
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-induced dual catalysis for N-α C(sp3)–H amination and alkenylation of N-alkyl benzamides†
Long-Hai
Li
,
Xin-Tao
Gu
,
Min
Shi
 * and
Yin
Wei
*
* and
Yin
Wei
*
State Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Science, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: mshi@mail.sioc.ac.cn; weiyin@sioc.ac.cn
First published on 12th October 2022
Abstract
The amination and alkenylation of the C(sp3)–H bond at the N-α position of secondary benzamides were both realized in this work by using N-hydroxyphthalimide (NHPI) imidate esters as substrates under a dual catalysis involving a photoredox catalyst and hydrogen atom transfer (HAT) catalyst. The developed methods significantly extended the scope of applications of the N-α position C(sp3)–H bond functionalization with regard to secondary N-alkylamides. More importantly, new reaction models in photoredox catalysis have been established. Based on corresponding experiments and density functional theory (DFT) calculations on the critical reaction steps combined with information reported previously, we proposed a synergistic photo- and organocatalytic reaction process for the C(sp3)–H bond functionalization and also clarified the occurrence of a chain process in the reaction pathway.
Introduction
Amides constitute one of the most important moieties in contemporary chemistry.1,2 They are one of the essential components in the process of maintaining life systems. Peptide bonds in proteins are amide bonds, which are widely found in many natural products, and are also one of the most common bonds in modern drug molecules. There are various methods available for constructing amide bonds,3–6 but new construction methods, especially greener and more efficient atom-economic ones, are still being investigated. The functionalization of amides is also important. With the rise of visible-light-catalyzed chemical transformations, the use of visible-light-induced photocatalysis to functionalize C–H bonds within amides is one of the hotspots in contemporary organic synthetic chemistry research. In general, there are two possible intermediates for the functionalization of the C–H bond at the N-α position of an amide: N-α alkyl radicals7–25 and acyliminium cations26 (Scheme 1A). The amide N-α alkyl radical is a nucleophilic free radical, and hence acceptors in this case are limited to electrophilic acceptors, such as electron-deficient olefins, azodicarboxylates, and electron-deficient aromatics, alkynes, high-valence iodine reagents, sulfonyl cyanides and some metal complexes. At the same time, the reaction of an acyliminium ion intermediate with attacking nucleophile has rarely been reported, probably due to the relatively harsh reaction conditions involved.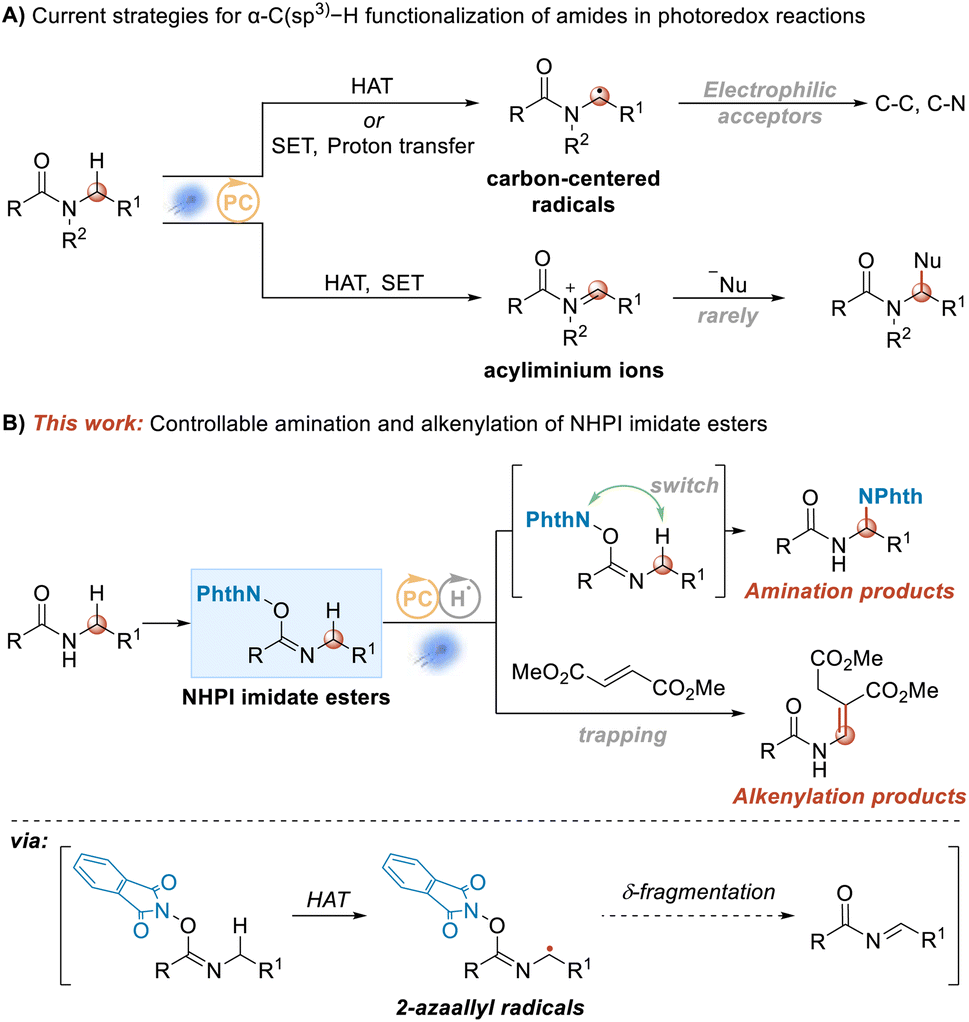 | ||
| Scheme 1 α-C(sp3)–H functionalization of amides in photoredox reactions. (A) Current strategies. (B) This work. | ||
Although there have been many related reports on the functionalization of the C–H bond at the N-α position of amides under visible-light photochemical catalysis, the research has mainly focused on the functionalization of the C–H bond at the N-α position of tertiary amides, and the research on the functionalization of the C–H bond at the N-α position of secondary amides has been investigated to a lesser degree.27–29 Therefore, development of a catalytic protocol to expand the N-α-position C(sp3)–H functionalization range of secondary amides is required; and meanwhile, it is also necessary to provide some new reaction modes on C(sp3)–H functionalization.
Recently, we introduced designed NHPI imidate ester substrates in visible-light-induced transformations as precursors of amidyl radicals.30 In our current work, also using NHPI imidate ester substrates, the α-position amination and alkenylation of amides were realized upon dual catalysis with a hydrogen atom transfer (HAT) catalyst and a photoredox catalyst through a 2-azaallyl radical (Scheme 1B). For the amination reaction, δ-fragmentation and subsequent rebound were shown to be involved. By introducing fumarate into the reaction system, the alkenylation products were provided instead of the amination products. Moreover, NHPI imidate ester substrates were generally readily prepared from the secondary amides in one pot, as shown in Scheme 2 and ESI.†
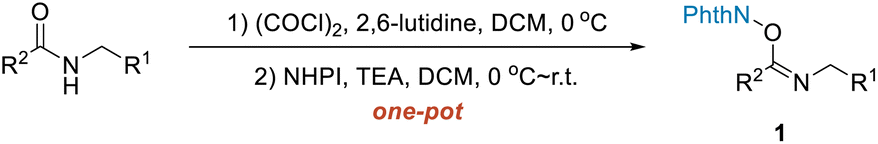 | ||
| Scheme 2 Preparation of starting NHPI imidate esters from secondary amides. | ||
Results and discussion
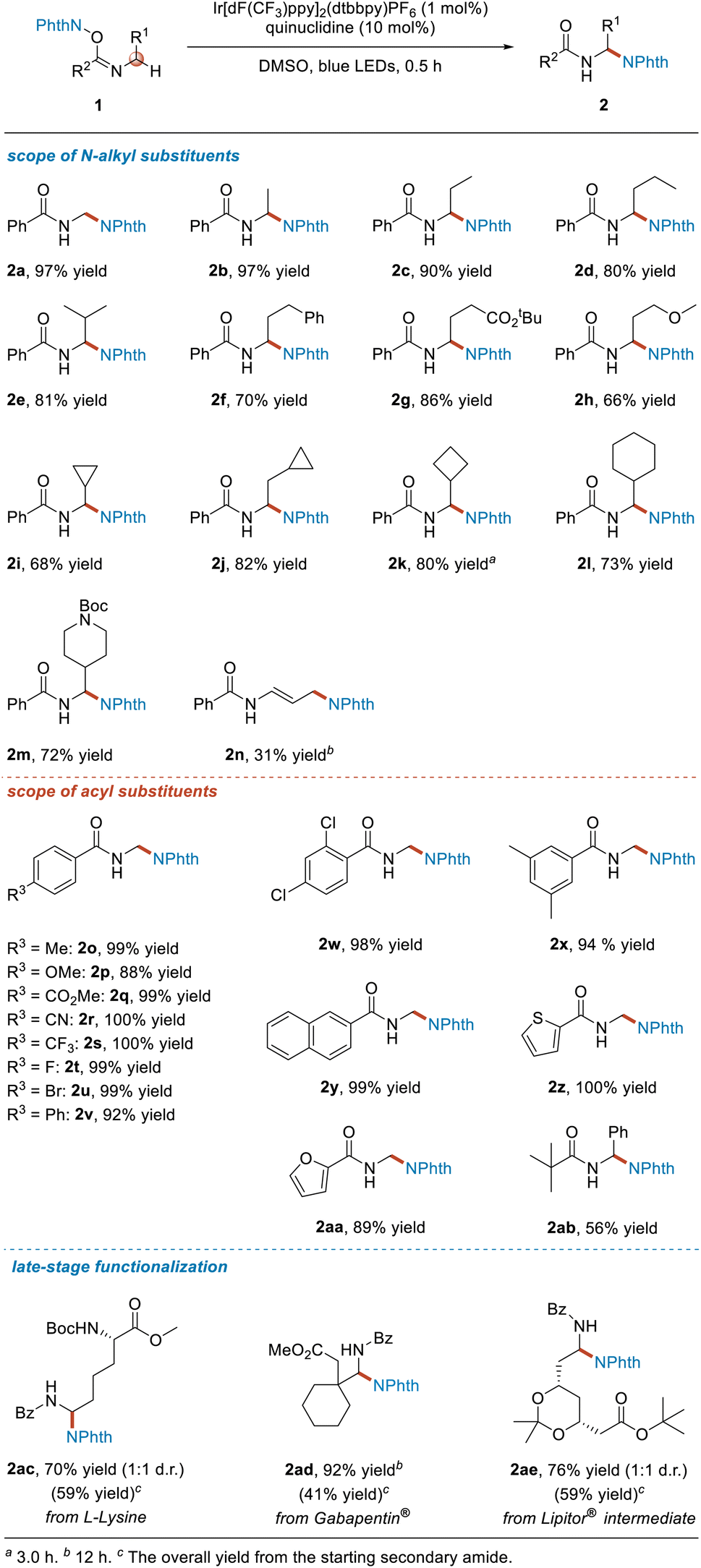
First we screened reaction conditions (see Table S1 in ESI†). The optimal conditions involved using 1 (0.20 mmol, 1.0 equiv.), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (0.002 mmol, 1.0 mol%), quinuclidine (0.02 mmol, 10 mol%), and DMSO as solvent and irradiation with blue LEDs at room temperature for 0.5 h (Table 1).
|
Next, we investigated the R1 substituent on substrates 1. When R1 was a hydrogen atom or a simple alkyl substituent, specifically a methyl, ethyl, propyl or isopropyl group, the target products 2a–2e were obtained in yields of more than 80%. As for phenethyl, 2-tert-butoxyethyl and 2-methoxyethyl groups, the desired products 2f–2h were obtained in yields of 66–86%. When R1 was a cycloalkyl substituent, whether a cyclopropyl, cyclopropylmethyl, cyclobutyl or cyclohexyl group, the reaction proceeded smoothly, affording the corresponding products 2i–2l in 68–82% yields. In addition, although it has been shown that the hydrogen atom at the N-α position of the NBoc moiety can undergo the HAT process with the quinuclidinium radical cation, the imidate ester 1m was still produced, giving the target product 2m efficiently and selectively. Interestingly, when allyl-substituted imidate ester 1n was used as the substrate, a distal aminated product was obtained instead of the normal α-functionalized product.
We then turned our attention to investigating the influence of the R2 substituent on substrates 1. The results indicated that for the para-substituted phenyl imidate esters, regardless of whether the substituent on the phenyl group was an electron-donating substituent, an electron-withdrawing substituent or a halogen atom, the reactions proceeded very well, furnishing the target products 2o–2v in excellent yields. Moreover, disubstitution of the phenyl group showed no significant impact on the reaction outcome, and the desired products 2w and 2x were obtained in 98% and 94% yields, respectively. Use of other types of aromatic substituted substrates, namely with a 2-naphthyl, 2-thienyl or 2-furanyl moiety, was also completely compatible with this reaction, delivering the target products 2y–2aa in good yields ranging from 89% to >99%. Furthermore, when R2 was an alkyl group, the reaction also proceeded smoothly, affording the product 2ab in 56% yield under the same reaction conditions.
Next, we set out to use the developed method for late-stage functionalization of some natural products containing amine groups and biologically active drug molecules or intermediates (Table 1). L-Lysine is one of the essential amino acids for humans. The NHPI imidate ester 1ac derived from L-lysine selectively provided the aminated product 2ac with a yield of 70% under the standard reaction conditions. Gabapentin® is an antiepileptic and anxiolytic drug. By converting it into the corresponding NHPI imidate ester 1ad, the corresponding product 2ad was efficiently and selectively obtained in 92% yield. In addition, a key intermediate used to synthesize the active molecule atorvastatin in the lipid-lowering drug Lipitor® could also be functionalized using this newly developed method, and such functionalization selectively afforded the α-aminated product 2ae in 76% yield. Moreover, 2ac, 2ad and 2ae were obtained in moderate yields from the starting secondary amides.
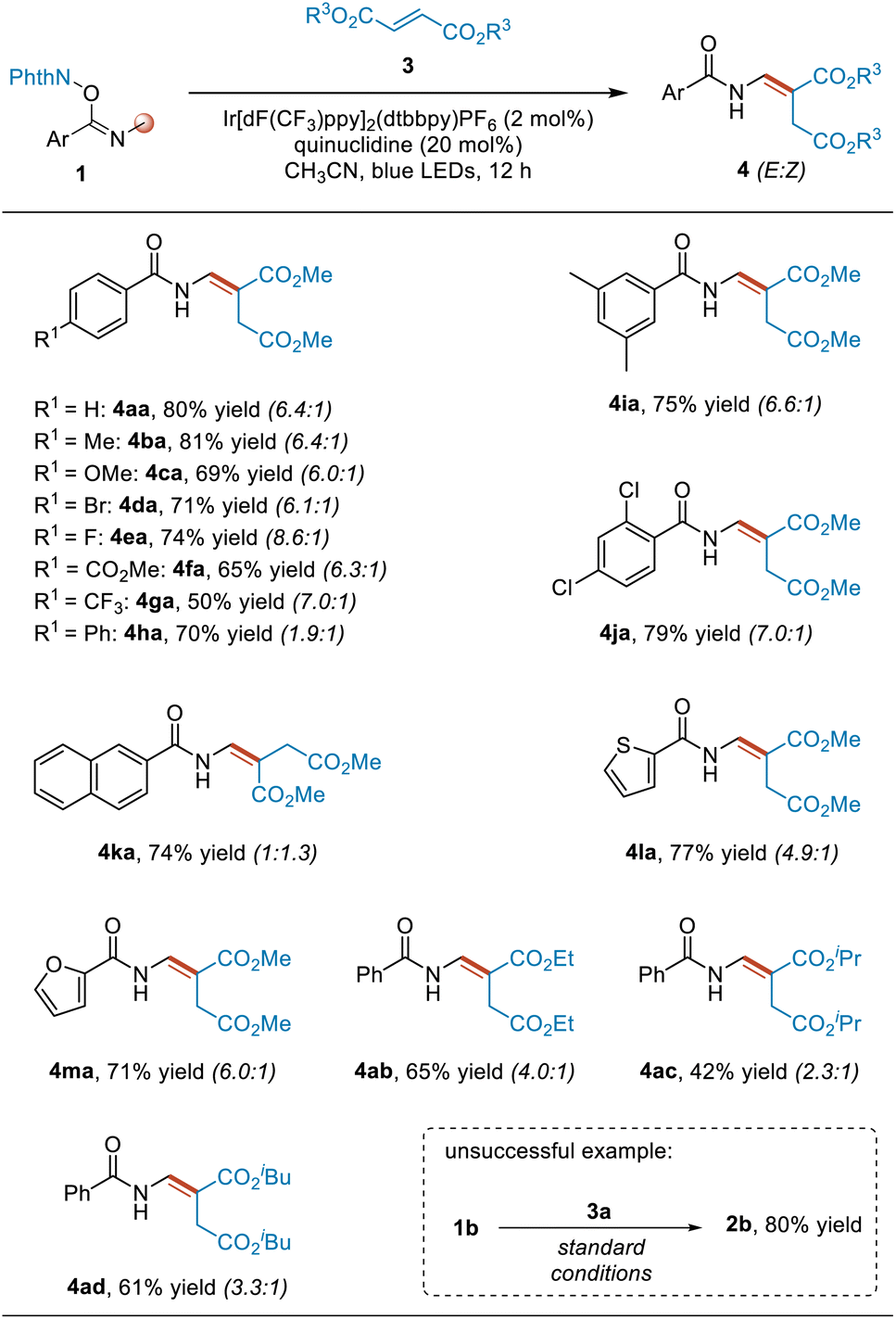
By introducing dimethyl fumarate to the above amination reaction system, the reaction changed the pathway to undergo alkenylation of benzamides, producing enamine products (Table 2). To study the applicability of this reaction, we first investigated the substituents on the phenyl group in substrates 1. Different para- and meta-substituted aromatic imidate esters served as coupling partners to provide the corresponding alkenylation products 4aa–4ja in usually good yields and ≥6.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z selectivity levels, except for the para-phenyl-substituted substrate. In the case of this substrate, the E/Z selectivity of the corresponding product 4ha was significantly reduced to 1.9:1, presumably due to steric and electronic effects. Next, we examined other aromatic substituents. When the substituent of the substrate was a naphthyl, 2-thienyl or 2-furanyl moiety, the reaction also proceeded smoothly, affording the desired products 4ka–4ma in good yields. Lastly, we investigated the fumarate 3 having other substituents. Diethyl, diisopropyl and diisobutyl fumarates were all found to react smoothly with substrate 1a, giving the desired products 4ab, 4ac and 4ad in yields of 65%, 42% and 61%, respectively, with relatively low E/Z selectivity levels. Not also that increasing the steric hindrance of the substituent in the fumarate was found to be overall associated with a gradual decrease in the Z/E selectivity of the product. In addition to the N-Me derivatives, an N-Et derivative, namely 1b, was also used to test this reaction. However, the desired alkenylation product was not obtained, and instead the rearranged product 2b was obtained. Besides dimethyl fumarate, other alkenes such as benzyl acrylate and methyl 2-benzyl acrylate were also used to react with 1a in an attempt to capture the radical intermediate A, but both mainly gave the rearranged product 2a. As for (3,3,3-trifluoroprop-1-en-2-yl)benzene, the rearranged product was not obtained at this time, and another product, namely N-(4,4-difluoro-3-phenylbut-3-en-1-yl)benzamide S2a, was isolated (for details, see pages S6 and S7 in ESI†).
:1 E/Z selectivity levels, except for the para-phenyl-substituted substrate. In the case of this substrate, the E/Z selectivity of the corresponding product 4ha was significantly reduced to 1.9:1, presumably due to steric and electronic effects. Next, we examined other aromatic substituents. When the substituent of the substrate was a naphthyl, 2-thienyl or 2-furanyl moiety, the reaction also proceeded smoothly, affording the desired products 4ka–4ma in good yields. Lastly, we investigated the fumarate 3 having other substituents. Diethyl, diisopropyl and diisobutyl fumarates were all found to react smoothly with substrate 1a, giving the desired products 4ab, 4ac and 4ad in yields of 65%, 42% and 61%, respectively, with relatively low E/Z selectivity levels. Not also that increasing the steric hindrance of the substituent in the fumarate was found to be overall associated with a gradual decrease in the Z/E selectivity of the product. In addition to the N-Me derivatives, an N-Et derivative, namely 1b, was also used to test this reaction. However, the desired alkenylation product was not obtained, and instead the rearranged product 2b was obtained. Besides dimethyl fumarate, other alkenes such as benzyl acrylate and methyl 2-benzyl acrylate were also used to react with 1a in an attempt to capture the radical intermediate A, but both mainly gave the rearranged product 2a. As for (3,3,3-trifluoroprop-1-en-2-yl)benzene, the rearranged product was not obtained at this time, and another product, namely N-(4,4-difluoro-3-phenylbut-3-en-1-yl)benzamide S2a, was isolated (for details, see pages S6 and S7 in ESI†).
|
During the investigation of the above amination reaction, we unexpectedly found that if using NHPI imine esters 1af and 1ag as substrates, the enamine products 5 and 6 were formed as isomeric mixtures rather than the addition products (Scheme 3A). These results, upon being compared with the normal amination reactions, were concluded to be consistent with the envisaged process shown in Scheme 1B, suggesting that the reaction may proceed through acylimine intermediates (Scheme 3B). When the phenyl or ethoxyacyl group was present, the corresponding acylimine intermediates were more acidic, resulting in intramolecular enamine isomerization (Scheme 3B, path b) taking place more easily than the intermolecular attack by the phthalimide anion (Scheme 3B, path a). Thus, the phenomenon of terminal amination of 1n shown in Table 1 could also be explained. To further determine whether the −NPhth rebound process is intermolecular or intramolecular, substrate 1ah was synthesized and a crossover experiment with 1a was performed (Scheme 3C). We obtained the mixture of products 2ah, 2a and crossover products S2ah and S2aah; they were detected and identified using LCMS. The results indicated the −NPhth rebound process to be intermolecular.
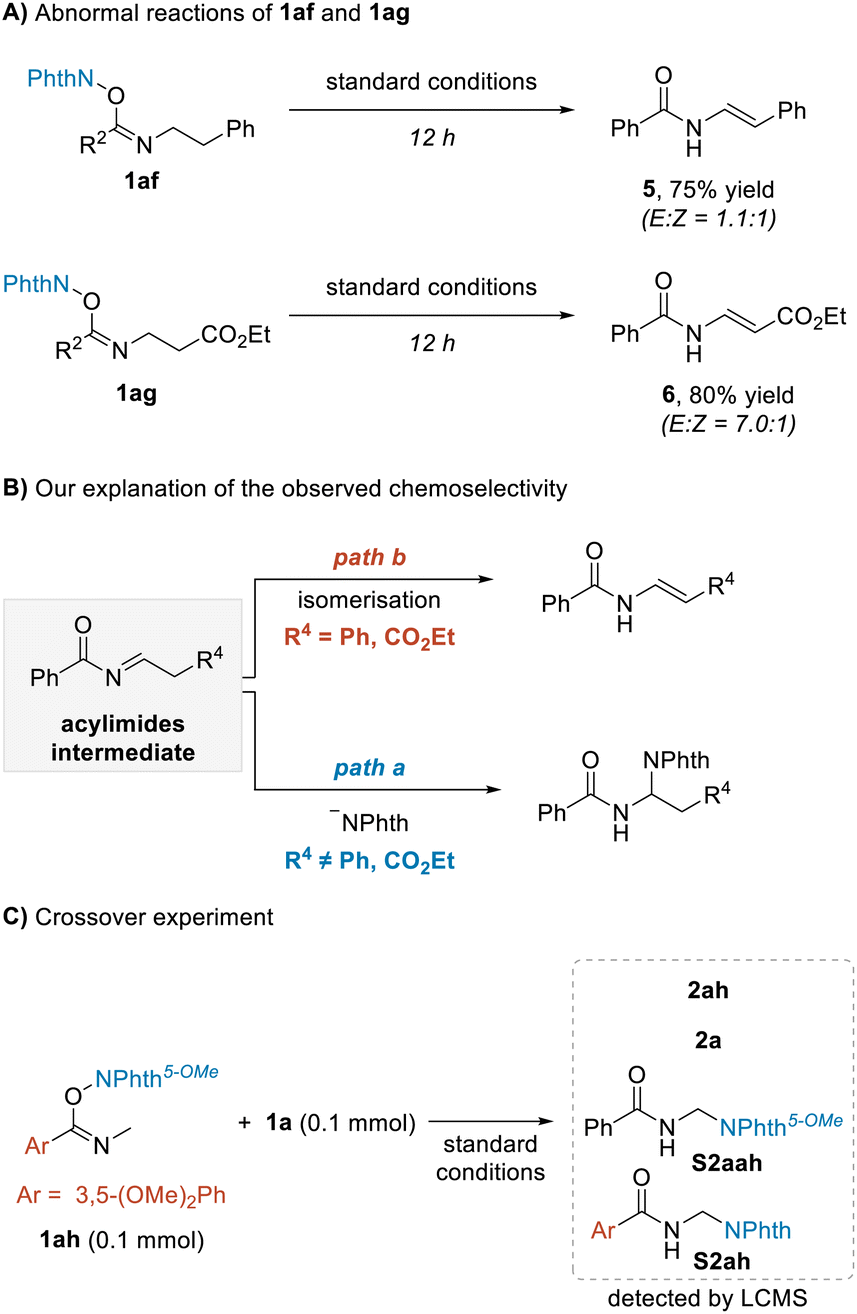 | ||
| Scheme 3 Preliminary mechanism analysis. (A) Abnormal reactions of 1af and 1ag. (B) Our explanation of the observed chemoselectivity. (C) Crossover experiment. | ||
On the basis of the above experimental results and the previously reported processes, we proposed a mechanism for the amination reactions, as shown in Scheme 4. First, according to this proposal, the photocatalyst Ir(III) enters into an excited state Ir(III)* 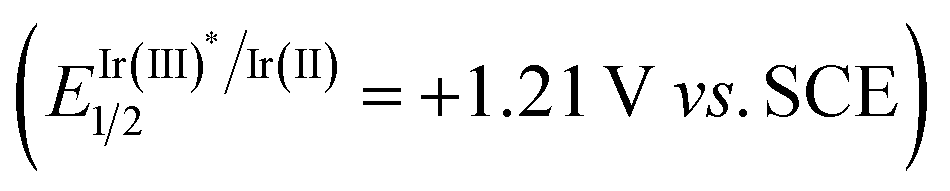 31 under visible-light irradiation, and then undergoes an SET process with quinuclidine (Eoxp = +1.10 V vs. SCE)32,33 to give the quinuclidinium radical cation. After a HAT process with NHPI imine ester 1a, the radical intermediate A is formed; and A undergoes a δ-fragmentation, due to the conjugation property of the 2-azaallyl radical, to result in acylimine B and phthalimide radical. Finally according to the proposed mechanism, the phthalimide anion is provided from phthalimide radical through another SET process, which undergoes a nucleophilic addition to the formed acylimine B, affording the target product 2a after protonation.
31 under visible-light irradiation, and then undergoes an SET process with quinuclidine (Eoxp = +1.10 V vs. SCE)32,33 to give the quinuclidinium radical cation. After a HAT process with NHPI imine ester 1a, the radical intermediate A is formed; and A undergoes a δ-fragmentation, due to the conjugation property of the 2-azaallyl radical, to result in acylimine B and phthalimide radical. Finally according to the proposed mechanism, the phthalimide anion is provided from phthalimide radical through another SET process, which undergoes a nucleophilic addition to the formed acylimine B, affording the target product 2a after protonation.
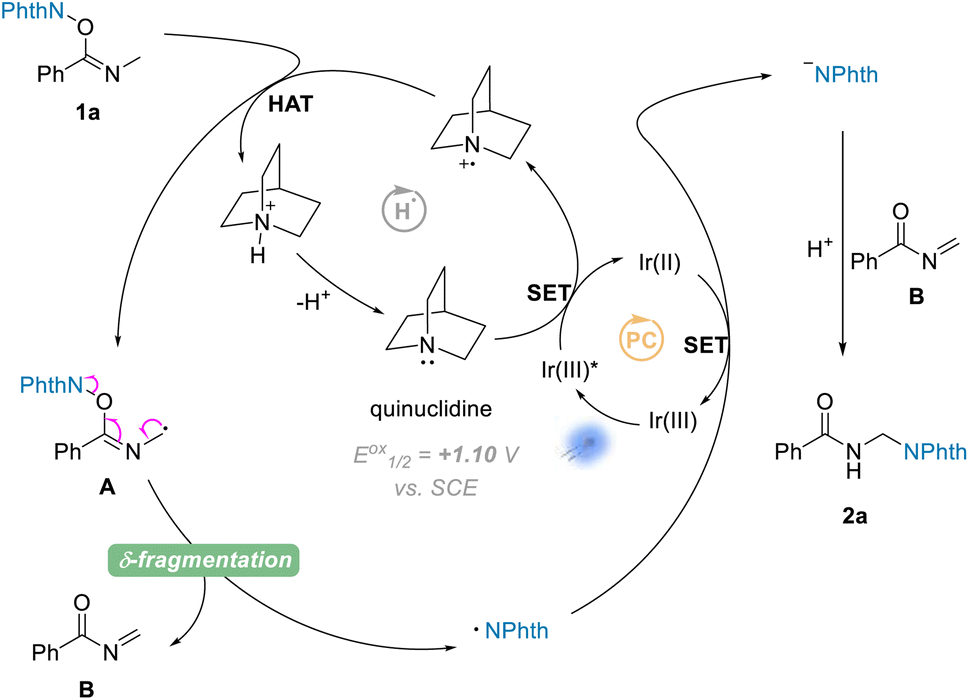 | ||
| Scheme 4 Proposed photoredox catalysis cycle. | ||
Although the HAT process between NHPI imidate 1 and the quinuclidinium radical cation has not yet been revealed, our density functional theory (DFT) calculations showed that this reaction process could take place efficiently under the standard reaction conditions (Scheme 5A). The calculation results demonstrated that the HAT from the N-α position of NHPI imide ester 1a to quinuclidinium radical cation can take place by overcoming an energy barrier of 22.0 kcal mol−1 to generate the radical intermediate A. The free energy change ΔGrxn,298 was calculated to be −10.8 kcal mol−1, indicating this HAT process to be thermodynamically favorable. In addition, the calculations showed the subsequent fragmentation process to also be a thermodynamically favorable pathway.
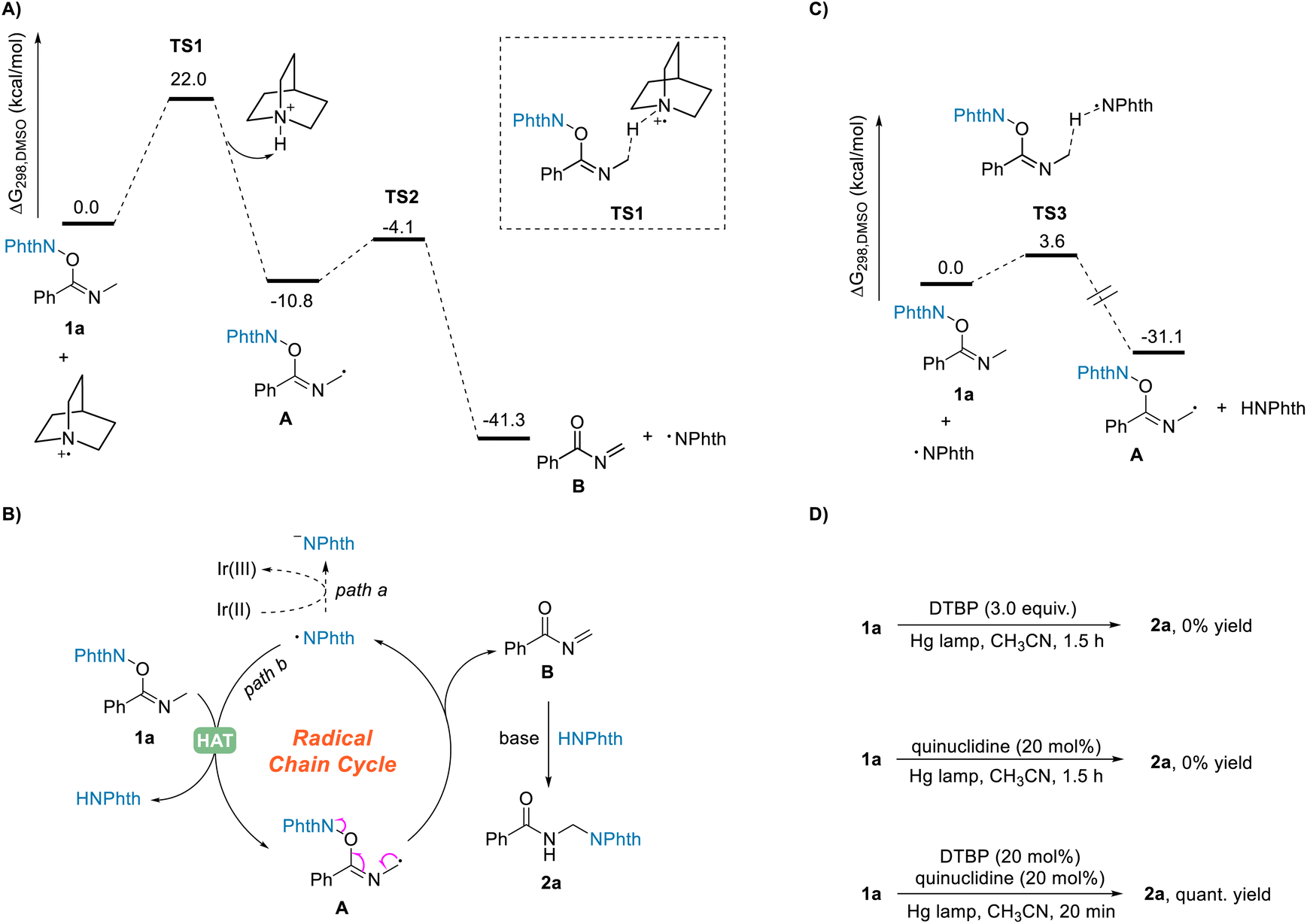 | ||
| Scheme 5 Mechanistic investigation of the amination reaction. (A) DFT calculations for the HAT process between substrate 1a and quinuclidinium radical cation, and the subsequent δ-fragmentation. (B) Proposed radical chain cycle. (C) DFT calculations for the HAT process between substrate 1a and phthalimide radical. (D) Results of irradiation experiments using a high-pressure mercury lamp. | ||
In a further in-depth study of the reaction, by using a reported method,34 we measured the quantum yield of the reaction Φ to be 20, suggesting the occurrence of radical chain processes in this transformation.35 A speculated radical chain cycle is shown in Scheme 5B. Phthalimide radical generated in the fragmentation process of intermediate A shown in Scheme 4 could not only undergo the SET process with Ir(II) to obtain the phthalimide anion (Scheme 5B, path a), it could also undergo the HAT process (Scheme 5B, path b) with substrate 1a to give intermediate A and phthalimide. After another fragmentation process, the phthalimide radical would be produced again, with the acylimine intermediate B formed at the same time. Finally, the phthalimide can couple with the acylimine intermediate B as well to obtain the target product 2a using quinuclidine as a base. This mechanistic hypothesis was supported by the DFT calculations. As shown in Scheme 5C, the energy barrier for the HAT process between the phthalimide radical and the substrate 1a was calculated to be only 3.6 kcal mol−1. Moreover, to further confirm the radical chain process, we also carried out irradiation experiments using a high-pressure mercury lamp (Scheme 5D). Di-tert-butyl peroxide (DTBP) can generate tert-butoxy radical under such irradiation, and the formed tert-butoxy radical can undergo an SET process with quinuclidine to obtain quinuclidinium radical cation.36 The target product was found to not be obtained upon irradiation with DTBP or quinuclidine alone, but when catalytic amounts of DTBP (20 mol%) and quinuclidine (20 mol%) were present together at the same time, the desired product 2a was obtained in a quantitative yield within a very short period of time (20 minutes). Although this result further clarified the occurrence of the radical chain process, a light on–off experiment indicated the need for constant irradiation of visible light35 (see page S113 in the ESI†). Furthermore, a kinetic isotope labeling experiment gave a kH/kD of 1.26, i.e., an atypical value, but apparently still possible in this reaction involving the HAT process (see page S115 in the ESI†).37
On the basis of above experimental results and the previously reported processes, we proposed a plausible mechanism for the alkenylation of N-alkyl benzamides, as shown in Scheme 6A. Through the same HAT process described in Scheme 4, the obtained intermediate A according to the proposed mechanism is captured by fumarate 3a to generate an intermediate C, which furnishes intermediate F after single-electron reduction (Scheme 6A, path a). As the newly formed NHPI imidate ester, intermediate F undergoes another dual photocatalysis process to afford acylimine intermediate G, and finally isomerizes upon visible-light irradiation to give product 4aa. We also proposed another possible reaction pathway as shown in Scheme 6A (path b). In path b, this process goes through a radical–radical coupling reaction between intermediate A and radical anion D (3a, Erep/2 = −1.38 V vs. SCE38) generated by the photoredox process (EIr(III)/Ir(II)1/2 = −1.37 V vs. SCE39) to obtain intermediate F. Subsequently, we embarked on DFT studies to gain insight into the reaction mechanism for the alkenylation of N-alkyl benzamides. As shown in Scheme 6B, the addition of intermediate A to fumarate 3a would, according to the DFT calculations, be required to overcome a high reaction energy barrier of 25.4 kcal mol−1. Compared with the rapid δ-fragmentation of intermediate A, this process would be difficult to perform selectively. Therefore, we turned to investigating the second proposed reaction pathway and found the radical–radical coupling of intermediate A with intermediate D to be more kinetically favorable (7.1 kcal mol−1) than the δ-fragmentation process (9.1 kcal mol−1), indicating path b to be a more reasonable pathway for the formation of 4aa.
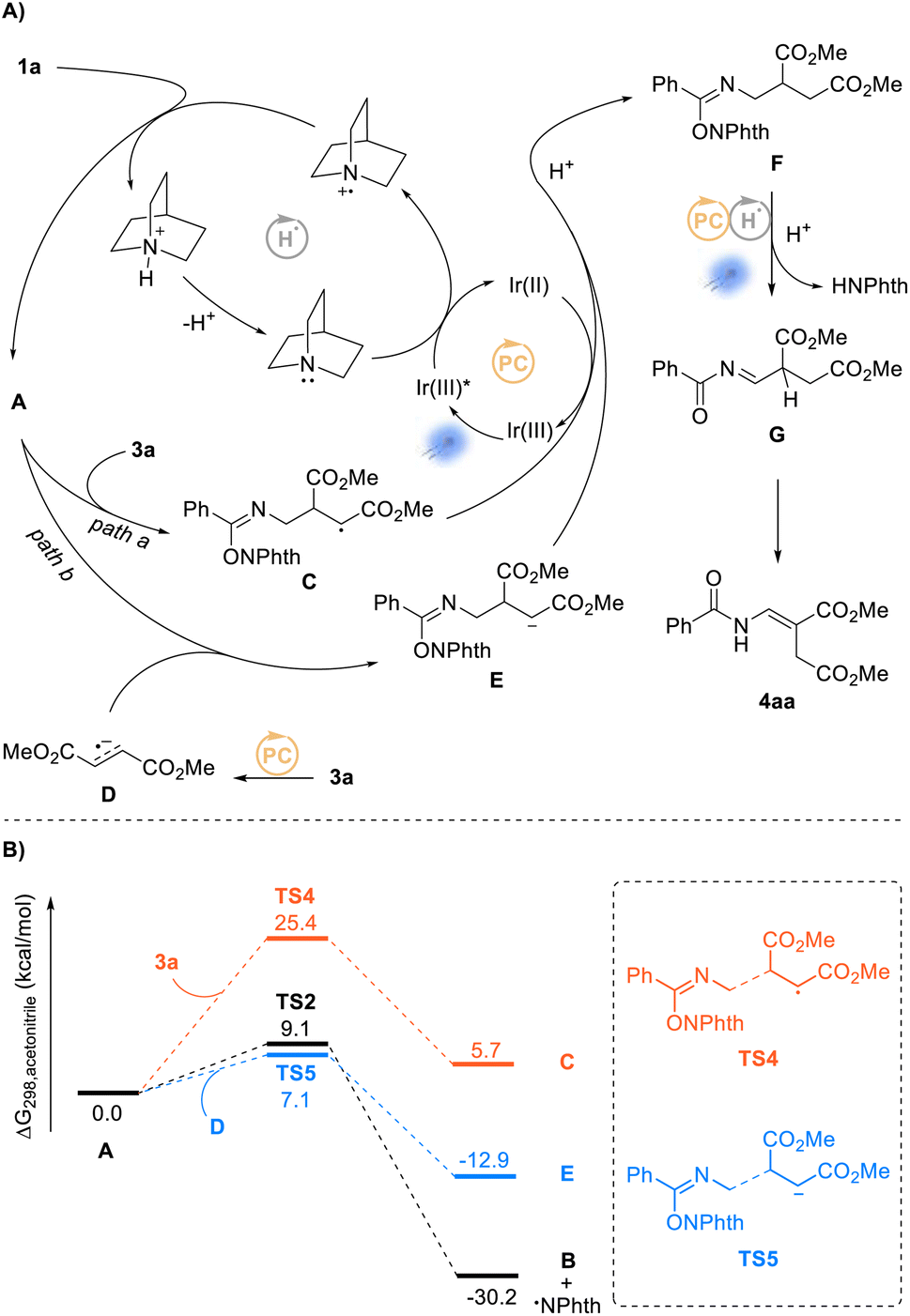 | ||
| Scheme 6 Mechanistic investigation of the alkenylation reaction. (A) Proposed photoredox catalysis cycle. (B) DFT calculations of the critical reaction steps between intermediate A and substrate 3a and the radical–radical coupling step. | ||
Conclusions
In summary, when using NHPI imidate esters as substrates and by introducing the hydrogen atom transfer (HAT) catalyst quinuclidine into the photoredox catalysis, the amination and alkenylation of the C(sp3)–H bond at the N-α position of secondary benzamides were realized under mild reaction conditions. These reactions significantly extended the scope of applications of N-α position C(sp3)–H bond functionalization with regard to the secondary N-alkylamides. Most importantly, new reaction models for 2-azaallylic radicals, namely the generation of 2-azaallyl radical intermediate and subsequent δ-fragmentation to furnish the amination and alkenylation products under photoredox catalysis, have been established. Further investigations are undergoing in our laboratory.Data availability
Experimental and computational data have been made available as ESI.†Author contributions
Long-Hai Li discovered the reactions. Min Shi and Long-Hai Li designed the experiments. Long-Hai Li and Xin-Tao Gu performed the experiments and analyzed the experimental results. Min Shi and Long-Hai Li wrote the manuscript. Yin Wei performed and described the computations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21372250, 21121062, 21302203, 21772037, 21772226, 21861132014, 91956115 and 22171078).References
- G. Arthur, The Amide Linkage: Selected Structural Aspects in Chemistry, Biochemistry, and Materials Science, Wiley-Interscience, 2000 Search PubMed.
- T. Wieland and M. Bodanszky, The World of Peptides: A Brief History of Peptide Chemistry, Springer, 1991 Search PubMed.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027–1032 CrossRef CAS PubMed.
- N. Ahmed, V. Chudasama, A. Maruani and A. Shamsabadi, Org. Biomol. Chem., 2020, 18, 6258–6264 RSC.
- Q. An, Y. Chen, W. Liu, H. Pan, X. Wang, Z. Wang, K. Zhang and Z. Zuo, J. Am. Chem. Soc., 2020, 142, 6216–6226 CrossRef CAS PubMed.
- G. N. Papadopoulos, M. G. Kokotou, N. Spiliopoulou, N. F. Nikitas, E. Voutyritsa, D. I. Tzaras, N. Kaplaneris and C. G. Kokotos, ChemSusChem, 2020, 13, 5934–5944 CrossRef CAS PubMed.
- B. Abadie, D. Jardel, G. Pozzi, P. Toullec and J.-M. Vincent, Chem.–Eur. J., 2019, 25, 16120–16127 CrossRef CAS PubMed.
- L. K. G. Ackerman, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059–14063 CrossRef CAS PubMed.
- L. Cavallo, M. Rueping, B. Maity, C. Zhu, H. Yue, L. Huang, M. Harb and Y. Minenkov, J. Am. Chem. Soc., 2020, 142, 16942–16952 CrossRef PubMed.
- J. Dong, Q. Xia, X. Lv, C. Yan, H. Song, Y. Liu and Q. Wang, Org. Lett., 2018, 20, 5661–5665 CrossRef CAS PubMed.
- R. Grainger, T. D. Heightman, S. V. Ley, F. Lima and C. N. Johnson, Chem. Sci., 2019, 10, 2264–2271 RSC.
- J. Hou, A. Ee, H. Cao, H.-W. Ong, J.-H. Xu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 17220–17224 CrossRef CAS PubMed.
- K. Matsumoto, M. Nakajima and T. Nemoto, J. Org. Chem., 2020, 85, 11802–11811 CrossRef CAS PubMed.
- J. B. McManus, N. P. R. Onuska and D. A. Nicewicz, J. Am. Chem. Soc., 2018, 140, 9056–9060 CrossRef CAS PubMed.
- C. M. Morton, Q. Zhu, H. Ripberger, L. Troian-Gautier, Z. S. D. Toa, R. R. Knowles and E. J. Alexanian, J. Am. Chem. Soc., 2019, 141, 13253–13260 CrossRef CAS PubMed.
- R. Sakamoto, T. Yoshii, H. Takada and K. Maruoka, Org. Lett., 2018, 20, 2080–2083 CrossRef CAS PubMed.
- Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef CAS PubMed.
- M.-X. Sun, Y.-F. Wang, B.-H. Xu, X.-Q. Ma and S.-J. Zhang, Org. Biomol. Chem., 2018, 16, 1971–1975 RSC.
- H. Tian, Q. Xia, Q. Wang, J. Dong, Y. Liu and Q. Wang, Org. Lett., 2019, 21, 4585–4589 CrossRef CAS PubMed.
- N. A. Till, R. T. Smith and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 5701–5705 CrossRef CAS PubMed.
- T. Wakaki, K. Sakai, T. Enomoto, M. Kondo, S. Masaoka, K. Oisaki and M. Kanai, Chem.–Eur. J., 2018, 24, 8051–8055 CrossRef CAS PubMed.
- J. B. McManus, N. P. R. Onuska and D. A. Nicewicz, J. Am. Chem. Soc., 2018, 140, 9056–9060 CrossRef CAS PubMed.
- (a) C. Dai, F. Meschini, J. M. R. Narayanam and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 4425–4431 CrossRef CAS PubMed; (b) R.-X. Liu, Y. Wei and M. Shi, Chin. J. Chem., 2020, 38, 947–951 CrossRef CAS.
- M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu and T. Rovis, Angew. Chem., Int. Ed., 2019, 58, 4002–4006 CrossRef CAS PubMed.
- J. Ye, I. Kalvet, F. Schoenebeck and T. Rovis, Nat. Chem., 2018, 10, 1037–1041 CrossRef CAS PubMed.
- A. W. Rand, H. Yin, L. Xu, J. Giacoboni, R. Martin-Montero, C. Romano, J. Montgomery and R. Martin, ACS Catal., 2020, 10, 4671–4676 CrossRef CAS.
- L.-H. Li, Y. Wei and M. Shi, Org. Chem. Front., 2021, 8, 1935–1940 RSC.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- W. Z. Liu and F. G. Bordwell, J. Org. Chem., 1996, 61, 4778–4783 CrossRef CAS PubMed.
- S. F. Nelsen and P. J. Hintz, J. Am. Chem. Soc., 1972, 94, 7114–7117 CrossRef CAS.
- U. Megerle, R. Lechner, B. König and E. Riedle, Photochem. Photobiol. Sci., 2010, 9, 1400–1406 CrossRef CAS PubMed.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- D. Griller, J. A. Howard, P. R. Marriott and J. C. Scaiano, J. Am. Chem. Soc., 1981, 103, 619–623 CrossRef CAS.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed.
- T. van Leeuwen, L. Buzzetti, L. A. Perego and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 4953–4957 CrossRef CAS PubMed.
- J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds. See DOI: https://doi.org/10.1039/d2sc03385b |
| This journal is © The Royal Society of Chemistry 2022 |