
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic generation of a non-heme Fe(III)-hydroperoxo species with O2 in water for the oxygen atom transfer reaction†
Eva
Pugliese
 a,
Nhat Tam
Vo
a,
Alain
Boussac
b,
Frédéric
Banse
a,
Yasmina
Mekmouche
c,
Jalila
Simaan
c,
Thierry
Tron
c,
Philipp
Gotico
b,
Marie
Sircoglou
a,
Zakaria
Halime
a,
Winfried
Leibl
*b and
Ally
Aukauloo
*ab
a,
Nhat Tam
Vo
a,
Alain
Boussac
b,
Frédéric
Banse
a,
Yasmina
Mekmouche
c,
Jalila
Simaan
c,
Thierry
Tron
c,
Philipp
Gotico
b,
Marie
Sircoglou
a,
Zakaria
Halime
a,
Winfried
Leibl
*b and
Ally
Aukauloo
*ab
aInstitut de Chimie Moléculaire et des Matériaux d'Orsay, UMR 8182, Université Paris-Saclay, CNRS, F-91405 Orsay Cedex, France. E-mail: ally.aukauloo@universite-paris-saclay.fr
bUniversité Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), F-91191 Gif-sur-Yvette, France. E-mail: winfried.leibl@cea.fr
cAix Marseille Université, Centrale Marseille, CNRS, ISM2 UMR 7313, 13397, Marseille, France
First published on 28th September 2022
Abstract
Coupling a photoredox module and a bio-inspired non-heme model to activate O2 for the oxygen atom transfer (OAT) reaction requires a vigorous investigation to shed light on the multiple competing electron transfer steps, charge accumulation and annihilation processes, and the activation of O2 at the catalytic unit. We found that the efficient oxidative quenching mechanism between a [Ru(bpy)3]2+ chromophore and a reversible electron mediator, methyl viologen (MV2+), to form the reducing species methyl viologen radical (MV˙+) can convey an electron to O2 to form the superoxide radical and reset an Fe(III) species in a catalytic cycle to the Fe(II) state in an aqueous solution. The formation of the Fe(III)-hydroperoxo (FeIII–OOH) intermediate can evolve to a highly oxidized iron-oxo species to perform the OAT reaction to an alkene substrate. Such a strategy allows us to bypass the challenging task of charge accumulation at the molecular catalytic unit for the two-electron activation of O2. The FeIII–OOH catalytic precursor was trapped and characterized by EPR spectroscopy pertaining to a metal assisted catalysis. Importantly, we found that the substrate itself can act as an electron donor to reset the photooxidized chromophore in the initial state closing the photocatalytic loop and hence excluding the use of a sacrificial electron donor. Laser Flash Photolysis (LFP) studies and spectroscopic monitoring during photocatalysis lend credence to the proposed catalytic cycle.
Introduction
The dioxygen molecule is undoubtedly the most environmentally compatible oxidant. Due to its triplet ground state, it does not readily oxidize diamagnetic organic compounds. Nature has developed both heme1–3 and non-heme enzymes4–6 to activate O2 through electron and proton transfers for essential oxidative metabolism and chemical transformations such as oxygen atom transfer (OAT) reactions as summarized in the following equation (S = substrate and SO = oxygenated substrate):| S + O2 + 2e− + 2H+ → SO + H2O |
Chemists have taken inspiration from biology to develop metal complexes that can bind and activate dioxygen in the presence of co-reductants or with added hazardous oxidants to form active metal–oxygen intermediates such as metal-peroxo or metal-oxo species capable of realizing OAT reactions.5,7–20 The electrochemical activation of O2 in the presence of a metal catalyst has also been pursued and is currently regaining much momentum. This approach relies on the electro-generation of superoxide or H2O2 at the surface of an electrode that ultimately reacts with the metal catalyst to form highly active metal-(hydro)peroxo species. This route too faces different challenges such as reducing the active metal-(hydro)peroxo intermediates or intermolecular reactions leading to unreactive bridged oxo metal complexes.6,21–30
An alternative route to develop more sustainable processes relies on the coupling of a photoredox module and a molecular transition metal catalyst for photocatalytic OAT reactions.31–43 The activation of O2 through light-induced electron transfer provides a unique opportunity to generate these reactive oxo and peroxo species in a benign and controlled manner.44–48 This quest comes along with more challenges on top of those of the classic chemical approaches. Indeed, light-induced electron transfer from the excited state of the photosensitizer module must be steered in a directional way towards the molecular catalyst for charge accumulation and O2 activation to further carry out multi-electron catalysis. Recent efforts in this exploration have paved the way for using light and O2 to oxygenate different substrates in a selective manner.46,49
König and coworkers reported the photocatalytic oxidation of ethyl benzenes to the corresponding ketone and benzyl alcohols to benzaldehydes with riboflavin tetraacetate (RFT), where O2 acts as the sacrificial electron acceptor to form H2O2 and regenerate RFT.50 The H2O2 formed was found to ultimately degrade RFT. In a subsequent study, Wolf and colleagues astutely took profit of this deleterious by-product in a dual catalytic system containing both the RFT and a non-heme iron(II) complex, [(TPA)FeII]2+ (TPA = tris(2-pyridylmethyl)amine). The latter acted as a fuse to protect the photoredox RFT by reacting with the in situ generated H2O2 to form the active iron(III)-hydroperoxo species, [(TPA)FeIII(OOH)]2+, that participates in the photocatalytic cycle for the oxygenation of alkyl benzene.51 However, insights into the photoredox activation of O2 at a metal complex are yet to be deciphered.
In this study, we used the well-known ruthenium(II) trisbipyridine, ([RuII(bpy)3]2+), as a photosensitizer because of its unique photophysical properties (a long life time of the excited state), its solubility in an aqueous medium, and the redox activity of the exited state together with the high oxidative potential of the oxidized form of the chromophore.52,53 Reversible electron acceptors such as methyl viologen (MV2+) are commonly used in light-induced electron transfer processes producing the oxidized photosensitizer, i.e., [RuIII(bpy)3]3+ from [RuII(bpy)3]2+ and the reduced methyl viologen radical (MV˙+) through an oxidative quenching mechanism.54,55 These photoreactions are normally carried out in the absence of O2 as the latter can interfere with the excited triplet state of the chromophore and importantly, can snatch an electron from MV˙+ to form O2˙− leading to subsequent reactions.56 We have been interested in taking profit of the reducing power of photogenerated MV˙+ (E° = −653 mV vs. Ag/AgCl)57 to activate concomitantly an O2 molecule to form the superoxide anion (O2˙−) and resetting a non-heme Fe(III) intermediate at the closure of an OAT catalytic cycle to the corresponding Fe(II) form. The chemical reaction between the photogenerated Fe(II) form and O2˙− should under the right pH conditions reinject the active iron(III)-hydroperoxo intermediate, FeIII–OOH, into the catalytic loop. Such an approach should allow us to bypass the challenging task of charge accumulation at the molecular catalytic unit for the two-electron activation of O2 for OAT reactions.
We report here the photocatalytic activity in an aqueous solution of a mixture of [RuII(bpy)3]2+/[(L5)FeII]2+/MV2+ in the presence of O2 as an oxygen atom source towards the OAT reaction to an alkene as a substrate as sketched in Scheme 1. To accomplish this task, we used an air-stable non-heme [(L5)FeII]2+ model catalyst (where L5 stands for N,N′,N′-tris(2-pyridylmethyl)ethane-1,2-diamine) (see Scheme 1). The choice of this complex was guided by the numerous studies done on the activation of O2 and H2O2.58–60 Photocatalytic runs demonstrated the oxygenation of styrene sulfonate in water to yield the corresponding epoxide or diol and the use of 18O2 indicated that O2 is the source of the inserted oxygen atom. Importantly, we have captured the Electron Paramagnetic Resonance (EPR) signals of both the high-spin [(L5)FeIII–η2O2]+ and the low-spin [(L5)FeIII–OOH]2+ species as observed for the chemically generated catalytically active forms under the right pH control.58,59 In what follows, we discuss the photoredox events that allow us to provide insights into the photocatalytic OAT reaction.
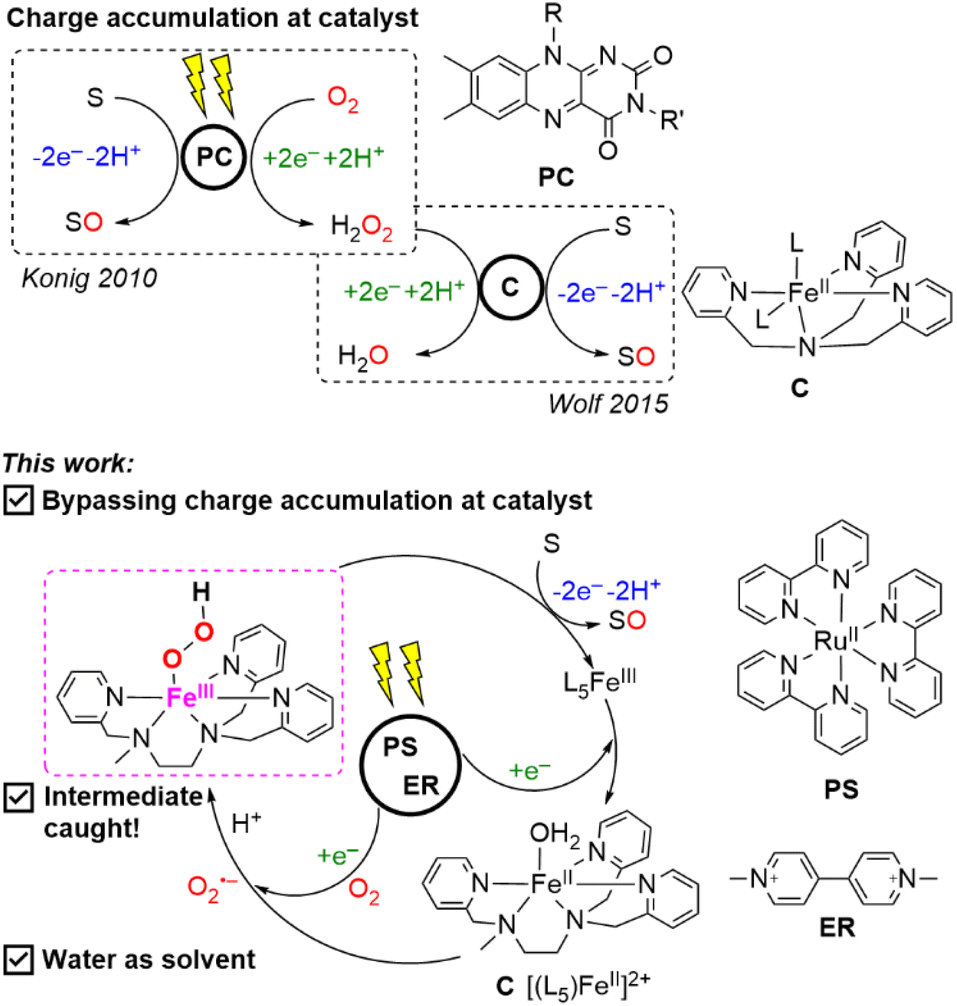 | ||
| Scheme 1 (Top) reported strategy of accumulating charges on a photocatalyst (PC) or catalyst (C) to perform oxygen atom transfer (S = substrate and SO = oxygenated substrate) in comparison to (bottom) the strategy employed in this paper of bypassing charge accumulation using stepwise charge transfers in the photosensitizer (PS), electron relay (ER), substrate, and catalyst. | ||
Results and discussion
Laser Flash Photolysis (LFP) experiments helped us to decipher the photochemical events and confirm the implication of both the methyl viologen and the iron catalyst in the activation process of O2 for the OAT reaction. Under our experimental conditions at pH 4, the catalyst is initially mainly present in its [(L5)FeII–OH2]2+ form (herein denoted as [(L5)FeII]2+).(i) Under anaerobic conditions and in the absence of the iron complex, the emission of the [RuII(bpy)3]2+* excited state was notably quenched by the addition of MV2+ (eqn (2)) with a shortening of the lifetime of the excited state from 600 ns to 270 ns.
| [RuII(bpy)3]2+ + hv → [RuII(bpy)3]2+* | (1) |
| [RuII(bpy)3]2+* + MV2+ → [RuIII(bpy)3]3+ + MV˙+ | (2) |
This event can be monitored by the appearance of the characteristic absorption bands of MV˙+ (390 and 605 nm) and the disappearance of the MLCT band of [RuII(bpy)3]2+ at 450 nm attesting the formation of the highly oxidizing [RuIII(bpy)3]3+ (Fig. S1†). Both species disappeared by charge recombination (eqn (2′)) on the ms time scale (Fig. S2,† blue and red traces).
| [RuIII(bpy)3]3+ + MV˙+ → [RuII(bpy)3]2+ + MV2+ | (2′) |
(ii) In the presence of O2, we observed an accelerated decay of MV˙+ compared to anaerobic conditions. This demonstrates rapid electron transfer from MV˙+ to O2 forming O2˙− (eqn (3), Fig. S2,† orange trace).
| MV˙+ + O2 → MV2+ + O2˙− | (3) |
The formation of superoxide radicals via MV˙+ in aqueous solution has been described in a recent study under similar conditions.61 It should be noted that the fast quenching of the [RuII(bpy)3]2+* excited triplet state by MV2+ (present at a concentration nearly two orders of magnitude higher than the concentration of O2) minimizes the formation of singlet oxygen via triplet–triplet energy transfer from [RuII(bpy)3]2+* to 3O2. Back electron transfer from O2˙− to [RuIII(bpy)3]3+ (eqn (4)) occurs with similar kinetics to those between MV˙+ and [RuIII(bpy)3]3+ in the absence of O2 (Fig. S2,† cyan trace).
| [RuIII(bpy)3]3+ + O2˙− → [RuII(bpy)3]2+ + O2 | (4) |
(iii) Excitation of [RuII(bpy)3]2+/MV2+ in the absence of O2 but in the presence of 4-styrenesulfonate as the substrate (herein denoted as S) leads to the faster recovery of [RuII(bpy)3]2+ than with MV˙+ only (Fig. S3†). This observation can be explained by the intermolecular oxidation of S by [RuIII(bpy)3]3+ to generate an alkenyl radical (eqn (5)) as certified when S was treated by chemically prepared [RuIII(bpy)3]3+ (Fig. S14†).
| [RuIII(bpy)3]3+ + S → [RuII(bpy)3]2+ + S˙+ | (5) |
(iv) No modification of the [RuII(bpy)3]2+* excited state lifetime was noticed in the presence of the iron complex only (Fig. S4†). This indicates that there is no direct activation of the iron complex from the excited triplet state of the photosensitizer neither through electron nor energy transfer processes. The excitation of a mixture of [RuII(bpy)3]2+/MV2+/[(L5)FeII]2+ under an inert atmosphere shows the rapid formation of a reduced MV˙+ radical (390 and 605 nm) together with photo-oxidized [RuIII(bpy)3]3+ (bleaching at 450 nm) (Fig. 1A). The classic recombination pathway was validated by the synchronous resetting of these two species to their resting state (Fig. 1B, solid red and blue traces).
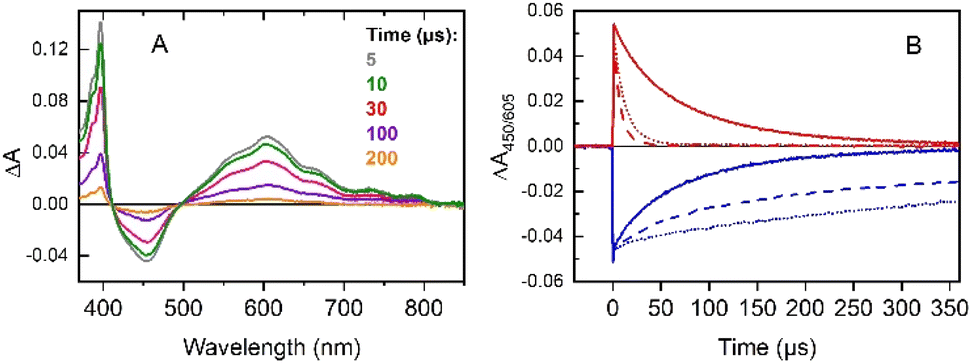 | ||
| Fig. 1 (A) Transient absorption spectra from an Ar-saturated Britton and Robinson (B&R) pH 4 buffer solution of [RuII(bpy)3]2+ (30 μM), MV2+ (20 mM), and [(L5)FeII]2+ (100 μM) at the indicated delay times after excitation. (B) Time-resolved absorption changes at 605 nm (red) and 450 nm (blue) in argon-saturated pH 4 buffer (solid), under aerobic conditions (dash), and after renewed excitation under anaerobic conditions of the previous solution (dot). | ||
We can thus conclude that neither oxidation of the [(L5)FeII]2+ catalyst by the photo-produced [RuIII(bpy)3]3+ nor its reduction by MV˙+ occurs in a degassed solution (Fig. 1A). The first process is related to the oxidation of the [(L5)FeII]2+ catalyst by the photo-produced [RuIII(bpy)3]3+. Although thermodynamically feasible, it was not detected probably because this reaction is slower than back electron transfer from the MV˙+ radical. A possible reason why the oxidation of [(L5)FeII]2+ is slow could be due to its oxidation being controlled by deprotonation, which is not favorable at pH 4. The absence of the second process, i.e., the reduction of the [(L5)FeII]2+catalyst by MV˙+, is easily explained by the fact that no reduction wave is accessible for the FeII complex in cyclic voltammetry (Fig. S6†) when scanning down to −1.0 V vs. SCE. As such, the reactions occurring under these conditions are identical to those in the absence of the [(L5)FeII]2+ catalyst (see (i)).
(v) Under aerobic conditions, we observed the fast disappearance of MV˙+ (absorption at 605 nm), confirming a rapid electron transfer from the MV˙+ radical to O2 to form the superoxide O2˙− anion in aqueous solution (eqn (3), Fig. 1B, dashed red trace).62,63 In addition, the recovery of the [RuII(bpy)3]2+ absorption band at 450 nm from the [RuIII(bpy)3]3+ formed is significantly slowed down (Fig. S5†), indicating that back electron transfer from MV˙+ or O2˙− no longer occurs. This observation can be explained by fast oxidation of MV˙+ by O2 as observed (eqn (3)), followed by a preferential reaction of O2˙− with the [(L5)FeII]2+catalyst (eqn (6)), the concentration of which is much higher than the concentration of [RuIII(bpy)3]3+.
| [(L5)FeII]2+ + O2˙− + H+ → [(L5)FeIII–OOH]2+ | (6) |
Interestingly, in a sequential experiment on a sample first excited in the presence of O2 and then degassed with argon, the original kinetics observed under anaerobic conditions were not recovered (Fig. 1B, dotted traces). As we can notice, the decay of the MV˙+ radical was nearly as fast as in the presence of O2 and the recovery of [RuII(bpy)3]2+ was very slow. This finding may be attributed to a modification of the [(L5)FeIII–OOH]2+ complex possibly relaxing to an [(L5)FeIII]3+ species that reacts with the MV˙+ radical (eqn (7)) under these new anaerobic conditions.
| MV˙+ + [(L5)FeIII]3+ → MV2+ + [(L5)FeII]2+ | (7) |
The rate constants determined for the different bimolecular interactions are summarized in Table 1. As discussed later, other reactions can also be undergoing to slow down the back electron transfer.
| Eqn | Reaction | Rate constant (M−1 s−1) |
|---|---|---|
| a The rate of quenching of the [RuII(bpy)3]2+* excited state by MV2+ is an apparent rate including the effect of escape yield of about 20%. The bimolecular rate constant for reaction (6) is difficult to estimate given the weak signal of [(L5)FeIII–OOH]2+ under the LFP conditions. | ||
| 2 | [RuII(bpy)3]2+* + MV2+ → [RuIII(bpy)3]3+ + MV˙+ | 0.45 × 109 |
| 2′ | [RuIII(bpy)3]3+ + MV˙+ → [RuII(bpy)3]2+ + MV2+ | 3.1 × 109 |
| 3 | MV˙+ + O2 → MV2+ + O2˙− | 0.6 × 109 |
| 4 | [RuIII(bpy)3]3+ + O2˙− → [RuII(bpy)3]2+ + O2 | 3 × 109 |
| 5 | [RuIII(bpy)3]3+ + S → [RuII(bpy)3]2+ + S˙+ | 8 × 104 |
| 7 | MV˙+ + [(L5)FeIII]3+ → MV2+ + [(L5)FeII]2+ | 0.9 × 109 |
Upon irradiating the triptych mixture of [RuII(bpy)3]2+/MV2+/[(L5)FeII]2+ in the presence of styrene sulfonate in an oxygenated aqueous Britton and Robinson (B&R) pH 4 buffer, two oxygenated products namely diol and benzaldehyde derivatives were detected with an overall TON of ca. 70 (Table S1 and Fig. S8†). When the pH of the buffer was fixed at 6, the corresponding epoxide was detected instead of the diol indicating that the ring opening under low pH conditions occurs. The efficiency of the observed photocatalytic reactivity for OAT is comparable with values reported for molecular iron complexes with H2O2 as the oxidant.64 An important control before pursuing further investigation was an 18O2 isotopic labelling experiment. The presence of an 18O atom in the high resolution mass spectrometry (HRMS) detected products confirms O2 as the origin of the oxygen atom under our photocatalytic reaction conditions (Fig. S9 and S10†). Control experiments with only the photosensitizer and the substrate under aerobic conditions did not give any oxidized substrate under irradiation, henceforth, excluding the intervention of singlet O2 in the observed photocatalytic activity. This experiment also excludes the formation of any decomposed fragments of the photosensitizer that could be responsible for the observed reactivity. The absence of MV2+ in the photocatalytic run resulted in no oxygenation reaction, providing solid evidence that the reversible electron carrier is a pre-requisite to carry out the OAT reaction. Photocatalysis in the absence of the iron catalyst or in the presence of FeCl3 instead of the molecular complex led to the formation of traces of the aldehyde supporting the fact that the molecular complex is a necessity for the activation of O2 and that no prominent radical chemistry is underway (Table S1 and Fig. S8†).
The difficulty in providing insights into a photocatalytic cycle is to capture and characterize any reactive intermediate under light irradiation. This is presumably due to the many events taking place concomitantly under the photocatalytic conditions. One way of solving this issue is to probe the electronic features of the chemically produced active species. According to previous studies,59,65 the [(L5)FeIII–OOH]2+ species can be followed by a band at 537 nm (in methanol) when chemically generated with an excess of H2O2, which is a precursor to active iron-oxo species. With the target to capture the signature of the light-driven formation of the [(L5)FeIII–OOH]2+ species, we followed the changes in the absorption spectrum of the catalyst during the illumination of a mixture of [RuII(bpy)3]2+/MV2+/[(L5)FeII]2+ under aerobic conditions (Fig. 2).
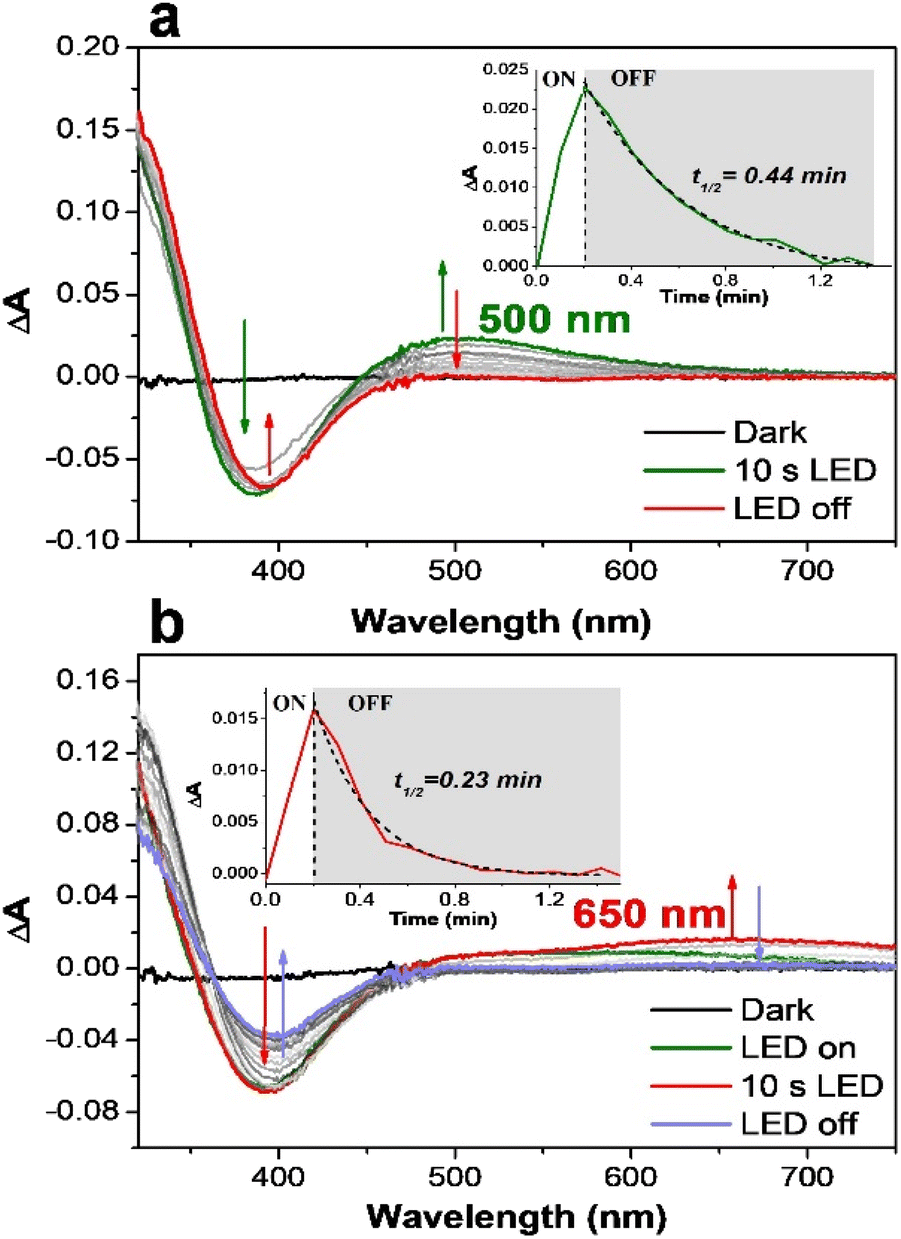 | ||
| Fig. 2 (a) Spectral evolution of the absorption of a B&R pH 4 buffer solution containing [RuII(bpy)3]2+ (30 μM), MV2+ (10 mM), EDTA (20 mM), and [(L5)FeII]2+ (100 μM) upon irradiation (100 W m−2 for 10 s) under aerobic conditions and no stirring. The difference of absorbance from the solution in the dark and during irradiation. Inset: kinetic profile of the 500 nm band. (b) The same experiment at pH 7.6. Inset: kinetic profile of the 650 nm band. | ||
Initially, we opted to use ethylenediaminetetraacetic acid (EDTA) as an electron source. At pH 4 and in the presence of EDTA (Fig. 2a), the loss of the absorption band at 390 nm, characteristic of [(L5)FeII]2+ (Fig. S11†), is observed together with the appearance of an absorption band around 500 nm, typical of the [(L5)FeIII–OOH]2+ intermediate.58 The difference spectrum during irradiation therefore represents all features of a light-induced conversion of [(L5)FeII]2+ to [(L5)FeIII–OOH]2+ (eqn (6)).
The quick decay of the 500 nm band indicates that such a species is very unstable (<1 min) under our experimental conditions. When the same experiment was performed at pH 6, the bleaching of [(L5)FeII]2+ is correlated with the appearance of a clear signature of the deprotonated peroxo form [(L5)FeIII–η2O2]+ with a broad absorption band at around 650 nm (Fig. 2b). EPR spectroscopy was used to further characterize the photo-generated FeIII–O2 species.
Prior to irradiation, the EPR spectrum of the [RuII(bpy)3]2+/MV2+/[(L5)FeII]2+ mixture (Fig. S15,† black trace) shows signals with g values at 4.41 and 4.04 that are assigned to a high spin (HS) [(L5)FeIII–X]2+ species (X being an exogenous ligand such as OH− based on the electrochemical studies in Fig. S6† or an anion from the buffer) and a signal at 4.27 likely due to a trace of Fe3+ impurities.
Upon irradiation of the above mixture in the presence of EDTA as an electron donor and under anaerobic conditions, the signals at 4.41 and 4.04 faded out with the appearance of a signal at g = 2.00 very likely originating from the MV˙+ organic radical (Fig. S15†). This result clearly shows that the HS Fe(III) species can be reduced by MV˙+. The EPR spectrum after light irradiation for 1 s under an aerobic atmosphere of an aqueous solution containing the triptych mixture buffered at pH 4 and an electron donor (EDTA) showed signals with g values of around 2.21, 2.15 and 1.97, a set of values corresponding to a typical low spin iron(III)-hydroperoxo species (red spectrum in Fig. 3) as described previously.58 The morphology of this set of signals supports the presence of different rotational isomers of the FeIII–OOH species. We may also note that concomitantly the signals at 4.41 and 4.04 also gain in intensity upon irradiation (Fig. S16†). This can be reasonably attributed to the formation of the same high-spin FeIII–X species present in the initial iron(II) complex that may result from the degradation of the FeIII–OOH intermediate. Furthermore, we found that upon prolonged irradiation time (20 s) both the signals for the FeIII–OOH intermediate and the HS FeIII–X species disappear indicating that MV˙+ can further reduce the FeIII–OOH catalytic precursor and the HS FeIII–X species to an EPR silent Fe(II) form (Fig. S16†). Such photo-events may also occur to explain the slow recovery of [RuII(bpy)3]2+ we discussed above.
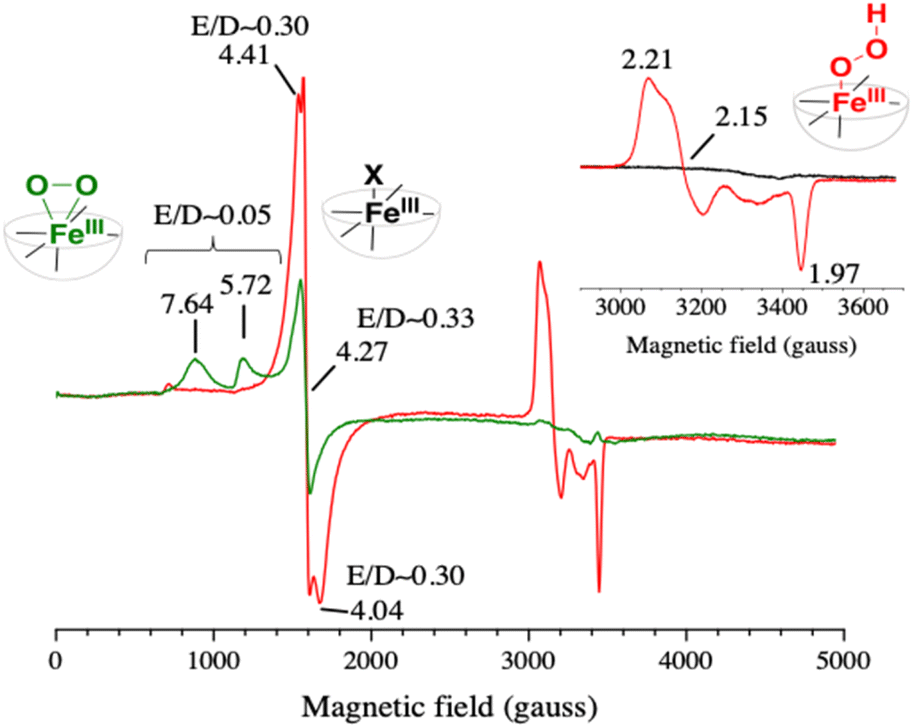 | ||
| Fig. 3 EPR spectra of a B&R pH 4 buffer solution containing [RuII(bpy)3]2+ (30 μM), MV2+ (20 mM), [(L5)FeII]2+ (100 μM) and 20 mM EDTA (red) or 200 mM TEOA (green) irradiated for 1 s with a 100 W m−2 blue LED. The spectra were recorded at 15 K with a modulation amplitude of 25 gauss and a microwave power of 20 mW (5 mW for the inset). The microwave frequency was ∼9.49 GHz. In red, the EPR spectrum of the photo-generated low spin [(L5)FeIII–OOH]2+ species, and in green the EPR spectrum of the photo-generated high spin [(L5)FeIII–η2O2]+ species; expanded view shown in Fig. S17.† In the inset, the black spectrum was recorded before the illumination. | ||
When the photogeneration was performed in the presence of an excess of triethanolamine (TEOA) (buffer at pH 4 and 200 mM TEOA, resulting in a pH value of 7.26), the EPR spectrum exhibits this time a high spin species with resonances at g = 7.64 and 5.72 (green spectrum in Fig. 3), which indicates the formation of an iron(III)-peroxo [(L5)FeIII–η2O2]+ intermediate with S = 5/2 (eqn (6′), see Scheme 2),58 while the signal at 4.27 for the Fe3+ impurity still persists (Fig. S15†).
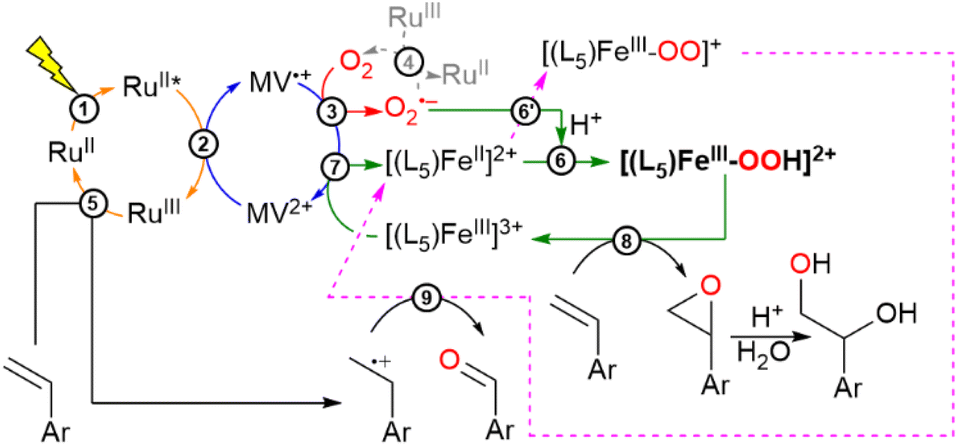 | ||
| Scheme 2 Photocatalytic OAT to an alkene substrate in the absence of a sacrificial electron donor using light, O2 as the oxygen source, [(L5)FeII]2+ as the catalyst, [RuII(bpy)3]2+ as the photosensitizer (written as RuII for simplification) and MV2+ as the electron relay. Reaction numbers refer to equation numbers in the discussion. | ||
The same experiment with TEOA as the donor was also performed in the visible absorption and it confirmed both the formation of the characteristic band of the iron(III)-peroxo species at 650 nm and the accumulation of MV˙+ once the O2 was locally consumed (Fig. S13†). Based on these results, we can conclusively support that the photochemically produced [(L5)FeIII–OOH]2+ and [L5FeIII–η2O2]+ species match the electronic signatures of the chemically generated ones. When conducted with 4-styrene sulfonate as the electron donor to [RuIII(bpy)3]3+, the observed spectral changes were not as notable as with EDTA, which is a better electron donor. However, the same trend of spectral modifications in the absorption spectra was observed (Fig. S12†). The low spin FeIII–OOH species was also evidenced albeit with lower intensities.
At this point, gathering all the photophysical events we can reasonably propose a photocatalytic cycle to explain the OAT reaction, as summarized in Scheme 2. It is noteworthy that in the photocatalytic runs no sacrificial electron donor was needed to perform the OAT reaction as described below. The first photophysical event concerns the generation of the charge separated state described by MV˙+–[RuIII(bpy)3]3+ (eqn (2)). From here, MV˙+ reacts with O2 to form O2˙− (eqn (3)), leaving behind an oxidizing equivalent of [RuIII(bpy)3]3+. The O2˙− can react with the starting Fe(II) complex to form after protonation [(L5)FeIII–OOH]2+ (eqn (6)) in the catalytic cycle, while [RuIII(bpy)3]3+ can oxidize the alkene substrate to form an alkenyl radical species (eqn (5)). According to in-depth mechanistic studies albeit in organic solvents, [(L5)FeIII–OOH]2+ can evolve to an iron-oxo species that enables the oxygen atom transfer reaction to the alkene to form an epoxide or a diol derivative as reported.8,66,67 Isotopic labelling experiments carried out in the presence of 18O2 produced the corresponding labeled and unlabeled epoxide at pH 6, suggesting the involvement of a high-valent iron-oxo species prone to O-atom exchange with the water solvent molecules. At pH 4, the diol derivative is detected with a double 18O, a mixed 18O and 16O and a double 16O content (Fig. S9 and Scheme S1†). Such an observation can be explained by the cis-hydroxylation of the alkene substrate by an iron-oxo-hydroxo intermediate or the formation of the epoxide, followed by a ring opening process with unlabeled water molecules (eqn (8), see Scheme 2).
The detected aldehyde may derive from the reaction of the alkenyl radical with the reactive [L5FeIII–η2O2]+ to form an unstable dioxetane ring that after cleavage leads to the corresponding aldehyde (eqn (9), see Scheme 2; further details in Scheme S1†).8 An important mechanistic aspect concerns the evolution of [(L5)FeIII–OOH]2+ leading to either Fe(V)O or Fe(IV)O following a heterolytic or homolytic cleavage scheme of the O–O bond of the catalytic precursor in an aqueous medium.20 These proposed mechanisms are based on the abundant work in this field and are recalled in the ESI (Scheme S1).† Accordingly, under these scenarios, either Fe(III) or Fe(II) would result after the OAT reaction to be refurnished in the catalytic cycle. Studies from Que and colleagues have shown that the presence of water would enhance the heterolytic bond cleavage in a molecular FeIII–OOH intermediate.20 In this present study, we can only speculate on such an issue as none of these high-valent iron oxo species have been characterized under our experimental conditions. Interestingly, from the EPR spectrum of the photogenerated low spin [(L5)FeIII–OOH]2+, we also observed a gain in the intensity of the signals of the high spin Fe(III) species that we argued to stem from the degradation of the [(L5)FeIII–OOH]2+ intermediate (Fig. S16†). Henceforth, it is more likely that the active species is actually Fe(V)O that would yield an Fe(III) species after the OAT reaction to re-enter the catalytic loop to be reduced by MV˙+ (eqn (7)). This electron transfer process is thermodynamically feasible as confirmed by the redox properties of the iron complex in an aqueous medium, where two observed cathodic peaks are assigned tentatively to the couples ([(L5)FeIII–OH2]3+/[(L5)FeII–OH2]2+) = 0.13 V vs. SCE and ([(L5)FeIII–OH]2+/[(L5)FeII–OH]+) = −0.17 V vs. SCE (Fig. S6†) compared to E0(MV2+/MV˙+) = −0.69 V vs. SCE. Further investigation on this segment of the catalytic cycle will be needed to clarify the actual nature of the catalytically active iron-oxo species in an aqueous medium.
Conclusions
In this study, we have shown a scenario where sharing the two-electron activation of O2 on both O2 and a non-heme Fe(III) catalyst allows us to avoid the charge accumulation process. EPR spectroscopy and LFP data backed the metal centered activity with the unprecedented interception of a light-driven formation of an FeIII–OOH species in the OAT reaction that rules out an auto-oxidation pathway. Another attractive feature of this study concerns the elimination of sacrificial electron donors that still prohibit any value-added synthetic procedure.68,69 For the overall efficiency of the photocatalytic system, it is important that productive reaction steps are competitive with respect to alternative short-circuit reactions. Two such “crossroads” can be easily identified as the preferential reduction of the resulting [(L5)FeIII–OH]2+ state vs. reduction of the catalytically active [(L5)FeIII–OOH]2+ state and the preferential oxidation by Ru(III) of the substrate vs. oxidation of the [(L5)FeII]2+ state. Another point that needs to be further investigated concerns the reactivity of the generated alkenyl radical.70–73 An alternative route to preclude such a pathway may reside in tandem oxidation reactions with different organic substrates. These challenges are currently being addressed.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Eva Pugliese: formal analysis and investigation. Nhat Tam Vo: formal analysis and investigation. Alain Boussac: formal analysis and investigation. Fréderic Banse: formal analysis. Yasmina Mekmouche: investigation. Jalila Simaan: investigation. Thierry Tron: investigation and funding acquisition. Philipp Gotico: formal analysis and investigation. Marie Sircoglou: formal analysis. Zakaria Halime: formal analysis, investigation, and data curation. Winfried Leibl: formal analysis, investigation, funding acquisition, data curation, and writing of the original draft. Ally Aukauloo: lead formal analysis, funding acquisition, writing of the original draft, and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
French National Research Agency ANR Multiplet (ANR-15-CE07-0021-01), E.P. thanks ANR LOCO, grant No: ANR-19-CE05-0020-02. We thank LABEX CHARMMMAT, grant No: ANR-11-LABX-0039).Notes and references
- P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef PubMed.
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef PubMed.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef PubMed.
- C. Krebs, D. Galonić Fujimori, C. T. Walsh and J. M. Bollinger, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS PubMed.
- J. P. T. Zaragoza and D. P. Goldberg, Dioxygen-dependent Heme Enzymes, The Royal Society of Chemistry, 2019, 1–36 Search PubMed.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed.
- S. Fukuzumi, K.-B. Cho, Y.-M. Lee, S. Hong and W. Nam, Chem. Soc. Rev., 2020, 49, 8988–9027 RSC.
- W. N. Oloo and L. Que, Acc. Chem. Res., 2015, 48, 2612–2621 CrossRef CAS PubMed.
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem., 2017, 1, 1–20 CrossRef.
- W. Nam, Acc. Chem. Res., 2007, 40, 465 CrossRef CAS.
- S. Friedle, E. Reisner and S. J. Lippard, Chem. Soc. Rev., 2010, 39, 2768–2779 RSC.
- J. T. Groves, J. Inorg. Biochem., 2006, 100, 434–447 CrossRef CAS.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS.
- F. Li, J. England and L. Que, J. Am. Chem. Soc., 2010, 132, 2134–2135 CrossRef CAS.
- Y.-M. Lee, S. Hong, Y. Morimoto, W. Shin, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2010, 132, 10668–10670 CrossRef CAS PubMed.
- J. Yoon, S. A. Wilson, Y. K. Jang, M. S. Seo, K. Nehru, B. Hedman, K. O. Hodgson, E. Bill, E. I. Solomon and W. Nam, Angew. Chem., Int. Ed., 2009, 48, 1257–1260 CrossRef CAS PubMed.
- L. Que, Acc. Chem. Res., 2007, 40, 493–500 CrossRef CAS.
- A. Thibon, J. England, M. Martinho, V. G. Young Jr, J. R. Frisch, R. Guillot, J.-J. Girerd, E. Münck, L. Que Jr and F. Banse, Angew. Chem., Int. Ed., 2008, 47, 7064–7067 CrossRef CAS PubMed.
- J. Cho, S. Jeon, S. A. Wilson, L. V. Liu, E. A. Kang, J. J. Braymer, M. H. Lim, B. Hedman, K. O. Hodgson, J. S. Valentine, E. I. Solomon and W. Nam, Nature, 2011, 478, 502–505 CrossRef PubMed.
- W. N. Oloo, A. J. Fielding and L. Que, J. Am. Chem. Soc., 2013, 135, 6438–6441 CrossRef PubMed.
- J.-J. Girerd, F. Banse and A. J. Simaan, in Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations, ed. B. Meunier, Springer, Berlin, Heidelberg, 2000, 145–177 Search PubMed.
- M. Martinho, P. Dorlet, E. Rivière, A. Thibon, C. Ribal, F. Banse and J.-J. Girerd, Chem.–Eur. J., 2008, 14, 3182–3188 CrossRef.
- S. E. Creager, S. A. Raybuck and R. W. Murray, J. Am. Chem. Soc., 1986, 108, 4225–4227 CrossRef.
- M. Mukherjee and A. Dey, ACS Cent. Sci., 2019, 5, 671–682 CrossRef.
- N. Kostopoulos, C. Achaibou, J.-M. Noël, F. Kanoufi, M. Robert, C. Fave and E. Anxolabéhère-Mallart, Inorg. Chem., 2020, 59, 11577–11583 CrossRef PubMed.
- J.-M. Noel, N. Kostopoulos, C. Achaibou, C. Fave, E. Anxolabéhère-Mallart and F. Kanoufi, Angew. Chem., Int. Ed., 2020, 59, 16376–16380 CrossRef PubMed.
- N. Ségaud, E. Anxolabéhère-Mallart, K. Sénéchal-David, L. Acosta-Rueda, M. Robert and F. Banse, Chem. Sci., 2014, 6, 639–647 RSC.
- R. Oliveira, W. Zouari, C. Herrero, F. Banse, B. Schöllhorn, C. Fave and E. Anxolabéhère-Mallart, Inorg. Chem., 2016, 55, 12204–12210 CrossRef CAS.
- E. Anxolabéhère-Mallart and F. Banse, Curr. Opin. Electrochem., 2019, 15, 118–124 CrossRef.
- A. L. Robinson, J.-N. Rebilly, R. Guillot, C. Herrero, H. Maisonneuve and F. Banse, Chem.–Eur. J., 2022, 28, e202200217 CrossRef CAS PubMed.
- M. E. Ener, Y.-T. Lee, J. R. Winkler, H. B. Gray and L. Cheruzel, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18783–18786 CrossRef.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef PubMed.
- S. Fukuzumi, T. Kishi, H. Kotani, Y.-M. Lee and W. Nam, Nat. Chem., 2011, 3, 38–41 CrossRef.
- X. Wu, X. Yang, Y.-M. Lee, W. Nam and L. Sun, Chem. Commun., 2015, 51, 4013–4016 RSC.
- H. Kotani, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 3249–3251 CrossRef.
- S. Fukuzumi, S. Kuroda and T. Tanaka, J. Am. Chem. Soc., 1985, 107, 3020–3027 CrossRef.
- J. Berglund, T. Pascher, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 1997, 119, 2464–2469 CrossRef CAS.
- W. Zhang, E. Fernández-Fueyo, Y. Ni, M. van Schie, J. Gacs, R. Renirie, R. Wever, F. G. Mutti, D. Rother, M. Alcalde and F. Hollmann, Nat. Catal., 2018, 1, 55–62 CrossRef CAS PubMed.
- C. Herrero, A. Quaranta, M. Sircoglou, K. Sénéchal-David, A. Baron, I. M. Marín, C. Buron, J.-P. Baltaze, W. Leibl, A. Aukauloo and F. Banse, Chem. Sci., 2015, 6, 2323–2327 RSC.
- C. Panda, J. Debgupta, D. Díaz Díaz, K. K. Singh, S. Sen Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273–12282 CrossRef CAS.
- B. Chandra, K. K. Singh and S. S. Gupta, Chem. Sci., 2017, 8, 7545–7551 RSC.
- C. Herrero, A. Quaranta, R. Ricoux, A. Trehoux, A. Mahammed, Z. Gross, F. Banse and J.-P. Mahy, Dalton Trans., 2015, 45, 706–710 RSC.
- B. Mühldorf, U. Lennert and R. Wolf, Phys. Sci. Rev., 2019, 4, 20180030 Search PubMed.
- F. Avenier, C. Herrero, W. Leibl, A. Desbois, R. Guillot, J.-P. Mahy and A. Aukauloo, Angew. Chem., Int. Ed., 2013, 52, 3634–3637 CrossRef PubMed.
- J. Chen and W. R. Browne, Coord. Chem. Rev., 2018, 374, 15–35 CrossRef.
- J. Rosenthal, T. D. Luckett, J. M. Hodgkiss and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 6546–6547 CrossRef.
- S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Nat. Chem., 2014, 6, 934–940 CrossRef.
- J. Jung, K. Ohkubo, D. P. Goldberg and S. Fukuzumi, J. Phys. Chem. A, 2014, 118, 6223–6229 CrossRef PubMed.
- R. Cibulka, R. Vasold and B. König, Chem.–Eur. J., 2004, 10, 6223–6231 CrossRef PubMed.
- B. Mühldorf and R. Wolf, Angew. Chem., Int. Ed., 2016, 55, 427–430 CrossRef PubMed.
- H. D. Gafney and A. W. Adamson, J. Am. Chem. Soc., 1972, 94, 8238–8239 CrossRef CAS.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- H. Sun, A. Yoshimura and M. Z. Hoffman, J. Phys. Chem., 1994, 98, 5058–5064 CrossRef CAS.
- M. Z. Hoffman, D. R. Prasad, G. Jones and V. Malba, J. Am. Chem. Soc., 1983, 105, 6360–6362 CrossRef CAS.
- K. Z. Ismail and S. G. Weber, Biosens. Bioelectron., 1991, 6, 699–705 CrossRef CAS PubMed.
- J. J. Orgill, C. Chen, C. R. Schirmer, J. L. Anderson and R. S. Lewis, Biochem. Eng. J., 2015, 94, 15–21 CrossRef CAS.
- A. J. Simaan, S. Döpner, F. Banse, S. Bourcier, G. Bouchoux, A. Boussac, P. Hildebrandt and J.-J. Girerd, Eur. J. Inorg. Chem., 2000, 2000, 1627–1633 CrossRef.
- A. Bohn, C. Chinaux-Chaix, K. Cheaib, R. Guillot, C. Herrero, K. Sénéchal-David, J.-N. Rebilly and F. Banse, Dalton Trans., 2019, 48, 17045–17051 RSC.
- A. Bohn, PhD thesis, Université Paris Saclay (COmUE), 2018, accessible at https://tel.archives-ouvertes.fr/tel-02018125.
- R. Farran, Y. Mekmouche, N. T. Vo, C. Herrero, A. Quaranta, M. Sircoglou, F. Banse, P. Rousselot-Pailley, A. J. Simaan, A. Aukauloo, T. Tron and W. Leibl, iScience, 2021, 24(4), 102378 CrossRef CAS.
- D. T. Sawyer and J. S. Valentine, Acc. Chem. Res., 1981, 14, 393–400 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- G. Olivo, O. Cussó, M. Borrell and M. Costas, J. Biol. Inorg Chem., 2017, 22, 425–452 CrossRef CAS PubMed.
- I. Bernal, I. M. Jensen, K. B. Jensen, C. J. McKenzie, H. Toftlund and J.-P. Tuchagues, J. Chem. Soc., Dalton Trans., 1995, 3667–3675 RSC.
- I. Gamba, Z. Codolà, J. Lloret-Fillol and M. Costas, Coord. Chem. Rev., 2017, 334, 2–24 CrossRef CAS.
- Y. Feng, J. England and L. Que, ACS Catal., 2011, 1, 1035–1042 CrossRef CAS.
- N. T. Vo, Y. Mekmouche, T. Tron, R. Guillot, F. Banse, Z. Halime, M. Sircoglou, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2019, 58, 16023–16027 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- L. M. Smith, H. M. Aitken and M. L. Coote, Acc. Chem. Res., 2018, 51, 2006–2013 CrossRef CAS.
- Y.-F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640–1653 CrossRef PubMed.
- L. J. Johnston and N. P. Schepp, J. Am. Chem. Soc., 1993, 115, 6564–6571 CrossRef.
- L. J. Johnston and N. P. Schepp, Pure Appl. Chem., 1995, 67, 71–78 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03129a |
| This journal is © The Royal Society of Chemistry 2022 |