
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA stimulus-responsive hexahedron DNA framework facilitates targeted and direct delivery of native anticancer proteins into cancer cells†
Wenjiao
Zhou
a,
Fang
Yang
b,
Shunmei
Li
b,
Ruo
Yuan
b and
Yun
Xiang
 *b
*b
aSchool of Chemistry and Chemical Engineering, Chongqing University of Technology, Chongqing 400054, P. R. China
bKey Laboratory of Luminescence Analysis and Molecular Sensing, Ministry of Education, School of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, P. R. China. E-mail: yunatswu@swu.edu.cn
First published on 13th August 2022
Abstract
The targeted and direct intracellular delivery of proteins plays critical roles in biological research and disease treatments, yet remains highly challenging. Current solutions to such a challenge are limited by the modification of proteins that may potentially alter protein functions inside cells or the lack of targeting capability. Herein, we develop a stimulus-responsive and bivalent aptamer hexahedron DNA framework (HDF) for the targeted and direct delivery of native therapeutic proteins into cancer cells. The unmodified proteins are caged inside the HDF nanostructures assembled from six programmable single stranded DNAs to protect the proteins from degradation by cathepsins and enhance their targeting capability and delivery efficiency with the nanostructure-integrated aptamers. In addition, the protein drugs can be selectively released from the HDF nanostructures by the intracellular ATP molecules to induce tumor cell apoptosis, highlighting their promising application potential for cell biology and precise protein medicines.
Introduction
Introducing exogenous active proteins into living cells plays essential roles in cell biological research and the treatment of human diseases, such as diabetes and cancers.1–3 The highly specific activity and low toxicity of protein therapeutics can potentially address some side effects caused by traditional small molecule chemical drugs.1,4 Despite that protein therapeutics exhibit such fascinating advantages, their bioavailability and therapeutic efficacy are highly dependent upon their specific and successful delivery into target cells. However, the instability and large size of proteins and the permeability of cell membranes significantly mitigate their delivery efficiency. Therefore, effective deliver systems are needed to ferry proteins into cells to fully realize their therapeutic potential.Various nanocarriers, including polymers,5–8 lipids,9,10 micelles,11 cell penetrating peptides12,13 and nanoparticles,14–16 have been developed to deliver protein therapeutics into cells via electrostatic adsorption or covalent conjugation of proteins to the carriers. The efficient cell uptake and ease of surface functionalization of these nanocarriers indeed facilitate their ability to load and transport proteins across the cell membranes. Nevertheless, these nanocarriers (e.g., nanomaterials) are prone to adsorb serum proteins via non-specific protein–nanomaterial interactions because of the high surface-to-volume ratio of the nanomaterials,9,14,17 and the attachment of proteins to the surfaces of the nanomaterials is not amenable to protect the proteins from degradation by lysosomal cathepsins during endocytosis. Besides, the activity of conjugated proteins can also decrease by 20–80% to cause significantly reduced effectiveness for protein therapy. Moreover, the cytotoxicity of nanomaterials9,18 and recent findings that nanoparticles and carbon nanotubes can accelerate both extravasation and intravasation of cancer cells for metastasis19,20 have triggered the development of new platforms for the delivery of native proteins into cells.
DNA nanostructures, nanoscale materials assembled from DNA strands, have recently received increasing attention as effective nanocarriers for intracellular biosensing21–26 and small molecule/genetic drug delivery27–33 applications, owing to their controllable sizes, structural stability, excellent biocompatibility and cell permeability. For instance, DNA nanoflowers synthesized via rolling circle amplification were employed to wrap and deliver proteins into cells,34 while the Ding group reported a DNA origami-based DNA rectangular nanostructure delivery platform by covalently conjugating proteins to nanostructures.35 These reports clearly unveiled the potential of DNA nanostructures as promising therapeutic delivery alternatives. Despite the preliminary success, further efforts are still highly demanded for enabling DNA nanostructures with targeted capability and the delivery of unmodified proteins. Polyhedron DNA frameworks36–41 assembled from simple DNA strands with exquisite designs can provide excellent structural flexibility with controllable external DNA skeletons and inner cavities to match proteins with different sizes, making them useful delivery platforms. Here, we report a bivalent aptamer-integrated hexahedron DNA framework (HDF) for targeted and direct delivery of native therapeutic proteins into cancer cells. Such a HDF nanostructure is assembled from six partially complementary single-stranded DNAs, two of which are inserted with the protein tyrosine kinase 7 (PTK 7) specific sgc8 aptamer, while one of which is connected with the ATP responsive aptamer. Therapeutic proteins can be encapsulated inside the cavity of HDF nanostructures, which show programmed configuration switch in response to an intracellular ATP stimulus. The compactness and rigidity of the HDF nanostructure protects the protein from being degraded by lysosomal cathepsins. Moreover, the aptamers can enhance its targeting capability and delivery efficiency, and the ATP molecules inside the cells can trigger selective and controlled release of the proteins, making this HDF nanostructure an efficient and precise delivery platform for protein therapeutics.
Results and discussion
RNase A with molecular dimensions of 2.2 nm × 2.8 nm × 3.8 nm (ref. 42) is one of the widely reported therapeutic proteins that can degrade its substrate RNA molecules residing in the cytosol upon cell uptake to induce cell apoptosis. For a proof-of-concept demonstration, RNase A is selected as the model anticancer protein and bivalent sgc8-HDF is designed as the carrier. As illustrated in Fig. 1A, such a sgc8-HDF nanostructure is assembled from six partially complementary ssDNAs with one ATP aptamer- (S5) and two sgc8 aptamer-containing strands (S3 and S6). According to the design of the lengths of the sides (20-bp) and the bottom strands (30-bp), the corresponding interior cavity volume and triangular pore in-circle diameter of the sgc8-HDF nanostructure are approximately calculated to be 89 nm3 and 1.9 nm, respectively, which is suitable to accommodate RNase A inside the cavity and to prevent its leakage. As illustrated in Fig. 1A, RNase A encapsulated sgc8-HDF (RNase A@sgc8-HDF) can therefore be prepared by incubating RNase A with the open sgc8-HDF precursor and the sealing strand (S5). Moreover, the RNase A@sgc8-HDF nanostructure can be readily transported into the target HeLa cancer cells via an endocytosis approach upon specific binding between the sgc8 aptamers and the over-expressed PTK7 receptors on the cell surfaces (Fig. 1B). Due to the enhanced stability of the DNA framework nanostructure, sgc8-HDF is expected to escape from the lysosome and the ATP molecules with elevated concentration inside the HeLa cells further bind the ATP aptamer sequences of RNase A@sgc8-HDF to open the hexahedron structure and release RNase A, which subsequently recognizes and degrades the intracellular RNA to induce cell apoptosis. | ||
| Fig. 1 Schematic illustration of the preparation of RNase A@sgc8-HDF and its delivery into cancer cells. (A) The encapsulation of RNase A by a bivalent sgc8 aptamer-integrated HDF for the formation of the RNase A@sgc8-HDF nanostructure. (B) Targeted delivery of RNase A@sgc8-HDF into HeLa cells and ATP-controlled release of native RNase A to degrade intracellular RNA for triggering cell apoptosis. | ||
Toward the preparation of the RNase A@sgc8-HDF nanostructure, the stepwise assembly formation of sgc8-HDF and the encapsulation of RNase A were characterized by native PAGE. As shown by the gel image in Fig. 2, the six distinct bands from Lanes 1 to 6 represent the corresponding sgc8-HDF assembly strands, respectively. The band mobility gradually decreases along with the successive addition of the assembly strands for the formation of sgc8-HDF (Lanes 7–11), due to the increase in the molecular weight and size from the assembled components. After incubating RNase A with the encapsulation strand (S5) and the open sgc8-HDF precursor, a further decrease in band mobility is observed in Lane 12, owing to the facts that the encapsulation of RNase A in the cavity of sgc8-HDF increases its molecular weight and the mobility of the RNase A@sgc8-HDF nanostructure is reduced by the PAGE conditions (pH 8.0) with positively charged RNase A (with an approximate pI of 9.3) as well.
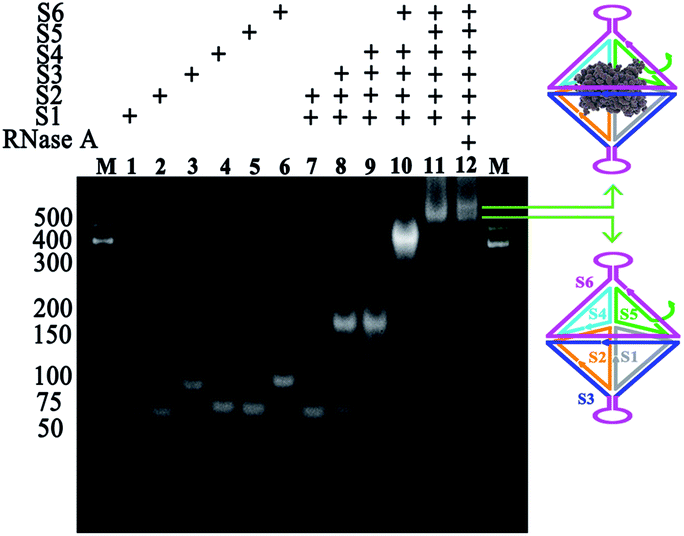 | ||
| Fig. 2 Gel characterization of the preparation of RNase A@sgc8-HDF. 5% Native PAGE characterization for the assembly formation of sgc8-HDF and the encapsulation of RNase A. Lane 1–6: S1–S6; Lane 7: S1 + S2; Lane 8: S1 + S2 + S3; Lane 9: S1 + S2 + S3 + S4; Lane 10: S1 + S2 + S3 + S4 + S6; Lane 11: S1 + S2 + S3 + S4 + S5 + S6; Lane 12: S1 + S2 + S3 + S4 + S5 + S6 + RNase A. | ||
Experiments were next performed to verify the controlled release of RNase A in response to external ATP triggers by labeling S6 in RNase A@ sgc8-HDF with the FAM/Dabcyl pair to make it in an initially fluorescence-quenched state (Fig. 3A). Because RNase A is a ribonuclease that can degrade single-stranded RNA, a fluorogenic RNA substrate sequence (15-nt) labeled with a fluorophore (Cy5) at the 5′-end and a quencher (BHQ2) at the 3′-end was used as the signal molecule to indicate the ATP-triggered release of RNase A, and the corresponding fluorescence spectra are displayed in Fig. 3A. It can be seen that the RNA substrate sequences alone in buffer show a small fluorescence emission peak from Cy5 at 670 nm (curve a), due to the efficient fluorescence quenching of Cy5 by BHQ2. When the RNA substrate sequences were mixed with RNase A@sgc8-HDF, a minor increase in the fluorescent signal is observed at 670 nm (curve b vs. a), indicating that a very small part of the RNA substrate sequences permeates through the triangular pores and is degraded by RNase A encapsulated inside the sgc8-HDF nanostructures. Besides, the low fluorescence from FAM at 520 nM, which is quenched by Dabcyl, also verifies that the sgc8-HDF nanostructure has a closed structure upon the hybridizations between the encapsulation strand S5 and the open sgc8-HDF precursor, and such a hybridization brings Dabcyl into close proximity to FAM to quench its fluorescence. However, when the mixture of RNase A@sgc8-HDF and the RNA substrate sequences was incubated with 5 mM ATP, significant recovery of fluorescence at both 520 nm and 670 nm can be observed (curve c vs. b). This is because of the responsive formation of the ATP/aptamer tertiary structure to open the side of sgc8-HDF to release RNase A. The opening of the RNase A@sgc8-HDF nanostructure then leads to the separation of FAM from Dabcyl, thereby recovering the fluorescence at 520 nm. In the meantime, RNase A can be released upon the opening of the sgc8-HDF nanostructure to degrade the RNA substrate sequences to show significant fluorescence at 670 nm. To evaluate the stability of the sgc8-HDF nanostructure, which is crucial for the successful delivery of RNase A inside the cells, sgc8-HDF was incubated with the cell growth medium containing 10% FBS and HeLa cell lysate, respectively, for different time scales by using a 30-bp dsDNA strand as the control, and the mixture were further analyzed by native PAGE. According to the results shown in Fig. 3B, the bands corresponding to the dsDNA almost disappear after 2 h of incubation with 10% FBS or the HeLa cell lysate while sgc8-HDF remains essentially stable even after 24 h of incubation in both media as evidenced by the existence of their representative bands. The high stability of sgc8-HDF, which is assumably due to the compact structure of the DNA nanostructure against nuclease digestion,43 makes it a promising potential delivery platform.
 | ||
| Fig. 3 ATP-controlled release of RNase A and the stability of the sgc8-HDF nanostructure. (A) Fluorescence spectra of different reaction mixtures: (a) RNA substrate sequence; (b) RNase A@sgc8-HDF + RNA substrate sequence; (c) RNase A@sgc8-HDF + ATP + RNA substrate sequence. The concentrations of RNase A@sgc8-HDF, ATP and the RNA substrate sequence were 50 nM, 5 mM and 300 nM, respectively. (B) 5% native PAGE characterization of the stability of the sgc8-HDF (200 nM) and the 30-bp dsDNA (200 nM) sequence incubated with 10% FBS and HeLa cell lysate, respectively. | ||
To investigate the cellular uptake of RNase A@sgc8-HDF, RNase A was labeled with the AF488 fluorescent dye while the sgc8-HDF nanostructure was free of any fluorescent labels to avoid interference. For this study, HeLa cells with highly expressed PTK7 receptors on the cell surfaces were selected as the representative cell model. As shown in Fig. 4 by the CLSM images, no visible fluorescence signals can be observed inside HeLa cells after incubating them with AF488-RNase A for 6 h at 37 °C. This is basically due to the permeability resistance of the cell membranes to prevent AF488-RNase A from entering the cells. However, the encapsulation of AF488-RNase A inside the HDF nanostructure (without the sgc8 aptamers) can facilitate its delivery as evidenced by the green fluorescence signals in the cells because the DNA nanostructures are prone to be transported into tumor cells via a well-established endocytosis pathway.44 Importantly, when AF488-RNase A@sgc8-HDF was used, significantly enhanced green fluorescence signals are observed in HeLa cells while obviously reduced ones are obtained with cells treated by the free sgc8 aptamers (PTK7 inhibitor), demonstrating that the incorporation of the sgc8 aptamers indeed enhances the delivery efficiency of AF488-RNase A@sgc8-HDF via the specific binding between the sgc8 aptamers and the PTK7 receptors on HeLa cell surfaces.
 | ||
| Fig. 4 Cellular uptake of AF488-RNase A@sgc8-HDF. CLSM images of HeLa cells (PTK7-positive) incubated with different molecules for 5 h. Scale bar = 20 μm. | ||
The endocytic uptake and delivery of AF488-RNase A@sgc8-HDF into the cytosol of HeLa cells were also investigated with a culture time of 3 and 6 h, respectively. As displayed in Fig. 5, after being cultured with HeLa cells for 3 h, the AF488-RNase A@sgc8-HDF nanostructures (green) are mainly trapped inside lysosomes (red) as shown by the yellow fluorescence from the co-localization. However, after extending the culture time to 6 h, a large number of green fluorescent signals in the cytoplasm and a significantly decreased overlap between AF488-RNase A@sgc8-HDF and lysosomes are observed, strongly indicating the escape of many AF488-RNase A@sgc8-HDF from lysosomes. These results confirm that AF488-RNase A@sgc8-HDF can be internalized into the target HeLa cells with a PTK7 receptor-dependent endolysosomal pathway, followed by escaping from the endolysosomal compartment into the cytoplasm.
 | ||
| Fig. 5 Subcellular distribution of AF488-RNase A@sgc8-HDF. CLSM images of the endocytic uptake of AF488-RNase A@sgc8-HDF by HeLa cells for 3 and 6 h, respectively. HeLa cells were stained with Hoechst 33342 (blue, cell nuclei) and LysoTracker Red DND-99 (red, lysosomes). Scale bar = 20 μm. | ||
After demonstrating the successful delivery of the nanostructures into the target cells, specific intracellular ATP-triggered opening of RNase A@sgc8-HDF (note: the HDF was labeled with the FAM/Dabcyl pair) was next verified by culturing it with HeLa cells, followed by CLSM imaging. As shown in Fig. 6, the visible green fluorescent signals located inside HeLa cells, owing to the separation of FAM from Dabcyl, indicate that RNase A@sgc8-HDF is opened by the ATP triggers, which is in accordance with the results in Fig. 3. However, when the aptamer sequence responsive to ATP in the nanostructure was replaced by the non-responsive one (RNase A@sgc8-mHDF), no visible green fluorescence signals are observed inside HeLa cells. These comparisons clearly indicate that the opening of the RNase A@sgc8-HDF nanostructure is highly dependent upon the ATP molecules inside the cells.
 | ||
| Fig. 6 Intracellular ATP-controlled release of RNase A from RNase A@sgc8-HDF. CLSM images of HeLa cells cultured with RNase A@sgc8-HDF and RNase A@sgc8-mHDF for 6 h. Scale bar = 20 μm. | ||
The therapeutic potential of the RNase A@sgc8-HDF nanostructure was finally evaluated by using RNase A, sgc8-HDF, and RNase A@HDF as the control molecules. After culturing HeLa cells with these molecules in the concentration range from 0.1 μM to 2 μM at 37 °C for 48 h, the viability of the cells was analyzed using MTT assays. According to Fig. 7A, RNase A and sgc8-HDF show almost no obvious cytotoxic effects (>95% cell viability) to HeLa cells when cultured for 48 h with the concentrations of the molecules up to 2 μM, which can be attributed to either the difficulty of RNase A to penetrate the cell membranes to enter the cells or the absence of RNase A in the sgc8-HDF nanostructure. In contrast, decreasing cell viability of HeLa cells is observed with increasing concentrations of the RNase A@HDF and RNase A@sgc8-HDF nanostructures from 0 to 2 μM, and the cell viability decreases to 48% and 21%, respectively, at 2 μM, indicating an enhanced therapeutic potency of RNase A@sgc8-HDF over RNase A@HDF, which is associated with the improved delivery efficiency by the aptamers as demonstrated previously. From the CLSM images in Fig. 7B, the culturing of RNase A (2 μM) or sgc8-HDF (2 μM) with HeLa cells causes no obvious cell apoptosis as indicated by the green signals from NucGreen® Dead stained cells, while RNase A@HDF (2 μM) and RNase A@sgc8-HDF (2 μM) lead to significant cell apoptosis with an increased potency by RNase A@sgc8-HDF. These observations agree well with the MTT assay, which reveals the potency of RNase A@sgc8-HDF for inducing cancer cell apoptosis with high efficiency.
 | ||
| Fig. 7 Evaluation of the therapeutic efficacy of the RNase A@sgc8-HDF nanostructure to cancer cells. (A) MTT assays for the cell viability of HeLa cells and (B) CLSM images of the corresponding HeLa cells cultured with different therapeutic molecules at 37 °C for 48 h. NucBlue® Live stained the nuclei of all cells in blue, while NucGreen® Dead stained only the nuclei of dead cells in green. Scale bar = 100 μm. | ||
Experimental
Reagents and materials
Tris–HCl, MgCl2, NaCl, EDTA, tris-borate, acrylamide/bis-acrylamide, N,N,N′,N′-tetramethylethylenediamine (TEMED) and the UNIQ-10 Spin Column DNA Gel Extraction Kit were obtained from Sangon Biotech Co., Ltd (Shanghai, China). ATP was bought from Worthington Biochemicals (Lakewood, NJ, USA). Gel-Red was ordered from Beyotime Biotechnology (Shanghai, China). Bovine pancreatic ribonuclease A (RNase A) was obtained from Sigma Aldrich (St. Louis, MO). Hoechst 33342, LysoTracker Red DND-99, Alexa Fluor™ 488 NHS ester (AF488-NHS), 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and the ReadyProbes™ Cell Viability Imaging Kit (blue/green) were ordered from Invitrogen Biotechnology Co., Ltd (Shanghai, China). Human cervical carcinoma cells (HeLa) were obtained from the cell bank of the type culture collection of the Chinese Academy of Sciences (Shanghai, China). All high-performance liquid chromatography (HPLC)-purified oligonucleotides were custom-synthesized by Sangon Biotech and the detailed sequences were as follows: Strand 1 (S1): 5′-GATTC AGACT TAGAC ACGAT CGTTC GACAT TAGCC ATGCT TCAGC CATAA CCTGG GAGCG TACAG CTTGT AAG-3′; Strand 2 (S2): 5′-CCACT ATGCA TTGTG GATGA TACCG CGTAC TATTC TCCAT AAAGT CTGAA TCCTT ACAAG CAGAG TTGAC GGA-3′; Strand 3 (S3): 5′-CGGAG AAGGG CGAAC AGCAT AGTGG TCCGT CAACT CATCT AACTG CTGCG CCGCC GGGAA AATAC TGTAC GGTTA GAGTA CGCTC CCAGG TTATG GCACT AACCT ACCCG TAC-3′; Strand 3′ without the sgc8 aptamer (S3′): 5′-CGGAG AAGGG CGAAC AGCAT AGTGG TCCGT CAACT CAGTA CGCTC CCAGG TTATG GCACT AACCT ACCCG TAC-3′; Strand 4 (S4): 5′-CGGTA TCATC CACAA AATGA ACTGC ATCTC TGATC TAGAA CCTTA GCTGC GCGGA TGAAT GGAGA ATAGT ACG-3′; Strand 5 (S5): 5′-GAAGC ATGGC TAATG TCGAA CGATC GTGTC ACATC CGCGC AGCTA AGGTT CAACC TGGGG GAGTA TTGCG GAGGA AGGT-3′; Strand 6 (S6): 5′-CAGGT ATCTA ACTGC TGCGC CGCCG GGAAA ATACT GTACG GTTAG AAGAT CAGAG ATGCA GTTCA TAGTT CGCCC TTCTC CGGTA CGGGT AGGTT AGATC CGCAA TACTC CCC-3′; Strand 6′ without the sgc8 aptamer (S6′): 5′-CAGGT AAGAT CAGAG ATGCA GTTCA TAGTT CGCCC TTCTC CGGTA CGGGT AGGTT AGATC CGCAA TACTC CCC-3′; FAM/Dabcyl labeled Strand 6* (S6*): 5′-Dabcyl-CAGGT ATCTA ACTGC TGCGC CGCCG GGAAA ATACT GTACG GTTAG AAGAT CAGAG ATGCA GTTCA TAGTT CGCCC TTCTC CGGTA CGGGT AGGTT AGATC CGCAA TACTC CCC-FAM-3′; PTK7 inhibitor: 5′-ATCTA ACTGC TGCGC CGCCG GGAAA ATACT GTACG TTAGA-3′; RNA substrate: 5′-(Cy5)rArArArArUrArArUrArArUrArArArA(BHQ2)-3′; dsDNA: 5′-AATAA GGTAA ACGAA TACGG AATTG CTAAT-3′; 5′-ATTAG CAATT CCGTA TTCGT TTACC TTATT-3′.Apparatus
A Hitachi F-7000 fluorescence spectrometer (Japan) was used to record the fluorescence spectra at room temperature in all experiments. All gel images were obtained using a Bio-Rad Gel Doc XR + System (Bio-Rad, Hercules, CA). The confocal laser scanning microscopy (CLSM) imaging studies and the MTT assays were, respectively, performed on a Zeiss LSM 900 confocal scanning system (Carl Zeiss Co., Ltd Germany) with an objective lens of 63× and a RT 6000 microplate reader.Synthesis of AF488-RNase A
AF488-RNase A was synthesized according previously published protocols.45 In brief, 100 μL of AF488-NHS (10 mg mL−1) in DMSO was added to 1 mL of sodium bicarbonate buffer (0.1 mM and pH 8.3) containing RNase A (10 mg mL−1). The mixture was then incubated at room temperature for 1 h with gently inverting it several times every 10 min to fully mix the two reactants and increase the labeling efficiency. After that, the mixture was purified by using Amicon Ultra Centrifugal Filters (3 k) to collect the AF488-RNase A conjugate, and the concentration of AF488-RNase A was determined by UV-Vis spectroscopy.Native Polyacrylamide Gel Electrophoresis (PAGE)
The freshly prepared samples were mixed with a 6 × DNA loading buffer (40% (v/v) sucrose solution, 0.25% (w/v) xylene cyanol FF and 0.25% (w/v) bromophenol blue) at a volume ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and loaded into native polyacrylamide gels. After the electrophoresis was performed at 100 V in 0.5 × TBE/Mg2+ (10 mM) buffer for 90 min, the gels were stained with Gel-Red and imaged immediately.
:1 and loaded into native polyacrylamide gels. After the electrophoresis was performed at 100 V in 0.5 × TBE/Mg2+ (10 mM) buffer for 90 min, the gels were stained with Gel-Red and imaged immediately.
Preparation of sgc8-HDF
The preparation of sgc8-HDF: sgc8-HDF was prepared by following reported procedures with slight modifications.46,47 The six DNA oligonucleotides S1, S2, S3, S4, S5 and S6 were mixed in an equal final concentration of 1 μM in Tris–HCl buffer (50 mM, 10 mM MgCl2, and pH 8.0) and annealed by heating to 95 °C, followed by cooling to 4 °C within 30 s to form sgc8-HDF.Preparation of RNase A@sgc8-HDF
The open sgc8-HDF precursor structure for capping RNase A was first prepared by the same procedures described above. The DNA sequences of S1, S2, S3, S4 and S6 each at a concentration of 1 μM were mixed in Tris–HCl buffer (50 mM, 10 mM MgCl2, and pH 8.0), heated to 95 °C for 15 min, and quickly cooled down to 4 °C within 30 seconds to form the open sgc8-HDF precursor. Subsequently, concentrated RNase A (0.2 mg mL−1) was added and incubated in Tris–HCl buffer for 15 min at 37 °C, followed by the addition of the encapsulation strand S5 at a final concentration of 1 μM and further incubation at 37 °C for 2 h. RNase A@sgc8-HDF was finally obtained by reconstituting it in Tris–HCl buffer via purification with PAGE gel and the UNIQ-10 Spin Column DNA Gel Extraction Kit.ATP controlled release of RNase A from RNase A@sgc8-HDF
The mixture of the RNA substrate sequence (300 nM, labeled with Cy5 and BHQ2) and RNase A@sgc8-HDF (50 nM, labeled with FAM and Dabcyl) was incubated with/without ATP (5 mM) in Tris–HCl buffer at 37 °C for 1 h. After that, the fluorescence emissions of the solutions were measured at 520 nm (excited at 490 nm) and 670 nm (excited at 649 nm), respectively.Cell culture
HeLa cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C in a humidified incubator containing 5% CO2.Stability assay
The stability of the sgc8-HDF nanostructure (without fluorescent labels) in biological media was monitored using 5% native polyacrylamide gel electrophoresis. Before loading into the gels, the sgc8-HDF samples were incubated in the cell growth medium containing 10% fetal bovine serum (10% FBS) and HeLa cell lysate at 37 °C for 0, 2, 4, 8, 12 and 24 h, respectively. 30-bp dsDNA incubated under the same conditions was used as the control.Cellular uptake and subcellular distribution of AF488-RNase A@sgc8-HDF
HeLa cells were planted on a 35 mm × 35 mm Petri dish (MatTek, U.S.A.) with a 10 mm well at a density of 2 × 104 cells per well and cultured overnight at 37 °C under a 5% CO2 environment. After washing with PBS three times, the cells were incubated with 1 mL of fresh culture medium containing AF488-RNase A, sgc8-HDF, AF488-RNase A@HDF or AF488-RNase A@sgc8-HDF each at a concentration of 100 nM, respectively, for 6 h for cellular uptake studies. The cells were then washed with PBS, and the cell nuclei were stained using the Hoechst 33342 fluorescent DNA dye (1 μg mL−1) at 37 °C for 20 min. After washing with PBS, CLSM imaging was performed. For the subcellular localization study, the cells were incubated with 1 mL of culture medium containing AF488-RNase A@sgc8-HDF (100 nM) for 3 and 6 h, respectively. Then, cellular lysosomes and cell nuclei were respectively stained with LysoTracker Red DND-99 (75 nM) and Hoechst 33342 (75 nM), followed by three times of washing with PBS before CLSM imaging.Cell viability study
The viability of the cells was evaluated using a standard MTT assay. Briefly, HeLa cells were seeded into a 96 well plate at a density of 5000 cells per well and cultured for 24 h under humidified conditions at 37 °C in 5% CO2. Subsequently, the culture medium was removed and the fresh culture medium containing RNase A, sgc8-HDF, RNase A@HDF or RNase A@sgc8-HDF was added and incubated for 6 h at different concentrations from 0.1 μM to 2 μM. After that, the medium was replaced by 100 μL of fresh medium and incubated for 42 h, followed by three times of washing with PBS buffer. Then, 20 μL of MTT reagent (5 mg mL−1) supplemented with fresh media was added into each well and incubated at 37 °C for an additional 4 h. Finally, after removing the MTT, 150 μL of DMSO was added and the cells were shaken on an orbital shaker for 15 min. This was followed by measuring the absorbance of the solutions at 490 using a RT 6000 microplate reader.Cell apoptosis induced by RNase A@sgc8-HDF
For cell apoptosis studies, the ReadyProbes® Cell Viability Imaging Kit (blue/green, Invitrogen Biotechnology Co., Ltd Shanghai, China) was used to determine the viability of cells, according to the manufacturer's protocol. Briefly, HeLa cells (2 × 104 cells per well) were planted in a 35 mm Petri dish and cultured at 37 °C for 24 h in a 5% CO2 incubator, followed by three times of washing with PBS. Subsequently, 1 mL of fresh medium containing RNase A, sgc8-HDF, RNase A@HDF or RNase A@sgc8-HDF at a concentration of 2 μM was, respectively, added and incubated at 37 °C for 6 h. After replacing with fresh medium and incubating the cells for additional 42 h, 2 drops of NucBlue Live and NucGreen Dead reagents were added and incubated for 30 min to stain the cells. Finally, the cells were washed three times with PBS and the apoptotic cells were monitored by CLSM with an objective lens of 20×.Conclusions
We have shown that the targeted and direct delivery of native anticancer proteins into tumor cells can be realized by using aptamer-incorporated sgc8-HDF nanostructures assembled from six programmable ssDNA strands. The RNase A therapeutic can be encapsulated inside the nanostructure and transported into the target cells with high efficiency. The controlled release of native RNase A was specifically triggered by the ATP molecules inside the cells, leading to effective apoptosis of the target cancer cells. By manipulating the lengths of the DNA assembly strands, the cavity dimensions of the nanostructure can be tuned to accommodate proteins with different sizes. In connection with specific cell targeting ligands (e.g., aptamers and peptides) and intracellular responsive species, such a DNA nanostructure can be a versatile and robust delivery platform for the direct delivery of various native proteins into the target cells.Although the successful delivery of native proteins has been realized by bivalent aptamer HDF nanostructures, their in vivo application to avoid macrophage clearance remains a challenge. Future work on the remodeling of the DNA nanostructure to tune its charge and hydrophilicity can potentially address such a challenge, offering our delivery system appealing applications in disease treatments and genome editing.
Data availability
The data that support the findings of this study are available in the main manuscript and also from the authors upon reasonable request.Author contributions
W. Z. and Y. X. conceived the idea. W. Z., F. Y. and S. L performed the experiments. W. Z., Y. X. and R. Y. analyzed the data. W. Z., Y. X. and R. Y. wrote the paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22174112 and 22004010) and Chongqing Research Program of Basic Research and Frontier Technology (cstc2020jcyj-msxmX0478).Notes and references
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- A. M. Scott, J. D. Wolchok and L. J. Old, Nat. Rev. Cancer, 2012, 12, 278–287 CrossRef CAS PubMed.
- D. V. Goeddel, D. G. Kleid, F. Bolivar, H. L. Heyneker, D. G. Yansura, R. Crea, T. Hirose, A. Kraszewski, K. Itakura and A. D. Riggs, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 106–110 CrossRef CAS.
- G. R. Devi, Cancer Gene Ther., 2006, 13, 819–829 CrossRef CAS PubMed.
- S. Pottanam-Chali and B. J. Ravoo, Angew. Chem., Int. Ed., 2020, 59, 2962–2972 CrossRef CAS PubMed.
- B. Liu, M. Ianosi-Irimie and S. Thayumanavan, ACS Nano, 2019, 13, 9408–9420 CrossRef CAS PubMed.
- C. Y. Liu, T. Wan, H. Wang, S. Zhang, Y. Ping and Y. Y. Cheng, Sci. Adv., 2019, 5, eaaw8922 CrossRef CAS PubMed.
- J. L. Lin, Z. K. Wang, Z. Y. Xu, L. Wei, Y. C. Zhang, H. Wang, D. W. Zhang, W. Zhou, Y. B. Zhang, Y. Liu and Z. T. Li, J. Am. Chem. Soc., 2020, 142, 3577–3582 CrossRef CAS PubMed.
- M. Ray, Y. W. Lee, F. Scaletti, R. Q. Yu and V. M. Rotello, Nanomedicine, 2017, 12, 941–952 CrossRef CAS PubMed.
- T. Ho, H. S. Kim, Y. Chen, Y. Li, M. W. LaMere, C. Chen, H. Wang, J. Gong, C. D. Palumbo, J. M. Ashton, H. Kim, Q. B. Xu, M. W. Becker and K. W. Leong, Sci. Adv., 2021, 7, eabg3217 CrossRef CAS PubMed.
- Y. Y. Jiang, H. X. Lu, F. Chen, M. Callari, M. Pourgholami, D. L. Morris and M. H. Stenzel, Biomacromolecules, 2016, 17, 808–817 CrossRef CAS PubMed.
- S. Mandal, G. Mann, G. Satish and A. Brik, Angew. Chem., Int. Ed., 2021, 60, 7333–7343 CrossRef CAS.
- S. Y. Yu, H. Yang, T. D. Li, H. F. Pan, S. L. Ren, G. X. Luo, J. L. Jiang, L. Q. Yu, B. B. Chen, Y. L. Zhang, S. J. Wang, R. Tian, T. Y. Zhang, S. Y. Zhang, Y. X. Chen, Q. Yuan, S. X. Ge, J. Zhang and N. S. Xia, Nat. Commun., 2021, 12, 5131 CrossRef CAS PubMed.
- H. F. Lu, S. X. Xu, Z. C. Guo, M. H. Zhao and Z. Liu, ACS Nano, 2021, 15, 18214–18225 CrossRef CAS PubMed.
- P. Y. Yuan, H. L. Zhang, L. H. Qian, X. Mao, S. B. Du, C. M. Yu, B. Peng and S. Q. Yao, Angew. Chem., Int. Ed., 2017, 56, 12481–12485 CrossRef CAS PubMed.
- C. Carrillo-Carrion, A. I. Bocanegra, B. Arnaiz, N. Feliu, D. C. Zhu and W. J. Parak, ACS Nano, 2019, 13, 4631–4639 CrossRef CAS PubMed.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. W. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. M. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS PubMed.
- M. Wang, J. A. Zuris, F. T. Meng, H. Rees, S. Sun, P. Deng, Y. Han, X. Gao, D. Pouli, Q. Wu, I. Georgakoudi, D. R. Liu and Q. B. Xu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2868–2873 CrossRef CAS PubMed.
- F. Peng, M. I. Setyawati, J. K. Tee, X. G. Ding, J. P. Wang, M. E. Nga, H. K. Ho and D. T. Leong, Nat. Nanotechnol., 2019, 14, 279–286 CrossRef CAS PubMed.
- X. F. Lu, Y. Zhu, R. Bai, Z. S. Wu, W. C. Qian, L. Y. Yang, R. Cai, H. Yan, T. Li, V. Pandey, Y. Liu, P. E. Lobie, C. Y. Chen and T. Zhu, Nat. Nanotechnol., 2019, 14, 719–727 CrossRef CAS PubMed.
- L. F. Zhou, A. R. Chandrasekaran, J. A. Punnoose, G. Bonenfant, S. Charles, O. Levchenko, P. Badu, C. Cavaliere, C. T. Pager and K. Halvorsen, Sci. Adv., 2020, 6, eabc6246 CrossRef CAS PubMed.
- Z. H. Di, J. Zhao, H. Q. Chu, W. T. Xue, Y. L. Zhao and L. L. Li, Adv. Mater., 2019, 31, 1901885 CrossRef PubMed.
- L. Zhou, M. X. Gao, W. L. Fu, Y. X. Wang, D. Luo, K. Chang and M. Chen, Sci. Adv., 2020, 6, eabb0695 CrossRef CAS PubMed.
- L. He, D. Q. Lu, H. Liang, S. T. Xie, C. Luo, M. M. Hu, L. J. Xu, X. B. Zhang and W. H. Tan, ACS Nano, 2017, 11, 4060–4066 CrossRef CAS PubMed.
- L. He, D. Q. Lu, H. Liang, S. T. Xie, X. B. Zhang, Q. L. Liu, Q. Yuan and W. H. Tan, J. Am. Chem. Soc., 2018, 140, 258–263 CrossRef CAS PubMed.
- R. Z. Peng, X. F. Zheng, Y. F. Lyu, L. J. Xu, X. B. Zhang, G. L. Ke, Q. L. Liu, C. J. You, S. Y. Huan and W. H. Tan, J. Am. Chem. Soc., 2018, 140, 9793–9796 CrossRef CAS PubMed.
- K. E. Bujold, J. C. C. Hsu and H. F. Sleiman, J. Am. Chem. Soc., 2016, 138, 14030–14038 CrossRef CAS PubMed.
- M. Y. Li, C. L. Wang, Z. H. Di, H. Li, J. F. Zhang, W. T. Xue, M. P. Zhao, K. Zhang, Y. L. Zhao and L. L. Li, Angew. Chem., Int. Ed., 2019, 58, 1350–1354 CrossRef CAS PubMed.
- Q. B. Mou, Y. Ma, G. F. Pan, B. Xue, D. Y. Yan, C. Zhang and X. Y. Zhu, Angew. Chem., Int. Ed., 2017, 56, 12528–12532 CrossRef CAS PubMed.
- J. B. Liu, L. L. Song, S. L. Liu, Q. Jiang, Q. Liu, N. Li, Z. G. Wang and B. Q. Ding, Nano Lett., 2018, 18, 3328–3334 CrossRef CAS PubMed.
- J. Zhang, Y. Y. Guo, F. Ding, G. F. Pan, X. Y. Zhu and C. Zhang, Angew. Chem., Int. Ed., 2019, 131, 13932–13936 CrossRef.
- T. T. Wu, J. B. Liu, M. M. Liu, S. L. Liu, S. Zhao, R. Tian, D. S. Wei, Y. Z. Liu, Y. Zhao, H. H. Xiao and B. Q. Ding, Angew. Chem., Int. Ed., 2019, 58, 14224–14228 CrossRef CAS PubMed.
- B. Liu, F. Hu, J. F. Zhang, C. L. Wang and L. L. Li, Angew. Chem., Int. Ed., 2019, 131, 8896–8900 CrossRef.
- E. Kim, L. Zwi-Dantsis, N. Reznikov, C. S. Hansel, S. Agarwal and M. M. Stevens, Adv. Mater., 2017, 29, 1701086 CrossRef.
- S. Zhao, F. Y. Duan, S. L. Liu, T. T. Wu, Y. X. Shang, R. Tian, J. B. Liu, Z. G. Wang, Q. Jiang and B. Q. Ding, ACS Appl. Mater. Interfaces, 2019, 11, 11112–11118 CrossRef CAS PubMed.
- P. Chidchob, D. Offenbartl-Stiegert, D. McCarthy, X. Luo, J. N. Li, S. Howorka and H. F. Sleiman, J. Am. Chem. Soc., 2019, 141, 1100–1108 CrossRef CAS PubMed.
- Y. Ma, Z. H. Wang, Y. X. Ma, Z. H. Han, M. Zhang, H. Y. Chen and Y. Q. Gu, Angew. Chem., Int. Ed., 2018, 130, 5487–5491 CrossRef.
- C. M. Platnich, A. A. Hariri, J. F. Rahbani, J. B. Gordon, H. F. Sleiman and G. Cosa, ACS Nano, 2018, 12, 12836–12846 CrossRef CAS PubMed.
- P. Zhang, A. Fischer, Y. Ouyang, J. Wang, Y. S. Sohn, R. Nechushtai, E. Pikarsky, C. Fan and I. Willner, Chem. Sci., 2021, 12, 14473 RSC.
- W. Fu, L. Ma, Y. Ju, J. Xu, H. Li, S. Shi, T. Zhang, R. Zhou, J. Zhu, R. Xu, C. You and Y. Lin, Adv. Funct. Mater., 2021, 2101435 CrossRef CAS.
- C. M. Erben, R. P. Goodman and A. J. Turberfield, J. Am. Chem. Soc., 2007, 129, 6992–6993 CrossRef CAS PubMed.
- G. Kartha, J. Bello and D. Harker, Nature, 1967, 213, 862 CrossRef CAS PubMed.
- A. S. Walsh, H. F. Yin, C. M. Erben, M. J. A. Wood and A. J. Turberfield, ACS Nano, 2011, 5, 5427–5432 CrossRef CAS PubMed.
- K. W. Ren, Y. Liu, J. Wu, Y. Zhang, J. Zhu, M. Yang and H. X. Ju, Nat. Commun., 2016, 7, 13580 CrossRef CAS PubMed.
- S. Navarro, E. Boix, C. M. Cuchillo and M. V. Nogués, J. Neuroimmunol., 2010, 227, 60–70 CrossRef CAS PubMed.
- R. P. Goodman, I. A. T. Schaap, C. F. Tardin, C. M. Erben, R. M. Berry, C. F. Schmidt and A. J. Turberfield, Science, 2005, 310, 1661–1665 CrossRef CAS.
- C. M. Erben, R. P. Goodman and A. J. Turberfield, J. Am. Chem. Soc., 2007, 129, 6992–6993 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02858a |
| This journal is © The Royal Society of Chemistry 2022 |