
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-driven biocatalytic oxidation
Chul-Ho
Yun
 a,
Jinhyun
Kim
b,
Frank
Hollmann
c and
Chan Beum
Park
*b
a,
Jinhyun
Kim
b,
Frank
Hollmann
c and
Chan Beum
Park
*b
aSchool of Biological Sciences and Technology, Chonnam National University, 77 Yongbong-ro, Gwangju 61186, Korea
bDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), 335 Science Road, Daejeon 34141, Korea. E-mail: parkcb@kaist.ac.kr
cDepartment of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629HZ Delft, The Netherlands
First published on 30th September 2022
Abstract
Enzymes are the catalyst of choice for highly selective reactions, offering nature-inspired approaches for sustainable chemical synthesis. Oxidative enzymes (e.g., monooxygenases, peroxygenases, oxidases, or dehydrogenases) catalyze a variety of enantioselective oxyfunctionalization and dehydrogenation reactions under mild conditions. To sustain the catalytic cycles of these enzymes, constant supply with or withdrawal of reducing equivalents (electrons) is required. Being redox by nature, photocatalysis appears a ‘natural choice’ to accomplish the electron-relay role, and many photoenzymatic oxidation reactions have been developed in the past years. In this contribution, we critically summarize the current developments in photoredoxbiocatalysis, highlight some promising concepts but also discuss the current limitations.
1. Introduction
Oxidation is an essential reaction for chemical synthesis. Historically, oxidation reactions (also on an industrial scale) utilize high-valent Cr- and Mn-salts, generating enormous amounts of environmentally questionable wastes. In the last decades, the research focus has shifted towards more acceptable stoichiometric oxidants, such as O2 or H2O2.1 Next to organocatalytic2 and transition-metal-catalyzed3 approaches, biocatalytic oxidation methods are also gaining interest, and a broad range of enzymes are available nowadays to perform selective oxidation reactions under very mild reaction conditions (Fig. 1).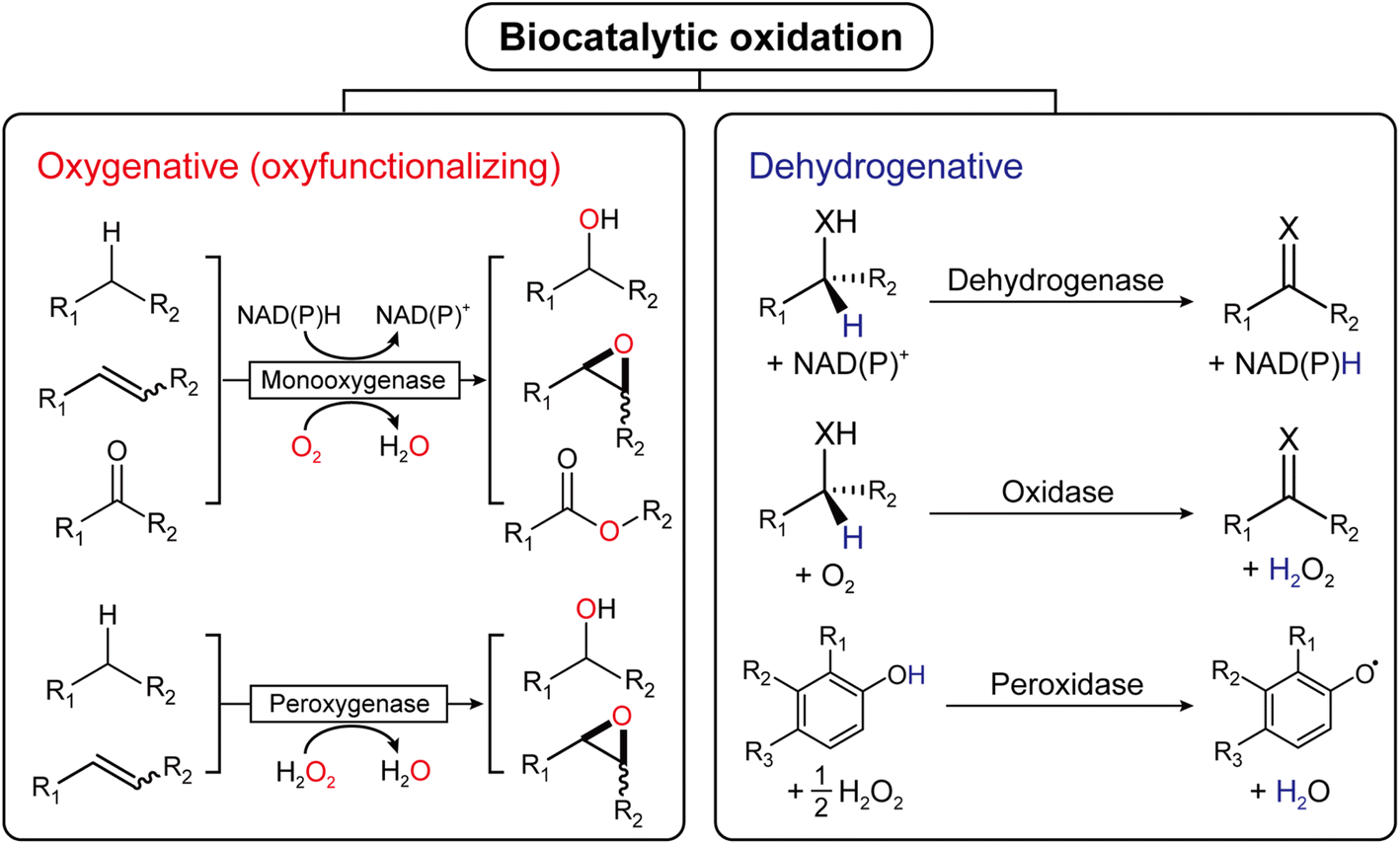 | ||
| Fig. 1 Biocatalytic oxidation is categorized into either oxygenative (oxyfunctionalizing) or dehydrogenative reactions. | ||
Mechanistically, biocatalytic oxidation reactions fall either into the ‘oxyfunctionalizing’ or the ‘dehydrogenative’ branch.4 Oxyfunctionalizing reactions have been traditionally catalyzed by so-called monooxygenases. These enzymes reductively activate O2 of which one O-atom is inserted into the starting material while the other O-atom is used to form H2O as a final product. More recently, ‘unspecific peroxygenases’ (UPOs) are receiving increasing interest as alternatives to monooxygenases. UPOs, in contrast to monooxygenases, directly utilize partially reduced oxygen (in the form of hydrogen peroxide, H2O2).5 Dehydrogenative oxidation reactions typically imply the oxidation of alcohols or amines to the corresponding ketones (imines), also aldehyde oxidations to the corresponding carboxylic acids fall into this category. The reducing equivalents liberated in these transformations are transferred either to nicotinamide cofactors (dehydrogenases) or to molecular oxygen (oxidases).4
As redox catalysts, all above-mentioned enzymes rely on the supply with redox equivalents to maintain their catalytic cycles. Counter-intuitively (at first sight) monooxygenases necessitate supply with reducing equivalents, which are—more or less directly—delivered to the monooxygenases' active sites from the reduced nicotinamide cofactors [NAD(P)H].6 Considering the overall four electron reduction of O2 catalyzed by monooxygenases and the fact that only two electrons stem from the starting material, this dependence on reducing equivalents, however, becomes clear. Established enzymatic methods to in situ regenerate the reduced nicotinamide cofactors comprise, amongst others, formate dehydrogenase-catalyzed oxidation of formic acid or glucose dehydrogenase-catalyzed oxidation of glucose.
Envisioning the use of clean solar power as energy source, photocatalysis has emerged as an alternative to the above-mentioned enzymatic regeneration systems.7–10 The promise of photocatalytic regeneration systems lies with access to a broader range of stoichiometric sources or acceptors of redox equivalents and simplified reaction schemes. Many photocatalysts—such as organic dyes, metal nanoparticles, and semiconductor quantum dots—have been tested to activate redox enzymes via the transfer of photoinduced electrons. In these systems, sacrificial electron donors [e.g., triethanolamine (TEOA), ethylenediaminetetraacetic acid (EDTA), ascorbic acid, or even water] have been used to recycle a cofactor (or a mediator) to sustain the biocatalytic reaction cycle. The reducing equivalents are transferred either directly to the oxidoreductase's active site or indirectly via the natural nicotinamide cofactors.
Alternatively, in recent years, so-called peroxyzymes (i.e., enzymes using H2O2 as a stoichiometric oxidizing agent) have gained considerable interest in the biocatalysis community. Particularly, peroxygenases [E.C. 1.11.2.] (https://www.iubmb.qmul.ac.uk/enzyme/EC1/11/2/) are promising alternatives to well-known P450 monooxygenases because of the significantly simpler catalytic cycle. Nevertheless, the poor robustness of heme-enzymes against H2O2 necessitates its provision in controlled amounts to balance stability and reactivity. This offers enormous possibilities for photocatalysis to solve this issue.
Finally, NAD(P)+-dependent oxidation reactions are also an attractive target for photocatalysis as simple photocatalytic NAD(P)H oxidation reactions are at hand.
More detailed information about the enzymes discussed in this contribution (e.g., genbank/Uniprot ID, heterologous expression conditions, and other technical details) have been summarized in Table 1.
| Classification of oxidative enzymes | Representative enzyme | Genbank ID (Uniprot ID) | Expression conditions (microbial host, culture media, antibiotics, additives, expression temperature, and culture time) | Ref. |
|---|---|---|---|---|
| BVMO | Phenylacetone monooxygenase (PAMO-P3 variant) from bacterium Thermobifida fusca | CP000088 (Q47PU3) | E. coli TOP10 cells, TB (terrific broth) medium with 0.1% L-arabinose, 100 μg mL−1 carbenicilline, 37 °C | 11 |
| P450 | CYP102A1 (heme domain) from bacterium Bacillus megaterium | WP_034650526 (P14779.2) | E. coli XL1-blue cells, Luria–Bertani (LB) medium, ampicillin (100 μg mL−1), 0.4 mM IPTG (isopropyl β-D-1-thiogalactopyranoside), and 0.5 mM, δ-ALA (δ-aminolevulinic acid), 30 °C for 6 h | 12 |
| Human CYP2E1 | NM_000773 (P05181.1) | E. coli DH5α cells, LB medium with 20 g glucose per L, ampicillin (100 μg mL−1), 1 mM IPTG, and 0.5 mM δ-ALA, 30 °C for 48 h | 13 and 14 | |
| CYP152A1 (P450Bsb) from bacterium Bacillus subtilis subsp. Subtilis str. 168 | BSU02100 (O31440.1) | E. coli BL21 (DE3) cells, LB medium, kanamycin (30 μg mL−1), 1 mM IPTG, 30 °C for 4 h | 15 | |
| CYP152A2 (P450CLA) from bacterium Clostridium acetobutylicum ATCC 824 | CAC3330 (Q97DZ0) | E. coli BL21 (DE3) cells, LB medium, kanamycin (30 μg mL−1), 1 mM IPTG, 30 °C for 4 h | 15 | |
| CYP152L1 from bacterium Jeotgalicoccus sp. 8456 | WP_198687844 (E9NSU2) | E. coli C43 cells, TB medium, kanamycin (50 μg mL−1). 0.1 mM IPTG, 1 mM δ-ALA, 30 °C for 20 h | 16 | |
| UPO | AaeUPO from mushroom Agrocybe aegerita (wild-type, PaDa-I mutant, SoLo mutant) | FM872457 (B9W4V6) | Saccharomyces cerevisiae, minimal medium, chloramphenicol (25 μg mL−1), 25 °C for 72 h | 17 and 18 |
| Pichia pastoris, basal salts medium, PTM1 trace salts (4.35 mL L−1) and antifoam 204 (0.25 mL L−1), methanol (0.5%, v/v), 25 °C | ||||
| CfuUPO from Caldariomyces fumago | X04486 (P04963) | CfuUPO is obtained by fermentation of C. fumago on synthetic medium, filtration of the mycelium, and subsequent chromatography. The strain was obtained from the International Mycological Institute, Surrey, UK | 19 | |
| LPMO | AA9E from fungus Thielavia terrestris (TtAA9E) | XP_003657336.1 (G2RGE5) | The preculture and production culture in P. pastoris SMD1168H were both done in YPD medium. The strain was grown for production for 4 days. Purified T. terrestris LPMO (TtLPMO9E, previously TtGH61E) was donated from Novozymes A/S (Denmark) | 20 |
| LPMO from Streptomyces coelicolor (ScAA10C) | NP_625478.1 (Q9RJY2) | Fresh colonies were inoculated in LB-ampicillin (50 μg mL−1) media and grown at 37 °C (CelS2) for 20 h | 21 and 22 | |
| LPMO from fungus Thielavia terrestris (copper saturated TtAA9) | GCA_900343105.1 | Thielavia terrestris LPMO (TtAA9) was provided as a broth from Novozymes A/S (Denmark). The TtAA9 was saturated with Cu(I) chloride under anaerobic conditions and on ice for 2 h, filtered, purified by chromatography, and concentrated by ultra-filtration | 23 | |
| Dehydrogenase | Alcohol dehydrogenase (ADH) from Saccharomyces cerevisiae (ScADH) | AAA34411 (P00331) | ScADH gene was transformed into S. cerevisiae 302–21#2 strain that does not produce active alcohol dehydrogenase, and cultivated in YEPD medium and antimycin A (1 μg mL−1) at 30 °C for 24 h | 24 and 25 |
| ADH from Thermus sp. ATN1 (TADH) | EU681191 (B2ZRE3) | TADH was produced by transforming pASZ2 (PET) into E. coli BL21 (DE3) pLysS cells and cultivation of the recombinant E. coli using the autoinduction system | 26 | |
| ADH from horse liver (HLADH) | M64864 (P00327) | Commercially available | 26 | |
| Formate dehydrogenase from Candida boidinii (CbFDH) | KM454879 | CbFDH plasmids were transformed into E. coli BL21 (DE3) pLysS cells and grown in LB medium with ampicillin (100 μg mL−1) and 1 mM IPTG at 37 °C | 27 and 28 |
2. Light-driven monooxygenase catalysis
Monooxygenases catalyze the insertion of one oxygen atom (O) from molecular oxygen (O2) into an organic molecule. For photochemical activation of a monooxygenase, photoinduced electrons should be transferred directly or indirectly to the enzyme's active site (Fig. 2). During direct transfer of photoinduced electrons, a photocatalyst and a monooxygenase having a redox prosthetic group (e.g., heme, flavin), interact in a relationship between an electron donor and an acceptor, respectively. The excited state of the photocatalyst is quenched by the oxidation of a sacrificial electron donor (e.g., EDTA, TEOA) to sustain the turnover of the photocatalyst.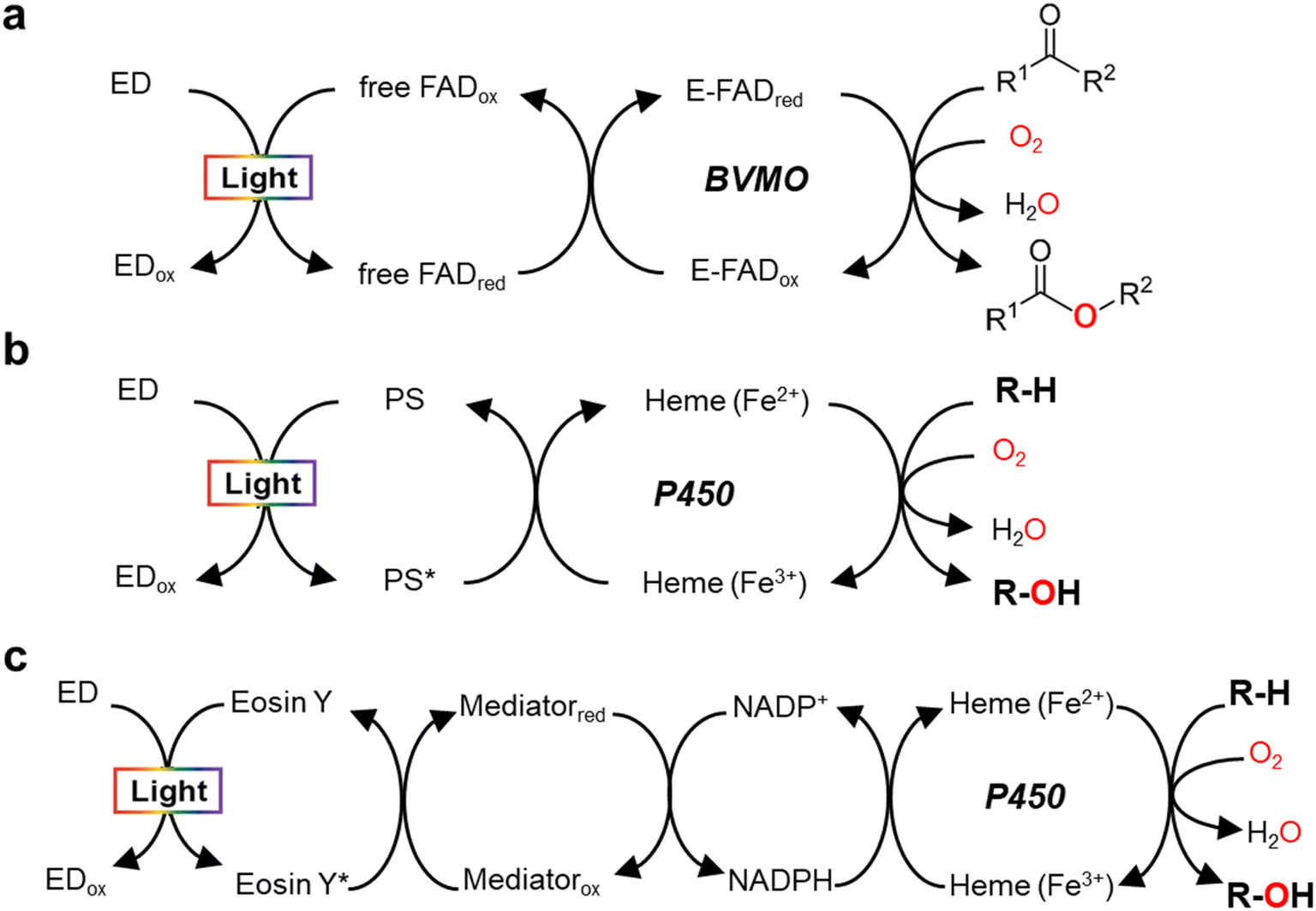 | ||
| Fig. 2 Light-driven monooxygenase catalysis via direct activation by electron transfer to active site (heme or flavin) (a and b) and indirect route by photoregeneration of NAD(P)H (c). (a) Light-driven direct electron transfer to enzyme-bound FAD (E-FAD) of the Baeyer–Villiger monooxygenase (BVMO, e.g. PAMO) using a flavin as a photosensitizer (PS) and EDTA as an sacrificial electron donor (ED).29 (b) Direct electron transfer from activated PS [e.g., eosin Y,31 flavin,12,13 Ru(II)36], which are made by light and ED (TEOA, EDTA, or diethyldithiocarbamate), to P450 heme. (c) The catalytic turnover of P450 is indirectly accomplished by photochemical reduction of NADPH through a cascaded electron delivery in a photocatalytic system having an ED (TEOA), a PS (eosin Y), and an electron mediator M [Cp*Rh(bpy)H2O]2+.40 | ||
In 2007, Hollmann et al. devised and implemented experimentally a light-driven catalytic scheme for flavin-dependent Baeyer–Villiger monooxygenase (BVMO, e.g. PAMO-P3)29 (Fig. 2a). Photochemical reduction of free flavin molecules (a photocatalyst) using EDTA produces reduced ones, which then transfer electrons to the FAD cofactor bound to the PAMO-P3 active site. Overall, visible light promotes direct reductive regeneration of the PAMO-P3-bound FAD; thus, the scheme does not require costly NADPH (or a complicated NADPH regeneration system). Here, the light-driven platform of PAMO-P3 for Baeyer–Villiger reactions of ketone substrates showed 48–93% conversion of ketone substrates with excellent enantioselectivity, which were comparable to conventional NADPH-based process (albeit at reduced catalytic rates). However, already then two major limitations had been pointed out: oxidative uncoupling of the regeneration reaction from enzymatic O2-activation and the slow electron transfer between free and enzyme-cofactors.30
A whole-cell-based, light-driven P450 platform has been devised using eosin Y (EY) as a photocatalyst and EDTA as an electron donor31 (Fig. 2b). EY could easily enter into the Escherichia coli cytoplasm and then bind to the P450's heme domain. The catalytic reaction of P450 was mediated via the direct transfer of light-induced electrons from the photoactivated EY to the heme domain of P450 under visible light. Photochemical activation of the P450 catalysis has been successfully demonstrated using many variants of CYP102A1 and human P450s for the bioconversion of various substrates. A possible contribution of peroxygenase activity in the whole-cell P450 photocatalysis however cannot be excluded, which needs further investigation.
Besides, several other light-driven whole-cell systems have been reported: (1) Rieske oxygenase using 5(6)-carboxyeosin as a photosensitizer and MES buffer as electron donor;32 (2) photoautotrophic cyanobacterium Synechocystis expressing the heterologous P450 (ref. 33); (3) BVMO reactions in metabolically engineered cyanobacteria;34 (4) microbial photosynthesis works for O2 generation for biocatalytic oxyfunctionalizations.35
In addition to the photosensitizing dyes (e.g., flavin,12,13 EY31), ruthenium (Ru)36 complexes have been used as a photocatalyst to reduce P450 heme iron (Fig. 2b). Ru(II) has been widely applied for single electron transfer in metal-containing hydrogenases and photochemical enzymatic reduction of small molecules by nitrogenases.37 The Cheruzel group attached a Ru(II)-diimine complex specifically close to the enzyme active site of CYP102A1 via an engineered mutant (L407C) for direct electron transfer from photoreduced Ru(I) to the P450 heme iron using diethyldithiocarbamate as a sacrificial electron donor.36 The Ru(II)-CYP102A1 platform catalyzed light-driven reaction of lauric acid hydroxylation, with an initial reaction rate of 125 min−1 and a total turnover number (TTN = maximum product concentration/enzyme concentration) of 935.36 Furthermore, the Ru(II)-CYP102A1 platform was also applied to the hydroxylation reaction of 10-undecenoic acid38 to produce (R)-9-hydroxy-10-undecenoic acid and the O-dealkylation reaction of nitrophenolate-based substrates as chromogenic substrates.39
In 2019, Le et al. reported that a flavin/EDTA/light platform can support the catalytic activities of CYP102A1 heme domain through direct delivery of photoexcited electrons to the P450 heme iron and indirect H2O2 generation for peroxygenase activity as well12 (Fig. 2b). Without the requirement of reductase and NADPH, the presence of only light and EDTA mediates the direct electron transfer from reduced flavins to the heme iron. The reduced heme can drive the hydroxylation of the organic substrate (R–H). On the other hand, the photoreduced flavin reacts quickly with O2 to produce H2O2. Hence, again, a contribution of the H2O2 shunt pathway cannot be fully excluded.12 For human CYP2E1, in contrast, the flavin/EDTA/light system works only via direct transfer of light-induced electrons to the P450 heme iron, not by the H2O2-mediated peroxide shunt.13
Indirect transfer of photoinduced electrons to monooxygenases has been extensively accomplished through photochemical reduction of biological cofactors [e.g., NAD(P)H, FADH2]. Here, the redox mediators function as a freely diffusing, remote communicator between a photocatalyst and a monooxygenase. For example, photochemical regeneration of NADPH for CYP102A1 (Y51F/F87A variant)-catalyzed O-dealkylation reaction was performed using EY as a photosensitizing dye, TEOA as an electron donor, and [Cp*Rh(bpy)H2O] as a selective NAD(P)H regeneration catalyst (Fig. 2c). The P450 sustainably maintained its catalytic turnover with continuous photoregeneration of NADPH. Thus, visible light-driven recycling of NADPH provides a new route for P450 monooxygenase catalysis.40 Later on, natural sunlight-driven platform for P450 catalysis has been demonstrated at preparative scale using P450s immobilized on poly(3-hydroxybutyrate) [P(3-HB)] scaffold. Through photochemical regeneration of NADPH using EY as a photocatalyst, the P450–P(3HB) complex could successfully catalyze O-dealkylation reactions. In addition, a P450-catalyzed reaction was accomplished under natural sunlight for four consecutive days using a solar-tracking module.41 This study hints at practical applicability of natural sunlight for driving photobiocatalysis, which can be replaced by LED light during night.
Interestingly, ferredoxin (Fd)-mediated transfer of electrons in photosystem I (PSI) has been adopted for indirect transfer of photoinduced electrons to P450 heme. Jensen and coworkers demonstrated that the heme domain of plant CYP79A1, which is involved in tyrosine oxidation, can be photochemically reduced by the PSI using Fd as a mediator.42,43 Upon photoactivation, the Fd-mediated electron transfer chain readily provides the required electrons to support the P450 catalysis.
In addition to abiotic photochemical and photoelectrochemical approaches, cyanobacteria have been reported to activate oxidative redox enzymes (e.g., BVMO,34 P450,33 alkane monooxygenase44) by producing O2 gas and reducing equivalents.
3. Photobiocatalytic oxyfunctionalization via H2O2-dependent pathway
H2O2-Dependent pathways are recently gaining increasing attention—instead of the above-mentioned reductive regeneration of the enzymes' active sites—for photobiocatalytic oxyfunctionalization. The catalytically active species in monooxygenases contain or are derived from an intermediate comprising activated oxygen in the oxidation state of H2O2.4 The latter has been derived from molecular oxygen via reductive activation within the enzyme active site via more or less complex and vulnerable mechanisms.6 Therefore, simplifying the catalytic mechanism and reaction schemes using already reduced oxygen in form of H2O2 is a very appealing approach. As H2O2, however, also acts as a strong oxidative inactivator of many enzymes, its stoichiometric application is not feasible and photochemical in situ generation methods providing H2O2 in appropriate rates represent a promising approach.Most prominent examples for H2O2-utilizing enzymes are the heme-dependent P450 monooxygenases and peroxygenases as well as the Cu-dependent lytic polysaccharide monooxygenases (LPMOs). Further enzymes such as hydrolases following the perhydrolase pathway,45,46 vanadium-dependent haloperoxidases47 or artificial peroxyzymes48 are worth mentioning but have not been used together with a photochemical H2O2 generation system yet.
3.1 Unspecific peroxygenases (UPOs)
UPOs catalyze a wide range of selective oxyfunctionalization reactions on inert C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. The catalytic cycle of peroxygenases represents the well-known H2O2-shunt shortcut on P450 monooxygenases (Fig. 3). P450 monooxygenases are mostly rather inefficient in utilizing the H2O2-shunt pathway, which has been assigned to the lack of a catalytic base (glutamate) facilitating the conversion of Cpd-0 to Cpd-I. This catalytically relevant amino acid is present in peroxygenases but not in P450 monooxygenases49 (Fig. 4).
C bonds. The catalytic cycle of peroxygenases represents the well-known H2O2-shunt shortcut on P450 monooxygenases (Fig. 3). P450 monooxygenases are mostly rather inefficient in utilizing the H2O2-shunt pathway, which has been assigned to the lack of a catalytic base (glutamate) facilitating the conversion of Cpd-0 to Cpd-I. This catalytically relevant amino acid is present in peroxygenases but not in P450 monooxygenases49 (Fig. 4).
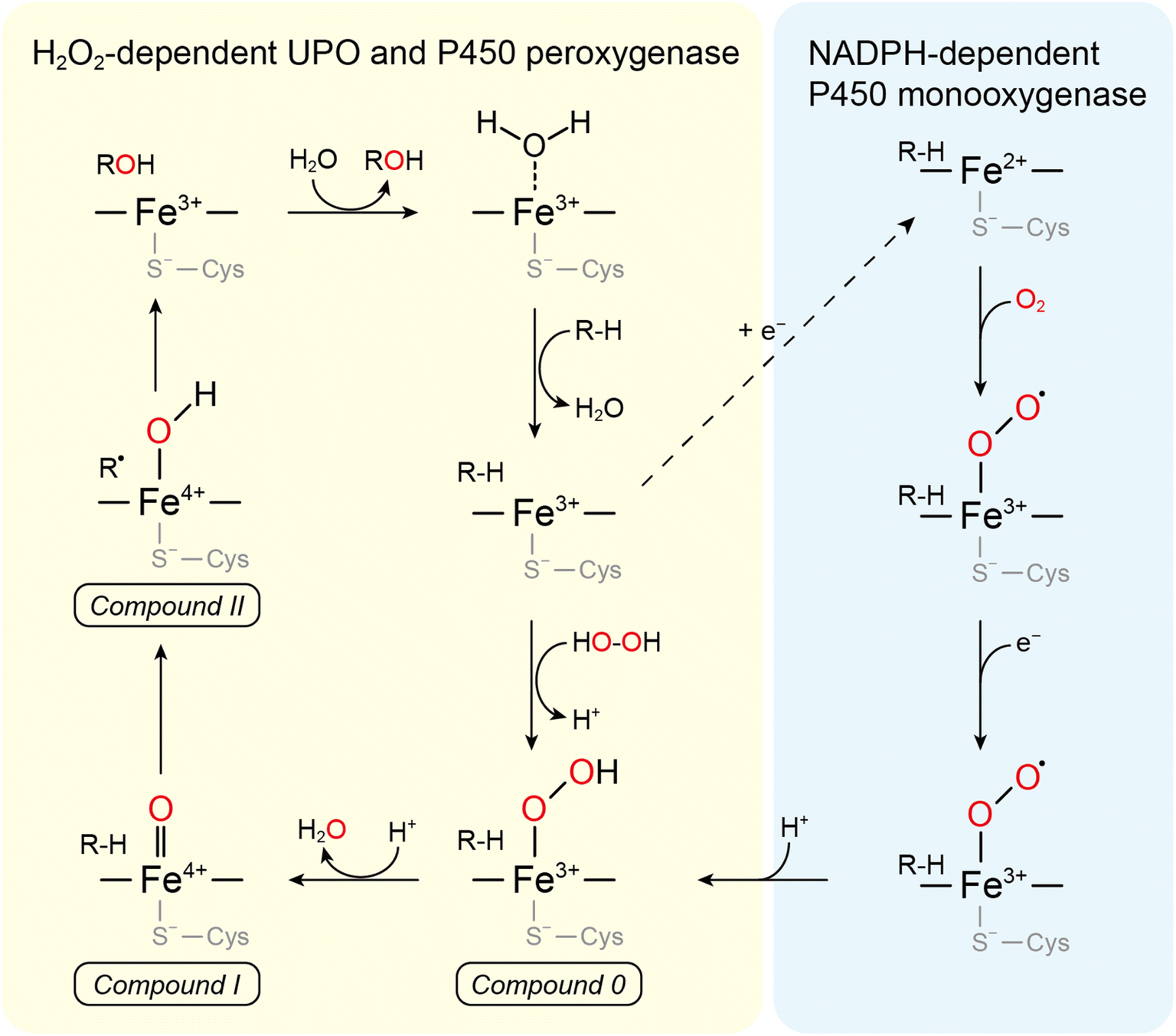 | ||
| Fig. 3 Comparison between the mechanism of P450s and UPOs for C-hydroxylation. The left cycle describes the H2O2-supported “peroxide shunt” shared by UPOs and P450 peroxygenases and the right one is the “classical” NADPH-dependent P450 monooxygenase route. Both cycles share compounds 0 (ferric peroxo-complex), I (oxo-ferryl cation radical complex), and II (ferryl hydroxide complex). | ||
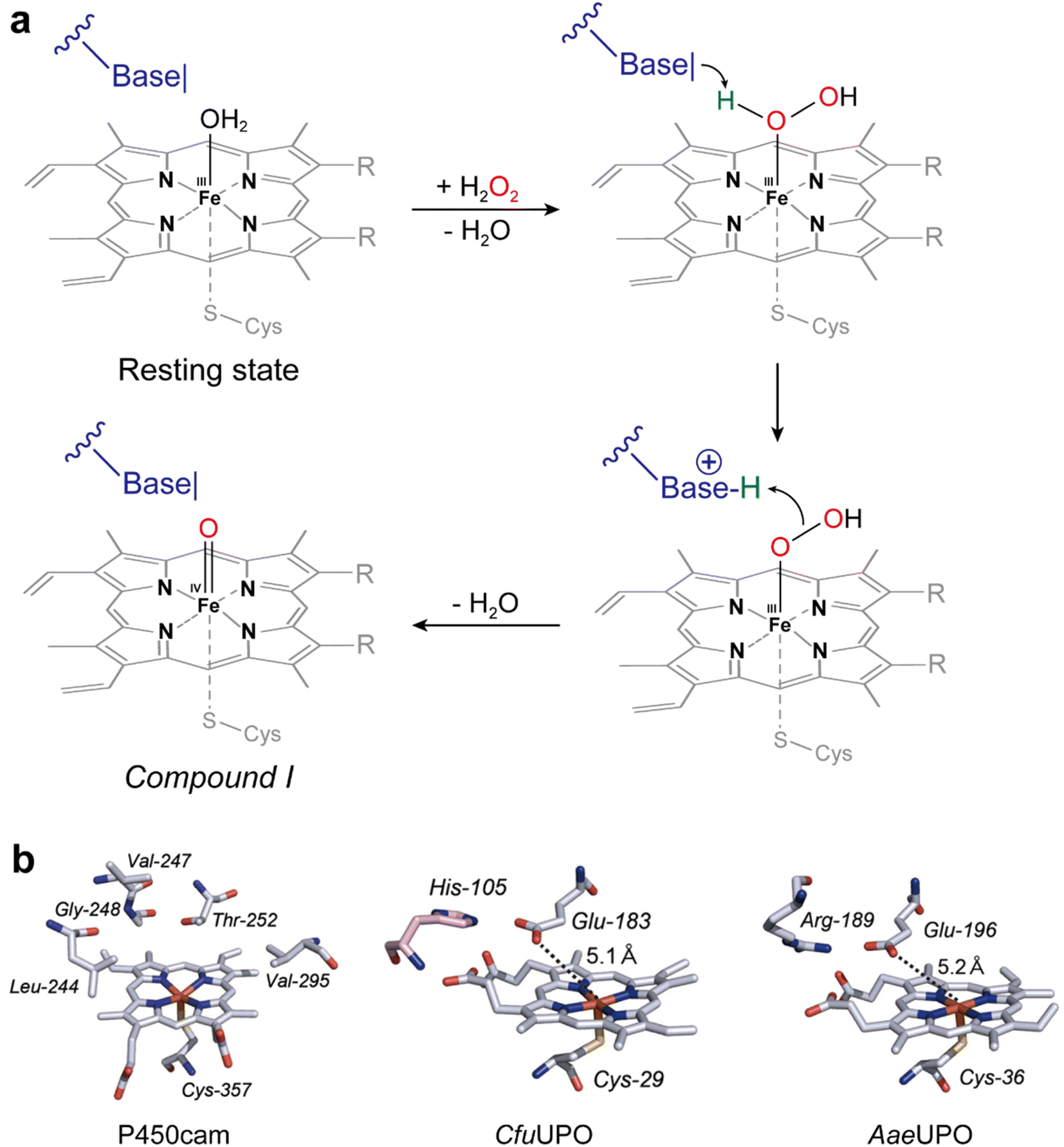 | ||
| Fig. 4 (a) Acid–base-catalysis as key difference between P450 monooxygenases and peroxygenases. (b) Active-site architectures of P450cam (POB code 2CPP), CfuCPO (PDB code 1CPO) and AaeUPO (PDB code 2YP1). Adapted from ref. 49 with permission from Springer. | ||
The first reported peroxygenase stems from Leptoxyphium fumago (previously Caldariomyces fumago)50 and found some popularity as oxyfunctionalization catalyst.51 However, poor activity and difficulties in the recombinant expression of this enzyme have limited its broad applicability in organic synthesis. This situation changed in 2004 with Hofrichter and coworkers reporting the UPO (initially termed as haloperoxidase) from Agrocybe aegerita (AaeUPO).52 Later, Alcalde and coworkers succeeded in its recombinant expression and engineering17,18 and more recently it's large-scale production.53 These pioneering contributions have paved the way to the engineering and identification of new, promising peroxygenases.5,52 Even though only a handful of UPOs have so far been functionally expressed and evaluated with respect to their catalytic properties, the synthetic potential is enormous.4,48,55
3.2 Cytochrome P450 peroxygenases
P450 monooxygenases and peroxygenases are structurally and mechanistically related. Hence, in principle, the H2O2-shunt pathway represents a doable alternative pathway to simplify the catalytic mechanism of P450 monooxygenases (Fig. 3). Unfortunately, however, most P450 monooxygenases exhibit very low activity with H2O2 (whereas the oxidative degradation of the catalytic heme moiety with H2O2 remains a challenge) resulting in comparably few catalytic cycles of P450 monooxygenases with H2O2. Protein engineering principally offers a solution to this issue56 which however yet remains to be fully exploited. The so-called P450 peroxygenases (bacterial CYP152 family) represent a subclass of the P450 monooxygenases, which in addition to the natural cycle also accept H2O2.57 Interestingly, these enzymes are all involved in fatty acid metabolism.The first reported P450 peroxygenases—CYP152B1 and CYP152A2—hydroxylate fatty acids exclusively in α-position.15,58,59 In contrast, CYP152A1 shows a preference for the β-position of fatty acids.15,59,60 On the other hand, CYP152L isozymes mainly perform oxidative fatty acid decarboxylation, producing terminal alkenes, along with α- and β-hydroxylation reactions.61,62 CYP152N1 catalyzes oxidative decarboxylation reaction of fatty acids to produce one-carbon shorter fatty acids.63 Thus, CYP152 peroxygenase reactions for alkene production are useful for the production of biofuels and fine chemicals.62,64
Finally, also the decoy-molecule—dual function small molecule—approach is worth mentioning.65 Using modified imidazoles as decoy molecules, Cong and coworkers successfully introduced H2O2-reactivity with some ‘conventional’ P450 monooxygenases. The imidazole is believed to partially replace the missing catalytic bases present in peroxygenases.
3.3 Lytic polysaccharide monooxygenases
LPMOs are key enzymes that catalyze degradation and decomposition of recalcitrant polysaccharides (e.g., cellulose, lignin, and chitin) by fungi and bacteria.66–68 Their active sites have a single copper ion, which is coordinated by an N-terminal histidine side chain and a further histidine residue to arrange the Cu-histidine brace.69 LPMOs are widespread in nature and classified as seven sequence-distinct families (i.e., 9–11 and 13–16) of the auxiliary activities according to the Carbohydrate Active enZymes (CAZy) database (https://www.cazy.org/Auxiliary-Activities.html).70Mechanistically, LPMOs catalyze the reductive activation of O2 for oxidative cleavage of the glycosidic bonds of polysaccharides via hydroxylation at either the C1 or the C4 position, followed by the cleavage of the scissile glycosidic bond and the formation of aldonic acids or 4-keto sugars at the oxidized chain ends.68 In addition to using O2 as an oxidant, LPMOs can utilize H2O2 as a cosubstrate to perform peroxygenation reactions on polysaccharides, catalyzing similar hydroxylation reactions with O2, which then lead to the spontaneous bond cleavage of the polysaccharides.68,71 Details of the catalytic mechanism remain to be fully elucidated.68
Nowadays, LPMOs are receiving a lot of attention because of their potential as an efficient decomposer of biomass.68,72,73 According to Bissaro et al.,71 LPMOs prefer H2O2 over O2 (via reductive activation) for hydroxylation of microcrystalline α-cellulose substrate (i.e., Avicel). The biocatalytic performance and stability could be controlled by regulating the supply of H2O2 in the absence of O2, suggesting that LPMOs possess more efficient peroxygenase activity than the monooxygenase activity (Table 2).
| LPMO | Photosensitizer | Reductant of O2 | Light source | Substrate | Product | Ref. |
|---|---|---|---|---|---|---|
| AA9E from Thielavia terrestris (TtAA9E) | Chlorophyllin (Chl) with ascorbic acid (AscA) | Photo-excited Chl | 150–200 μmol photons s−1 m −2 | Phosphoric acid-swollen cellulose (PASC), Avicel microcrystalline cellulose, lignin, xyloglucan | Diverse oligosacharides, gluconic acid, D-glucose, D-cellobiose, cello-oligosaccharides, xylogluco-oligosaccharides | 20 |
| LPMO from Streptomyces coelicolor (ScAA10C) | Chl with AscA, (or V–TiO2) | Photo-excited Chl (or H2O) | 150 W, xenon lamp visible light (I = 25% Imax, 42 W cm−2) | Crystalline cellulose (Avicel) | Aldonic acid, GlcGlc1A, (Glc)2Glc1A | 21 and 22 |
| LPMO from Thielavia terrestris (copper saturated TtAA9) | Chl with AscA | Photo-excited Chl | White LEDs (200 μmol photons m−2 s−1) | Microcrystalline cellulose (Avicel) | Gluconic acid (GlcA) | 23 |
| Phosphoric acid-swollen cellulose (PASC) | GlcA | |||||
| Cellulose nanofibrils (CNF) | GlcA |
4. Photocatalytic platforms for in situ H2O2 generation
H2O2 represents a ‘double-edged sword’ enabling simplified reaction schemes on the one hand but also, on the other hand, inactivates enzymes. To balance both reactivities, in situ generation of H2O2 has proven suitable. Next to enzymatic,74,75 chemical76,77and electrochemical78–80 H2O2 generation schemes, also photo(electro)catalytic systems have been developed. The light-driven methods require a proper photocatalyst that, when excited by light, oxidises an electron donor and transfers the reducing equivalents to oxygen, producing H2O2. The steady-state H2O2 concentration is intrinsically in an equilibrium between photocatalytic generation and degradation.81 In this section, we discuss organic or inorganic photocatalytic platforms for light-driven H2O2 generation and biocatalytic oxyfunctionalization.4.1 Flavins
Flavin molecules, such as flavin adenine mononucleotide (FMN), flavin adenine dinucleotide (FAD) and riboflavin, can produce H2O2 photochemically at the expense of electron donors and molecular oxygen. For example, FMN extracts an electron from EDTA after being activated by light, then the reduced FMN generates H2O2via O2 reduction.82 The flavin-based in situ H2O2 generation strategy was first designed for driving CfuCPO-83 and AaeUPO-catalyzed84 oxyfunctionalization reactions (Table 3). Even though in these early works, the benefits of photochemical in situ H2O2 generation could be demonstrated, EDTA as sacrificial electron donor exhibits several disadvantages impeding it widespread use. Particularly, the degradation products, eventually formaldehyde and ethylene diamine, are not desirable from a practical and an environmental point of view. Other sacrificial electron donors such as ascorbic acid, formate, hydrazine or phosphite can substitute for EDTA but still need to be further explored.| Method | AaeUPO (concentration) | Reagents, light sources, and remarks | TTN (ee%) | STY (g L−1 day−1) | Ref. |
|---|---|---|---|---|---|
| a Abbreviations: AaeUPO: UPO from Agrocybe aegerita, WT: wild-type, PaDa-I or SoLo: recombinant mutant of AaeUPO, TTN: total turnover number, ee: enantiometric excess (%), STY: space time yield, AOx: alcohol oxidase from Pichia pastoris, FDM: formaldehyde dismutase from Pseudomonas putida F61, FDH: formate dehydrogenase from Candida boidinii, 3HB6H: 3-hydroxybenzoat-6-hydroxylase from Rhodococcus jostii, YgjM: homologue of old yellow enzyme from Bacillus subtilis, SO: sulfite oxidases SO from Arabidopsis thaliana. | |||||
| Stoichiometric addition | WT (40 nM) | H2O2 (1 mM), 1 mM ethylbenzene | 6000 (97) | 1.8 | 84 |
| WT (600 nM) | t-BuOOH (60 mM), 1 mM ethylbenzene | 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (95–98) 000 (95–98) |
40 | 85 | |
| Enzymatic | WT (100 nM) | AOx (60 nM), FDM (295 nM), FDH (1 μM), NAD+ (1.6 mM), 3HB6H (11 μM), MeOH (5 mM), 50 mM ethylbenzene | 468500 (>99) |
17.6 | 86 |
| AOx (60 nM), MeOH (5 mM), 15 mM ethylbenzene | 291500 (>99) |
11.0 | 86 | ||
| PaDa-I (25 nM) | FDH (250 nM), NAD+ (0.5 mM), YqjM (2.67 μM), MeOH (200 mM), sodium formate (75 mM), 10 mM ethylbenzene | 390000 (>96) |
5.2 | 87 | |
| PaDa-I or SoLo (500 nM) | SO (200 nM), calcium sulfite (100 mM), O2, 50 mM ethylbenzene | 30800 (98) |
19 | 88 | |
| PaDa-I (4 μM) | Soluble hydroxylase (95 nM), H2, FMN (200 μM), two-phase system: 0.75 mL ethylbenzene, 0.75 mL KPi buffer (50 mM, pH 7.5), 7 mL excess of headspace filled with gas (40% O2 and 60% H2) | 11250 (99) |
3.2 | 89 | |
| Photochemical | WT (40 nM) | FMN (50 μM), EDTA (1 mM), O2, 1 mM ethylbenzene. Visible light (Philips 7748XHP 200W, white light bulb) | 11470 (>97) |
3.5 | 84 |
| PaDa-I (150 nM) | Au–TiO2 (10 mg mL−1), MeOH (250 mM), O2, 15 mM ethylbenzene, white light bulb (Philips 7748XHP 205W) | 79000 (>98) |
0.48 | 90 | |
| PaDa-I (150 nM) | Rutile–Au–TiO2 (30 mg mL−1), H2O, O2, 10 mM ethylbenzene, visible light (λ > 400 nm) (Philips 7748XHP 150 W, white light bulb) | 37000 (98) |
0.17 | 91 | |
| Carbon nanodot (CND) (1 mg mL−1), FMN (0.1 mM), H2O, O2, 10 mM ethylbenzene, visible light (Philips 7748XHP 150 W, white light bulb) | 100000 (98) |
2.4 | |||
| WT (200 nM) | TiO2 (2 mg mL−1), MeOH (5%, v/v), O2, 10 mM ethylbenzene, 365 nm LED (either M365LP1, Thorlabs or LEDMOD365.1050.V2, Omicron) | 220000 |
2.6 | 92 | |
| PaDa-I (100 nM) | Graphitic carbon nitride (g-C3N4) (5 mg mL−1), MeOH or formic acid (250 mM), 50 mM ethylbenzene, white light bulb (Osram 200 W light bulb) | 60000 (98) |
0.59 | 93 | |
| PaDa-I (100 nM) | Wavelength-complementary photosensitizers (5 μM FMN, 10 μM methylene blue, 10 μM phenosafranin), MeOH (200 mM), NAD+ (0.4 mM), 4.8 μM FDH, O2, 10 mM ethylbenzene, Commercial LEDs (24 W) or LIGHTINGCURE LC8 L9566 (Hamamatsu) | 100000 (>95) |
7.3 | 94 | |
| PaDa-I (100 nM) | Nitrogen-doped carbon nanodots (N-CNDs) (5 mg mL−1), O2, 50 mM ethylbenzene, white light bulb (Philips 7748XHP 150 W) | 90000 |
0.56 | 95 | |
| PaDa-I (150 nM) | Au–TiO2 (5 mg mL−1), MeOH (250 mM), O2, 60 mM ethylbenzene, LED (405 nm), SMBB405V-1100-02Z, 125 mA | 372000 |
7.8 | 96 | |
| PaDa-I (100 nM) | Anthraquinone-2-sulfonate (0.5 mM), MeOH (40%), O2, 50 mM ethylbenzene, visible light (λ > 400 mm) (white light bulb, Philips 7748XHP 150W) | ∼150000 (34) |
0.46 | 97 | |
| PaDa-I (25 nM) | Carbon nitride photocatalyst (CN-OA-m) (2 mg mL−1), MeOH (250 mM), O2, 10 mM ethylbenzene, green light (528 nm) (Kessil PR160-525 LED) | 284000 (99) |
0.87 | 98 | |
| PaDa-I (50 nM) | Kraft lignin (or lignosulfate) (8 mg mL−1), O2, 10 mM ethylbenzene, visible light (λ > 400 nm, photon flux: 1.74 μE cm−2 s−1), a xenon arc lamp (with an infrared water filter and 400 nm longpass filter) | 72600 (81000) (>99) |
0.0819 (0.0914) | 99 | |
| Photoelectro-chemical | PaDa-I (100 nM) | Flavin-SWNT (single walled carbon nanotubes) electrode, U, O2, 100 mM ethylbenzene, U– power source and electrodes: WMPG100 potentiostat, 450 W Xe lamp with a 420 nm cut-off filter | 123900 |
1.0 | 100 |
| Electrochemical | PaDa-I (50 nM) | U, O2, acetone (3.3%, v/v), 139 mM ethylbenzene, U: −10 (TTN) or −30 (STY) mA cm−2 by multichannel potentiostat/galvanostat G300™ (projected area: Aelectrode = 2 cm2) | 400000 |
25 | 78 |
| Chemical | PaDa-I (15 U mLRM−1) | Metal catalyst (0.1 mg mLRM−1) of 0.5% Au–0.5% Pd/TiO2, 10 mM ethylbenzene, under 2 bar of 80% H2 and 20% air from a high-pressure gas reservoir | 25900 (98) |
1.5 | 76 |
Nevertheless, mostly due to its simplicity, the flavin/EDTA/light system has also been applied for some P450 peroxygenase reactions including CYP152A1, CYP152A2, and CYP152L1 (Fig. 5). The P450 fatty acid peroxygenases hydroxylate (or decarboxylate) saturated fatty acids (C12–C20)101–103 (Fig. 5a and b) The catalysis of CYP152L1 using this photochemical method was also applied to the conversion of ω-hydroxylated fatty acids (C12, C15, C16), obtained from palm kernel oil, into the corresponding alkenols104 (Fig. 5c). In the flavin/EDTA/light system, a peroxygenase activity of bacterial CYP102A1 heme domain were confirmed to catalyse the hydroxylation of 4-nitrophenol and lauric acid12 (Fig. 5d and e).
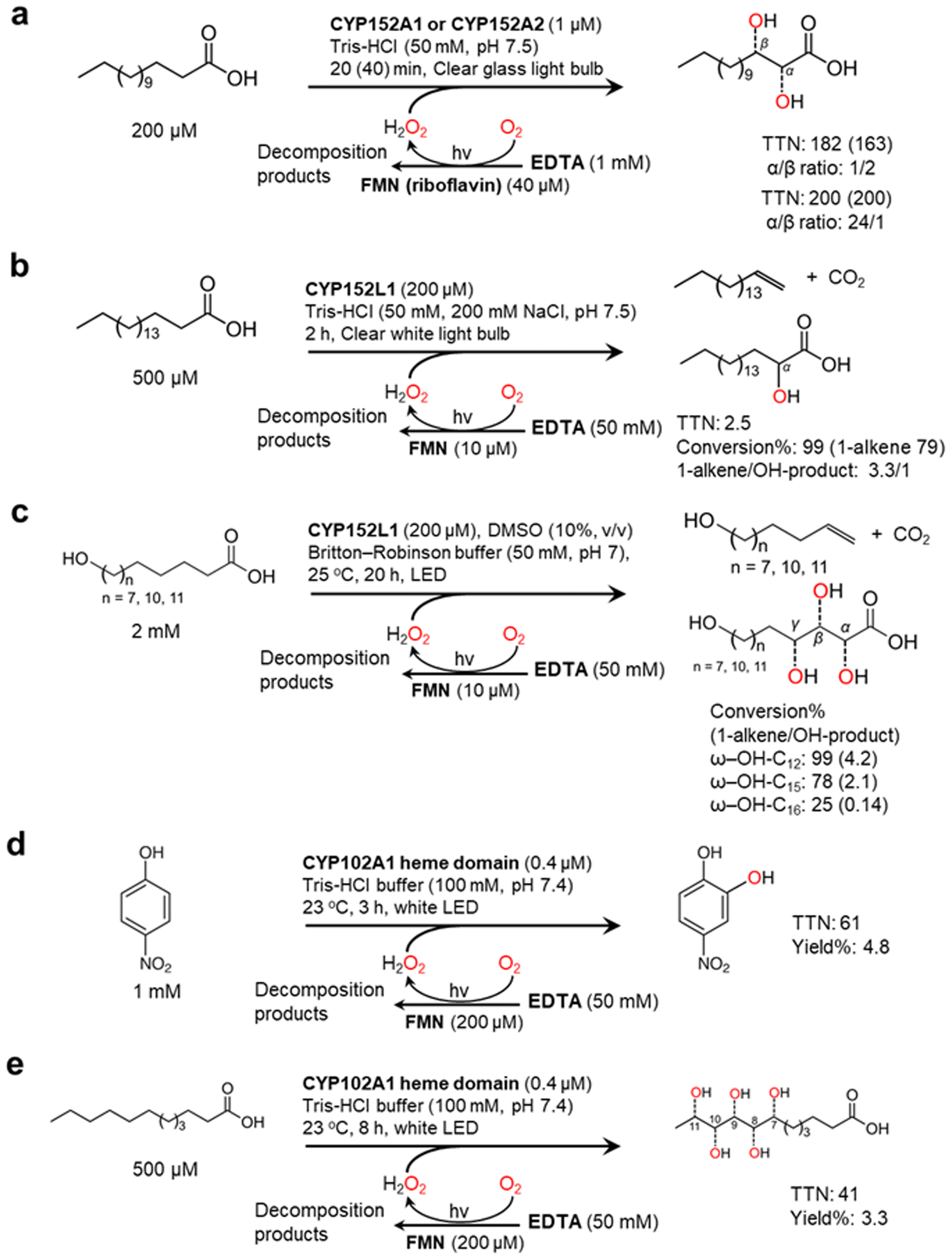 | ||
| Fig. 5 Photochemically driving P450 peroxygenases using FMN as a photocatalyst and EDTA as an electron donor. Hydroxylation of fatty acids101–103 (a and b) and ω-hydroxylated fatty acids104 (c) is catalyzed by CYP152 peroxygenases. CYP102A1 heme domain also shows peroxygenase activity towards 4-nitrophenol (d) and lauric acid (e).12 | ||
Next to waste generation, another drawback of flavins as photocatalysts is the rather narrow wavelength spectrum of flavins, only partially covering the spectrum of sunlight. In view of future sunlight-driven processes Willot et al. explored wavelength-complementary photosensitizers such as FMN (yellow), phenosafranine (green), and methylene blue (red) to utilize the different wavelengths of visible light more efficiently.94 In these experiments, co-catalysis by formate dehydrogenase and the nicotinamide cofactors enabled replacement of EDTA by formate.
Choi et al. demonstrated photoelectrocatalytic formation of H2O2 driven by flavin-hybridized single-walled carbon nanotube (SWNT) photoelectrode100 (Table 3). Lumichrome, a flavin derivative, was used as a photosensitizer. When flavin-hybridized SWNTs were exposed to visible light, flavins allowed a significant anodic shift of the O2 reduction potential. Because of flavin's high reactivity toward O2, photoactivated flavins boost electron delivery from the SWNT cathode to O2 for more efficient H2O2 production. The flavin-functionalized SWNT photoelectrode system enabled AaeUPO to hydroxylate ethylbenzene to (R)-1-phenylethanol at a TTN and STY of 123900 (95% ee) and 1.0 (g L−1 d−1), respectively. However, when 2-phenoxypropionic acid was used as a substrate, the TTN and STY was only 5900 and 0.13 (g L−1 d−1), respectively.
4.2 Titanium dioxide
Zhang et al. reported the use of gold-loaded rutile titanium dioxide (Au–TiO2) and methanol to drive AaeUPO-catalyzed oxyfunctionalization of various alkane substrates90 (Table 3 and Fig. 6a). Through the methanol-driven reductive O2 activation, Au–TiO2 provided H2O2 in concentrations with which the enzyme remained active and stable. Later, the same authors, demonstrated that also water may serve as sacrificial electron donor.91 Compared to the use of methanol, reaction rates and robustness of the overall reaction were significantly impaired. This may be attributed to higher rate of the photocatalytic oxidation of alcohols compared to water. Furthermore, alcohols act as radical scavengers thereby prolonging the enzyme lifetime.92 The radical inactivation of the biocatalyst by surface-borne by hydroxyl radicals (HO˙) can also be alleviated by spatial separation of the photo- and biocatalyst. Under optimized reaction conditions maximum TTN for the biocatalyst of 220000 and space time yields (STY) of 2.6 g L−1 d−1 together with photonic efficiencies of 13.6% were achieved.
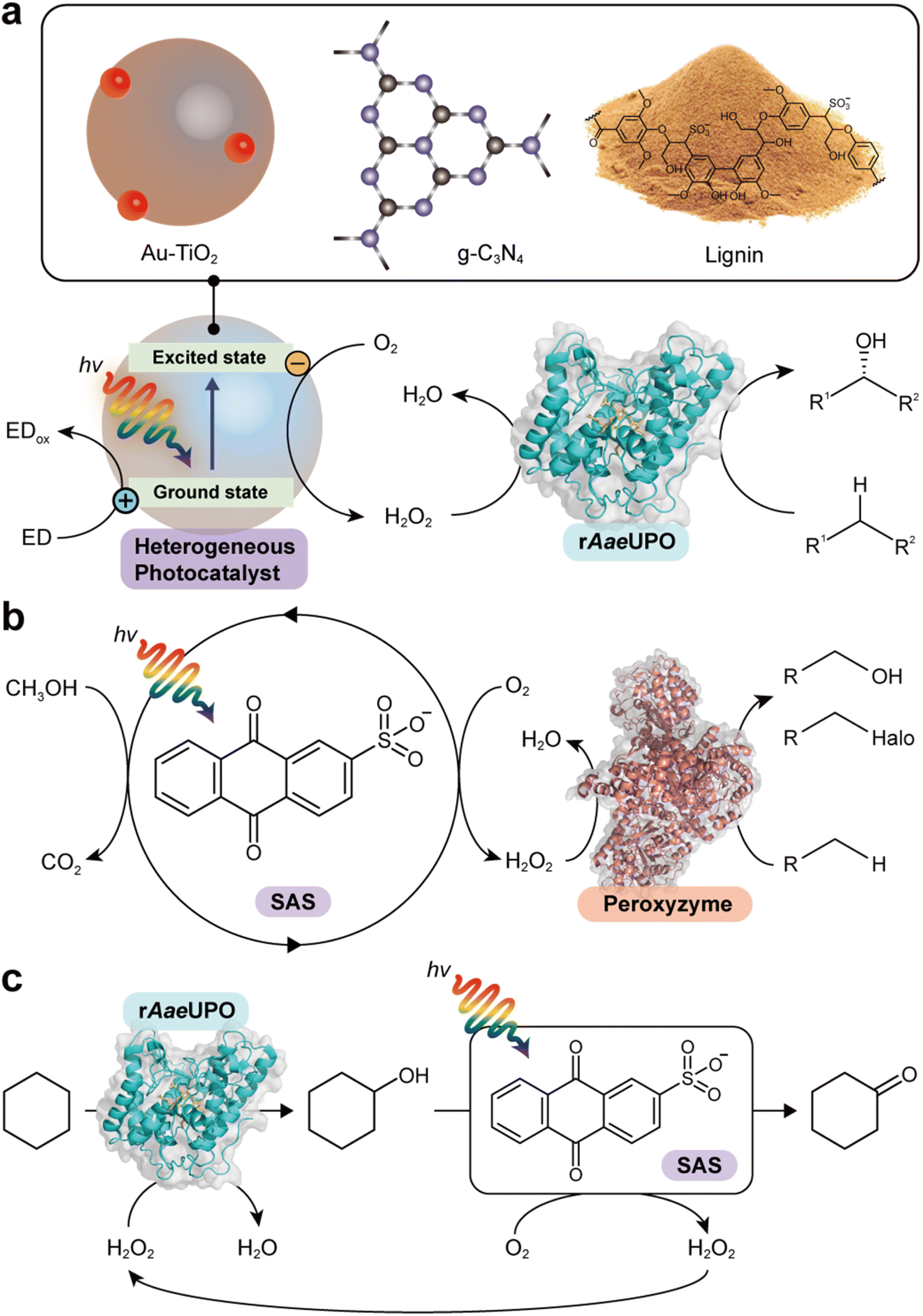 | ||
| Fig. 6 Photocatalytic formation of H2O2 and selective oxidation reactions catalyzed by rAaeUPO and peroxyzyme. Representative photocatalyts include Au–TiO2,90 g-C3N4,93 lignin99 (a), and anthraquinone (SAS)97,106 (b). (c) Photocatalytic oxidation of cyclohexane to cyclohexanone. The SAS-catalyzed aerobic overoxidation of cyclohexanol to cyclohexanone produces H2O2 as a byproduct, which itself is used to support the rAaeUPO-catalyzed hydroxylation of cyclohexane.97 | ||
4.3 Carbon nitrides
Carbon nitrides are known to catalyse light-induced water oxidation yielding O2 and H2, they can also be used for the oxidation of alcohols to the corresponding carbonyl groups.105 Instead of transferring electrons obtained from water- or alcohol oxidation to protons also O2 can serve as electron acceptor eventually yielding H2O2. Graphitic carbon nitride (g-C3N4) for example is a good alternative to the TiO2-based photocatalysts93 (Table 3 and Fig. 6a). Again, however, surface-bourne hydroxyl radicals impair the stability of the biocatalyst, which can be overcome by spatial separation of the bio- and photo catalyst. Using different wavelengths can tune the redox potential of a carbon nitride photocatalyst (CN-OA-m) and thereby influence the selectivity of the photoenzymatic cascade.984.4 Lignin
Lignin, a major component that accounts for 20–30% of plants, is a heterogeneous polymer mostly consisting of aromatic subunits. This year Kim et al. identified lignin as a multifunctional photocatalyst for such as in situ H2O2 formation through H2O oxidation and O2 reduction.99 Particularly appealing is the intrinsic hydroxyl radial-scavenging activity enabling more robust photoenzymatic cascade reactions without the need for spatial separation of both catalysts (Fig. 6a). This very promising combination will certainly yield interesting photoenzymatic reaction schemes.4.5 Anthraquinone
Water-soluble anthraquinone 2-sulfonate (SAS) is a simple, robust, and promising organo-photocatalyst enabling simpler sacrificial electron donors than EDTA such as primary alcohols.97,106 This H2O2 generation photocatalyst has been demonstrated with peroxygenase- and haloperoxidase-catalyzed oxidation reactions97 (Fig. 6b). The ability of SAS to also oxidize secondary alcohols to ketones enabled a truly chemoenzymatic reaction converting cyclohexane into cyclohexanone with the first step being peroxygenase-catalyzed and promoted by the H2O2 obtained from the second, SAS-photocatalytic cyclohexanol-oxidation step97 (Fig. 6c).4.6 γ-Ray-induced H2O2 formation
γ-Rays induce (non-catalytic) water splitting and significant formation of H2O2 in aqueous media exposed to γ-irradiation.107 This phenomenon can be exploited to promote peroxygenase-catalysed oxyfunctionalization reactions,108 potentially opening up new possibilities of making use of nuclear wastes. For example, a proper dose rate of gamma radiation (12.9 Gy min−1) could provide suitable H2O2 concentration (0.1 mM at steady state with an initial rate of 0.1 mM h−1 for H2O2 formation) that enables significantly increased product formation while minimizing the inactivation of rAaeUPO.108 Again, inactivation of the biocatalysts by the in situ formed reactive oxygen species represented a major impediment en route to preparative application, which may be overcome by spatial separation of the biocatalyst from the radiation source (e.g. in a flow chemistry setup).5. Photoenzymatic oxyfunctionalization: current state of the art
The peroxygenase from Agrocybe aegerita (AaeUPO) represents the most frequently used representative of novel peroxygenases capable of selective oxyfunctionalization chemistry.48,54,75,109 Therefore, it is not very astonishing that it also dominates the following section of practical examples of photoenzymatic oxyfunctionalization reactions.
Table 3 and Fig. 7 summarize the current state of the art of photoenzymatic oxyfunctionalization reactions. Whereas possible, a particular focus was put on the TTN of the biocatalyst as this number also gives the reader an impression of the biocatalyst usage. Today, still enzymes are generally perceived as expensive, which per se is not true. The costs of enzyme production are subject to economy of scale which means that the price for the production of an enzyme exponentially decrease with the scale of its fermentation.110 If produced on small scale (i.e. <1 m3) production costs for an enzyme can easily surpass 10000 € kg−1 or more. This is typically the case with enzymes commercialized by SMEs and general chemical vendors and probably motivates scientists for statements like ‘enzymes are expensive’. If, however, a biocatalyst is produced on large scale (i.e. ≫10 m3) the production costs drop to values in the range of 250–1000 € kg−1, prices that are difficult to beat by traditional chemical catalysts (especially of rare transition metal catalysts or catalysts obtained from complex multi-step syntheses).
 | ||
| Fig. 7 Highest-record photobiocatalytic hydroxylation reactions of non-activated C–H bonds. All the photobiocatalysis were catalyzed by rAaeUPO (PaDa-I mutant) except (f) which was catalyzed by wild-type AaeUPO. TTN (nmol product per nmol enzyme) and enantioselectivity (ee%) for (R)-isoform were shown. References: (a);96 (b), (m), (o)–(r);90 (c)–(e), (g), (i)–(l), (n);98 (f);84 (h).99 | ||
Furthermore, the cost contribution of an enzyme to the cost of the desired product directly correlate with its catalytic performance (or TTN). Hence, to attain a cost contribution of less than 10 € molProduct−1 (acceptable for the production of active pharmaceutical ingredients) enzymes produced at 250 or 10000 € kgenzyme−1 would have to perform at least 1200 or 50000, respectively. Consequently, reaching a cost contribution of less than 0.1 € molProduct−1 requires TTN of more than 120000 or 5000000, respectively. Of course, many other factors contribute to the final production costs of a chemical compound but the TTN at least gives a first orientation about the economic potential of an enzymatic process.
A range of photobiocatalytic oxyfunctionalization reactions, especially using AaeUPO, have been reported (Table 3). The reaction- and product scope attainable matches the scope of AaeUPO reactions reported using conventional in situ H2O2 generation systems and/or simple addition of H2O2.5 The same is true concerning TNs reported for AaeUPO in these photobiocatalytic reactions that fall in the range of 17500 to 372000. In line with the above-discussed issues limitations of P450 monooxygenases following the H2O2 shunt pathway, the TNs observed with those enzymes typically fall back by one to two orders of magnitude behind those reported with peroxygenases (Fig. 5 and 7).
One common feature of most photoenzymatic reactions reported so far is the very low product concentrations, generally being in the lower millimolar range. Obviously, this is a result of the poor water solubility of the (mostly hydrophobic) starting materials of interest. Such low product titres, however, significantly impair their economic viability and also result in enormous amounts of waste water.111 One possibility is to apply water-miscible organic cosolvents to increase the solubility of the hydrophobic reagents. Particularly, peroxygenases appear very suitable for such harsh reaction conditions as e.g. AaeUPO can withstand very high concentrations of typical organic cosolvents.112 Kara and coworkers recently reported g-C3N4–adsorbed AaeUPO in a microaqueous reaction system comprised of cyclohexane as the main liquid phase.95 This approach, provided it is further optimized, may address this substrate-solubility issue at least for liquid starting materials. Another option to increase the overall reagent concentration is to make use of the so-called two-liquid-phase-system approach (2LPS).113 In the 2LPS approach, a nonpolar organic phase serves substrate reservoir and product sink as well with the reagents partitioning between the organic layer and the aqueous, catalyst-containing, aqueous phase. Phase transfer limitations, as often observed in such heterogeneous reaction media, can be alleviated by emulsion formation and thereby maximization of the interphase area as demonstrated by Churakova et al. in the photoenzymatic sulfoxidation thioanisol.114
6. Light-driven biocatalytic dehydrogenation
6.1 Photo(electro)chemical oxidation of NAD(P)H
NAD(P)+-dependent dehydrogenases catalyse a range of synthetically relevant oxidation reactions such as the regio- and/or stereoselective oxidation of alcohols to the corresponding aldehydes or ketones. In the catalytic cycle NAD(P)+ is reduced to NAD(P)H. For economic reasons, the latter needs to be recycled back into its oxidised form. From a thermodynamic point-of-view, O2 is an attractive electron acceptor because the redox potential of O2 reduction [Ered(O2/O2˙−) = 0.33 V vs. normal hydrogen electrode (NHE), Ered(O2/H2O2) = 0.27 V vs. NHE at pH 7]115,116 is more positive than that of NADH oxidation [Eox(NAD+/NADH) = −0.32 V vs. NHE at pH 7.0].117 Furthermore, only H2O2 (which can easily be dismutated into O2 and H2O) or H2O are formed as by-products. NAD(P)H, however, is comparably stable against direct oxidation with O2 necessitating catalytic acceleration of the reaction. Amongst the enzymatic methods, NAD(P)H oxidases have been established.118 Also non-enzymatic catalysts such as ABTS,119,120 quinoids,121 flavins26,122,123 or organic dyes124 such as Congo Red24 have been reported for the aerobic reoxidation of NAD(P)H. The sometimes rather sluggish NAD(P)H oxidation kinetics of these reagents can be accelerated by illumination.6.2 Light-driven biocatalytic dehydrogenation reactions
The Park group reported a representative example for photobiocatalytic oxidation reactions,24 in which β-sheet-rich amyloid nanofibers performed NAD+ regeneration in couple with enzymatic alcohol oxidation under an aerobic environment (Fig. 8a). Note that in contrast to many studies on photo(electro)catalytic NADH recycling,125,126 the reverse reaction (i.e., NAD(P)+ production) has been rarely investigated despite the importance of the process. A solvatochromic Congo red (CR) was hybridized into an amyloid-derived peptide [e.g., Fmoc-diphenylalanine (Fmoc-FF)] through π–π interaction between aromatic functional groups of the Fmoc-FF and CR. This interaction made CR photocatalytic because the CR's donor–acceptor couple in the β-sheet-rich nanostructure became highly planarized, which restrained nonradiative relaxation via CR's twisted intramolecular charge-transfer. As a result, CR performed O2-fueled regeneration of NAD+ under visible light by (i) transferring CR's photoexcited electrons (in a locally excited state) to O2 and (ii) delivering NADH's electrons to CR. The regenerated NAD+ activated alcohol dehydrogenase (ADH) from Saccharomyces cerevisiae (ScADH) to oxidize ethanol to acetaldehyde [ScADH's total turnover number (TTNScADH): 57692, TTNNADH: 3, TTNCR: 30, yield: 60%].
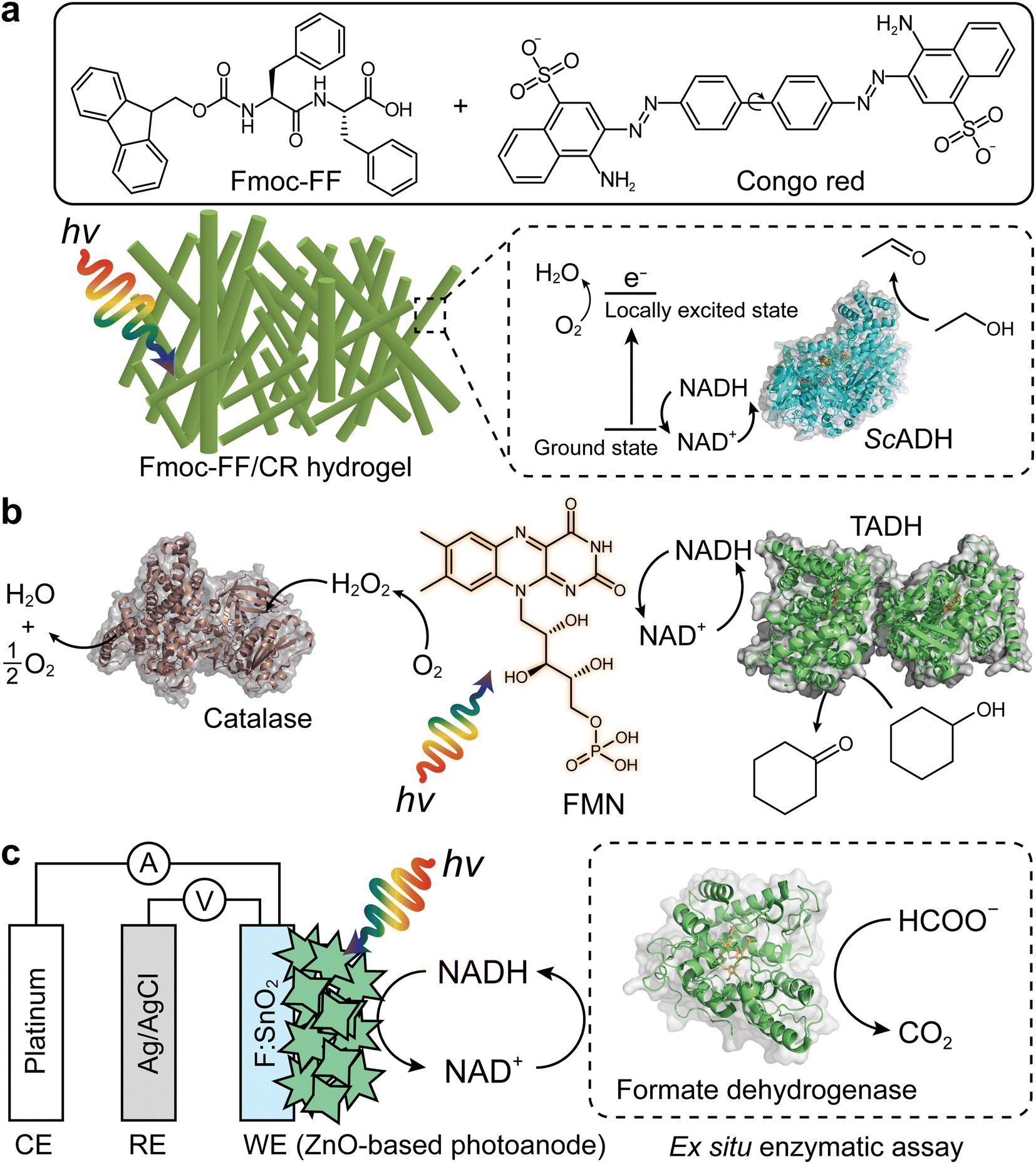 | ||
| Fig. 8 Photo(electro)chemical regeneration of NAD(P)+ for biocatalytic oxidation reactions. (a) Fmoc-FF/CR-driven NAD+ recycling for biocatalytic oxidation of alcohol under visible light.24 (b) Photoenzymatic synthesis of cyclohexanone through coupling of flavin mononucleotide-sensitized regeneration of NAD+ and TADH-driven oxidation.26 (c) ZnO-Based photoelectrochemical oxidation of NADH to NAD+.27 The formation of NAD+ was confirmed by ex situ enzymatic assay. WE: working electrode. RE: reference electrode. CE: counter electrode. | ||
Another example of photocatalyst—using O2 as an electron acceptor—are flavin molecules,26,123 such as flavin mononucleotide (FMN), flavin adenine dinucleotide, and riboflavin. The Hollmann group demonstrated the use of flavins to oxidize NAD(P)H under light conditions26 (Fig. 8b). Flavin photoexcitation made flavin's redox potential more positive, facilitating its NAD(P)+ regeneration in the presence of O2. The flavin-sensitised regeneration of NAD(P)+ activated ADH from Thermus sp. ATN1 (TADH) to catalyse the oxidation of cyclohexanone to cyclohexanol [TTNNADH: 170, TTNFMN: 340, TTNTADH: 4359, FMN's turnover frequency (TOFFMN): 38 h−1, TOFTADH: 498 h−1]. In the photoenzymatic reaction, catalase was used to dismutate H2O2 to O2 and H2O. The photocatalytic NAD+ regeneration system was also applied to make ADH from horse liver (HLADH) catalyse the oxidation of diols e.g., 1,4-butanediol,26 1,5-pentanediol,26meso-3-methyl-1,5-pentanediol123 to enantiopure lactones via hemiacetal intermediates [yield: 85–100%, TTNNADH: 8.5–200, TTNFMN: 85–400, TTNHLADH: 1150].
One disadvantage of O2 as terminal electron acceptor is its low solubility in aqueous media necessitating efficient external aeration. Photoelectrochemical (PEC) systems can overcome this issue because mechanistically (photo)anodes serve as electron acceptors.127–130 Ottone et al.27 reported ZnO materials for PEC regeneration of NAD+ (Fig. 8c). Dual simulated solar irradiation (180 mW cm−2) and applied voltage (1.41 V vs. NHE at pH 7) on porous ZnO and flower-like ZnO (ZnOF) photoanodes drove the oxidation of NADH to NAD+ with conversions of 34.7% and 58.7%, respectively. An ex situ enzymatic assay using formate dehydrogenase confirmed that enzymatically active form of NAD+—formed by the ZnOF film—was 94%, not 100%. The minor loss should originate from dimerization and/or fragmentation of NADH due to direct oxidation. Taken together, multiple photochemical and photoelectrochemical systems open up sustainable enzymatic oxidation reactions through light-powered NAD(P)+ regeneration.
7. Conclusions and perspectives
Photobiocatalytic oxidation is an emerging synthetic route but still in infancy. For example, only a few cases of light-driven monooxygenase catalysis have been reported,12,13,29,31,36 and their catalytic activities are quite low (TTN < 1000). For operation of H2O2-accepting enzymes, light-driven in situ H2O2 generation is advantageous because it uses a clean light energy source and does not need any cofactors and additional enzymes. But the limited number of UPOs and P450 peroxygenases poses an obstacle to broader applications. Genome mining of new UPOs and heterologous expression in a suitable heterologous host have been tried, but the number of characterized UPOs remains limited. While the current target for genome mining mostly focused on fungal sources, its range should be expanded to other kingdoms, such as bacteria and plants, to obtain a diverse spectrum of UPOs. For directed evolution to achieve desired activity and stability, high-throughput screening (HTS) system should be applied to select mutants having the enhanced properties from the enzyme library. Information on 3D structures of diverse UPOs are essential for enzyme engineering, only a few structures of UPOs are available. On the other hand, the P450 peroxygenases (CYP152 family) catalyze only hydroxylation and decarboxylation reactions of fatty acids. More genome mining of new P450 peroxygenases is necessary for the expansion of substrate scope and their applications.Light-driven reaction systems compete with more traditional approaches and today still the light-driven approaches appear to be less efficient compared to the latter. The direct photochemical regeneration of PAMO for example allowed for only a fraction of catalytic turnovers for the enzyme as compared to the reaction system utilizing NADPH together with an enzymatic regeneration system (96 TTNs as compared to 9471).29,131 Similarly, for the CYP152L1-catalyzed oxidative decarboxylation and hydroxylation of ω-OH lauric acid, the conversion achievable by a light-driven system (FMN/EDTA/light) was less than 10% of that by a secondary enzymatic system (CamAB/NADH/FDH/formic acid).104 Obviously, direct photochemical regeneration systems need significant improvement in terms of activity and robustness. Particularly, the interaction of the non-natural redox partner with the enzyme active site has to be engineered. Furthermore, in case of O2-dependent enzymes, the oxygen dilemma is still pending a promising solution.6 On the other hand, for ADH-catalyzed oxidation reactions, the aerobic regeneration of NAD+ by photochemical system (FMN/light) (TTNNAD of 170)26 appeared to be comparable in terms of cofactor recycling to the NADH oxidase system (TTNNAD of 109).132 Since most reports on light-driven biocatalytic oxidation have focused so far on the demonstration of proof-of-concepts, further engineering studies on the optimization of reaction conditions should enhance their performances.
We should also consider technical issues for practical applications of photobioreactors. TiO2-based photocatalysts rely on a limited wavelength range (to near UV and blue light), thus leaving a significant spectrum of solar energy unexploited. In this context, a recent study using multiple wavelength-complementary photocatalysts provides insight into more efficient use of the polychromatic light energy.94 Regarding the light sources, white light bulbs or light-emitting diodes (LEDs) have been widely used for photochemically driving several redox enzymes7 including UPOs (Table 3), P450 peroxygenases (Fig. 5), and dehydrogenases (Fig. 8). A versatile and advanced photoreactor platform is desired for HTS, preparative-scale batch reactions, and continuous processing, like the ‘Photochemistry LED Illuminator’ that identifies the optimal wavelength and power for specific photochemical transformation.96
Whole cell-based approaches are attractive for the scale-up and industrial applications of light-driven biocatalytic oxidation (Fig. 9). Future efforts are needed for de novo synthesis of photocatalysts, peroxygenase engineering through rational design and/or directed evolution, and reaction optimization in a one-pot cascade process. The whole-cell platform should allow for high substrate concentration, consequently high productivities can be obtained. Therefore, engineering of strains by integration of desired peroxygenase genes into the genome will also accelerate the development of improved bacterial strains for massive production of desired chemicals.
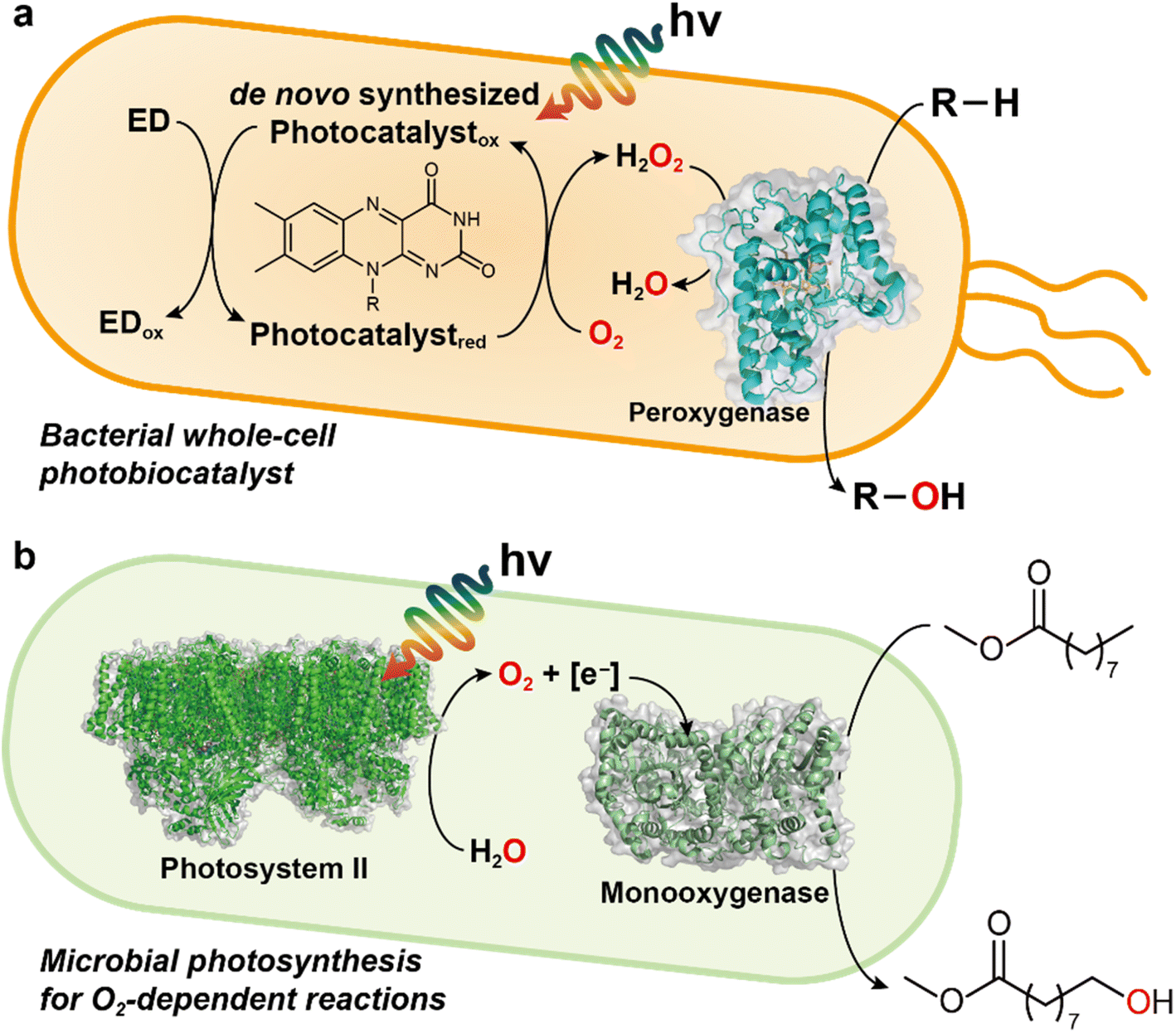 | ||
| Fig. 9 Examples of light-driven biocatalytic oxidation in microorganisms. (a) Photoenzymatic, whole-cell biocatalyst expressing peroxygenase genes. De novo synthesized photocatalysts (e.g., FMN, FAD) quickly react with O2 to yield H2O2 in the presence of electron donor (ED) and light. The peroxygenase can hydroxylate the substrate (R–H) by using H2O2 as an oxygen donor. EDTA or TEOA can be used as an ED and other EDs such as methanol, formate, or simple amino acids may serve as alternatives. (b) Photosystem-assisted monooxygenase reaction. Photosystem II oxidizes water for producing O2 and reducing equivalents (e.g., NADH). These molecules are consumed for monooxygenase-driven catalysis (e.g., oxyfunctionalization). | ||
Future research should focus on preparative oxyfunctionalization chemistry of peroxygenases. In a recent review, Dong et al.4 provided guidelines to assess the catalytic activity and productivity for the practical usefulness of enzymes for the production of desired products. According to the enzyme performance requirements of a given product category of pharma, fine chemical, speciality chemical, and bulk chemicals, required minimal productivity (TTN) for an enzyme is 4000, 26666, 160000, and 800000, respectively.4,109 Meanwhile, minimally required STY (g L−1 d−1) values for fine chemical production and pharmaceuticals are 2.4 and 0.024, respectively.133 Based on AaeUPO's performance by photochemically driven UPO catalysis, the requirements up to ‘speciality chemical’ (TTN of 160000) are satisfied for some cases of UPO-catalyzed reactions (Table 3 and Fig. 7). For wide expansion of UPO's scope to the production of “bulk chemicals,” further development of new UPOs, highly active and stable proteins, and whole-cell platforms are necessary.
When combined with the given knowledge and advances of photocatalysis, the unique catalytic activities and structures of monooxygenases, peroxygenases, and dehydrogenases promise a future route for greener synthesis using solar energy. Current challenges of the limited number of redox enzymes, narrow substrate range, and low stability are expected to be overcome by genome mining of new enzymes, protein engineering, and the development of whole-cell biocatalysts. A sustainable photobioreactor with optimized light source, robust photocatalyst, and waste-free electron donor is needed for driving preparative chemistry of peroxygenases and dehydrogenases.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) (Grant numbers: NRF-2015R1A3A2066191, NRF-2021R1A2C2092565, NRF-2019H1A2A1075810), Republic of Korea.References
- E. Roduner, W. Kaim, B. Sarkar, V. B. Urlacher, J. Pleiss, R. Gläser, W.-D. Einicke, G. A. Sprenger, U. Beifuß, E. Klemm, C. Liebner, H. Hieronymus, S.-F. Hsu, B. Plietker and S. Laschat, ChemCatChem, 2013, 5, 82–112 CrossRef CAS.
- M. P. van der Helm, B. Klemm and R. Eelkema, Nat. Rev. Chem., 2019, 3, 491–508 CrossRef CAS.
- X. Ye and C.-H. Tan, Chem. Sci., 2021, 12, 533–539 RSC.
- J. Dong, E. Fernández-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS.
- M. Hobisch, D. Holtmann, P. G. de Santos, M. Alcalde, F. Hollmann and S. Kara, Biotechnol. Adv., 2021, 51, 107615 CrossRef CAS PubMed.
- D. Holtmann and F. Hollmann, ChemBioChem, 2016, 17, 1391–1398 CrossRef CAS.
- S. H. Lee, D. S. Choi, S. K. Kuk and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 7958–7985 CrossRef CAS PubMed.
- L. Schmermund, V. Jurkas, F. F. Ozgen, G. D. Barone, H. C. Büchsenschütz, C. K. Winkler, S. Schmidt, R. Kourist and W. Kroutil, ACS Catal., 2019, 9, 4115–4144 CrossRef CAS.
- M. C. R. Rauch, M. M. E. Huijbers, M. Pabst, C. E. Paul, M. Pešić, I. W. C. E. Arends and F. Hollmann, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140303 CrossRef CAS PubMed.
- F. F. Özgen, M. E. Runda and S. Schmidt, ChemBioChem, 2021, 22, 790–806 CrossRef.
- M. Bocola, F. Schulz, F. Leca, A. Vogel, M. W. Fraaije and M. T. Reetz, Adv. Synth. Catal., 2005, 347, 979–986 CrossRef CAS.
- T. K. Le, J. H. Park, D. S. Choi, G. Y. Lee, W. S. Choi, K. J. Jeong, C. B. Park and C.-H. Yun, Green Chem., 2019, 21, 515–525 RSC.
- T. K. Le, J. Kim, N. A. Nguyen, T. H. H. Nguyen, E. G. Sun, S. M. Yee, H. S. Kang, S. J. Yeom, C. B. Park and C.-H. Yun, ChemSusChem, 2021, 14, 3054–3058 CrossRef CAS PubMed.
- E. M. J. Gillam, Z. Y. Guo and F. P. Guengerich, Arch. Biochem. Biophys., 1994, 312, 59–66 CrossRef CAS PubMed.
- M. Girhard, S. Schuster, M. Dietrich, P. Dürre and V. B. Urlacher, Biochem. Biophys. Res. Commun., 2007, 362, 114–119 CrossRef CAS.
- A. Dennig, M. Kuhn, S. Tassoti, A. Thiessenhusen, S. Gilch, T. Bulter, T. Haas, M. Hall and K. Faber, Angew. Chem., Int. Ed., 2015, 54, 8819–8822 CrossRef CAS.
- P. Molina-Espeja, E. Garcia-Ruiz, D. Gonzalez-Perez, R. Ullrich, M. Hofrichter and M. Alcalde, Appl. Environ. Microbiol., 2014, 80, 3496–3507 CrossRef.
- P. Molina-Espeja, S. Ma, D. M. Mate, R. Ludwig and M. Alcalde, Enzyme Microb. Technol., 2015, 73–74, 29–33 CrossRef CAS.
- M. P. J. Deurzen, B. W. van Groen, F. Rantwijk and R. A. van Sheldon, Biocatalysis, 1994, 10, 247–255 CrossRef.
- D. Cannella, K. B. Möllers, N.-U. Frigaard, P. E. Jensen, M. J. Bjerrum, K. S. Johansen and C. Felby, Nat. Commun., 2016, 7, 11134 CrossRef CAS PubMed.
- B. Bissaro, Z. Forsberg, Y. Ni, F. Hollmann, G. Vaaje-Kolstad and V. G. H. Eijsink, Green Chem., 2016, 18, 5357–5366 RSC.
- B. Bissaro, E. Kommedal, Å. K. Røhr and V. G. H. Eijsink, Nat. Commun., 2020, 11, 890 CrossRef CAS.
- B. M. Blossom, D. A. Russo, R. K. Singh, B. van Oort, M. B. Keller, T. I. Simonsen, A. Perzon, L. F. Gamon, M. J. Davies, D. Cannella, R. Croce, P. E. Jensen, M. J. Bjerrum and C. Felby, ACS Sustainable Chem. Eng., 2020, 8, 9301–9310 CrossRef CAS.
- G. Son, J. Kim and C. B. Park, ACS Appl. Energy Mater., 2020, 3, 1215–1221 CrossRef CAS.
- A. J. Ganzhorn, D. W. Green, A. D. Hershey, R. M. Gould and B. V. Plapp, J. Biol. Chem., 1987, 262, 3754–3761 CrossRef CAS.
- S. Gargiulo, I. W. C. E. Arends and F. Hollmann, ChemCatChem, 2010, 3, 338–342 CrossRef.
- C. Ottone, D. Pugliese, M. Laurenti, S. Hernández, V. Cauda, P. Grez and L. Wilson, ACS Appl. Mater. Interfaces, 2021, 13, 10719–10727 CrossRef CAS.
- Q. Guo, L. Gakhar, K. Wickersham, K. Francis, A. Vardi-Kilshtain, D. T. Major, C. M. Cheatum and A. Kohen, Biochemistry, 2016, 55, 2760–2771 CrossRef CAS PubMed.
- F. Hollmann, A. Taglieber, F. Schulz and M. T. Reetz, Angew. Chem., Int. Ed., 2007, 46, 2903–2906 CrossRef CAS PubMed.
- A. Taglieber, F. Schulz, F. Hollmann, M. Rusek and M. T. Reetz, ChemBioChem, 2008, 9, 565–572 CrossRef CAS.
- J. H. Park, S. H. Lee, G. S. Cha, D. S. Choi, D. H. Nam, J. H. Lee, J.-K. Lee, C.-H. Yun, K. J. Jeong and C. B. Park, Angew. Chem., Int. Ed., 2015, 54, 969–973 CrossRef CAS PubMed.
- F. F. Özgen, M. E. Runda, B. O. Burek, P. Wied, J. Z. Bloh, R. Kourist and S. Schmidt, Angew. Chem., Int. Ed., 2020, 59, 3982–3987 CrossRef.
- F. Mascia, S. B. Pereira, C. C. Pacheco, P. Oliveira, J. Solarczek, A. Schallmey, R. Kourist, V. Alphand and P. Tamagnini, Green Chem., 2022, 24, 6156–6167 RSC.
- E. Erdem, L. Malihan-Yap, L. Assil-Companioni, H. Grimm, G. D. Barone, C. Serveau-Avesque, A. Amouric, K. Duquesne, V. de Berardinis, Y. Allahverdiyeva, V. Alphand and R. Kourist, ACS Catal., 2022, 12, 66–72 CrossRef CAS PubMed.
- A. Hoschek, A. Schmid and B. Bühler, ChemCatChem, 2018, 10, 5366–5371 CrossRef CAS.
- N. H. Tran, D. Nguyen, S. Dwaraknath, S. Mahadevan, G. Chavez, A. Nguyen, T. Dao, S. Mullen, T. A. Nguyen and L. E. Cheruzel, J. Am. Chem. Soc., 2013, 135, 14484–14487 CrossRef CAS PubMed.
- Q. Lam, A. Cortez, T. T. Nguyen, M. Kato and L. Cheruzel, J. Inorg. Biochem., 2016, 158, 86–91 CrossRef CAS PubMed.
- M. Kato, D. Nguyen, M. Gonzalez, A. Cortez, S. E. Mullen and L. E. Cheruzel, Biorog. Med. Chem., 2014, 22, 5687–5691 CrossRef CAS.
- Q. Lam, M. Kato and L. Cheruzel, Biochim. Biophys. Acta, 2016, 1857, 589–597 CrossRef CAS.
- S. H. Lee, Y. C. Kwon, D. M. Kim and C. B. Park, Biotechnol. Bioeng., 2013, 110, 383–390 CrossRef CAS PubMed.
- J. H. Lee, D. H. Nam, S. H. Lee, J. H. Park, C. B. Park and K. J. Jeong, J. Ind. Eng. Chem., 2016, 33, 28–32 CrossRef CAS.
- K. Jensen, P. E. Jensen and B. L. Møller, ACS Chem. Biol., 2011, 6, 533–539 CrossRef CAS.
- S. B. Mellor, A. Z. Nielsen, M. Burow, M. S. Motawia, D. Jakubauskas, B. L. Moller and P. E. Jensen, ACS Chem. Biol., 2016, 11, 1862–1869 CrossRef CAS PubMed.
- A. Hoschek, B. Bühler and A. Schmid, Angew. Chem., Int. Ed., 2017, 56, 15146–15149 CrossRef CAS PubMed.
- Z. Zhao, D. Lan, X. Tan, F. Hollmann, U. T. Bornscheuer, B. Yang and Y. Wang, ACS Catal., 2019, 9, 2916–2921 CrossRef CAS.
- F. Björkling, S. E. Godtfredsen and O. Kirk, Chem. Commun., 1990, 19, 1301–1303 RSC.
- G. T. Höfler, A. But and F. Hollmann, Org. Biomol. Chem., 2019, 17, 9267–9274 RSC.
- M.-C. Sigmund and G. J. Poelarends, Nat. Catal., 2020, 3, 690–702 CrossRef CAS.
- O. Shoji and Y. Watanabe, J. Biol. Inorg Chem., 2014, 19, 529–539 CrossRef CAS PubMed.
- P. D. Shaw and L. P. Hager, J. Biol. Chem., 1961, 236, 1626–1630 CrossRef CAS.
- F. van Rantwijk and R. A. Sheldon, Curr. Opin. Biotechnol., 2000, 11, 554–564 CrossRef CAS.
- R. Ullrich, J. Nuske, K. Scheibner, J. Spantzel and M. Hofrichter, Appl. Environ. Microbiol., 2004, 70, 4575–4581 CrossRef CAS PubMed.
- F. Tonin, F. Tieves, S. Willot, A. van Troost, R. van Oosten, S. Breestraat, S. van Pelt, M. Alcalde and F. Hollmann, Org. Process Res. Dev., 2021, 25, 1414–1418 CrossRef CAS PubMed.
- A. Beltrán-Nogal, I. Sánchez-Moreno, D. Méndez-Sánchez, P. G. de Santos, F. Hollmann and M. Alcalde, Curr. Opin. Struct. Biol., 2022, 73, 102342 CrossRef PubMed.
- C. Aranda, J. Carro, A. González-Benjumea, E. D. Babot, A. Olmedo, D. Linde, A. T. Martínez and A. Gutiérrez, Biotechnol. Adv., 2021, 51, 107703 CrossRef CAS.
- H. Joo, Z. Lin and F. H. Arnold, Nature, 1999, 399, 670–673 CrossRef CAS PubMed.
- A. W. Munro, K. J. McLean, J. L. Grant and T. M. Makris, Biochem. Soc. Trans., 2018, 46, 183–196 CrossRef CAS PubMed.
- O. Fujishiro, O. Shoji, S. Nagano, H. Sugimoto, Y. Shiro and Y. Watanabe, J. Biol. Chem., 2011, 286, 29941–29950 CrossRef PubMed.
- C. E. Paul, E. Churakova, E. Maurits, M. Girhard, V. B. Urlacher and F. Hollmann, Bioorg. Med. Chem., 2014, 22, 5692–5696 CrossRef CAS PubMed.
- L. Hammerer, M. Friess, J. Cerne, M. Fuchs, G. Steinkellner, K. Gruber, K. Vanhessche, T. Plocek, C. K. Winkler and W. Kroutil, ChemCatChem, 2019, 22, 5642–5649 CrossRef.
- J. Belcher, K. J. McLean, S. Matthews, L. S. Woodward, K. Fisher, S. E. J. Rigby, D. R. Nelson, D. Potts, M. T. Baynham, D. A. Parker, D. Leys and A. W. Munro, J. Biol. Chem., 2014, 289, 6535–6550 CrossRef CAS.
- Y. Jiang, Z. Li, C. Wang, Y. J. Zhou, H. Xu and S. Li, Biotechnol. Biofuels, 2019, 12, 79 CrossRef PubMed.
- H. Onoda, O. Shoji, K. Suzuki, H. Sugimoto, Y. Shiro and Y. Watanabe, Catal. Sci. Technol., 2018, 8, 434–442 RSC.
- M. A. Rude, T. S. Baron, S. Brubaker, M. Alibhai, S. B. Del Cardayre and A. Schirmer, Appl. Environ. Microbiol., 2011, 77, 1718–1727 CrossRef CAS PubMed.
- N. Ma, Z. Chen, J. Chen, J. Chen, C. Wang, H. Zhou, L. Yao, O. Shoji, Y. Watanabe and Z. Cong, Angew. Chem., Int. Ed., 2018, 57, 7628–7633 CrossRef CAS PubMed.
- Z. Forsberg, M. Sorlie, D. Petrovic, G. Courtade, F. L. Aachmann, G. Vaaje-Kolstad, B. Bissaro, A. K. Rohr and V. G. H. Eijsink, Curr. Opin. Struct. Biol., 2019, 59, 54–64 CrossRef CAS PubMed.
- P. Chylenski, B. Bissaro, M. Sorlie, A. K. Rohr, A. Varnai, S. J. Horn and V. G. H. Eijsink, ACS Catal., 2019, 9, 4970–4991 CrossRef CAS.
- B. Wang, Z. Wang, G. J. Davies, P. H. Walton and C. Rovira, ACS Catal., 2020, 10, 12760–12769 CrossRef CAS.
- L. Ciano, G. J. Davies, W. B. Tolman and P. H. Walton, Nat. Catal., 2018, 1, 571–577 CrossRef CAS.
- B. L. Cantarel, P. M. Coutinho, C. Rancurel, T. Bernard, V. Lombard and B. Henrissat, Nucleic Acids Res., 2009, 37, D233–D238 CrossRef CAS PubMed.
- B. Bissaro, Å. K. Røhr, G. Müller, P. Chylenski, M. Skaugen, Z. Forsberg, S. J. Horn, G. Vaaje-Kolstad and V. G. H. Eijsink, Nat. Chem. Biol., 2017, 13, 1123–1128 CrossRef CAS PubMed.
- Y. Gaber, B. Rashad, R. Hussein, M. Abdelgawad, N. S. Ali, T. Dishisha and A. Várnaie, Biotechnol. Adv., 2020, 43, 107583 CrossRef CAS PubMed.
- V. G. H. Eijsink, D. Petrovic, Z. Forsberg, S. Mekasha, Å. K. Røhr, A. Várnai, B. Bissaro and G. Vaaje-Kolstad, Biotechnol. Biofuels, 2019, 12, 58 CrossRef PubMed.
- H. L. Wapshott-Stehli and A. M. Grunden, Enzyme Microb. Technol., 2021, 145, 109744 CrossRef CAS PubMed.
- B. O. Burek, S. Bormann, F. Hollmann, J. Z. Bloh and D. Holtmann, Green Chem., 2019, 21, 3232–3249 RSC.
- S. J. Freakley, S. Kochius, J. van Marwijk, C. Fenner, R. J. Lewis, K. Baldenius, S. S. Marais, D. J. Opperman, S. T. L. Harrison, M. Alcalde, M. S. Smit and G. J. Hutchings, Nat. Commun., 2019, 10, 4178 CrossRef PubMed.
- F. Hollmann and A. Schimid, J. Inorg. Biochem., 2009, 103, 313–315 CrossRef CAS PubMed.
- A. E. W. Horst, S. Bormann, J. Meyer, M. Steinhagen, R. Ludwig, A. Drews, M. Ansorge-Schumacher and D. Holtmann, J. Mol. Catal. B: Enzym., 2016, 133, S137–S142 CrossRef.
- T. Krieg, S. Huttmann, K.-M. Mangold, J. Schrader and D. Holtmann, Green Chem., 2011, 13, 2686–2689 RSC.
- L. Getrey, T. Krieg, F. Hollmann, J. Schrader and D. Holtmann, Green Chem., 2014, 16, 1104–1108 RSC.
- C. Kormann, D. W. D. W. Bahnemann and M. R. Hoffmann, Environ. Sci. Technol., 1988, 22, 798–806 CrossRef CAS PubMed.
- W. R. Frisell, C. W. Chung and C. G. Mackenzie, J. Biol. Chem., 1959, 234, 1297–1302 CrossRef CAS PubMed.
- D. I. Perez, M. M. Grau, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2009, 6848–6850 RSC.
- E. Churakova, M. Kluge, R. Ullrich, I. Arends, M. Hofrichter and F. Hollmann, Angew. Chem., Int. Ed., 2011, 50, 10716–10719 CrossRef CAS PubMed.
- E. Fernández-Fueyo, Y. Ni, A. G. Baraibara, M. Alcalde, L. M. van Langen and F. Hollmann, J. Mol. Catal. B: Enzym., 2016, 134, 347–352 CrossRef.
- Y. Ni, E. Fernandez-Fueyo, A. G. Baraibar, R. Ullrich, M. Hofrichter, H. Yanase, M. Alcalde, W. J. H. van Berkel and F. Hollmann, Angew. Chem., Int. Ed., 2016, 55, 798–801 CrossRef CAS PubMed.
- M. Pesic, S. J.-P. Willot, E. Fernández-Fueyo, F. Tieves, M. Alcalde and F. Hollmann, Z. Naturforsch., C: J. Biosci., 2019, 74, 101–104 CrossRef CAS PubMed.
- M. M. C. H. van Schie, A. T. Kaczmarek, F. Tieves, P. G. de Santos, C. E. Paul, I. W. C. E. Arends, M. Alcalde, G. Schwarz and F. Hollmann, ChemCatChem, 2020, 12, 3186–3189 CrossRef CAS.
- A. Al-Shameri, S. J.-P. Willot, C. E. Paul, F. Hollmann and L. Lauterbach, Chem. Commun., 2020, 56, 9667 RSC.
- W. Zhang, B. O. Burek, E. Fernández-Fueyo, M. Alcalde, J. Z. Bloh and F. Hollmann, Angew. Chem., Int. Ed., 2017, 56, 15451–15455 CrossRef CAS PubMed.
- W. Zhang, E. Fernandez-Fueyo, Y. Ni, M. van Schie, J. Gacs, R. Renirie, R. Wever, F. G. Mutti, D. Rother, M. Alcalde and F. Hollmann, Nat. Catal., 2018, 1, 55–62 CrossRef CAS PubMed.
- B. O. Burek, S. R. de Boer, F. Tieves, W. Zhang, M. van Schie, S. Bormann, M. Alcalde, D. Holtmann, F. Hollmann, D. W. Bahnemann and J. Z. Bloh, ChemCatChem, 2019, 11, 3093–3100 CrossRef CAS.
- M. M. C. H. van Schie, W. Zhang, F. Tieves, D. S. Choi, C. B. Park, B. O. Burek, J. Z. Bloh, I. W. C. E. Arends, C. E. Paul, M. Alcalde and F. Hollmann, ACS Catal., 2019, 9, 7409–7417 CrossRef CAS.
- S. J. P. Willot, E. Fernandez-Fueyo, F. Tieves, M. Pesic, M. Alcalde, I. W. C. E. Arends, C. B. Park and F. Hollmann, ACS Catal., 2019, 9, 890–894 CrossRef CAS PubMed.
- M. Hobisch, M. M. C. H. van Schie, J. Kim, K. R. Andersen, M. Alcalde, R. Kourist, C. B. Park, F. Hollmann and S. Kara, ChemCatChem, 2020, 12, 4009–4013 CrossRef CAS.
- H. E. Bonfield, K. Mercer, A. Diaz-Rodriguez, G. C. Cook, B. S. J. McKay, P. Slade, G. M. Taylor, W. X. Ooi, J. D. Williams, J. P. M. Roberts, J. A. Murphy, L. Schmermund, W. Kroutil, T. Mielke, J. Cartwright, G. Grogan and L. J. Edwards, ChemPhotoChem, 2020, 4, 45–51 CrossRef CAS.
- B. Yuan, D. Mahor, Q. Fei, R. Wever, M. Alcalde, W. Zhang and F. Hollmann, ACS Catal., 2020, 10, 8277–8284 CrossRef CAS.
- L. Schmermund, S. Reischauer, S. Bierbaumer, C. K. Winkler, A. Diaz-Rodriguez, L. J. Edwards, S. Kara, T. Mielke, J. Cartwright, G. Grogan, B. Pieber and W. Kroutil, Angew. Chem., Int. Ed., 2021, 60, 6965–6969 CrossRef CAS PubMed.
- J. Kim, T. V. T. Nguyen, Y. H. Kim, F. Hollmann and C. B. Park, Nat. Synth., 2022, 1, 217–226 CrossRef.
- D. S. Choi, Y. Ni, E. Fernandez-Fueyo, M. Lee, F. Hollmann and C. B. Park, ACS Catal., 2017, 7, 1563–1567 CrossRef CAS.
- M. Girhard, E. Kunigk, S. Tihovsky, V. V. Shumyantseva and V. B. Urlacher, Biotechnol. Appl. Biochem., 2013, 60, 111–118 CrossRef CAS PubMed.
- I. Zachos, S. K. Gaßmeyer, D. Bauer, V. Sieber, F. Hollmann and R. Kourist, Chem. Commun., 2015, 51, 1918–1921 RSC.
- K. Koninger, M. Grote, I. Zachos, F. Hollmann and R. Kourist, J. Visualized Exp., 2016, 111, e53439 Search PubMed.
- S. Bojarra, D. Reichert, M. Grote, A. G. Baraibar, A. Dennig, B. Nidetzky, C. Mugge and R. Kourist, ChemCatChem, 2018, 10, 1192–1201 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- W. Zhang, J. Gacs, I. W. C. E. Arends and F. Hollmann, ChemCatChem, 2017, 9, 3821–3826 CrossRef CAS.
- S. Le Caër, Water, 2011, 3, 235–253 CrossRef.
- W. Zhang, H. Liu, M. M. C. H. van Schie, P.-L. Hagedoorn, M. Alcalde, A. G. Denkova, K. Djanashvili and F. Hollmann, ACS Catal., 2020, 10, 14195–14200 CrossRef CAS PubMed.
- S. Bormann, A. G. Baraibar, Y. Ni, D. Holtmann and F. Hollmann, Catal. Sci. Technol., 2015, 5, 2038–2052 RSC.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2011, 15, 266–274 CrossRef CAS.
- Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2014, 6, 930–943 CrossRef CAS.
- T. Hilberath, A. van Troost, M. Alcalde and F. Hollmann, Front. Catal., 2022, 2, 882992 CrossRef.
- M. M. C. H. van Schie, J.-D. Spöring, M. Bocola, P. D. de María and D. Rother, Green Chem., 2021, 23, 3191–3206 RSC.
- E. Churakova, I. W. C. E. Arends and F. Hollmann, ChemCatChem, 2013, 5, 565–568 CrossRef CAS.
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. P. de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS.
- J. Kim, S. H. Lee, F. Tieves, C. E. Paul, F. Hollmann and C. B. Park, Sci. Adv., 2019, 5, eaax0501 CrossRef CAS PubMed.
- J. Kim and C. B. Park, Curr. Opin. Chem. Biol., 2019, 49, 122–129 CrossRef CAS PubMed.
- F. Hollmann, I. W. C. E. Arends and K. Buehler, ChemCatChem, 2010, 2, 762–782 CrossRef CAS.
- I. Schröder, E. Steckhan and A. Liese, J. Electroanal. Chem., 2003, 541, 109–115 CrossRef.
- S. Aksu, I. W. C. E. Arends and F. Hollmann, Adv. Synth. Catal., 2009, 351, 1211–1216 CrossRef CAS.
- G. Hilt, T. Jarbawi, W. R. Heineman and E. Steckhan, Eur. J. Chem., 1997, 3, 79–88 CrossRef CAS.
- J. B. Jones and K. E. Taylor, J. Chem. Soc., Chem. Commun., 1973, 205–206 RSC.
- M. Rauch, S. Schmidt, I. W. C. E. Arends, K. Oppelt, S. Kara and F. Hollmann, Green Chem., 2017, 19, 376–379 RSC.
- P. Könst, S. Kara, S. Kochius, D. Holtmann, I. W. C. E. Arends, R. Ludwig and F. Hollmann, ChemCatChem, 2013, 5, 3027–3032 CrossRef.
- J. Kim, S. H. Lee, F. Tieves, D. S. Choi, F. Hollmann, C. E. Paul and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 13825–13828 CrossRef CAS PubMed.
- D. Wang, J. Kim and C. B. Park, ACS Appl. Mater. Interfaces, 2021, 13, 58522–58531 CrossRef CAS PubMed.
- J. Kim, Y. W. Lee, E.-G. Choi, P. Boonmongkolras, B. W. Jeon, H. Lee, S. T. Kim, S. K. Kuk, Y. H. Kim, B. Shin and C. B. Park, J. Mater. Chem. A, 2020, 8, 8496–8502 RSC.
- J. Kim, Y. Um, S. Han, T. Hilberath, Y. H. Kim, F. Hollmann and C. B. Park, ACS Appl. Mater. Interfaces, 2022, 14, 11465–11473 CrossRef CAS PubMed.
- S. K. Kuk, J. Jang, J. Kim, Y. Lee, Y. S. Kim, B. Koo, Y. W. Lee, J. W. Ko, B. Shin, J.-K. Lee and C. B. Park, ChemSusChem, 2020, 13, 2940–2944 CrossRef CAS PubMed.
- J. Kim, J. Jang, T. Hilberath, F. Hollmann and C. B. Park, Nat. Synth., 2022, 1, 776–786 CrossRef.
- F. Schulz, F. Leca, F. Hollmann and M. T. Reetz, Beilstein J. Org. Chem., 2005, 1, 10 Search PubMed.
- B. R. Riebel, P. R. Gibbs, W. B. Wellborn and A. S. Bommarius, Adv. Synth. Catal., 2003, 345, 707–712 CrossRef CAS.
- M. K. Julsing, S. Cornelissen, B. Buhler and A. Schmid, Curr. Opin. Chem. Biol., 2008, 12, 177–186 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |