
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTranscriptome-wide profiling of N6-methyladenosine via a selective chemical labeling method†
Yalun
Xie‡
 a,
Shaoqing
Han‡
a,
Qiming
Li
b,
Zhentian
Fang
a,
Wei
Yang
a,
Qi
Wei
a,
Yafen
Wang
a,
Yu
Zhou
b,
Xiaocheng
Weng
a and
Xiang
Zhou
*a
a,
Shaoqing
Han‡
a,
Qiming
Li
b,
Zhentian
Fang
a,
Wei
Yang
a,
Qi
Wei
a,
Yafen
Wang
a,
Yu
Zhou
b,
Xiaocheng
Weng
a and
Xiang
Zhou
*a
aCollege of Chemistry and Molecular Sciences, Key Laboratory of Biomedical Polymers of Ministry of Education, Wuhan University, Wuhan, China. E-mail: xzhou@whu.edu.cn
bCollege of Life Science, Wuhan University, Wuhan, Hubei, China
First published on 5th October 2022
Abstract
Studies of chemical modifications on RNA have ushered in the field of epitranscriptomics. N6-Methyladenosine (m6A) is the most typical RNA modification and is indispensable for basic biological processes. This study presents a chemical pulldown method (m6A-ORL-Seq) for transcriptome-wide profiling of m6A. Moreover, chemical labeling results in a specific reverse transcription (RT) truncation signature. This study has identified four thousand high-confidence m6A sites at single-base resolution in the human transcriptome. Unlike previously reported methods based on m6A-antibody or m6A-sensitive enzymes, the antibody/enzyme-free chemical method provides a new perspective for single-base m6A detection at the transcriptome level.
Introduction
More than 163 chemical modifications have been identified in RNA, these modifications bearing varied chemical groups regulate gene expressions in multiple ways.1 Among them, N6-methyladenosine (m6A) is the most abundant post-transcriptional modification in messenger RNA (mRNA) and noncoding RNA (ncRNA) in eukaryotes.2 The discovery of m6A-associated proteins (m6A writers, readers, and erasers) has revealed that m6A is a crucial regulator in mRNA splicing, export, translation, decay, and structure.3–11 Furthermore, aberrant levels of m6A have been closely associated with tumor development.12,13 To interpret further the biological functions of m6A, detection and precise location of m6A in transcriptome is essential.Combined with m6A specific antibodies and RNA-Seq, two independent works (m6A-Seq or MeRIP-Seq) reported the map of m6A methylomes among mammals and determined a highly conserved m6A motif (DRACH, D = G/A/U; R = G/A; H = A/C/U) that was enriched in 3′ UTRs and near stop codons.14,15 However, this most used approach has several fated drawbacks, including low resolution (>200 nt), binding to particular RNA sequences, large amounts of starting samples (>100 μg total RNA), and abundant false positives (limited specificity). Therefore, antibody-dependent or antibody-free enzyme-dependent methods have been subsequently developed. Crosslinking immunoprecipitation-based methods (PA-Seq,16 m6A-CLIP,17 and miCLIP18) improve resolution. The antibody-free enzymatic approaches (MAZTER-Seq,19 m6A-REF-Seq,20 DART-Seq,21 m6A-label-Seq,22 m6A-SEAL-Seq23 and m6A-SAC-Seq24) have also been developed for higher resolution and quantitative profiling. Meanwhile, third-generation sequencing techniques including single-molecule real-time (SMRT) sequencing25 and nanopore sequencing,26,27 enable long-read and single-molecule sequencing of m6A. However, the sequencing data derived from enzymatic approaches also contains abundant false positives resulting from sequence bias or RNA structure. MAZTER-Seq and m6A-REF-Seq use an m6A-sensitive RNA endoribonuclease (MazF) to facilitate m6A sequencing at single-base resolution. On the other hand, Luo et al.28 confirmed that a CCACAG motif and RNA secondary structure affects MazF cleavage, leading to widespread false positives. Although third-generation sequencing enables the direct detection of RNA modification, the high error probability limits its application to practical samples.29 Hence, an antibody/enzyme-free and selective chemical labeling method is urgently necessary to solve these problems.
Due to the inert reactivity of methyl group in m6A, chemical discrimination of m6A and A is a great challenge for chemist. To date, no antibody/enzyme-free chemical approach was developed for selective and efficient labeling of m6A in RNA. It is worth mentioning that Gaunt et al.30 recently reported a method based on metallophotocatalyst-assisted chemistry can label N6-methyladenosine (6mdA) in DNA. However, when this method was applied to the labeling of a 13 nt RNA oligo containing m6A, the yield was as low as 5% together with considerable RNA degradation, thus this method is difficult to be used for the study of m6A transcriptomics at the current stage. In this article, we report an unprecedented chemical method for selective labeling and transcriptome-wide profiling of m6A via three steps of chemical reactions, oxidation, reduction, functional labeling (m6A-ORL-Seq, Fig. 1).
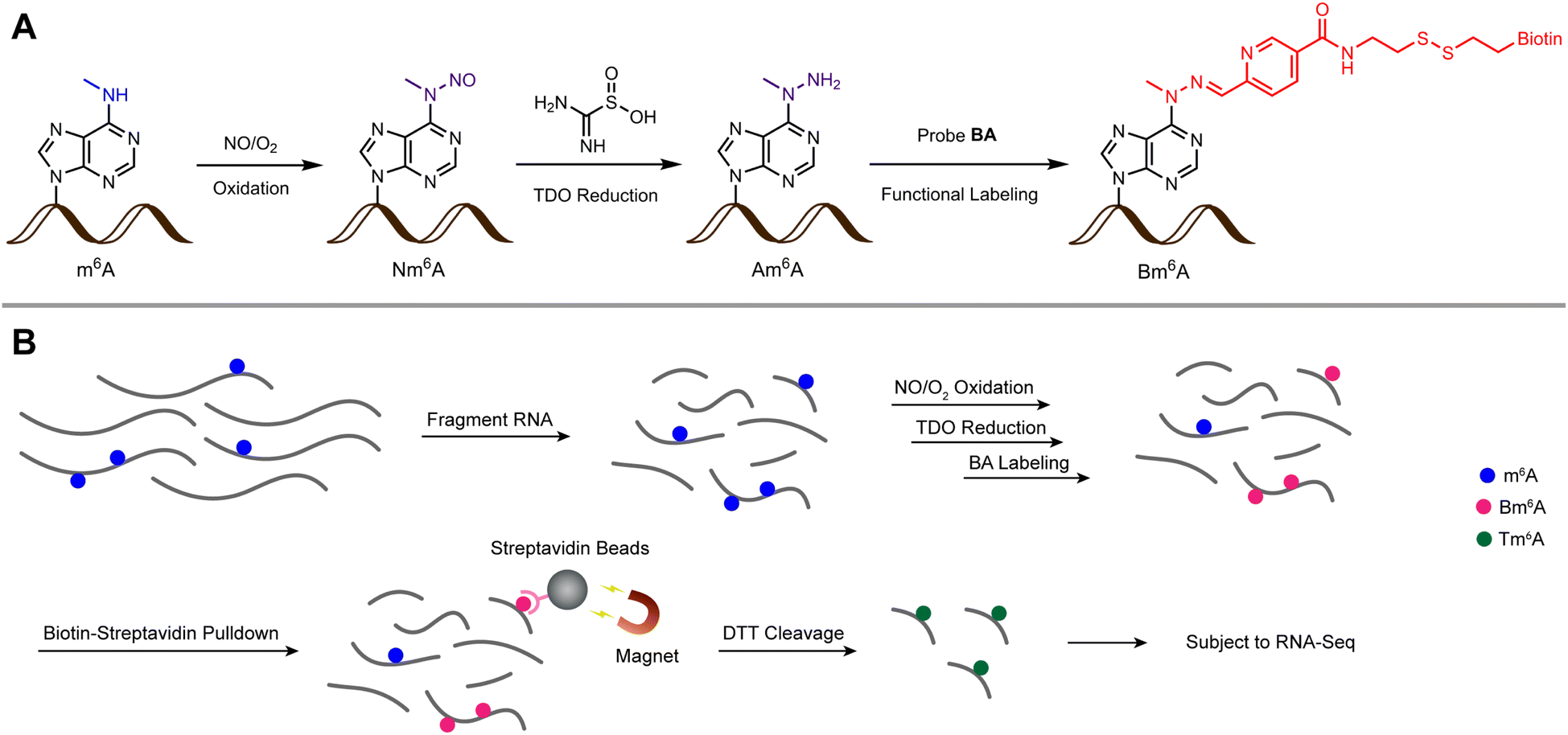 | ||
| Fig. 1 A chemical method to selective label and locate m6A in transcriptomes. (A) RNA m6A was stepwise converted into Nm6A, Am6A, and Bm6A through a three-step chemical labeling method. (B) A schematic illustration of m6A-ORL-Seq for transcriptome-wide profiling of m6A. Nm6A: N6-nitroso m6A; Am6A: N6-amino m6A; Bm6A: biotinylated m6A; Tm6A: thiolated m6A; TDO: thiourea dioxide; BA: biotinylated aryl aldehyde probe. | ||
Coincidentally, Hili et al.31 and Helm et al.32 reported sodium nitrite-based chemical methods for m6A detection. However, these two methods are not applicable to transcriptome-wide studies at the current stage and are also powerless for locating m6A sites of low modification level unless the combination with m6A immunoprecipitation. The detailed discussion compared to the method in this study is stated in ESI.† Meanwhile, Wu et al.33 reported sodium nitrite-based chemical methods for genomic 6 mA profiling.
Experimental
Synthesis of RNA oligo and model containing m6A
The RNA phosphoramidites were purchased from Wuhu Huaren Science and Technology Co., Ltd. 12/15/60 nt m6A-modified RNA was synthesized on 12-column DNA synthesizer (PolyGen). Universal CPG (39.5 μmol g−1, 1000 Å, Beijing DNAchem Biotechnology Co. Ltd.) bound RNA was firstly treated with a mixture of 28% aqueous ammonia and 40% aqueous methylamine (200 μL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), then concentrated in vacuo and incubated with a mixture of DMSO and Et3N·3HF (200 μL, 1:1) at 60 °C for 2.5 h. At last RNA oligonucleotides were purified using ethanol precipitation.
:1), then concentrated in vacuo and incubated with a mixture of DMSO and Et3N·3HF (200 μL, 1:1) at 60 °C for 2.5 h. At last RNA oligonucleotides were purified using ethanol precipitation.
N-Nitrosation (oxidation) of m6A to Nm6A, deamination of A/G/C to I/X/U in ribonucleosides
A 1.5 mL tube was mixed with 50 μM C, G, A, and m6A in 200 μL sodium acetate buffer (1.0 M, pH 4.5). In an ice bath, 3.00 sccm NO bubbling into the bottom of the tube (lid is open) for 3.0 min through a 22 gauge needle (this treatment must be handled in a standard laboratory chemical hood). Then cover the lid, the solution was standing at 4 °C for 4–30 h. After incubation, the solution was directly injected into HPLC and analyzed. A: 3% CH3CN in TEAA (100 mM, pH 7.5); B: 90% CH3CN in H2O; 35 °C, 1.0 mL min−1, 260 nm, 0–10–15–25 min, 0–0–5–100% B.N-Nitrosation (oxidation) of m6A to Nm6A in oligo/model RNA/total RNA/poly (A)+ RNA
A 1.5 mL tube was mixed with RNA in 200 μL sodium acetate buffer (3.0 M, pH 4.5, containing 2 mM EDTA). In an ice bath, 3.00 sccm NO bubbling into the bottom of the tube (lid is open) for 6.0 min through a 22 gauge needle (this treatment must be handled in a standard laboratory chemical hood). Then cover the lid, the solution was standing at 4 °C for 20 h. Then RNA was purified using ethanol precipitation or RNA Clean & Concentrator-5 (RCC, Zymo).TDO reduction of Nm6A to Am6A
For ribonucleosides: 100 μM Nm6A, 100 mM TDO, 300 mM Na2CO3/NaHCO3 (pH 10.0), 50 °C, 1000 rpm, 5 min. For RNA oligo, 5.0 μg oligo, 100 mM TDO, 300 mM Na2CO3/NaHCO3, 2 mM EDTA, pH 10.0, 50 °C, 1000 rpm, 10 min, total volume 50 μL. For total RNA and poly (A)+ RNA, 100 mM TDO, 300 mM Na2CO3/NaHCO3, 2 mM EDTA, pH 10.0, 50 °C, 1000 rpm, 10 min, total volume 30 μL.Biotinylation via probe BA labeling, Am6A to Bm6A in RNA
Oligo RNA/model RNA/total RNA/poly (A)+ RNA, 2.0 mM BA, 100 mM MES, 10% DMF, pH 5.5, 37 °C, 1000 rpm, 1.0 h, total volume 50 μL.Enzymatic digestion and LC-HRMS analysis
500 ng RNA oligonucleotide was digested with 1 U of nuclease P1 (Sigma-Aldrich) in digestion buffer (2 mM ZnCl2, 10 mM NaCl, 25 μL) at 37 °C for 2 h, followed by incubating with 1 U rSAP (NEB) in freshly prepared 100 mM NH4HCO3 at 37 °C for 2 h. The digested samples were centrifuged at 12000 rpm for 10 min, and 20 μL of the solution was loaded into LC-HRMS (Thermo Fisher Scientific LTQ Orbitrap Elite) analysis.
Human cell culture
HEK-293T cells were cultured in DMEM medium without sodium pyruvate (Dulbecco's Modified Eagle Medium, HyClone) supplemented with 10% FBS (Cegrogen) and 1% penicillin/streptomycin (Genview). The cells were maintained at 37 °C under a humidified atmosphere containing 5% CO2.Isolation of total RNA and poly (A)+ RNA from cells
Total RNA was isolated from HEK-293T with TRIzol (Invitrogen) according to the manufacture's protocol. Then genomic DNA was removed by TURBO DNase (Invitrogen). Poly (A)+ RNA was isolated by subjecting total RNA to oligo (dT) enrichment using oligo d(T)25 Magnetic Beads (NEB) according to the manufacture's protocol.RNA fragmentation
Total RNA or poly (A)+ RNA was fragmented using RNA fragmentation reagents (Thermo) at 70 °C. For 60–200 nt segments (m6A-ORL-Seq), using 15.0 min as incubating time; for 100–300 nt segments (MeRIP-Seq), using 8.0 min as incubating time.Dot blot assay
RNA samples were pipetted and dotted on a tailored Amersham Hybond-N+ membrane (GE Healthcare). After drying naturally, the samples were fixed on the membrane by UV light (254 nm) cross-linking at room temperature and washed with 1× TBST (0.1% Tween-20) three times. Next, the membrane was blocked with 5% BSA at 70 rpm, 37 °C for 2 h and washed with 1× TBST five times. After incubation with streptavidin–horseradish peroxidase (1:1500) (Thermo Scientific) at 70 rpm, 37 °C for 1 h and washing with 1× TBST four times, the biotinylated samples in the membrane were visualized by enhanced chemiluminescence (SuperSignal West Pico PLUS Chemiluminescent Substrate, Thermo Scientific) using a ChemiDoc XRS+ Imaging System (Bio-Rad). At last, the membrane was washed twice with 1× TBST and stained with methylene blue.
Protocol for m6A-ORL-Seq
5.0 μg model RNA (MALAT1-2577-m6A)/30.0 μg total RNA/5.0 μg poly (A)+ RNA was stepwise oxidized (N-nitrosation), TDO reduced and BA labeled. After each chemical treatment, RNA was purified using RCC. Purified biotinylated RNA (500 ng purified biotinylated model RNA), mixing with 50 μg Yeast tRNA (Thermo) and subjected to pulldown procedure. Non-treated RNA or biotinylated RNA was used as input. 25 μL Dynabeads MyOne Streptavidin C1 (Thermo) was washed twice by 200 μL solution A (100 mM NaOH, 50 mM NaCl) to remove RNase contamination, then washed twice with RNase-free (RNF) H2O. The beads were resuspended in 100 μL binding solution containing 10 μL of high-salt wash buffer (100 mM Tris,10 mM EDTA, 1.0 M NaCl, pH 7.5, 0.05% Tween 20) and 90 μl RNF H2O, and incubated with the biotinylated RNA for 1 h. The beads with biotinylated RNA were washed three times with 1.0 mL high salt wash buffer. Enriched RNA was cleaved from beads using 50 μL of 100 mM DTT at 37 °C, 1000 rpm for 15 min. After collecting the supernatant, the second 50 μl of 100 mM DTT cleavage was performed at 50 °C for 5 min. The twice-eluted RNA was combined and purified using RCC, subsequently quantified by Qubit 4. 50 ng RNA was used for the next library construction (detailed in part 10). Libraries were sequenced on the Illumina HiSeq X-Ten platform with a paired-end model (PE150).MeRIP-seq
MeRIP-Seq (10.0 μg poly (A)+ RNA) was performed using EpiMark® N6-Methyladenosine Enrichment Kit (NEB) according to the manufacture's protocol.Data preprocessing
The human hg38 genome and list of human transcripts v31 were downloaded from Gencode (https://www.gencodegenes.org/). Note that we include a barcode of random decamer (NNNNNNNNNN) ligated to the fragments during library construction. Raw FASTQ reads were trimmed to remove adaptor contamination, random hexamer and aligned to the reference genome using cutadapt (v1.18)34 and STAR 2.7.5a,35 respectively. Reads less than 30 in length were removed, and only the proper pair and uniquely mapped alignments were persisted for the downstream pipelines. Then aligned reads were used for peak calling by exomePeak. Overlapped peaks between different samples were found by the BEDtools intersect function. MetaPlotR package36 was used for creating metagene plots. HOMER was used to detect the sequence motif.Candidate m6A sites at base resolution were determined according to the following steps: (i) scanned the motif DRACH in the peaks, counting the number of DRACH at the head or terminal of a sequence read. (ii) Each DRACH motif on the reference genome must be no less than 5 truncated reads. (iii) The stop rate at A must be greater than 0.1.
SELECT validation37
The FTO demethylation (FTO+) was performed in a 50 μL scale reaction containing 1.0 μg total RNA or poly (A)+ RNA, purified FTO protein, 80 μM (NH4)2Fe(SO4)2, 2 mM L-ascorbic acid, 300 μM 2-ketoglutarate, 2 U/μL RNase inhibitor, 50 μg mL−1 BSA, 50 mM HEPES, pH 7.0. For the control reaction (FTO-EDTA), EDTA was added to 5 mM before the addition of the enzyme. The reactions were incubated at 37 °C for 1 h and subsequently quenched by EDTA (0.5 M, 2.0 μL). Then RNA was purified using RCC for next use.100 ng RNA sample (FTO+, FTO-EDTA) were added with Up primer (800 nM, 1.0 μL), down primer (800 nM, 1.0 μL) and dTTP (100 μM, 1.0 μL) in 17 μL 1 × CutSmart buffer (NEB). The RNA and primers were annealed by incubating mixture at a temperature gradient: 90 °C for 1 min, 80 °C for 1 min, 70 °C for 1 min, 60 °C for 1 min, 50 °C for 1 min, and then 40 °C for 6 min. Subsequently, adding 0.01 U Bst 2.0 DNA polymerase (NEB), 0.5 U SplintR ligase (NEB), and 10 nmol ATP to the former mixture to the final volume of 20 μL. The reaction mixture was incubated at 40 °C for 20 min, denatured at 80 °C for 20 min and kept at 4 °C. Then 20 μL qPCR reaction consisted of 2× Hieff qPCR SYBR Green Master Mix (Yeasen) 200 nM qPCRF primer, 200 nM qPCRR primer and 2.0 μL reaction mixture was performed on the following parameters: 95 °C, 5 min; (95 °C, 10 s; 60 °C, 35 s; data collection) × 40 cycles; 4 °C, hold.
Reverse transcription (RT) reactions for ODN-X
ODN-X (2 μL, 4 μM) was mixed with 1 μL of 2 μM 12 nt RT primer 5′-HEX-AGAATCATCGAA and denatured at 70 °C for 5 min, then cooled down slowly to 4 °C (1 °C per 10 s) and hold at 4 °C. After adding 5 U M-MuLV reverse transcriptase (NEB) or 5 U SS III reverse transcriptase (Invitrogen), 2 μL corresponding 5 × buffer (NEB M-MuLV buffer or 5 × FS buffer), 3 μL of 100 μM dNTPs and to the RNA-primer mix (For ODN-Tm6A, additional 1 μL of 100 mM DTT was added), the RT reactions were carried out at 42 °C for 30 min. Then 30 μL deionized formamide was immediately added to the reaction mixture and heated to 90 °C for 10 min, followed by 20% denaturing PAGE analysis.Results
Chemical labeling at the nucleoside level
Inspired by the design of N-nitrosation reactivity-based fluorescent probes,38,39 we noticed that m6A could transform into N-nitrosamine (Nm6A) via N-nitrosation. After attempts of several nitrosating reagents,40–42 the most direct reagent, nitric oxide (NO) aqueous solution, was eventually chosen to facilitate the N-nitrosation of m6A at a mild condition (4 °C, sodium acetate buffer, pH 4.5). While NO can also have the deaminating ability of other bases under aerobic conditions, which converts adenosine (A) into inosine (I), guanosine (G) into xanthosine (X), and cytosine (C) into uridine (U).43,44 Under optimized conditions, the conversion rate of m6A to Nm6A is 88%, A to I is 31%, G to X is 56%, C to U is 9.8% (Fig. 2A). Other abundant modifications in human transcriptome,45 pseudouridine (Ψ), 5-methylcytidine (m5C), N1-methyladenosine (m1A), N4-acetylcytidine (ac4C), and N7-methylguanosine (m7G) were also tested in the same manner (ESI Fig. S30†); m5C was similarly converted into m5U, and m7G was similarly converted into m7X. Next, a green reductant, thiourea dioxide (TDO),46 was screened out to reduce Nm6A into N6-amino m6A (Am6A, HPLC yield: 60%, Fig. 2B, ESI Fig. S1†) and m6A in a Na2CO3/NaHCO3 buffered solution (300 mM, pH = 10.0) at 50 °C. Then, 2-pyridinecarboxaldehyde was used to test the reactivity with Am6A, which was efficiently transformed to the hydrazone in water without any catalyst (ESI,† part 2). Thus, a functional probe, biotinylated aryl aldehyde bearing a disulfide linkage (probe BA, Fig. 2C), was chemically synthesized, and Am6A was quantitatively labeled with a biotin molecule (Bm6A) via bioorthogonal hydrazone ligation.47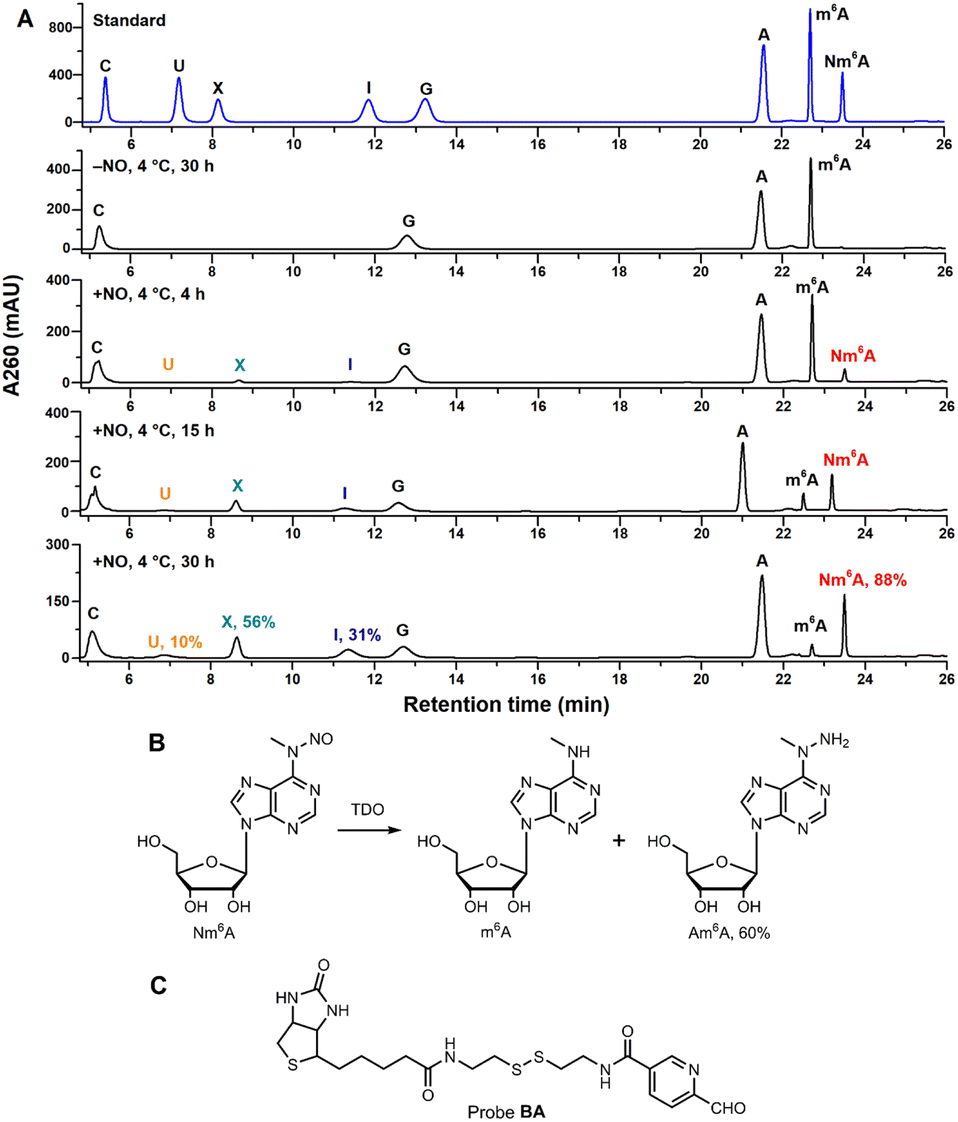 | ||
| Fig. 2 (A) HPLC analysis of A/m6A/G/C ribonucleosides under NO/O2 incubation. Conversion rates were determined by HPLC quantification at 260 nm using external standards. See ESI† for detailed conditions. The shifted retention times of A, m6A, and Nm6A in the fourth HPLC trace (15 h incubation) may be caused by the instability of instruments. (B) Nm6A nucleoside was converted into m6A and Am6A under TDO reduction. Reaction condition: 100 μM Nm6A, 100 mM TDO, 300 mM Na2CO3/NaHCO3, pH = 10.0, 50 °C, 5 min. (C) Chemical structure of probe BA. | ||
Test the feasibility of this chemical approach into RNA labeling
A 12 nt RNA oligo (12nt-m6A) containing only U and m6A was synthesized. Optimization determined that 12nt-m6A was quantitatively converted into Nm6A modified oligo (12nt-Nm6A) through prior NO bubbling into an acidic buffered solution (ESI, Fig. S4†). 1 nmol of 12nt-Nm6A was converted into 12nt-Am6A via TDO reduction under heat and alkaline condition with the yield of 46% (ESI, Fig. S6†). After HPLC purification, almost 90% of the purified 12nt-Nm6A was converted into biotinylated product (12nt-Bm6A) labeling with probe BA (Fig. 3A, ESI, Fig. S8†). Meanwhile, we applied this chemical approach to labeling another 15 nt RNA oligo containing A/U/G/C/m6A (15nt-m6A). Denaturing polyacrylamide gel electrophoresis (PAGE) analysis indicated that almost 50% of 15nt-m6A was converted into biotinylated product (Fig. 3B). In addition, the modified oligo after each chemical incubation step was enzymatically digested into ribonucleosides as further confirmed by LC-HRMS (ESI, Fig. S10–S12†).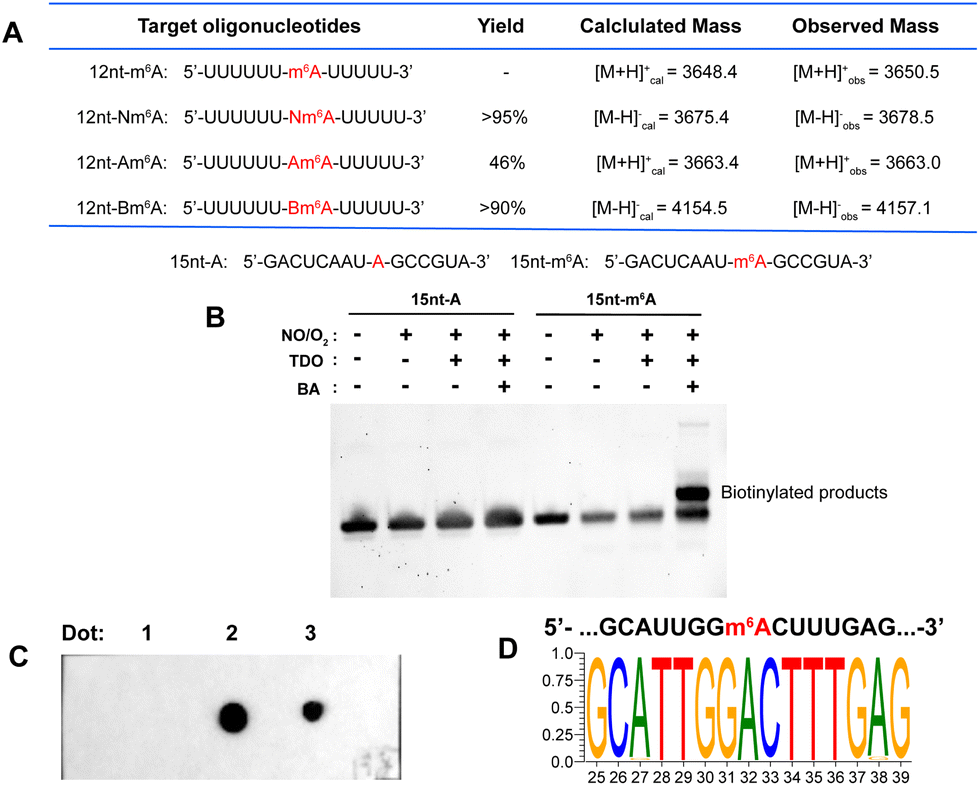 | ||
| Fig. 3 (A) A table of results of 12 nt RNA oligo tests. Yields were calculated by HPLC at 260 nm. See ESI† for more details. (B) Denaturing PAGE analysis of 15 nt RNA oligo. (C) Dot blot assay confirmed selective biotinylation of m6A in MALAT1-2577-m6A. Dot 1: three-step labeling products of MALAT1-2577-A; Dot 2: three-step labeling products of MALAT1-2577-m6A; Dot 3: a mixture containing three-step labeling products of MALAT1-2577-A and 5′-biotinylated DNA. The methylene blue staining result was presented in ESI, Fig. S15.† (D) Motif for the sequence of model RNA (MALAT1-2577-m6A) after m6A-ORL-Seq procedure. | ||
Before applying this method to the profiling of m6A methylomes, a comprehensive test was performed. Firstly, the optimal condition for N-nitrosation (the central step) was investigated by controlling the NO bubbling time and incubation time. Denaturing PAGE analysis and RNA quantification confirmed that the optimal N-nitrosation and TDO reduction conditions do not cause considerable RNA degradation (ESI, Fig. S13†). Then, total RNA was extracted from human embryonic kidney 293T (HEK-293T) and fragmented into 60–200 nt, followed by N-nitrosation. Then, the purified RNA was digested into ribonucleosides and further quantified using LC-MS/MS analysis (ESI, Fig. S14†), which determined 20 h as the optimal incubating time for the high ratio of Nm6A/I (avoiding excessive mutation of A) and sufficient Nm6A concentration (high labeling efficiency). As proof of concept, m6A-ORL was conducted to a 60 nt model RNA modified with an m6A modification (MALAT1-2577-m6A). After chemical labeling and purification, the dot blot assay proved the selective biotinylation of m6A in model RNA (Fig. 3C). Meanwhile, labeled model RNA was mixed with yeast tRNA and performed with a pulldown procedure. Afterward, the enriched RNA was subjected to RNA-Seq (Fig. 1B). Notably, under SuperScript III mediated reverse transcription (RT), labeled m6A (a thiol residue modified m6A, Tm6A, site 32) was read as A (Fig. 3D), and a major mutation of A to G (6.8%) was observed, since I was known to be read as G during RT.48 Dot blot assay further confirmed that m6A-ORL was capable of selective labeling m6A-containing RNA in HEK-293T total RNA (ESI, Fig. S16†). These results indicated m6A-ORL is promising for the transcriptome-wide analysis of m6A.
m6A-ORL-seq uncovers the transcriptome-wide m6A methylomes in HEK-293T
Next, m6A-ORL was performed on transcriptome-wide analysis of m6A in HEK-293T. First, 30 μg total RNA (m6A-ORL1) or 5.0 μg poly (A)+ RNA (m6A-ORL2) extracted from HEK-293T cells spiked with 0.01% spike-in RNAs (ESI,† page S29) were used to conduct three-step chemical reactions. The libraries were constructed following the procedure presented in ESI† and then subjected to high-throughput sequencing. Analyzing the count of spike-in RNAs confirmed m6A-ORL enable the enrichment of m6A fragment (ESI, Fig. S26†). A considerable amount of m6A peaks were identified in the human transcriptome with a canonical motif of DRACH (Fig. 4B). Remarkably, two prominent regions of m6A (MALAT1, ACTB) confirmed by miCLIP18 and m6A-REF-Seq20 were also detected by m6A-ORL-Seq (Fig. 4A). The mean normalized reads were high specifically distributed near the center of m6A peaks, further verifying the low background enrichment of m6A segments and by m6A-ORL (Fig. 4C). A pie chart presents the fraction of m6A peaks in each of five non-overlapping transcript segments, the results were consistent with the previously reported distribution of m6A (Fig. 4D).14–25 Furthermore, a comparative analysis between m6A-ORL-Seq and the gold-standard MeRIP-Seq were conducted. The overall enrichment fold of m6A-ORL peaks is higher than MeRIP (ESI, Fig. S28†). Upon observation, nearly 50% of m6A-ORL peaks overlapped with MeRIP and a similar distribution of m6A peaks (Fig. 4E and F). These results indicated our method had the same level of reliability as MeRIP. Despite this, we focused on 50% of the peaks that did not overlap with MeRIP. The metagene profile of these unique peaks revealed the enrichment of 5′ UTR and 3′ UTR regions, signifying the plausible N6,2′-O-dimethyladenosine (m6Am) and m6A sites that MeRIP could not detect (Fig. 4H). Significant m6A and m6Am motifs (BCA,18 B = G/U/C) were searched out (Fig. 4I). These results further indicated the sequencing data was comprised of the thorough information for human m6A methylome with nearly null noise. Compared to the reported m6Am peaks derived from m6Am-Seq,49 we found 24% of reported sites are overlapped with these unique peaks (Fig. 4J). Further base resolution analysis confirmed that considerable m6A and m6Am sites existed in m6A-ORL unique peaks (ESI, Fig. S17†). Meanwhile, m6A antibody was inclined to enrich >100 nt m6A-containing RNA fragments,14,15 while chemical labeling was more efficient to short fragments due to the steric hindrance. Overall, due to the novel chemistry's distinct attributes, m6A-ORL has a higher resolution (Fig. 4G) and provides a new perspective for m6A methylomes compared to MeRIP.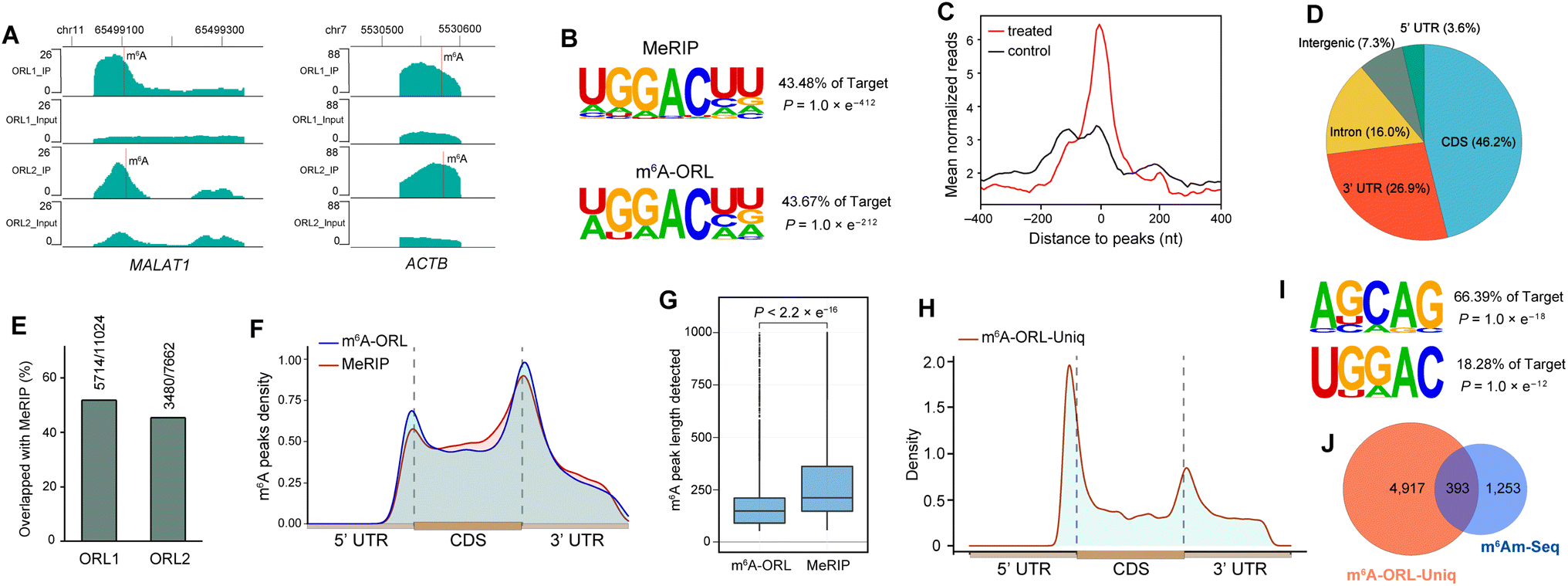 | ||
| Fig. 4 m6A-ORL-seq uncovers the transcriptome-wide m6A methylomes in HEK-293T. (A) m6A-ORL identified two known m6A sites in the human transcriptome. MALAT1 (chr11: 65499104) and ACTB (chr7: 5530572). (B) The motif search both yielded canonical DRACH in HEK-293T using MeRIP-Seq or m6A-ORL-Seq. (C) A density plot of the distribution of mean normalized reads around the center of m6A peaks. (D) Transcriptome-wide distribution of m6A peaks. (E) Overlap of m6A peaks identified by m6A-ORL and MeRIP. (F) Metagene profile of the m6A peaks identified by m6A-ORL and MeRIP. (G) The length of m6A peaks detected by m6A-ORL and MeRIP. (H) Metagene profile of the m6A-ORL-Uniq peaks. (I) Searched m6A and m6Am motif in the m6A-ORL-Uniq peaks. (J) Overlap of m6Am peaks between m6A-ORL-Uniq peaks and m6Am-Seq peaks. | ||
m6A-ORL detects m6A sites at single-base resolution
In the data analysis of m6A-ORL-Seq on model RNA, a specific signature of RT truncation was observed, marking the m6A site (site 32) at single-base resolution (Fig. 5A, ESI, Fig. S18†). Hence, it was speculated that the loss of hydrogen donor (replaced with a bulky group) at the N6 position caused an RT-stop (Fig. 5B). Then, several 15 nt RNA oligos containing N6-modified adenosine were synthesized using our recently developed method50 (ESI,† part 13) to verify this hypothesis. RT reactions were subsequently performed using SuperScript III (SS III, extensively used for RNA-Seq library construction) and M-MuLV reverse transcriptase (Fig. 5C). Denaturing PAGE indicated that the RT reactions were smoothly extended encountering with A or m6A while stopped at 12 nt where encountered an m6A derivatives (m6,6A, Bm6A, Tm6A). Based on RT truncation, a stringent threshold of stop rate (>10%) and truncated reads (>4) was set for high confidence. Then the DRACH motif was scanned in m6A-ORL peaks, identifying 4001 sites that stopped at A. To reduce the false positives, the regions of m6A peaks in the input sample were detected referring to the same threshold, 18 sites were screened out to be false positives. The low rate of false positives (0.4%) indicated the high specificity of stop sites. The filtered 3983 sites were considered high-confidence m6A sites identified by m6A-ORL-Seq. A credible m6A site (YTHDF2, chr1: 28743781) confirmed by miCLIP18 and m6A-label-Seq22 was chosen as a representative example. Similarly, a significant truncation at A in the GGACG motif was revealed by m6A-ORL-Seq (Fig. 5D). Statistics on frequency of sequence landscape revealed seven types of most frequent motif: GGACU (21%), GAACU (17%), AGACU (15.2%), UGACU (12.0%), AAACU (8.4%), GGACA (7.6%), and GGACC (6.1%), which is consistent with the most widely used method miCLIP.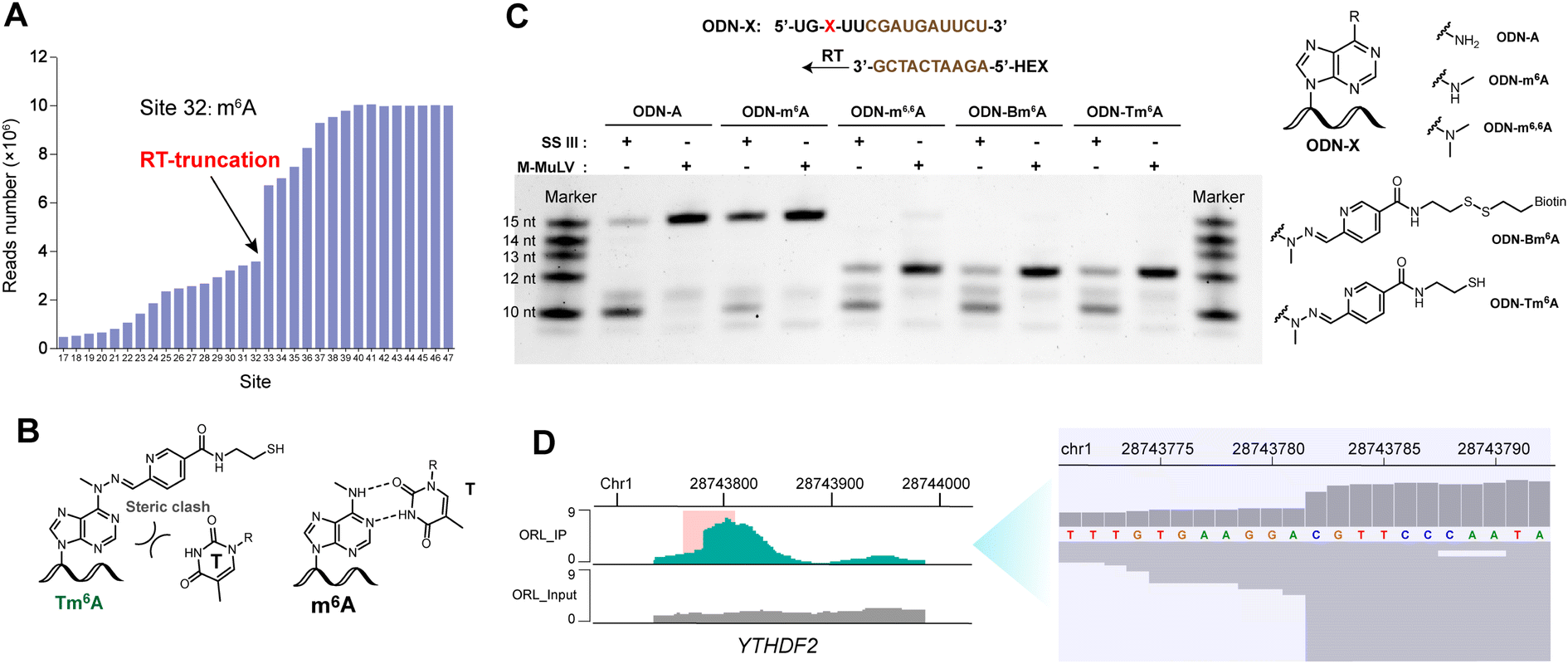 | ||
| Fig. 5 m6A-ORL results in a specific RT truncation signature. (A) Statistics on reads number of each site in model m6A-RNA revealed a specific RT-truncation at site 32. (B) A schematic drawing indicated that m6A pairs with T, Tm6A may induce RT-stop. (C) RT experiments using several 15 nt oligos containing modified adenosine. (D) m6A-ORL identified an m6A site (YTHDF2, chr1: 28743781) at single-base resolution. | ||
As belonging to chemical-assisted methods, the m6A sites identified by m6A-ORL-Seq were compared to m6A-label-Seq22 sites, m6A-SAC-Seq24 sites, and m6A-SEAL-Seq23 peaks (Fig. 6). It turns out m6A-ORL-Seq and m6A-SAC-Seq maintain higher consistency (Fig. 6A). The majority (77%) of m6A-ORL-Seq sites were located at m6A-SEAL-Seq peaks (Fig. 6B). To further assess the accuracy of these sites, a comprehensive comparison to the previously reported sites by different base resolution methods (miCLIP-CIMS,18 miCLIP-CITS,18 m6A-REF-Seq,20 DART-Seq,21 m6A-label-Seq,22 m6A-SAC-Seq24) was performed (ESI, Fig. S29†). The comparative results determined that the m6A sites identified by m6A-ORL had the highest overlapping rates except for DART-Seq, which indicates the m6A sites detected by m6A-ORL-Seq are the most reproducible and trustable. Altogether 2174 of 3983 high-confidence m6A were identified by these reported methods, and six sites derived from 1809 unique sites were further confirmed by SELECT (an independent m6A-sensitive ligation-based method)36 validation (ESI, Fig. S23†). These results proved m6A-ORL-Seq is highly reliable for transcriptome-wide detection of m6A.
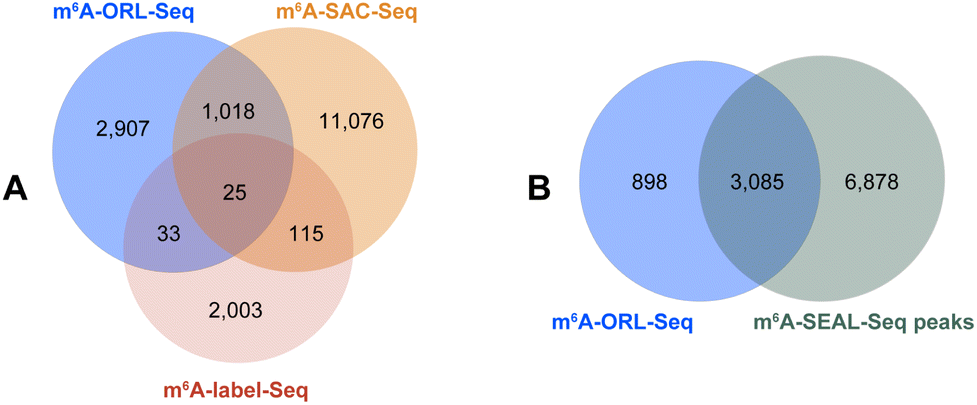 | ||
| Fig. 6 Overlapping extents between the m6A datasets identified by different chemical-assisted methods. (A) Overlap between m6A-ORL-Seq, iodine-mediated m6A-label-Seq and m6A-SAC-Seq. (B) Overlap between m6A-ORL-Seq sites and m6A-SEAL-Seq peaks. | ||
m6A-ORL can detects m6A sites of low methylation level
Counting spike-in RNAs in the sequencing data has indicated m6A-ORL-Seq can selectively enrich low m6A level fragments of low methylation level (ESI, Fig. S26†). To further confirm m6A-ORL can detects m6A sites of low methylation level, reported data which containing m6A stoichiometric information was referred to. MAZTER-Seq provided a feasible approach to m6A quantitative profiling (only for m6ACA motif).19 In their study, approximately 80000 m6A sites were identified and relatively quantified in HEK-293T transcriptome. Therefore, identified m6ACA sites were extracted from the dataset of each method and were aligned with the dataset of MAZTER-Seq. Then, 775 of miCLIP-CIMS sites (27%), 214 of miCLIP-CITS sites (18%), 1742 of DART-Seq sites (22%), 783 of m6A-REF-Seq sites (18%), 46 of m6A-label-Seq sites (8%), and 148 of m6A-ORL-Seq sites (27%) were mapped to the dataset of MAZTER-Seq. Stoichiometry distribution (ESI, Fig. S24†) revealed that most sites identified by miCLIP-CIMS and DART-Seq were methylated at low levels, which was consistent with the distribution of global m6A methylation levels.19 The methylation level distribution of sites detected by miCLIP-CITS and m6A-REF-Seq were relatively average. Considering the intrinsic characteristic of chemical labeling, m6A-label-Seq and m6A-ORL-Seq are apt to detect m6A of high methylation levels. However, m6A-label-Seq has no advantage in low methylation detection, m6A-ORL-Seq still provided considerable m6A sites of low methylation levels (20% of identified sites were methylated at the levels lower than 20%, ESI, Fig. S24†). m6A-SAC-Seq is a recently reported method which can also measure m6A methylation level.24 1043 of m6A-ORL-Seq sites (26.2%) was mapped to the dataset of m6A-SAC-Seq, indicating 52% of identified sites were methylated at the levels lower than 20% (ESI, Fig. S25†). These results confirmed that this chemical pulldown strategy enabled enrichment and detection of m6A sites at low methylation levels.
Discussion
Despite many methods being developed for m6A profiling, nature still cast a cloud over m6A code. The finding that little overlap between datasets seems worrisome (ESI, Fig. S29†), here we would share an opinion that technical noises and false positives resulting from the methodological flaws or sequencing depth are not the only major factor. (1) Aebersold et al.51 have revealed the effects of biological variation in HeLa cell lines across laboratories. Substantial heterogeneity was found between cell lines variants, and a progressive divergence was proceeded along with the times of cell passage. This variability could have a complex effect on the m6A methylation pattern. (2) m6A is a highly dynamic and extensive modification and the majority of m6A is methylated at low level, identified m6A sites might be a random subset of massive sites. Generally, all the current methods are limited in their ability to identify the low methylation sites in particular within lowly expressed genes. Meanwhile, many true methylated sites of low levels were probably filtered out for a high level of confidence. Schwartz et al.19 has revealed that many of m6A sites detected in only a single study are indeed methylated. Similar signs could also be found in DART-Seq. DART-Seq shows low overlap (<2.5%) with the methods listed in Fig. S29,† but has much higher overlap with MAZTER-Seq dataset. The majority of m6A sites identified by DART-Seq were methylated at low levels (ESI, Fig. S24†), which may cause the extremely low overlap with other methods. What's more, for m6A stoichiometry, a low correlation was found between two independent methods, MAZTER-Seq and m6A-SAC-Seq (ESI, Fig. S25†). This discrepancy could also be due to the biological variability and high complexity of m6A topologies.The strengths and major concerns of the current base resolution methods for m6A profiling (miCLIP/m6A-CLIP, DART-Seq, MAZTER-Seq/m6A-REF-Seq, m6A-label-Seq) were listed (ESI, Table S6†). Compared to these antibody/enzyme-dependent methods, m6A-ORL is the only entirely chemical method, it is not expensive, easy to scale up. Most importantly, m6A-ORL provides the most reproducible m6A sites with high-confidence compared to all the current methods. However, m6A-ORL-Seq inevitably induced mutations (mainly, A to G mutations) in bioinformatics analysis. Compared to the mutation rates (0.43–0.47%) and mapping rates (94–95%) of non-chemical treatment data, the mutation rates (2.4–3.2%) and mapping rates (82–90%) of m6A-ORL are acceptable. Meanwhile, the current protocol of m6A-ORL could be improved, for example, screening a more suitable nitrosating and reduction reagents were urgent for the lower mutation rates and higher labeling efficiency, thus the method could be further developed for relative and even absolute quantification of m6A levels (omitting the pulldown procedure). This unique chemistry enables the further application to chemical derivatization-assisted MS analysis52 for sensitive detection of m6A and other modifications. Similarly, m6A-ORL could also be used for the genomic profiling of 6mdA in DNA.
In principle other N-methyl modifications in RNA, such as N4-methylcytidine (m4C) and N2-methylguanosine (m2G), were probably N-nitrosated during chemical treatment. However, to the best of our knowledge, no reports confirmed the existence and abundance of m2G in the human cells were found, instead it was found in mice as a tRNA modification.53,54 m4C was found in human 12S rRNA,55 while the abundance remains unknown. In general, these tRNA/rRNA modifications would not interfere with the m6A profiling in mRNA and lncRNA where researchers are more concerned with.
In summary, this study reported a novel chemical pulldown approach for transcriptome-wide profiling of m6A. This method can further detect high-confidence m6A sites at base resolution. Furthermore, since the principle of m6A-ORL is quite different from all the previously reported methods based on antibody/enzyme-dependent methods, m6A-ORL provides a new perspective for m6A detection at the transcriptome level and we believe it will help to obtain new insights into RNA methylation.
Data availability
Sequencing data have been deposited into the Gene Expression Omnibus (GEO). The accession number is GSE185753 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE185753).Code availability
All custom codes used in this study are available at https://github.com/sqhan-whu/m6A-ORL-Seq.Author contributions
X. Z., X. W., and Y. Z. guided the research; Y. X. and X. Z. conceived the project; Y. X. designed and performed the experiments with the help of Z. F., W. Y., Q. W., and Y. W.; S. H. designed and performed the bioinformatics analysis with the help of Q. L.; Y. X. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support by the Natural Science Foundation of China (91753201 and 21721005 to X. Zhou). We thank Prof. Cai-Guang Yang (Shanghai Institute of Materia Medica, Chinese Academy of Sciences) for generously providing purified FTO protein and the human FTO gene (ΔN31) cloned vector pET28a, and Prof. Bi-Feng Yuan (Wuhan University) for providing LC-HRMS instructions and constructive suggestions.Notes and references
- P. Boccaletto, M. A. Machnicka, E. Purta, P. Piatkowski, B. Baginski, T. K. Wirecki, V. de Crécy-Lagard, R. Ross, P. A. Limbach, A. Kotter, M. Helm and J. M. Bujnicki, Nucleic Acids Res., 2018, 46, D303–D307 CrossRef CAS.
- Y. Yue, J. Liu and C. He, Genes Dev., 2015, 29, 1343–1355 CrossRef CAS PubMed.
- B. S. Zhao, I. A. Roundtree and C. He, Nat. Rev. Mol. Cell Biol., 2017, 18, 31–42 CrossRef CAS.
- Y. Yue, J. Liu and C. He, Genes Dev., 2015, 29, 1343–1355 CrossRef CAS PubMed.
- B. Wu, L. Li, Y. Huang, J. Ma and J. Min, Curr. Opin. Struct. Biol., 2017, 47, 67–76 CrossRef CAS.
- J. Liu, Y. Yue, D. Han, X. Wang, Y. Fu, L. Zhang, G. Jia, M. Yu, Z. Lu, X. Deng, Q. Dai, W. Chen and C. He, Nat. Chem. Biol., 2014, 10, 93–95 CrossRef CAS PubMed.
- D. Theler, C. Dominguez, M. Blatter, J. Boudet and F. H. Allain, Nucleic Acids Res., 2014, 42, 13911–13919 CrossRef CAS.
- G. Jia, Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y. G. Yang and C. He, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef CAS.
- S. D. Kasowitz, J. Ma, S. J. Anderson, N. A. Leu, Y. Xu, B. D. Gregory, R. M. Schultz and P. J. Wang, PLoS Genet., 2018, 14, e1007412 CrossRef.
- J. M. Fustin, M. Doi, Y. Yamaguchi, H. Hida, S. Nishimura, M. Yoshida, T. Isagawa, M. S. Morioka, H. Kakeya, I. Manabe and H. Okamura, Cell, 2013, 155, 793–806 CrossRef CAS.
- N. Liu, Q. Dai, G. Zheng, C. He, M. Parisien and T. Pan, Nature, 2015, 518, 560–564 CrossRef CAS PubMed.
- S. Panneerdoss, V. K. Eedunuri, P. Yadav, S. Timilsina, S. Rajamanickam, S. Viswanadhapalli, N. Abdelfattah, B. C. Onyeagucha, X. Cui, Z. Lai, T. A. Mohammad, Y. K. Gupta, T. H. Huang, Y. Huang, Y. Chen and M. K. Rao, Sci. Adv., 2018, 4, eaar8263 CrossRef CAS.
- R. Su, L. Dong, C. Li, S. Nachtergaele, M. Wunderlich, Y. Qing, X. Deng, Y. Wang, X. Weng, C. Hu, M. Yu, J. Skibbe, Q. Dai, D. Zou, T. Wu, K. Yu, H. Weng, H. Huang, K. Ferchen, X. Qin, B. Zhang, J. Qi, A. T. Sasaki, D. R. Plas, J. E. Bradner, M. Wei, G. Marcucci, X. Jiang, J. C. Mulloy, J. Jin, C. He and J. Chen, Cell, 2018, 172, 90–105 CrossRef CAS PubMed.
- D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, M. Salmon-Divon, L. Ungar, S. Osenberg, K. Cesarkas, J. Jacob-Hirsch, N. Amariglio, M. Kupiec, R. Sorek and G. Rechavi, Nature, 2012, 485, 201–206 CrossRef CAS PubMed.
- K. D. Meyer, Y. Saletore, P. Zumbo, O. Elemento, C. E. Mason and S. R. Jaffrey, Cell, 2012, 149, 1635–1646 CrossRef CAS PubMed.
- K. Chen, Z. Lu, X. Wang, Y. Fu, G. Z. Luo, N. Liu, D. Han, D. Dominissini, Q. Dai, T. Pan and C. He, Angew. Chem., Int. Ed., 2015, 54, 1587–1590 CrossRef CAS.
- S. Ke, E. A. Alemu, C. Mertens, E. C. Gantman, J. J. Fak, A. Mele, B. Haripal, I. Zucker-Scharff, M. J. Moore, C. Y. Park, C. B. Vågbø, A. Kusśnierczyk, A. Klungland, J. E. Darnell. Jr. and R. B. Darnell, Genes Dev., 2015, 29, 2037–2053 CrossRef CAS PubMed.
- B. Linder, A. V. Grozhik, A. O. Olarerin-George, C. Meydan, C. E. Mason and S. R. Jaffrey, Nat. Methods, 2015, 12, 767–772 CrossRef CAS.
- M. A. Garcia-Campos, S. Edelheit, U. Toth, M. Safra, R. Shachar, S. Viukov, R. Winkler, R. Nir, L. Lasman, A. Brandis, J. H. Hanna, W. Rossmanith and S. Schwartz, Cell, 2019, 178, 731–747 CrossRef CAS PubMed.
- Z. Zhang, L. Q. Chen, Y. L. Zhao, C. G. Yang, I. A. Roundtree, Z. Zhang, J. Ren, W. Xie, C. He and G. Z. Luo, Sci. Adv., 2019, 5, eaax0250 CrossRef CAS PubMed.
- K. D. Meyer, Nat. Methods, 2019, 16, 1275–1280 CrossRef CAS.
- X. Shu, J. Cao, M. Cheng, S. Xiang, M. Gao, T. Li, X. Ying, F. Wang, Y. Yue, Z. Lu, Q. Dai, X. Cui, L. Ma, Y. Wang, C. He, X. Feng and J. Liu, Nat. Chem. Biol., 2020, 16, 887–895 CrossRef CAS PubMed.
- Y. Wang, Y. Xiao, S. Dong, Q. Yu and G. Jia, Nat. Chem. Biol., 2020, 16, 896–903 CrossRef CAS.
- L. Hu, S. Liu, Y. Peng, R. Ge, R. Su, C. Senevirathne, B. T. Harada, Q. Dai, J. Wei, L. Zhang, Z. Hao, L. Luo, H. Wang, Y. Wang, M. Luo, M. Chen, J. Chen and C. He, Nat. Biotechnol., 2022, 40, 1210–1219 CrossRef CAS.
- I. D. Vilfan, Y.-C. Tsai, T. A. Clark, J. Wegener, Q. Dai, C. Yi, T. Pan, S. W. Turner and J. Korlach, J. Nanobiotechnology, 2013, 11, 8 CrossRef CAS PubMed.
- D. R. Garalde, E. A. Snell, D. Jachimowicz, B. Sipos, J. H. Lloyd, M. Bruce, N. Pantic, T. Admassu, P. James, A. Warland, M. Jordan, J. Ciccone, S. Serra, J. Keenan, S. Martin, L. McNeill, E. J. Wallace, L. Jayasinghe, C. Wright, J. Blasco, S. Young, D. Brocklebank, S. Juul, J. Clarke, A. J. Heron and D. J. Turner, Nat. Methods, 2018, 15, 201–206 CrossRef CAS.
- A. Leger, P. P. Amaral, L. Pandolfini, C. Capitanchik, F. Capraro, V. Miano, V. Migliori, P. Toolan-Kerr, T. Sideri, A. J. Enright, K. Tzelepis, F. J. van Werven, N. M. Luscombe, I. Barbieri, J. Ule, T. Fitzgerald, E. Birney, T. Leonardi and T. Kouzarides, Nat. Commun., 2021, 12, 7198 CrossRef CAS PubMed.
- Z. Zhang, T. Chen, H.-X. Chen, Y.-Y. Xie, L.-Q. Chen, Y.-L. Zhao, B.-D. Liu, L. Jin, W. Zhang, C. Liu, D.-Z. Ma, G.-S. Chai, Y. Zhang, W.-S. Zhao, W. H. Ng, J. Chen, G. Jia, J. Yang and G.-Z. Luo, Nat. Methods, 2021, 18, 1213–1222 CrossRef CAS.
- L.-Y. Zhao, J. Song, Y. Liu, C.-X. Song and C. Yi, Protein Cell, 2020, 11, 792–808 CrossRef CAS.
- M. Nappi, A. Hofer, S. Balasubramanian and M. J. Gaunt, J. Am. Chem. Soc., 2020, 142, 21484–21492 CrossRef CAS.
- Y. Mahdavi-Amiri, K. Chung Kim Chung and R. Hili, Chem. Sci., 2021, 12, 606–612 RSC.
- S. Werner, A. Galliot, F. Pichot, T. Kemmer, V. Marchand, M. V. Sednev, T. Lence, J.-Y. Roignant, J. König, C. Höbartner, Y. Motorin, A. Hildebrandt and M. Helm, Nucleic Acids Res., 2021, 49, e23 CrossRef CAS PubMed.
- X. Li, S. Guo, Y. Cui, Z. Zhang, X. Luo, M. T. Angelova, L. F. Landweber, Y. Wang and T. P. Wu, Genome Biol., 2022, 23, 122 CrossRef CAS.
- M. Martin, EMBnet J, 2011, 17, 10–12 CrossRef.
- A. Dobin, C. A. Davis, F. Schlesinger, J. Drenkow, C. Zaleski, S. Jha, P. Batut, M. Chaisson and T. R. Gingeras, Bioinformatics, 2013, 29, 15–21 CrossRef CAS.
- A. O. Olarerin-George and S. R. Jaffrey, Bioinformatics, 2017, 33, 1563–1564 CrossRef CAS.
- Y. Xiao, Y. Wang, Q. Tang, L. Wei, X. Zhang and G. Jia, Angew. Chem., Int. Ed., 2018, 57, 15995–16000 CrossRef CAS.
- Z. Mao, H. Jiang, Z. Li, C. Zhong, W. Zhang and Z. Liu, Chem. Sci., 2017, 8, 4533–4538 RSC.
- Z. Mao, H. Jiang, X. Song, W. Hu and Z. Liu, Anal. Chem., 2017, 89, 9620–9624 CrossRef CAS.
- M. P. V. Tato, L. Castedo and R. Riguera, Chem. Lett., 1985, 14, 623–626 CrossRef.
- M. A. Zolfigol, Synth. Commun., 1999, 29, 905–910 CrossRef CAS.
- J. A. Hrabie, J. R. Klose, D. A. Wink and L. K. Keefer, J. Org. Chem., 1993, 58, 1472–1476 CrossRef CAS.
- D. A. Wink, K. S. Kasprzak, C. M. Maragos, R. K. Elespuru, M. Misra, T. M. Dunams, T. A. Cebula, W. H. Koch, A. W. Andrews, J. S. Allen and L. K. Keefer, Science, 1991, 254, 1001–1003 CrossRef CAS.
- J. L. Caulfield, J. S. Wishnok and S. R. Tannenbaum, J. Biol. Chem., 1998, 273, 12689–12695 CrossRef CAS PubMed.
- D. Wiener and S. Schwartz, Nat. Rev. Genet., 2021, 22, 119–131 CrossRef CAS.
- P. Chaudhary, S. Gupta, P. Sureshbabu, S. Sabiah and J. Kandasamy, Green Chem., 2016, 18, 6215–6221 RSC.
- R. K. Lim and Q. Lin, Chem. Commun., 2010, 46, 1589–1600 RSC.
- C. Basilio, A. J. Wahba, P. Lengyel, J. F. Speyer and S. Ochoa, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 613–616 CrossRef CAS.
- H. Sun, K. Li, X. Zhang, J. Liu, M. Zhang, H. Meng and C. Yi, Nat. Commun., 2021, 12, 4778 CrossRef CAS.
- Y. Xie, Z. Fang, W. Yang, Z. He, K. Chen, P. Heng, B. Wang and X. Zhou, Bioconjugate Chem., 2022, 33, 353–362 CrossRef CAS PubMed.
- Y. Liu, Y. Mi, T. Mueller, S. Kreibich, E. G. Williams, A. Van Drogen, C. Borel, M. Frank, P.-L. Germain, I. Bludau, M. Mehnert, M. Seifert, M. Emmenlauer, I. Sorg, F. Bezrukov, F. S. Bena, H. Zhou, C. Dehio, G. Testa, J. Saez-Rodriguez, S. E. Antonarakis, W.-D. Hardt and R. Aebersold, Nat. Biotechnol., 2019, 37, 314–322 CrossRef CAS PubMed.
- Q.-Y. Cheng, J. Xiong, C.-J. Ma, Y. Dai, J.-H. Ding, F.-L. Liu, B.-F. Yuan and Y.-Q. Feng, Chem. Sci., 2020, 11, 1878–1891 RSC.
- Y. Zhang, X. Zhang, J. Shi, F. Tuorto, X. Li, Y. Liu, R. Liebers, L. Zhang, Y. Qu, J. Qian, M. Pahima, Y. Liu, M. Yan, Z. Cao, X. Lei, Y. Cao, H. Peng, S. Liu, Y. Wang, H. Zheng, R. Woolsey, D. Quilici, Q. Zhai, L. Li, T. Zhou, W. Yan, F. Lyko, Y. Zhang, Q. Zhou, E. Duan and Q. Chen, Nat. Cell Biol., 2018, 20, 535–540 CrossRef CAS.
- Q. Chen, M. Yan, Z. Cao, X. Li, Y. Zhang, J. Shi, G.-h. Feng, H. Peng, X. Zhang, Y. Zhang, J. Qian, E. Duan, Q. Zhai and Q. Zhou, Science, 2016, 351, 397–400 CrossRef CAS.
- L. V. Haute, A. G. Hendrick, A. R. D'Souza, C. A. Powell, P. Rebelo-Guiomar, M. E. Harbour, S. Ding, I. M. Fearnley, B. Andrews and M. Minczuk, Nucleic Acids Res., 2019, 47, 10267–10281 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, experimental procedures, methods, analytical characterization. Fig. S1–S31 and Tables S1–S6. See DOI: https://doi.org/10.1039/d2sc03181g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |