
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational development of molecularly imprinted nanoparticles for blocking PD-1/PD-L1 axis†
Zikuan
Gu‡
,
Shuxin
Xu‡
,
Zhanchen
Guo
and
Zhen
Liu
 *
*
State Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University 163 Xianlin Avenue, Nanjing 210023, China. E-mail: zhenliu@nju.edu.cn
First published on 30th August 2022
Abstract
Blocking the PD-1/PD-L1 immune checkpoint has emerged as a promising strategy in cancer immunotherapy, in which monoclonal antibodies are predominately used as inhibitors. Despite their remarkable success, monoclonal antibody-based therapeutics suffer from drawbacks due to the use of antibodies, such as high cost, low stability and high frequency of immune-related adverse effects. Therefore, novel anti-PD-1/PD-L1 therapeutics that can address these issues are of significant importance. Herein, we report a molecularly imprinted polymer (MIP) based PD-1 nano inhibitor for blocking the PD-1/PD-L1 axis. The anti-PD-1 nanoMIP was rationally designed and engineered by epitope imprinting using the N-terminal epitope of PD-1 as the binding site. The anti-PD-1 nanoMIP showed good specificity and high affinity towards PD-1, yielding a disassociation constant at the 10−8 M level, much better than that between PD-1 and PD-L1. Via steric hindrance, this inhibitor could effectively block PD-1/PD-L1 interaction. Besides, it could effectively reactivate T cells and reverse the chemoresistance of tumor cells. Therefore, this present study not only provides a novel and promising immune checkpoint blockade inhibitor but also boosts further development of MIPs for cancer immunotherapy.
Introduction
Cancer immunotherapy, which boosts the immunity to kill malignant cells, has emerged as an established pillar of cancer treatment.1 T lymphocytes play a vital role in the immune system and mediate the immune response to kill the malignant cells.2 Immune checkpoints are crucial signalling pathways that mediate the escape of tumour cells from immune-associated attack.3 The programmed cell death receptor 1 (PD-1) overexpressed on the surface of T lymphocytes can inhibit the function of T cells by binding the programmed cell death ligand 1 (PD-L1) overexpressed on the cancer cells. The interaction of PD-1 to PD-L1 facilitates cancer progression, therapy resistance and the inhibition of the functions of T cells, including cytolytic activity, proliferation, and cytokine release.4 Moreover, the interaction of PD-1 to PD-L1 contributes to the acquisition of resistance to conventional chemotherapeutic agents.5 Therefore, blocking the PD-1/PD-L1 immune checkpoint has been deemed as a promising strategy to overcome the immune escape, reactivate the anti-tumour immune response and reverse the chemoresistance of tumour cells.Antibody-based therapeutics targeting PD-1 or PD-L1 have been the mainstream in PD-1/PD-L1 pathway blockade.6 Anti-PD-1/PD-L1 monoclonal antibodies, such as nivolumab, atezolizumab and pembrolizumab, have been approved by the Food and Drug Adminstration (FDA) for the use in a broad range of cancers, including melanoma, non-small cell lung cancer, kidney cancer and bladder cancer.7 Despite the remarkable therapeutic success of antibody-based therapeutics, considerable drawbacks such as poor bioavailability, immune-related adverse events and the high cost of large-scale production also limit the wide use of antibodies.8 Furthermore, treatment with nivolumab has been reported to be associated with increased lipase and increased alanine aminotransferase,9 while other anti-PD-1/PD-L1 antibodies may potentially induce pneumonitis, hepatitis and neurotoxic effects.10 Besides, off target effect of antibodies has been reported to cause a range of side effects, such as rash, diarrhea, hypertension, etc,11 which are thought to be caused by the engagement of the receptor by the therapeutic antibodies in normal tissues.12 Therefore, novel anti-PD-1/PD-L1 strategies that can address these issues with enhanced efficiency are much needed.
Nanotechnology has emerged as a powerful weapon to arm conventional immunotherapies with numerous advantages including passive targeting to tumor, efficient cargo loading and controlled release.13 The applications of nanotechnology in anti-PD-1 therapy potentiate the effectiveness of checkpoint inhibitors and minimize their side effects, where nanomaterials serve as a carrier for the combined use of anti-PD-1/PD-L1 synergistic theraptic.14 However, these strategies rely on the existing molecular drugs and fail to get rid of the dependence to monoclonal antibodies.
Molecularly imprinted polymers (MIPs), which are chemically synthesized mimics of antibodies or artifical antibodies, harbour tailor-made binding sites complementary to the target molecules in shape, size and functional groups.15 MIPs provide unique strengths to address the limitations of antibodies, including ease in preparation, low cost and high stability, and so on. In recent years, MIPs have found wide applications in many areas, such as disease diagnosis,16 cell imaging,17 signaling pathway blocking, enzyme inhibiting18 and targeted drug delivery.19 However, to the best of our knowledge, applying MIPs in immunetherapy has not been reported yet so far.
Herein, we report molecularly imprinted nanoparticles (nanoMIP) for blocking the PD-1/PD-L1 interaction. The principle of PD-1 targeting and PD-1/PD-L1 blocking by nanoMIP specific to PD-1 protein (anti-PD-1 nanoMIP) is illustrated in Scheme 1. Similar to the PD-1-binding therapeutic monoclonal antibody nivolumab,20 the N-terminal epitope of PD-1 was designed as the binding site in this study. Though the N-terminal loop of PD-1 is not involved in binding with PD-L1, nivolumab can effectively block the PD-1/PD-L1 interaction by binding the N-terminal loop of PD-1. Through binding the N-terminal loop of PD-1, anti-PD-1 nanoMIP was thought to be able to block the interaction of PD-1/PD-L1 via steric hindrance and thereby be able to block the signaling pathway of PD-1/PD-L1. The engineered anti-PD-1 nanoMIP showed good specificity and high affinity towards PD-1, yielding a disassociation constant (Kd) at the 10−8 M level. The experiments at the cellular level indicated that the anti-PD-1 nanoMIP recovered the T cell function and reversed the chemoresistance to doxorubicin of the tumor cells. Such an MIP-based strategy can be extended to other immunotherapy-related signaling pathways potentially. As the first exploration on the potential of MIPs for immunotherapy, this study not only provides a new therapeutic for blocking PD-1/PD-L1 axis, but also opens a new avenue to the rational development of artificial antibodies for cancer immunotherapy.
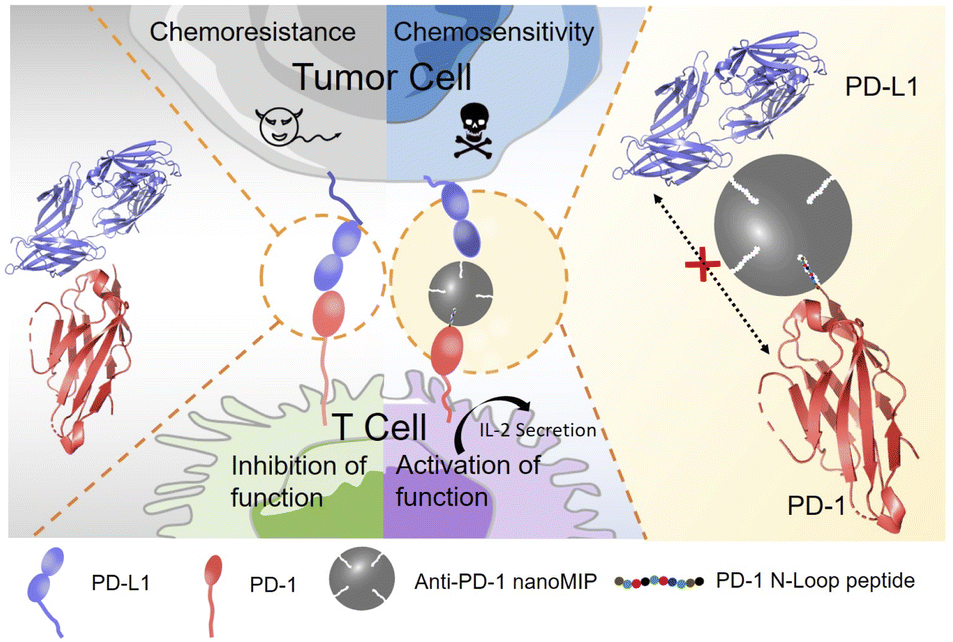 | ||
| Scheme 1 Schematic illustration of anti-PD-1 nanoMIP as a PD-1/PD-L1 antagonist. Anti-PD-1 nanoMIP blocks the interaction of PD-1 and PD-L1 by binding the PD-1 N-terminal epitope and steric hindrance, and the blockade actives the function of T cell and reverses the chemoresistance of tumor cells. | ||
Results and discussion
Synthesis of the anti-PD-1 nanoMIP
The extracellular domain of PD-1 comprises an N-terminal domain and immunoglobulin variable (IgV) domain. The N-terminal loop is a flexible structure without glycosylation.20 Though not involved in the interaction with PD-L1, the atomic interaction details at the binding interface of PD-1-nivolumab Fab complex (Fig. S1†) show that the N-terminal loop of PD-1 contributes the majority of hydrogen bonds (7 out of 13) within the nivolumab and PD-1 interface. The amino acids of N-terminal loop (S27, P28, D29 and R30) form seven hydrogen bonds with heavy chain and light chain of nivolumab, so the N-terminal epitope provides an ideal recognizing site for MIPs. Besides, the peptide sequence with more than 9 amino acids is specific enough to represent a unique code of unstructured domains for the identification of a specific protein.21 Based on this, the N-terminal loop of PD-1 was chosen as the recognizing epitope for the designing of anti-PD-1 nanoMIP. The principle and procedure of the imprinting approach are schematically illustrated in Scheme S1.† By using the newly established method called reverse microemulsion-confined epitope-oriented surface imprinting and cladding (ROSIC)22 with modification, we prepared anti-PD-1 nanoMIP. The peptide with 10 amino acids as same as the N-terminal loop of PD-1 was synthesized as the template molecule, which was further modified with a hydrophobic C13 fatty chain for facile template anchoring. For the modification of C13 fatty chain, an extra lysine (K) was added on the C-terminal of peptide sequence (Fig. S2†). Reverse microemulsion formed with surfactant and oil phase was constructed as a nanoscale reactor. Due to the presence of a C13 fatty chain on the template, the peptide was anchored at the aqueous/oil interface, with the templating peptide sequence protruding in the confined aqueous phase. Then, four silylating reagents were chosen as the functional monomers according the amino acid composition of the epitope, including 3-aminopropyl-triethoxysilane (APTES), 3-ureidopropyltri-ethoxysilane (UPTES), isobutyltriethoxysilane (IBTES), benzyltri-ethoxysilane (BnTES). Besides, tetraethyl orthosilicate (TEOS) was used as the cross-linker. Silylating reagents dissolved in the oil phase (cyclohexane in this study) diffused into the aqueous phase. Via the polymerization of functional monomers, a polymeric network formed around the peptide template. After polymerization, the synthesized nanoparticles were released from microemulsion. After extracting out the template with an appropriate eluent, anti-PD-1 nanoMIP with well-formed cavities complementary to PD-1 epitope in aspects of shape, size and functionalities was obtained. For comparison, corresponding non-imprinted polymer nanoparticles (nanoNIP) were prepared with the same procedure except that no template was used during the preparation.Optimization of monomer composition
Since the monomer composition determines the performance of prepared polymers to a great extent, the ratio of functional monomers was optimized in terms of imprinting factor (IF), which is determined by the ratio of the amount of PD-1 epitope captured by anti-PD-1 nanoMIP over that by nanoNIP. According to our previous study,22 functional monomers, which could provide non-covalent interaction with template peptide were elaboratedly arranged in terms of peptide sequence. As shown in Fig. 1A, the ratio of (TEOS)/(APTES)/(UPTES)/(IBTES)/(BnTES) at 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12:3 : 9:6 gave the best IF value (7.9). The monomer ratio yielding the highest IF value was chosen for further characterization. From transmission electron microscopic (TEM) images shown in Fig. 1B and S3,† the diameter of nanoMIP was found to be about 40 nm. Additionally, dynamic light scattering (DLS) analysis showed that the hydrodynamic size of anti-PD-1 nanoMIP and nanoNIP was about 63 nm (Fig. S4†). The larger size obtained by DLS may be attributed to the hydration of nanoparticles in aqueous solution. Moreover, the polymer dispersity index (PDI) values of anti-PD-1 nanoMIP and nanoNIP were 0.100 and 0.138 respectively, showing the prepared nanoparticles were homogeneous systems. Scanning TEM (STEM) mapping demonstrated that the anti-PD-1 nanoMIP contained the element Si, O, and N (Fig. 1C). Moreover, the X-ray photoelectron spectroscopy (XPS) spectrum also confirmed the presence of the element Si, O, C, and N (Fig. 1D) and the related surface atomic concentrations are summarized in Table S1.† The binding energy of N1s peak at 402 eV (Fig. S5B†) and O1s peak at 532 eV (Fig. S5C†) were respectively assigned to the amino group and silicon–oxygen bond, indicating the successful preparation of anti-PD-1 nanoMIP by polymerization of by functional monomers.
:12:3 : 9:6 gave the best IF value (7.9). The monomer ratio yielding the highest IF value was chosen for further characterization. From transmission electron microscopic (TEM) images shown in Fig. 1B and S3,† the diameter of nanoMIP was found to be about 40 nm. Additionally, dynamic light scattering (DLS) analysis showed that the hydrodynamic size of anti-PD-1 nanoMIP and nanoNIP was about 63 nm (Fig. S4†). The larger size obtained by DLS may be attributed to the hydration of nanoparticles in aqueous solution. Moreover, the polymer dispersity index (PDI) values of anti-PD-1 nanoMIP and nanoNIP were 0.100 and 0.138 respectively, showing the prepared nanoparticles were homogeneous systems. Scanning TEM (STEM) mapping demonstrated that the anti-PD-1 nanoMIP contained the element Si, O, and N (Fig. 1C). Moreover, the X-ray photoelectron spectroscopy (XPS) spectrum also confirmed the presence of the element Si, O, C, and N (Fig. 1D) and the related surface atomic concentrations are summarized in Table S1.† The binding energy of N1s peak at 402 eV (Fig. S5B†) and O1s peak at 532 eV (Fig. S5C†) were respectively assigned to the amino group and silicon–oxygen bond, indicating the successful preparation of anti-PD-1 nanoMIP by polymerization of by functional monomers.
 | ||
| Fig. 1 Characterization of anti-PD-1 nanoMIP. (A) The amount of the PD-1 epitope captured by anti-PD-1 nanoMIP and nanoNIP prepared with different ratios of the monomers; (B) TEM images for the imprinted nanoparticle with the optimal monomer ratio; (C) STEM Mapping for the imprinted nanoparticle with the optimal monomer ratio (Si: left upper; O: right upper; N: left down; Merge: right down); (D) XPS spectrum for the imprinted nanoparticles with the optimal monomer ratio. Error bars represent three parallel experiments. | ||
Performance and specificity test of anti-PD-1 nanoMIP
The PD-1 epitope peptide and recombinant PD-1 protein were employed as the targets to study the selectivity and affinity of anti-PD-1 nanoMIP at the peptide and protein levels, respectively. As shown in Fig. 2A and B, compared with interfering peptides and proteins, anti-PD-1 nanoMIP exhibited well acceptable specificity at both peptide and protein levels, yielding cross-reactivity ≤ 14.2% towards the interfering peptides and ≤ 19.7% towards the interfering proteins. The binding isotherm of the anti-PD-1 nanoMIP towards the PD-1 epitope peptide was then evaluated. For facile detection, PD-1 epitope peptide was labelled with the fluorescent dye fluorescein isothiocyanate isomer (FITC) (Fig. S2C†). A calibration curve of FITC-labelled PD-1 epitope peptide was firstly established (Fig. S6†). Based on the calibration curve, the adsorption isotherm and the saturated adsorption capacity of anti-PD-1 nanoMIP and nanoNIP to PD-1 epitope peptide (Fig. S7A†) were then determined. Compared with nanoNIP, anti-PD-1 nanoMIP exhibited much higher adsorption capacity toward the PD-1 epitope peptide (458 ng mg−1). Hill plot analysis gave a Kd value of 2.58 × 10−8 M (Fig. 2C). Additionally, the Kd of nanoNIP to PD-1 epitope peptide was about 6.98 × 10−7 M (Fig. S7B†), showing the affinity of anti-PD-1 nanoMIP to PD-1 epitope peptide is 27 times higher than that of nanoNIP. To further investigate the binding capability of anti-PD-1 nanoMIP to the PD-1 protein, we measured the binding affinity and kinetics of the anti-PD-1 nanoMIP to recombinant PD-1 protein by biolayer interferometry (BLI). Anti-PD-1 nanoMIP was firstly modified onto an aminopropylsilane (APS) probe, and then interacted with PD-1 protein in phosphate buffer (10 mM, pH 7.4) of different concentrations until getting saturation, followed by dissociation process in the same buffer solution. The binding responses are presented in Fig. 2D, showing a Kd value of 7.27 × 10−8 M. The Kd value on the peptide level and the protein level are in the same order of magnitude, which are both much lower than the Kd value between PD-1 and PD-L1 (8.2 μM).23 Such strong affinity endowed the anti-PD-1 nanoMIP the potential to block the PD-1/PD-L1 interaction.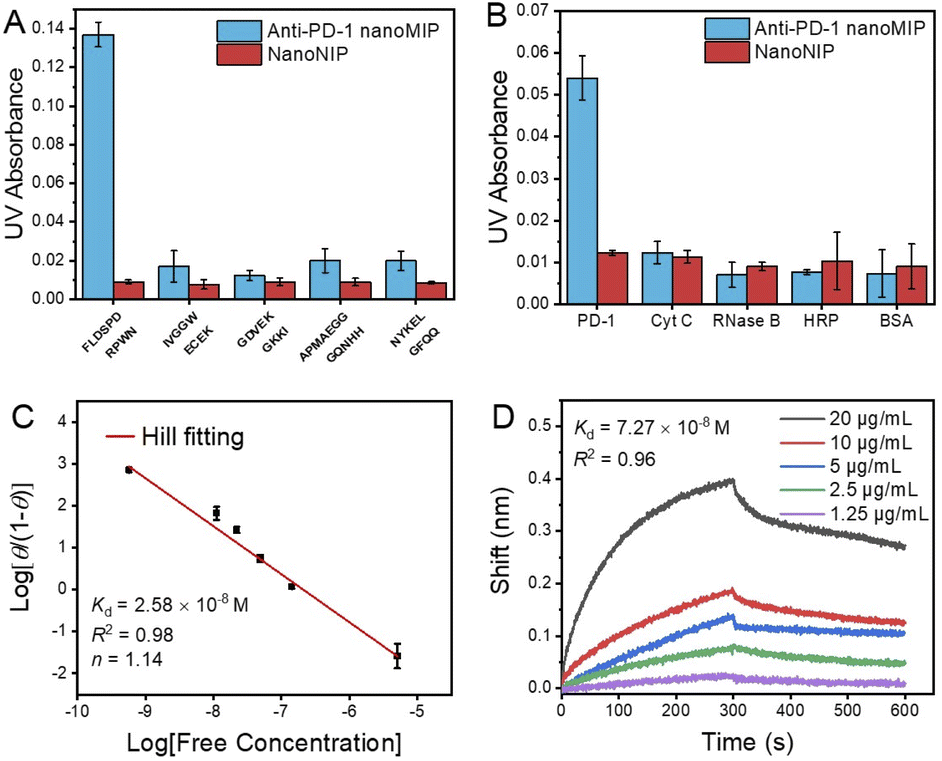 | ||
| Fig. 2 Specificity test of anti-PD-1 nanoMIP. The amounts of different peptides (A) and different proteins (B) captured by anti-PD-1 nanoMIP and nanoNIP; (C) binding isotherms of anti-PD-1 nanoMIP to PD-1 epitope peptide (Hill fitting: log[θ/(1−θ)] = logKd − nlog[free concentration]); (D) The BLI binding curves of anti-PD-1 nanoMIP towards recombinant PD-1 protein. Error bars represent three parallel experiments. | ||
Using anti-PD-1 nanoMIP as the PD-1/PD-L1 inhibitor
Next, we investigated the feasibility of using anti-PD-1 nanoMIP as the PD-1/PD-L1 inhibitor. Enzyme linked immunosorbent assay (ELISA) was used to investigate the blocking efficiency at the protein level. As illustrated in Fig. 3A, the ELISA wells were first coated with PD-1 protein, and anti-PD-1 nanoMIP particles were then added into the wells to bind PD-1 protein via recognizing its N-terminal loop. After washing, the amount of unbound PD-1 protein in each well was determined using biotin-conjugated PD-L1, followed by streptavidin-horseradish peroxidase (HRP) labelling. In control group, the signal of HRP colour reaction could be detected. In nanoNIP group, because the interaction of PD-1 to PD-L1 could not be blocked by nanoNIP, so the signal of HRP binding onto the 96-well plate could also be detected. In anti-PD-1 nanoMIP group, nanoMIP could bind PD-1 and block the binding between PD-1 to PD-L1, so the signal of HRP colour reaction decreased to a great extent. As shown in Fig. 3B, anti-PD-1 nanoMIP group showed a lower signal than that of the control group, exhibiting binding rate by 50.3%, 38.8%, 37.5%, after incubation with 6.25, 12.5 and 25 μg mL−1 anti-PD-1 nanoMIP, respectively. While there was no significant difference between nanoNIP group and control group. This competition experiment demonstrates that anti-PD-1 nanoMIP blocked the interaction between PD-1 and PD-L1, which can be attributed to the specific binding of anti-PD-1 nanoMIP to PD-1 protein.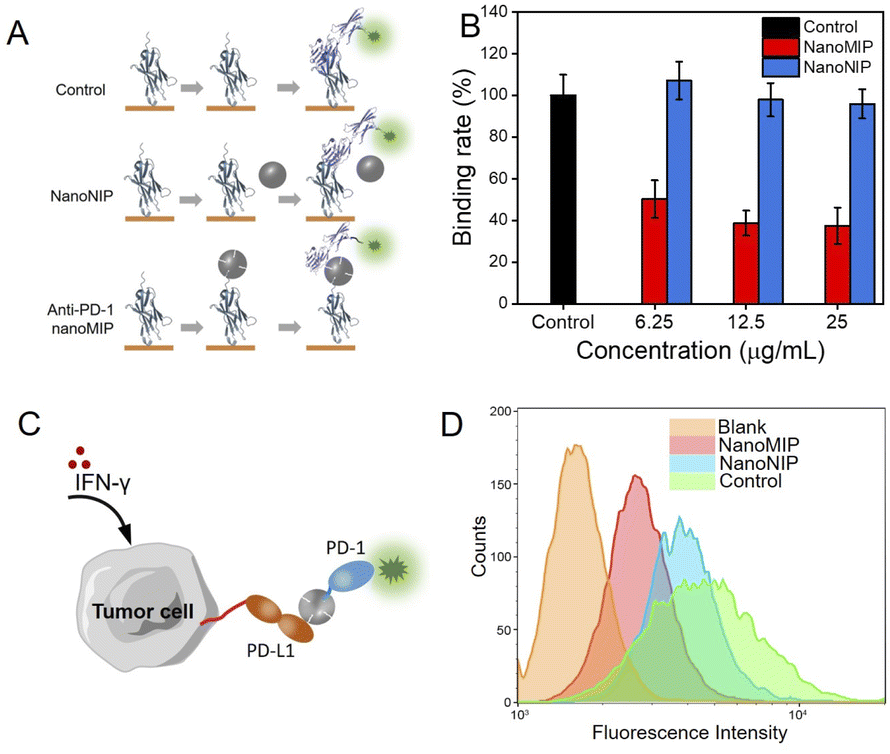 | ||
| Fig. 3 Anti-PD-1 nanoMIP blocked PD-1/PD-L1 interaction in protein and cellular level. (A) Schematic illustration of ELISA analysis of anti-PD-1 nanoMIP blocking the interaction of PD-1 and PD-L1; (B) ELISA result of blocking the PD-1 interaction with PD-L1; (C) Schematic illustration of flow cytometry analysis of anti-PD-1 nanoMIP blocking the PD-1/PD-L1 interaction on cell surface; (D) Flow cytometry of 786-O cells stained with PE-labelled PD-L1 with different pretreatments. Error bars represent three parallel experiments. | ||
The blocking efficacy of anti-PD-1 nanoMIP was further investigated at the cell level. 786-O cells were used as the model PD-L1-overexpressed tumour cell. 786-O cells were pre-treated with interferon-γ (IFN-γ) to simulate the expression of PD-L1. As shown in Fig. S8,† more PD-L1 molecules expressed on the 786-O cells were detected on the cells pre-treated with IFN-γ. To investigate whether anti-PD-1 nanoMIP could block the interaction of PD-1/PD-L1 on the cell surface, a flow cytometry assay was designed. As illustrated in Fig. 3C, P-phycoerythrin (PE)-labelled PD-1 protein was pre-mixed with anti-PD-1 nanoMIP before adding to 786-O cells. The amount of PE-labelled PD-1 binding on the 786-O cells was determined by flow cytometry assay. The results show that the PD-1/PD-L1 interaction led to a higher fluorescence signal for blank group (Fig. 3D& S9). When PE-labelled PD-1 was pre-incubated with anti-PD-1 nanoMIP before adding to 786-O cells, a much lower fluorescence intensity was detected, indicating that anti-PD-1 nanoMIP could bind PD-1 protein and prevent PD-1 from binding with PD-L1. As comparison, the group incubated with PE-labelled PD-1 pretreated with nanoNIP showed slight difference with the control group (no added anti-PD-1 nanoMIP and nanoNIP). This experiment demonstrated anti-PD-1 nanoMIP could effectively block the interaction of PD-1/PD-L1 at the cell level.
In vitro test of PD-1/PD-L1 inhibitory effect
Furthermore, the in vitro PD-1/PD-L1 inhibitory effect by anti-PD-1 nanoMIP was investigated via a co-cultured model of T cells and cancer cells. The principle is illustrated in Fig. 4A. Jurkat T cells secret interleukin-2 (IL-2) after dealing with phorbol-12-myristate-13-acetate (PMA)/phytohemagglutinin (PHA).7a When 786-O cells were co-cultured with Jurkat T cells, IL-2 secretion of Jurkat T cells decreased. The amount of IL-2 secreted by Jurkat T cells after co-cultured with 786-O cells was measured to assess the activation status of T cells. We also test the cytotoxicity of anti-PD-1 nanoMIP and nanoNIP to Jurkat T cells and 786-O cells evaluated by the 3-(4,5-dimethylthiazol-2-yl-)-2,5-diphenyltera-zolium bromide (MTT) assay. As shown in Fig. S10,† no obvious cytotoxicity was observed when the concentration of anti-PD-1 nanoMIP and nanoNIP was lower than 400 μg mL−1. Activated by PMA/PHA, Jurkat T cells treated with anti-PD-1 nanoMIP, nanoNIP or nivolumab were co-cultured with 786-O cells for 24 h. The IL-2 in supernatants of co-incubated cells was measured by the ELISA kit. As shown in Fig. 4B, compared with control group, anti-PD-1 nanoMIP could substantially increase the secretion of IL-2 which was 1.2 times higher than that of control groups, showing a considerate efficacy as nivolumab which is a commercial antibody. As comparison, nanoNIP hardly affected the IL-2 secretion. | ||
| Fig. 4 Anti-PD-1 nanoMIP recovered the function of T cells and reversed the chemoresistance of tumour cells. (A) Illustration of anti-PD-1 nanoMIP increasing interlukin-2 (IL-2) secretion by Jurkat T cells; (B) IL-2 secretion from Jurkat T cells with different treatments; (C) Illustration of anti-PD-1 nanoMIP reversing the chemoresistance; (D) Cancer cell viability after DOX treatment synergized with 5 μg mL−1 monoclonal antibody, 200 μg mL−1 anti-PD-1 nanoMIP and nanoNIP. (*p < 0.05, **p < 0.01, ***p < 0.001). Error bars represent three parallel experiments. | ||
Anti-PD-1 nanoMIP reverses the chemoresistance
To further validate the inhibitory effect of anti-PD-1 nanoMIP, the chemoresistance reversal of cancer cells was investigated. The chemosensitivity has been proven to be improved by anti-PD-1/PD-L1 in many clinical and basic researches.7 The co-cultured 786-O and Jurkat T cells model was employed to investigate the synergistic cytotoxic effect of doxorubicin (DOX) and anti-PD-1 nanoMIP (Fig. 4C). 786-O cells treated with different PD-1 antagonists were co-cultured with the Jurkat T cells, followed by incubation with DOX (5, 20 μM) to induce the cell death. As shown in Fig. 4D, blocking the signal of PD-1/PD-L1 with anti-PD-1 nanoMIP significantly increased the chemosensitivity of 786-O to DOX. The cell viability treated with anti-PD-1 nanoMIP decreased by ca. 36.5% and 44.7% with different concentration of DOX (5, 20 μM), as compared to the group treated with DOX and Jurkat T cells only (p < 0.01). Considering that only ca. 17.0% of cytotoxicity was observed when the concentration of free DOX was up to 80 μM (Fig. S11†), this synergistic effect is intriguing. Of note, anti-PD-1 nanoMIP was more effective than nivolumab with 19.7% and 24.2% cell viability reduction at 5 μM and 20 μM DOX. As compared, nanoNIP showed almost no effect on the reversal of chemoresistance. This result provides another layer of evidence that anti-PD-1 nanoMIP can effectively block the PD-1/PD-L1 immune checkpoint.Conclusions
In summary, based on the molecular imprinting methodology, we rationally designed and engineered an artificial PD-1/PD-L1 inhibitor. By recognizing the N-terminal loop of PD-1, anti-PD-1 nanoMIP could specifically bind PD-1 with higher affinity than that of PD-1 to PD-L1. Distinct from competing for the binding of PD-1 ligands, anti-PD-1 nanoMIP blocked the interaction of PD-1/PD-L1 by binding the N-terminal non-competing epitope via its nanoscale steric effect. The anti-PD-1 nanoMIP was experimentally demonstrated to be able to effectively reactivate T cells and reverse the chemoresistance of tumour cells by blocking PD-1/PD-L1 pathway, exhibiting better efficacy to commercial antibody. Due to the facile preparation routine, anti-PD-1 nanoMIP hold great potential to couple with various therapeutics for cancer immunotherapy. Thus, the present study not only provides a new therapeutic for blocking PD-1/PD-L1 axis, but also opens a new avenue to the rational development of artificial antibodies towards cancer immunotherapy.Data availability
The PDB accession code 5WT9 and the UniProt accession code Q15116 were used in this study. All other relevant data are available from the corresponding authors upon reasonable request.Author contributions
Z. Liu conceived the idea, supervised the project, and finalized the manuscript. Z. Gu and S. Xu designed and carried out the experiments, analysed the data and drafted the manuscript. Z. Guo assisted in material synthesis and characterization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of the Key Grant (21834003) from the National Natural Science Foundation of China and the Young Scientists Grant (22007046) from National Natural Science Foundation of China to XSX.Notes and references
- (a) D. M. Pardoll, Nat. Rev. Cancer, 2012, 12, 252–264 CrossRef CAS; (b) I. Mellman, G. Coukos and G. Dranoff, Nature, 2011, 480, 480–489 CrossRef CAS PubMed; (c) A. Ribas and J. D. Wolchok, Science, 2018, 359, 1350–1355 CrossRef CAS PubMed.
- (a) R. Kennedy and E. Celis, Immunol. Rev., 2008, 222, 129–144 CrossRef CAS PubMed; (b) J. Keene and J. Forman, J. Exp. Med., 1982, 155, 768–782 CrossRef CAS PubMed.
- (a) Y. Ishisa, Y. Agata, K. Shibahara and T. Honjo, EMBO J., 1992, 11, 3887–3895 CrossRef; (b) S. R. Gordon, R. L. Maute, B. W. Dulken, G. Hutter, B. M. George, M. N. McCracken, R. Gupta, J. M. Tsai, R. Sinha, D. Corey, A. M. Ring, A. J. Connolly and I. L. Weissman, Nature, 2017, 545, 495–499 CrossRef CAS PubMed; (c) E. A. Akbay, S. Koyama, J. Carretero, A. Altabef, J. H. Tchaicha, C. L. Christensen, O. R. Mikse, A. D. Cherniack, E. M. Beauchamp, T. J. Pugh, M. D. Wilkerson, P. E. Fecci, M. Butaney, J. B. Reibel, M. Soucheray, T. J. Cohoon, P. A. Janne, M. Meyerson, D. N. Hayes, G. I. Shapiro, T. Shimamura, L. M. Sholl, S. J. Rodig, G. J. Freeman, P. S. Hammerman, G. Dranoff and K. K. Wong, Cancer Discovery, 2013, 3, 1355–1363 CrossRef CAS PubMed.
- (a) T. Azuma, S. Yao, G. Zhu, A. S. Flies, S. J. Flies and L. P. Chen, Blood, 2008, 111, 3635–3643 CrossRef CAS; (b) S. L. Topalian, C. G. Drake and D. M. Pardoll, Curr. Opin. Immunol., 2012, 24, 207–212 CrossRef CAS PubMed.
- (a) M. Black, I. B. Barsoum, P. Truesdell, T. Cotechini, S. K. M. Goodfellow, M. Petorff, R. Siemens, M. Koti, A. W. B. Craig and C. H. Graham, Oncotarget, 2016, 7, 10557–10567 CrossRef PubMed; (b) S. W. Liu, S. Chen, W. G. Yuan, H. Y. Yan, K. W. Chen, D. J. Li and D. L. Li, Oncotarget, 2017, 8, 99901–99912 CrossRef PubMed.
- S. L. Topalian, F. S. Hodi, J. R. Brahmer, S. N. Gettinger, D. C. Smith, D. F. McDermott, J. D. Powderly, R. D. Carvajal, J. A. Sosman, M. B. Atkins, P. D. Leming, D. R. Spigel, S. J. Antonia, L. Horn, C. G. Drake, D. M. Pardoll, L. P. Chen, W. H. Sharfman, R. A. Anders, J. M. Taube, T. L. McMiller, H. Y. Xu, A. J. Korman, M. J. Kunkel, S. Agrawal, D. McDonald, G. D. Kollia, A. Gupta, J. M. Wigginton and M. Sznol, N. Engl. J. Med., 2012, 366, 2443–2454 CrossRef CAS PubMed.
- (a) H. Borghaei, L. P. Ares, L. Horn, D. R. Spigel, M. Steins, N. E. Ready, L. Q. Chow, E. E. Vokes, E. Felip, E. Holgado, F. Barlesi, M. Kohlhaufl, O. Arrieta, M. A. Burgio, J. Fayette, H. Lena, E. Poddubskaya, D. E. Gerber, S. N. Gettinger, C. M. Rudin, N. Rizvi, L. Crino, G. R. Blumenschein, Jr., S. J. Antonia, C. Dorange, C. T. Harbison, F. G. Finckenstein and J. R. Brahmer, N. Engl. J. Med., 2015, 373, 1627–1639 CrossRef CAS PubMed; (b) J. E. Rosenberg, J. H. Censits, T. Powles, M. S. Heijden, A. V. Balar, A. Necchi, N. Dawson, P. H. O'Donnell, A. Balmanoukian, Y. Loriot, S. Srinivas, M. M. Retz, P. Grivas, R. W. Joseph, M. D. Galsky, M. T. Fleming, D. P. Petrylak, J. L. Perez-Gracia, H. A. Burris, D. Castellano, C. Canil, J. Bellmunt, D. Bajorin, D. Nickles, R. Bourgon, G. M. Frampton, N. Cui, S. Mariathasan, O. Abidoye, G. D. Fine and R. Dreicer, Lancet, 2016, 387, 1909–1920 CrossRef CAS PubMed; (c) C. Robert, J. Schachter, G. V. Long, A. Arance, J. J. Grob, L. Mortier, A. Daud, M. S. Carlino, C. McNeil, M. Lotem, J. Larkin, P. Lorigan, B. Neyns, C. U. Blank, O. Hamid, C. Mateus, R. Shapira-Frommer, M. Kosh, H. Zhou, N. Ibrahim, S. Ebbinghaus and A. Ribas, N. Engl. J. Med., 2015, 372, 2521–2532 CrossRef CAS PubMed.
- M. R. Casals, J. R. Brahmer, M. K. Callahan, A. F. Chavez, N. Keegan, M. A. Khamashta, O. Lambotte, X. Mariette, A. Prat and M. E. S. Almazor, Nature Reviews Disease Primers, 2020, 6, 38 CrossRef PubMed.
- J. S. Weber, S. P. Angelo, D. Minor, F. S. Hodi, R. Gutzmer, B. Neyns, C. Hoeller, N. I. Khushalani, W. H. M. Jr, C. D. Lao, G. P. Linette, L. Tomas, P. Lorigan, K. F. Grossmann, J. C. Hassel, M. Maio, M. Sznol, P. A. Ascierto, P. Mohr, B. Chmielowski, A. Bryce, I. M. Svane, J. J. Grob, A. M. Krackhardt, C. Horak, A. Lambert, A. S. Yang and J. Larkin, Lancet Oncol., 2015, 16, 375–384 CrossRef CAS.
- D. Y. Wang, J. E. Salem, J. V. Cohen, S. Chandra, C. Menzer, F. Ye, S. L. Zhao, S. Das, K. E. Beckerman, L. Ha, W. K. Rathmell, K. K. Ancell, J. M. B. Pharmd, C. B. Pharmd, E. J. Davis, D. D. Chism, L. Horn, G. V. Long, M. S. Carlino, B. L. Vignes, Z. Eroglu, J. C. Hassel, A. M. Menzies, J. A. Sosman, R. J. Sullivan, J. J. Moslehi and D. B. Johnson, JAMA Oncology, 2018, 4, 1721–1728 CrossRef.
- S. Liu and R. Kurzrok, Cancer Treat. Rev., 2014, 40, 883–891 CrossRef CAS PubMed.
- (a) M. E. Lacouture, S. A. Boerner and P. M. LoRusso, Clin. Lung Cancer, 2006, 8, 36–42 CrossRef PubMed; (b) U. Rodeck, J. Cell. Physiol., 2009, 218, 32–34 CrossRef CAS PubMed.
- (a) M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed; (b) J. J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed; (c) I. I. Slowing, J. L. V. Escoto, C. W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- (a) W. J. Jeong, J. Bu, Y. X. Han, A. J. Drelich, A. Nair, P. Kral and S. Hong, J. Am. Chem. Soc., 2020, 142, 1832–1837 CrossRef CAS PubMed; (b) R. Meir, K. Shamalov, T. Sadan, M. Motiei, G. Yaari, C. J. Cohen and R. Popovtzer, ACS Nano, 2017, 11, 11127–11134 CrossRef CAS PubMed; (c) X. Li, X. P. Wang, A. Ito and N. M. Tsuji, Nat. Commun., 2020, 11, 3858 CrossRef PubMed.
- (a) G. Wulff, Angew. Chem., Int. Ed., 1995, 34, 1812–1832 CrossRef CAS; (b) H. Q. Shi, W. B. Tsai, M. D. Garrison, S. Ferrari and B. D. Ratner, Nature, 1999, 398, 593–597 CrossRef CAS PubMed; (c) Y. Hoshino, H. Koide, T. Urakami, H. Kanazawa, T. Kodama, N. Oku and K. J. Shea, J. Am. Chem. Soc., 2010, 132, 6644–6645 CrossRef CAS PubMed.
- (a) L. L. Zhou, Y. J. Wang, R. R. Xing, J. Chen, J. Liu, W. Li and Z. Liu, Biosens. Bioelectron., 2019, 145, 111729 CrossRef CAS PubMed; (b) J. Ye, Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2014, 53, 10386–10389 CrossRef CAS PubMed; (c) J. L. Urraca, C. S. A. Aureliano, E. Schillinger, H. Esselmann, J. Wiltfang and B. Sellergren, J. Am. Chem. Soc., 2011, 133, 9220–9223 CrossRef CAS PubMed.
- (a) D. Y. Yin, X. L. Li, Y. Y. Ma and Z. Liu, Chem. Commun., 2017, 53, 6716–6719 RSC; (b) S. S. Wang, Y. R. Wen, Y. J. Wang, Y. Y. Ma and Z. Liu, Anal. Chem., 2017, 89, 5646–5652 CrossRef CAS PubMed; (c) S. Shine, Z. E. Schich, A. Malakpour, W. Wan, N. Dizeyi, R. Mohammadi, K. Rurack, A. G. Wingren and B. Sellergen, J. Am. Chem. Soc., 2015, 137, 13908–13912 CrossRef.
- (a) Y. R. Dong, W. Li, Z. K. Gu, R. R. Xing, Y. Y. Ma, Q. Zhang and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 10621–1625 CrossRef CAS PubMed; (b) P. X. M. Rangel, E. Moroni, F. Merlier, L. A. Gheber, R. Vago, B. T. S. Bui and K. Haupt, Angew. Chem., Int. Ed., 2020, 59, 2816–2822 CrossRef PubMed; (c) J. J. Xu, H. H. Miao, L. H. Zhou, B. T. S. Bui, K. Haupt and G. Q. Pan, Angew. Chem., Int. Ed., 2021, 60, 24526–24533 CrossRef CAS.
- (a) Z. K. Gu, Y. R. Dong, S. X. Xu, L. S. Wang and Z. Liu, Angew. Chem., Int. Ed., 2020, 60, 2663–2667 CrossRef PubMed; (b) F. Canfarotta, L. Lezina, A. Guerreiro, J. Czulak, A. Petukhov, A. Daks, K. S. Kempisty, A. Poma, S. Piletsky and N. A. Barlev, Nano Lett., 2018, 18, 4641–4646 CrossRef CAS PubMed; (c) H. F. Lu, S. X. Xu, Z. C. Guo, M. H. Zhao and Z. Liu, ACS Nano, 2021, 15, 18214–18225 CrossRef CAS.
- S. G. Tan, H. Zhang, Y. Chai, H. Song, Z. Tong, Q. H. Wang, J. X. Qi, G. Wong, X. D. Zhu, W. J. Liu, S. Gao, Z. F. Wang, Y. Shi, F. Q. Yang, G. F. Gao and J. H. Yan, Nat. Commun., 2017, 8, 14369 CrossRef CAS PubMed.
- (a) H. Nishino, C. S. Huang and K. J. Shea, Angew. Chem., Int. Ed., 2006, 45, 2392–2396 CrossRef CAS; (b) S. Sudasarnam, Proteins, 1998, 30, 228–231 CrossRef; (c) I. A. Wilson, D. H. Haft, E. D. Getzoff, J. A. Tainer, R. A. Lerner and S. Brenner, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 5255–5269 CrossRef CAS PubMed.
- Z. C. Guo, R. R. Xing, M. H. Zhao, Y. Li, H. F. Lu and Z. Liu, Adv. Sci., 2021, 2101713 CrossRef CAS.
- X. X. Cheng, V. Veverka, A. Radhakrishnan, L. C. Waters, F. W. Muskett, S. H. Morgan, J. Huo, C. Yu, E. J. Evans, A. J. Leslie, M. Griffiths, C. Stubberfield, R. Griffin, A. J. Henry, A. Jansson, J. E. Ladbury, S. Ikemizu, M. D. Carr and S. J. Davis, J. Biol. Chem., 2013, 288, 11771–11785 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03412c |
| ‡ Equally contributed authors. |
| This journal is © The Royal Society of Chemistry 2022 |