
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Terminal cysteine mediated backbone-side chain cyclization for chemically enhanced phage display†
Mengmeng
Zheng
,
Fredrik
Haeffner
and
Jianmin
Gao
 *
*
Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, MA 02467, USA. E-mail: Jianmin.Gao@bc.edu
First published on 30th June 2022
Abstract
Phage display, an ingenious invention for evaluating peptide libraries, has been limited to natural peptides that are ribosomally assembled with proteinogenic amino acids. Recently, there has been growing interest in chemically modifying phage libraries to create nonnatural cyclic and multicyclic peptides, which are appealing for use as inhibitors of protein–protein interactions. While earlier reports largely focused on side-chain side-chain cyclization, we report herein a novel strategy for creating backbone-side chain cyclized peptide libraries on phage. Our strategy capitalizes on the unique reactivity of an N-terminal cysteine (NCys) with 2-cyanobenzothiazole (CBT) which, in conjugation with another thiol-reactive group, can elicit rapid cyclization between an NCys and an internal cysteine. The resulting library was screened against two model proteins, namely Keap1 and Sortase A. The screening readily revealed potent inhibitors for both proteins with certain Keap1 ligands reaching low nanomolar potency. The backbone-side chain cyclization strategy described herein presents a significant addition to the toolkit of creating nonnatural macrocyclic peptide libraries for phage display.
Introduction
Phage display,1 a powerful platform for constructing and screening peptide libraries, has been widely used in the discovery of functional peptides as molecular probes and enzyme inhibitors. Despite the wide popularity, phage display suffers from an intrinsic limitation. That is, only natural peptides (peptides of proteinogenic amino acids to be precise) can be displayed and evaluated utilizing this platform. Yet, it is highly desirable to incorporate non-natural structural motifs into phage libraries as certain designed functional groups or topologies (e.g., cyclization) have proven to enhance peptides' interactions with their biological target.2–5 Excitingly, clever strategies are being developed to enable peptide modification on phage, yielding non-natural peptide libraries that display macrocyclic structures6–10 and/or designed functionalities.11,12 Such chemically enhanced peptide libraries expand the chemical space of phage display and have proven advantageous over canonical peptide libraries. Despite this promise, the field is still in its infancy with only a handful of methods reported for constructing nonnatural phage libraries. Additional strategies are needed to access peptide libraries of diverse designs. Herein we report a novel strategy for phage modification, which utilizes the highly efficient conjugation of an N-terminal cysteine (NCys) with 2-cyanobenzothiazole (CBT)13 to give backbone-side chain cyclized libraries featuring a rigid luciferin linker (Fig. 1). | ||
| Fig. 1 Non-natural phage libraries displaying cyclic peptides. (A) Cyclization through Cys–Cys crosslinking. (B) Cyclization through conjugation of a cysteine and a nonnatural amino acid. (C) Backbone-side chain cyclization enabled by NCys–Cys crosslinking. | ||
Screening of this library readily revealed potent peptide ligands for two proteins, namely Keap1 and Sortase A, which are chosen as examples for those involved in protein–protein interactions and difficult-to-inhibit enzymes.
Results
Library design
In comparison to their linear counterparts, cyclic peptide libraries are especially appealing since less conformational entropy is lost upon target binding. Although phage libraries of disulphide-cyclized peptides are available, the disulphide crosslinks display sub-optimal stability under reducing conditions. Furthermore, it is desirable to incorporate non-natural structural motifs that can further enhance the conformational rigidity of the peptides.14 Efforts towards this end have resulted in several elegant publications in recent literature, which describe peptide libraries with symmetric aromatic crosslinkers inserted between cysteine side chains6,7 (Fig. 1A) or between a cysteine side chain and reactive tyrosine derivative (Fig. 1B).8 Inspired by these reports, we postulated that NCys–Cys crosslinking can be accomplished using an asymmetric crosslinker to give backbone-side chain cyclized peptides on phage (Fig. 1C). Note that backbone-side chain cyclized phage libraries are rare with just two papers that appeared in recent literature.15,16 Importantly, our proposed NCys–Cys crosslinking strategy would not need the incorporation of nonnatural amino acids, which is technically simpler than some reported methods.8,9NCys is known to conjugate with CBT (Fig. S1†) under physiologic conditions with fast reaction kinetics (k2: ∼10 M−1 s−1).13 To achieve backbone-side chain cyclization, we derivatized CBT with a reversible thiol-reactive group, namely α-cyanoacrylamide (M-a-23, Fig. 1C),17 to induce facile NCys–Cys crosslinking of CXnC peptides (n randomized residues flanked by a pair of cysteines). CXnC peptide libraries can be readily constructed on M13 phage. We chose a reversible thiol-reactive group for the design of M-a-23 to ensure exclusive NCys modification by CBT. Importantly, α-cyanoacrylamide exhibits fast thiol conjugation kinetics to elicit rapid peptide cyclization, which is desirable for phage library construction.17,18 For comparison, we evaluated a non-cyano-substituted analogue of M-a-23 for peptide cyclization, which was only observed after several days (Fig. S2†).
Synthetic details of M-a-23 can be found in the ESI.† With the compound in hand, we first tested it against several model peptides, which underwent complete conjugation within 2 h (Fig. 2A, S4, and S5†). Two peaks were recorded with the same molecule weight (17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 in ratio), presumably due to the diastereomeric peptides generated from the Michael addition (Fig. 2B). We did notice a small degree of double CBT modification in which the internal cysteine also conjugated with CBT to give a thioimidate (Fig. 2A and S3†).19,20 Fortunately, we found that the internal cysteine modification could be readily reversed by the addition of free cysteine, which competitively cleaved the thioimidate to recyclize the peptide (Fig. 2A). It is important to note that the cysteine treatment caused no effect to the M-a-23 cyclized peptides, despite the potential reversibility of the α-cyanoacrylamide–thiol addition. Furthermore, the cyclized peptide was found to be stable in a redox buffer containing 1 mM glutathione (GSH:GSSG, 9:1). Finally and importantly, we found that M-a-23 modified peptides resisted iodoacetamide labelling (see later discussion and Fig. S14† for details), lending strong support to the efficient and stable peptide cyclization elicited by M-a-23.
:3 in ratio), presumably due to the diastereomeric peptides generated from the Michael addition (Fig. 2B). We did notice a small degree of double CBT modification in which the internal cysteine also conjugated with CBT to give a thioimidate (Fig. 2A and S3†).19,20 Fortunately, we found that the internal cysteine modification could be readily reversed by the addition of free cysteine, which competitively cleaved the thioimidate to recyclize the peptide (Fig. 2A). It is important to note that the cysteine treatment caused no effect to the M-a-23 cyclized peptides, despite the potential reversibility of the α-cyanoacrylamide–thiol addition. Furthermore, the cyclized peptide was found to be stable in a redox buffer containing 1 mM glutathione (GSH:GSSG, 9:1). Finally and importantly, we found that M-a-23 modified peptides resisted iodoacetamide labelling (see later discussion and Fig. S14† for details), lending strong support to the efficient and stable peptide cyclization elicited by M-a-23.
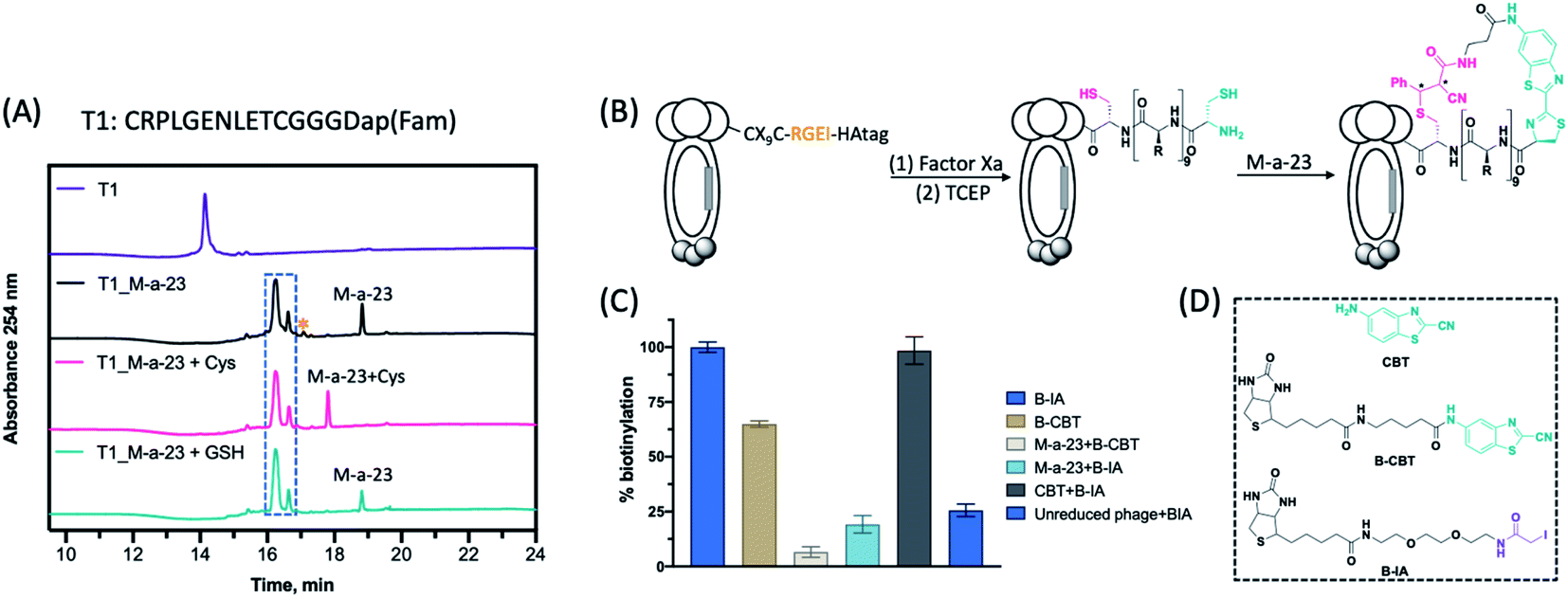 | ||
| Fig. 2 Construction of backbone-side chain cyclized phage libraries. (A) LC-MS analysis of peptide cyclization by M-a-23. Chromatograms show absorbance at 254 nm. Reaction of peptide T1 (40 μM) with M-a-23 (100 μM, 2.5 eq.) at rt for 2 h, followed by addition of Cys (250 μM) for 2 h, or incubated with GSH:GSSG (9:1, 1 mM) for 2 h. * denotes a second M-a-23 molecule reacting with the internal cysteine to give a thioimidate product. (B) Schematic illustration of the M-a-23 mediated peptide cyclization on phage. Factor Xa cleavage followed by TCEP reduction readily gives a CX9C library that harbours an N-terminal cysteine and an internal cysteine. Subsequent conjugation with M-a-23 triggers the formation of cyclic peptides. The newly generated chiral centres are marked with *. (C) ELISA assay to confirm phage modification with various CBT derivatives and controls. (D) Chemical structure of CBT, B-CBT and B-IA. | ||
To construct M-a-23 cyclized libraries, we prepared a CX9C phage library (sequence diversity: 4.5 × 108), which installs a Factor Xa cleavage site preceding the CX9C peptides (Fig. 2B). Subjecting this library to Factor Xa cleavage followed by TCEP reduction readily exposed a free NCys on the displayed peptides (Fig. S6 and S7†). The M-a-23 induced peptide cyclization on phage was validated using a streptavidin-mediated ELISA assay as well as a phage pulldown assay. These assays measure the extent of phage biotinylation after treatment with various combinations of reagents (Fig. 2D). As expected, treating a TCEP reduced CX9C phage library with B-IA elicited near quantitative phage pulldown (90%, Fig. S7†). Hence, the B-IA treated phage was used as a positive control in the ELISA assay. Treating the reduced CX9C library with B-CBT resulted in a high level of phage biotinylation (Fig. 2C and S7†). Specifically, B-CBT treated CX9C phage resulted in ∼80% pulldown, indicating efficient NCys modification by B-CBT. As expected, treating the reduced CX9C library with M-a-23 first and then by B-CBT resulted in little biotinylation and pulldown (Fig. 2C and S7†), confirming efficient phage modification by M-a-23. Importantly, a low level of biotinylation was observed when M-a-23 modified CX9C phage library was treated with B-IA (Fig. 2C). In contrast, treating the CX9C library with CBT first and then B-IA afforded a high level of phage biotinylation. These contrasting results indicate that the M-a-23 modified peptides on phage exist in cyclic forms. It is worth noting that, although efficient, M-a-23 did not elicit 100% phage modification, which would inevitably leave some unmodified phage in the library. However, we envision this is of little concern because our phage display protocol presents multiple copies of each unique phage variant (>1010 input phage vs. 4.5 × 108 sequence diversity). Considering the M-a-23 labelling efficiency of ∼80%, most copies for any unique phage variant will be converted into the M-a-23 cyclized form. In other words, despite the non-perfect labelling efficiency, each unique peptide sequence of the library will be well represented in its M-a-23 cyclized form for phage selection.
Screen for PPI inhibitors
Macrocyclic peptides have shown great promise as PPI inhibitors.2,5 To explore the potential of our luciferin-cyclized library for PPI inhibition, we carried out phage panning against Keap1 (Fig. 3A), an important protein that binds Nrf2 to upregulate cells' response to oxidative stress. Inhibiting the Keap1–Nrf2 interaction presents a promising strategy for therapeutics development for a number of diseases.8,21 For phage panning, we recombinantly expressed Keap1 with an N-terminal His-Tag, which was chemically biotinylated using biotin-NHS (Fig. S8†). The biotinylated protein was immobilized on streptavidin-coated magnetic beads, against which the M-a-23 cyclized CX9C library was screened. The enrichment of Keap1 binding phage was monitored through sequencing of randomly selected phage after each round of panning. Excitingly, a significantly increased output population was observed after two rounds of panning (Fig. S9†), and the sequencing results revealed several repeating sequences (Fig. 3B and S10†), which all display peptide segments that resemble the natural binding partner of Keap1.22 The peptides were synthesized through solid phase peptide synthesis with a fluorescein installed onto a C-terminal Dap residue preceded by a triple glycine linker. The fluorescein-labelled peptides were assessed for Keap1 binding through a fluorescence polarization assay, in which a peptide was mixed with Keap1 at varied concentrations. Plotting the recorded fluorescence polarization data against protein concentration gave a binding curve, from which a Kd value was extracted via curve fitting. | ||
| Fig. 3 Phage panning reveals potent binders of Keap1. (A) Crystal structure of Keap1 Kelch domain in complex with the Nrf2 domain (75QLDEETGEFL84, pdb 2FLU). (B) Enriched peptide sequences after two rounds of panning against Keap1. The corresponding Kd values for Keap1 binding are listed to the right. #Kd value cited from ref. 22. (C) Chemical structure of K5 derivatives. (D) Fluorescence polarization assay results of various K5 derivatives binding the target protein Keap1. *: FAM labelled peptide. (E) Competition assay of K5 derivatives binding to Keap1 recorded through competition with a Nrf2-derived peptide (Fam-βAla-DEETGEF). Each data point in (D) and (E) represents the mean value of three independent measurements. | ||
As shown by the tabulated Kd values (Fig. 3B and S11†), all peptide hits yielded from phage panning proved to be potent Keap1 binders. In particular, K4_M-a-23* and K5_M-a-23* were found to exhibit low nanomolar affinity, yielding a Kd value of 0.14 and 0.06 μM respectively. To evaluate the potential significance of the luciferin linker, we analysed the disulphide cyclized peptides for Keap1 binding as negative controls. Gratifyingly, the M-a-23 cyclized peptides were found to display greater potency than the disulphide controls. For example, the M-a-23 cyclized K6 gave a Kd of 1.0 μM for Keap1 binding, six times lower than that of the disulphide control (Kd: 5.9 μM) (Fig. S12†). Similarly, the M-a-23 cyclized K5 exhibited eight times higher affinity than the disulphide control as well (Kd: 0.06 vs. 0.48 μM) (Fig. 3C and D). The binding affinity differences determined by Keap1 titration were further confirmed using a competition assay, in which the non-fluorophore labelled K5_SS and K5_M-a-23 were titrated into a preformed Keap1-fluorescence substrate complex. The competition assay revealed four times greater potency for the M-a-23 cyclized K5 (Fig. 3E). We further compared the M-a-23 cyclized K5 to a linear control, for which the NCys of K5* was capped with CBT (Fig. 3C and D). This linear control peptide showed eight times lower affinity for Keap1 as well, indicating the importance of the M-a-23 induced cyclization for the peptide's binding to Keap1.
To gain structural insight into the peptide-Keap1 binding, we performed computational docking studies of using a structural analogue of K5_M-a-23, namely K5_M-a-23-2, in which the cyano group is omitted (Fig. 4A, see below for more details of this analogue). The docking poses showed that this luciferin-cyclized peptide adopts a binding geometry very similar to that of Nrf2 peptide (Fig. 4C), which is perhaps not surprising given the high structural similarity between these two peptides (Fig. 3B). Importantly, the docked structure revealed a direct contact between the protein and the phenyl group of the thiol-acrylamide adduct, which may explain the higher potency of K5_M-a-23 for Keap1 binding in comparison to the disulphide or linear controls.
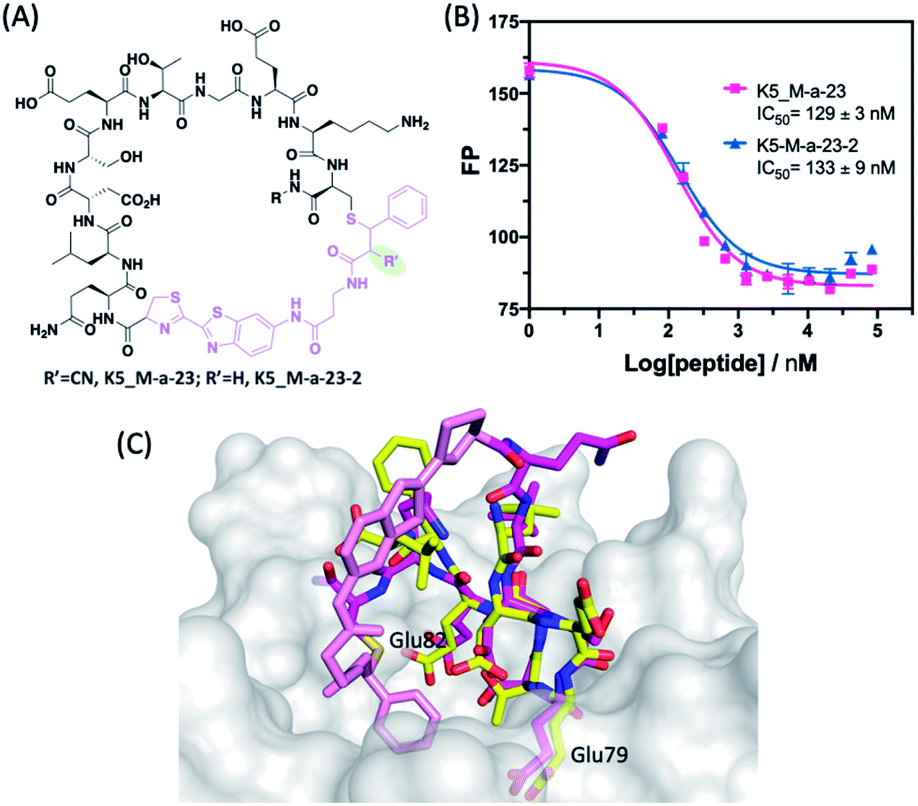 | ||
| Fig. 4 Irreversible peptide cyclization with M-a-23-2. (A) Chemical structure of peptides K5_M-a-23 and K5_M-a-23-2. (B) Results of the competition assay showing identical Keap1 binding affinity for K5_M-a-23 and K5_M-a-23-2. (C) Overlay of a docking pose of K5_M-a-23-2 with the natural peptide substrate of Keap1. The Keap1–Nrf2 complex structure is rendered using PDB file 5WFV. Details protocols for docking and superposition can be found in the ESI.† Colour scheme: Keap1, white; peptide ligand N: blue; O: red; Nrf2 C: yellow; K5_M-a-23-2 C: magenta; M-a-23-2 derived crosslinker: pink. | ||
Evaluating the robustness of the M-a-23 cyclized peptides
Considering the potential reversibility of the M-a-23 induced cyclization, we tested the peptide hits for Keap1 binding in a redox buffer mimicking intracellular conditions. Specifically, the Keap1 binding of K5_M-a-23* was examined in the presence of GSH:GSSG (9:1). Nearly identical binding curves were obtained in the presence and absence of glutathione (Fig. S13†). To quantitatively assess the kinetic stability of the M-a-23 cyclized peptides, we used iodoacetamide (IA), an irreversible cystine alkylation agent to capture the free internal cysteine in order to estimate the cyclic peptide ring-opening rate. Specifically, K5_M-a-23* was incubated with 500 μM IA in PBS buffer at room temperature, and the sample was monitored by LC-MS at different time point (Fig. S14†). The data showed that, after 2 h incubation, only 15% of the peptide was alkylated with IA. In contrast, a CBT modified linear control peptide was fully alkylated under the same experimental condition (Fig. S15†). Fitting the kinetic data of K5_M-a-23* disappearance over time yielded a pseudo-first order rate constant (kobs) with a value of 1.38 × 10−5 s−1, which corresponds to a half-life of 20 hours for the M-a-23 cyclized peptides. It is interesting to note that the dissociation rate (k−1) for a bimolecular α-cyanoacrylamide–cysteine conjugate is 1.5 × 10−2 s−1 (Fig. S16†). In comparison, the cyclic peptide ring opening is ∼1000 times slower than the bimolecular conjugate, owing to the intramolecular nature of the M-a-23 mediated peptide cyclization.
We further envisioned that removing the CN group in M-a-23 would completely eliminate the reversibility issue of the cysteine–α-cyanoacrylamide linkage. Towards this end, we evaluated M-a-23-2 cyclized peptides (Fig. 4A). Without the CN group, M-a-23-2 elicited much slowed peptide cyclization (Fig. S2†), which is unsuitable for phage library construction, however may be acceptable for the synthesis of cyclic peptides during hit optimization. Indeed, although much slower, M-a-23-2 did elicit complete cyclization of K5 after four days. Using the competition assay, we assessed the Keap1 binding of K5_M-a-23-2 in comparison to K5_M-a-23 in parallel. The results showed essentially identical binding curves and IC50 values (Fig. 4B). The tight binding of K5_M-a-23-2 is supported by a docking structure (Fig. 4C), in which the cyclic peptide displays a overlapping structures as the Nrf2 peptide. Overall these results suggest that the combined use of M-a-23 and M-a-23-2 for library screening and peptide hit synthesis respectively may be a powerful strategy to develop potent and stable macrocyclic peptide inhibitors.
Screening for inhibitors of protein-processing enzymes
Similar to proteins involved in protein–protein interactions, some protein processing enzymes including selected proteases and ligases have also frustrated small molecule drug discovery efforts since these enzymes often display large and shallow active sites. Comparatively, macrocyclic peptides, due to their large size, present an attractive modality for inhibiting such protein-processing enzymes.23,24 To test the potential of our M-a-23 cyclized phage library on this front, we have performed phage panning against Sortase A (SrtA) as a model target protein. SrtA is an important enzyme that covalently anchors the extracellular proteins of Staphylococcus aureus to its cell wall (Fig. 5A). SrtA inhibition is believed to be a powerful mechanism to curb staph aureus virulence without forcing bacterial evolution to acquire antibiotic resistance.25–27 We expressed SrtA with an AviTag on the N-terminus of the protein (Fig. S17†), which allows highly efficient and site-specific biotinylation of this target protein. Then phage panning was performed against target proteins after their immobilization on streptavidin-coated magnetic beads.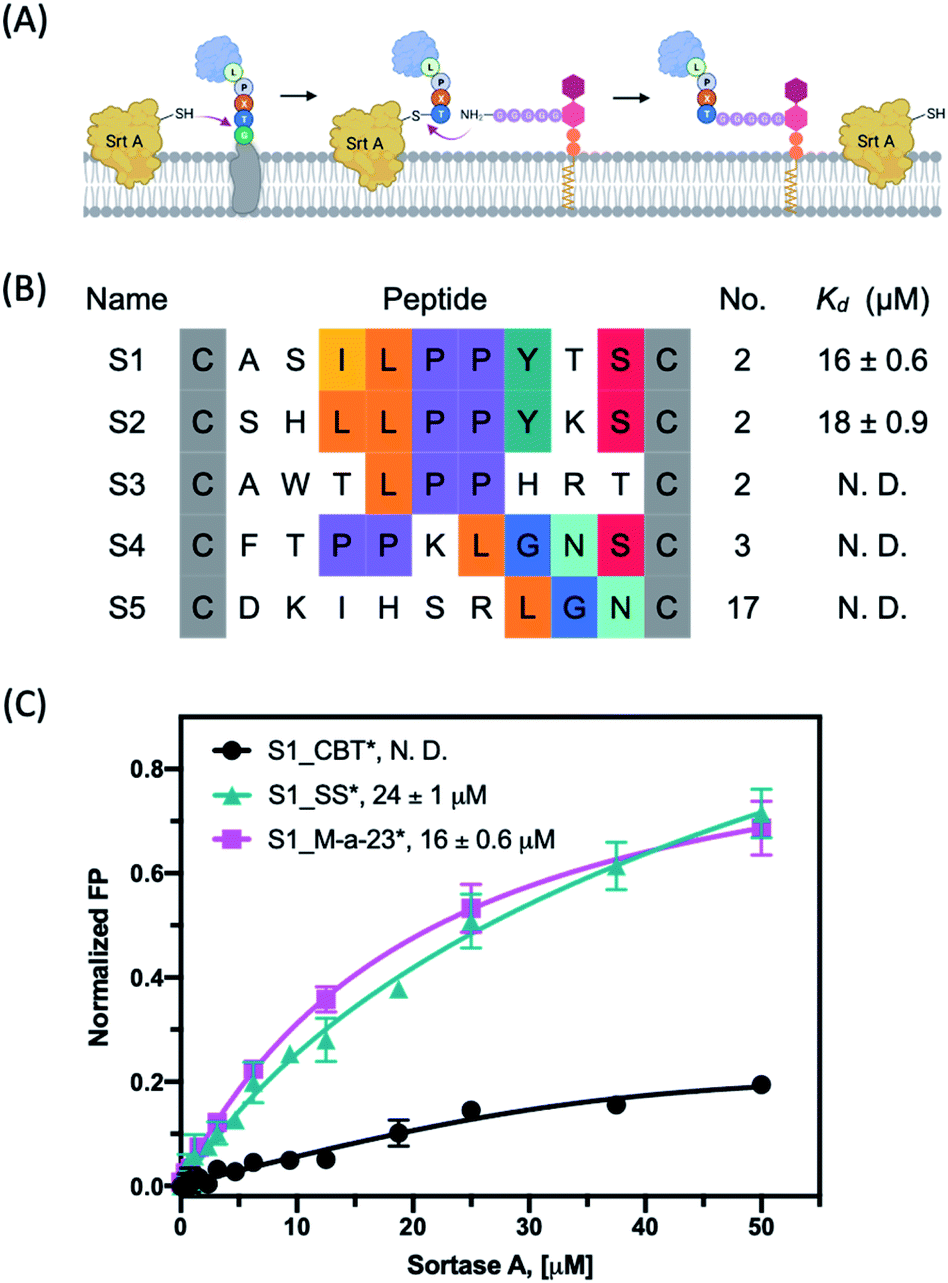 | ||
| Fig. 5 (A) Schematic representation of how extracellular proteins are anchored to the cell wall via Sortase A. Figure was created with http://Biorender.com. (B) The primary sequences after four rounds of panning against Sortase A. (C) Fluorescence polarization assay of various peptide hit S1 derivatives with target protein SrtA. Each data point presents the mean value of three independent measurements. *: FAM labeled peptide. | ||
Similar to what we observed with Keap1, panning of the M-a-23 cyclized CX9C library against SrtA led to the convergence of peptide sequences after four rounds of panning: multiple recurring sequences were found to harbour either an LPP or LGN signature (Fig. 5B and S18†). We note that LPP-containing sequences were also observed in previously reported cyclic peptide ligands of SrtA.26 These peptide hits were synthesized in pure form and characterized for SrtA binding. Interestingly, the peptides S1_M-a-23* and S2_M-a-23* showed low micromolar binding affinity to SrtA, which shared the LPPYXS motif (Fig. 5B). The remaining peptide sequences (S3–S5) did not show significant SrtA binding up to 50 μM and are deemed as false positive hits (Fig. S19†). In addition, the M-a-23 cyclized peptides were found to bind SrtA with either comparable or just slightly higher affinity in comparison to the disulphide cyclized peptides. For example, M-a-23 cyclized S2 gives a Kd value of 18 μM in contrast to 25 μM for the corresponding disulphide peptide (Fig. S19†). In other words, SrtA inhibition appears to benefit to a smaller degree by having a M-a-23 derived crosslinker compared to the Keap1 binding peptides. The underlying chemical basis for this observation needs to be further explored as molecular docking studies did not reveal compelling hypotheses (Fig. S20 and S21†). Nevertheless, the peptide cyclization was found to be critical for the peptide's binding to SrtA whereas a CBT capped control peptide showed a much reduced SrtA binding propensity under the same experimental conditions (Fig. 5C).
Conclusions
This contribution describes a novel strategy for constructing macrocyclic peptide libraries on bacteriophage, which enables facile screening for potent inhibitors for difficult-to-inhibit proteins. Specifically, we devised a CBT based crosslinker, namely M-a-23, that elicits facile crosslinking of an N-terminal cysteine (NCys) and internal cysteine to create backbone-side chain cyclized peptides on phage. Our design of M-a-23 installs an α-cyanoacrylamide, a fast and reversible thiol-reactive group, onto CBT, which is known to selectively modify N-terminal cysteines. The reversibility of α-cyanoacrylamide–thiol conjugation avoids competition with CBT for NCys modification. Importantly, M-a-23 cyclized peptides exhibit robust stability as the intramolecular α-cyanoacrylamide–thiol linkage show much slowed dissociation kinetics. There are several reports in literature describing phage-displayed macrocyclic peptide libraries. However, except two recent papers,15,16 all other reports describe side chain-side chain crosslinked peptide macrocycles. This contribution presents a rare example of backbone-side chain cyclized peptide libraries on phage. In comparison to the two recent reports of backbone-side chain cyclized libraries,15,16 our M-a-23 crosslinker enjoys the ease of synthesis, well-characterized reaction profiles and no need for a reduction step16 for stable cyclization. These characteristics make it appealing for ligand discovery for a variety of biological targets.Author contributions
M. Z. and J. G. conceived the project and composed the manuscript. M. Z. collected all experimental data. F. H. performed molecular docking studies and contributed to the composition of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support is provided by the National Institute of Health and the National Science Foundation through grants GM124231 and CHE-1904874, respectively.Notes and references
- G. P. Smith and V. A. Petrenko, Chem. Rev., 1997, 97, 391–410 CrossRef CAS
.
- T. Passioura, T. Katoh, Y. Goto and H. Suga, Annu. Rev. Biochem., 2014, 83, 727–752 CrossRef CAS
- R. Derda and S. Ng, Curr. Opin. Chem. Biol., 2019, 50, 128–137 CrossRef CAS PubMed
- C. Heinis and G. Winter, Curr. Opin. Chem. Biol., 2015, 26, 89–98 CrossRef CAS PubMed
- C. Sohrabi, A. Foster and A. Tavassoli, Nat. Rev. Chem., 2020, 4, 90–101 CrossRef CAS
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS
- M. R. Jafari, L. Deng, P. I. Kitov, S. Ng, W. L. Matochko, K. F. Tjhung, A. Zeberoff, A. Elias, J. S. Klassen and R. Derda, ACS Chem. Biol., 2014, 9, 443–450 CrossRef CAS PubMed
- A. E. Owens, J. A. Iannuzzelli, Y. Gu and R. Fasan, ACS Cent. Sci., 2020, 6, 368–381 CrossRef CAS PubMed
- X. S. Wang, P.-H. C. Chen, J. T. Hampton, J. M. Tharp, C. A. Reed, S. K. Das, D.-S. Wang, H. S. Hayatshahi, Y. Shen, J. Liu and W. R. Liu, Angew. Chem., Int. Ed., 2019, 58, 15904–15909 CrossRef CAS PubMed
- X. Zheng, W. Liu, Z. Liu, Y. Zhao and C. Wu, Bioconjugate Chem., 2020, 31, 2085–2091 CrossRef CAS
- K. A. McCarthy, M. A. Kelly, K. Li, S. Cambray, A. S. Hosseini, T. van Opijnen and J. Gao, J. Am. Chem. Soc., 2018, 140, 6137–6145 CrossRef CAS
- J. M. Tharp, J. T. Hampton, C. A. Reed, A. Ehnbom, P.-H. C. Chen, J. S. Morse, Y. Kurra, L. M. Pérez, S. Xu and W. R. Liu, Nat. Commun., 2020, 11, 1392 CrossRef CAS
- H. Ren, F. Xiao, K. Zhan, Y. P. Kim, H. Xie, Z. Xia and J. Rao, Angew. Chem., Int. Ed., 2009, 48, 9658–9662 CrossRef CAS
- V. Adebomi, R. D. Cohen, R. Wills, H. A. H. Chavers, G. E. Martin and M. Raj, Angew. Chem., Int. Ed., 2019, 58, 19073–19080 CrossRef CAS
- X. Zheng, Z. Li, W. Gao, X. Meng, X. Li, L. Y. P. Luk, Y. Zhao, Y.-H. Tsai and C. Wu, J. Am. Chem. Soc., 2020, 142, 5097–5103 CrossRef CAS PubMed
- T. R. Oppewal, I. D. Jansen, J. Hekelaar and C. Mayer, J. Am. Chem. Soc., 2022, 144, 3644–3652 CrossRef CAS PubMed
- I. M. Serafimova, M. A. Pufall, S. Krishnan, K. Duda, M. S. Cohen, R. L. Maglathlin, J. M. McFarland, R. M. Miller, M. Frodin and J. Taunton, Nat. Chem. Biol., 2012, 8, 471–476 CrossRef CAS
- P. A. Jackson, J. C. Widen, D. A. Harki and K. M. Brummond, J. Med. Chem., 2017, 60, 839–885 CrossRef CAS PubMed
- S. G. L. Keyser, A. Utz and C. R. Bertozzi, J. Org. Chem., 2018, 83, 7467–7479 CrossRef CAS PubMed
- W. Wang and J. Gao, J. Org. Chem., 2020, 85, 1756–1763 CrossRef CAS PubMed
- D. A. Abed, M. Goldstein, H. Albanyan, H. Jin and L. Hu, Acta Pharm. Sin. B, 2015, 5, 285–299 CrossRef PubMed
- Y. Chen, D. Inoyama, A. N. Kong, L. J. Beamer and L. Hu, Chem. Biol. Drug Des., 2011, 78, 1014–1021 CrossRef CAS
- A. Angelini, L. Cendron, S. Chen, J. Touati, G. Winter, G. Zanotti and C. Heinis, ACS Chem. Biol., 2012, 7, 817–821 CrossRef CAS PubMed
- P. Sarkar, Z. Li, W. Ren, S. Wang, S. Shao, J. Sun, X. Ren, N. G. Perkins, Z. Guo, C.-E. A. Chang, J. Song and M. Xue, J. Med. Chem., 2020, 63, 6979–6990 CrossRef CAS PubMed
- F. T. Hofmann, J. W. Szostak and F. P. Seebeck, J. Am. Chem. Soc., 2012, 134, 8038–8041 CrossRef CAS
- I. Rentero Rebollo, S. McCallin, D. Bertoldo, J. M. Entenza, P. Moreillon and C. Heinis, ACS Med. Chem. Lett., 2016, 7, 606–611 CrossRef CAS PubMed
- J. Zhang, H. Liu, K. Zhu, S. Gong, S. Dramsi, Y.-T. Wang, J. Li, F. Chen, R. Zhang, L. Zhou, L. Lan, H. Jiang, O. Schneewind, C. Luo and C.-G. Yang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13517–13522 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed protocols for synthesis and biochemical assays, and additional characterization of the peptide hits. See https://doi.org/10.1039/d2sc03241d |
| This journal is © The Royal Society of Chemistry 2022 |