PhysRevB.13.5188.
- M. Methfessel and A. T. Paxton, High-Precision Sampling for Brillouin-Zone Integration in Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40(6), 3616–3621, DOI:10.1103/PhysRevB.40.3616.
- A. Rendón-Calle, S. Builes and F. Calle-Vallejo, Substantial Improvement of Electrocatalytic Predictions by Systematic Assessment of Solvent Effects on Adsorption Energies, Appl. Catal. B, 2020, 276, 119147, DOI:10.1021/j.apcatb.2020.119147.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108(46), 17886–17892, DOI:10.1021/jp047349j.
- A. Bagger, L. Arnarson, M. H. Hansen, E. Spohr and J. Rossmeisl, Electrochemical CO Reduction: A Property of the Electrochemical Interface, J. Am. Chem. Soc., 2019, 141(4), 1506–1514, DOI:10.1021/jacs.8b08839.
- F. Calle-Vallejo, J. I. Martínez, J. M. García-Lastra, P. Sautet and D. Loffreda, Fast Prediction of Adsorption Properties for Platinum Nanocatalysts with Generalized Coordination Numbers, Angew. Chem., Int. Ed., 2014, 53, 8316–8319, DOI:10.1002/anie.201402958.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03527h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |