
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngendering reactivity at group 5-heteroatom multiple bonds via π-loading
Jade I. Fostvedt
 a,
Jocelyne Mendoza
a,
Sacy Lopez-Flores
b,
Diego Alcantar
a,
Robert G. Bergman
*a and
John Arnold
*a
a,
Jocelyne Mendoza
a,
Sacy Lopez-Flores
b,
Diego Alcantar
a,
Robert G. Bergman
*a and
John Arnold
*a
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: arnold@berkeley.edu; rbergman@berkeley.edu
bCollege of Letters & Science, University of California, Berkeley, CA 94720, USA
First published on 29th June 2022
Abstract
In this Perspective, we discuss the strategy of π-loading, i.e., coordination of two or more strongly π-donating ligands to a single metal center, as it applies to promoting reactivity at group 5 transition metal-imido groups. When multiple π-donor ligands compete to interact with the same symmetrically-available metal dπ orbitals, the energy of the imido-based frontier molecular orbitals increases, leading to amplified imido-based reactivity. This strategy is of particular relevance to group 5 metals, as mono(imido) complexes of these metals tend to be inert at the imido group. Electronic structure studies of group 5 bis(imido) complexes are presented, and examples of catalytically and stoichiometrically active group 5 bis(imido) and chalcogenido–imido complexes are reviewed. These examples are intended to encourage future work exploring π-loaded bis(imido) systems of the group 5 triad.
 Jade I. Fostvedt | Jade I. Fostvedt is a recent graduate of the University of California, Berkeley (PhD with Prof. John Arnold, 2022), and previously received her B.S. in chemistry from the University of South Dakota (2017). Her current research interests include design of tantalum systems for small molecule activation, as well as implementation and assessment of programs to promote equity among incoming undergraduate students in chemistry. She has a particular passion for teaching inorganic chemistry. |
 Jocelyne Mendoza | Jocelyne Mendoza is currently an undergraduate in the College of Chemistry at the University of California, Berkeley. She is interested in the synthesis of organic compounds for production of therapeutic methods and drugs, along with contributing to diversity efforts in the field of chemistry. |
 Sacy Lopez-Flores | Sacy Lopez-Flores is an undergraduate student in the College of Letters & Science at the University of California, Berkeley, and is majoring in Molecular and Cell Biology. She is currently studying the transition metals in the 5th group of the periodic table. She is interested in genetics and DNA in the human body system. |
 Diego Alcantar | Diego Alcantar is an undergraduate in the College of Chemistry at the University of California, Berkeley. He is currently studying group 5 transition metals. He has an interest in electrochemistry, particularly in the development of electrochemical cells. |
 Robert G. Bergman | Robert G. Bergman is Professor Emeritus and Professor of the Graduate School, recalled for research and part-time teaching in the College of Chemistry at the University of California, Berkeley. His current research is collaborative, focusing on synthetic and mechanistic studies of stoichiometric and catalytic reactions involving early metal complexes and supramolecular systems. |
 John Arnold | John Arnold is a Professor of Chemistry and Undergraduate Dean in the College of Chemistry at UC Berkeley. He is a graduate of the University of Salford (Applied Chemistry, 1982) and the University of California, San Diego (PhD with Prof. T. D. Tilley, 1986). He carried out postdoctoral work with Professor Sir Geoffrey Wilkinson at Imperial College, London from 1987 to 1988 before a short stint as a Royal Society University Research Fellow (1988 to 1989). He began his independent career at UC Berkeley as an assistant professor in 1989. |
1. Introduction
Imido complexes of early transition metals (groups 3–5) are key intermediates in the stoichiometric and catalytic syntheses of many nitrogen-containing organic compounds, such as pyrroles, carbodiimides, amines, imines, and guanidines.1,2 These transformations are mediated by elementary reactions of small molecule substrates with the metal–imido bond, primarily 1,2-additions of element–hydrogen bonds,3–5 [2 + 2] cycloadditions with unsaturated substrates,2,6,7 and imido group transfer.8 Early transition metal–imido complexes first gained attention for their remarkable reactivity in 1988, when both Wolczanski and Bergman reported the seminal examples of Zr imido-mediated C–H activation.9,10Since then, the chemistry of group 3 and 4 mono(imido) complexes has expanded rapidly,5,6,11–23 while the related chemistry of group 5 mono(imido) complexes has comparatively lagged.24–28 This discrepancy can be attributed to the increasing covalency of the bonding interaction between the group 5 metal nd and the imido 2p orbitals, relative to the more polar and reactive bonds formed between imido ligands and transition metals of groups 3 and 4.29,30 To address this issue, our group and others have employed a π-loading strategy to enhance reactivity at group 5 metal–imido bonds. This strategy involves coordination of two or more strongly π-bonding ligands (i.e., imido, oxo, nitrido, etc.; see Fig. 1 for depictions of common π-bonding ligands.) to a single metal center; these ligands compete to interact with the symmetrically available dπ orbitals on the metal center, destabilizing and polarizing metal-imido groups and thus engendering imido-based reactivity.29–32 The π-loading strategy has found great utility in the realm of early transition metal imido chemistry, in particular through combinations of an imido group and the orbitally related cyclopentadienyl (Cp) ligand.9,14,24,25,33,34
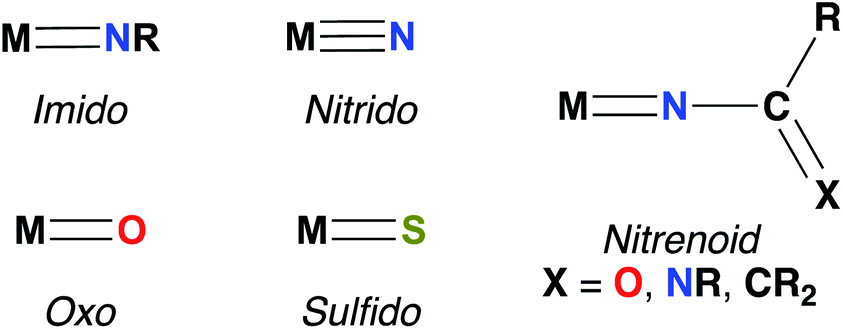 | ||
| Fig. 1 Common π-bonded metal-heteroatom species. | ||
The π-loading strategy has proven particularly successful in expanding imido-based reactivity in group 5 bis(imido) transition metal complexes, relative to their unreactive mono(imido) congeners.29,30 In this Perspective, we communicate how to implement π-loading to increase reactivity at group 5 transition metal–imido bonds, as well as highlight how these design principles can be extended to other π-donor ligands, such as oxos and sulfidos. This Perspective is organized as follows: we first detail the electronic structure of bis(imido) complexes, and outline synthetic routes to bis(imido) complexes. Second, we present examples of reactive π-loaded bis(imido) complexes and emerging research into π-loaded chalcogenido–imido and bis(chalcogenido) complexes. We end by summarizing trends in reactivity and provide predictions of promising classes of complexes for future researchers to target. This Perspective only covers the chemistry of π-loaded group 5 bis(imido) and chalcogenido–imido complexes; the extensive chemistry of group 3–6 mono- and bis(imido) complexes has been reviewed elsewhere.1–4,12,35–37
2. Structure of π-loaded bis(imido) complexes
A commonly employed approach to π-loading in group 5 transition metal complexes involves incorporation of the bent bis(imido) motif. Putative π-loaded tris(imido) complexes of Nb(V) and Ta(V) have also been reported, but crystallographic characterization of these complexes was tenuous, and their reactivity remains to be studied.31 The π-loading strategy is summarized in the qualitative molecular orbital diagram depicted in Fig. 2.29,38,39 On the left side of the diagram, the molecular orbitals (MOs) for the C2v-symmetric [M(NR)2]+ (M = V, Nb, Ta) fragment are shown. The relative ordering of the MOs depends on the angle between the two imido groups; for angles larger than ca. 130°, the energy of the 1b1 MO drops below that of the 1b2 MO.38 In other words, depending on the angle between the imido groups, the HOMO is either the 1b1 or 1b2 MO. That being said, Nimido–M–Nimido (M = V, Nb, Ta) bond angles that lie below the ideal angle of 120° are most common, often approaching 90°, as this geometry permits better orbital overlap between imido-based σ- and π-donor orbitals with metal atomic orbitals.39 In either case, the HOMO (1b1 or 1b2) consists of metal–imido bonding orbitals; this is consistent with the observation that bis(imido) complexes tend to react at one of the two metal-imido groups. Calculations have suggested that the nonbonding LUMO (2a1) is composed of a linear combination of metal dz2 and dx2–y2 orbitals, with large lobes oriented along the y-axis (effectively a dy2 orbital).38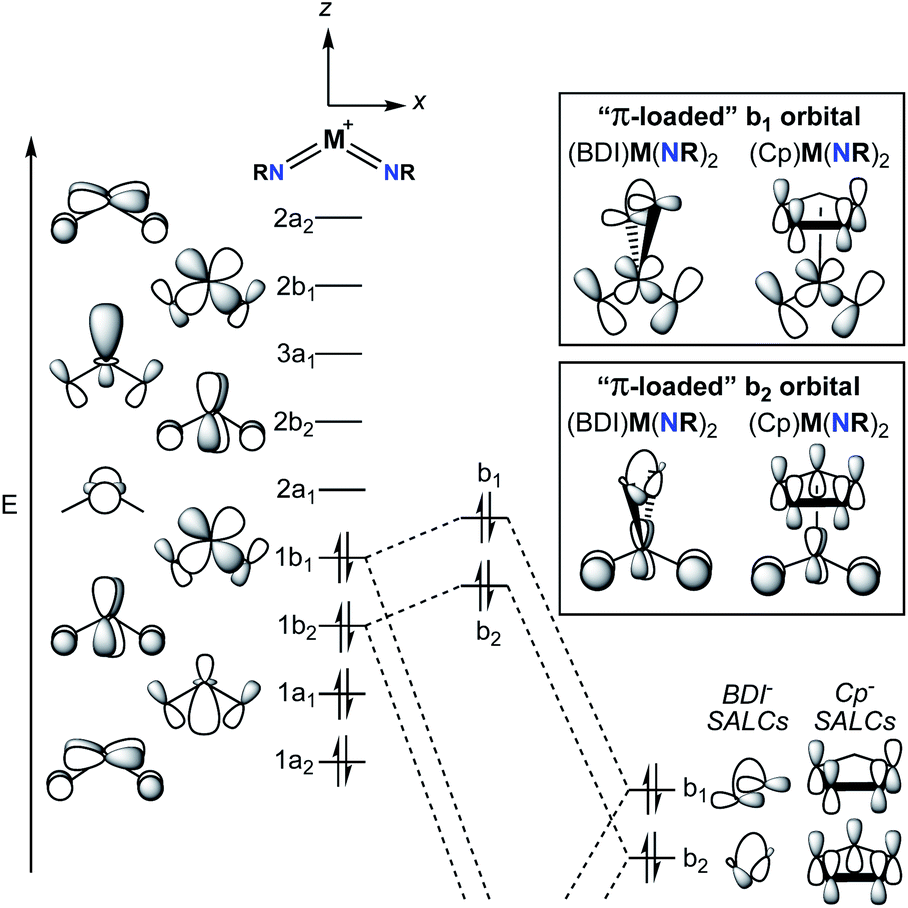 | ||
| Fig. 2 Molecular orbitals for the [M(NR)2]+ fragment (left, M = V, Nb, Ta), selected symmetry adapted linear combinations (SALCs) of molecular orbitals for a cyclopentadienyl (Cp) ancillary ligand and a β-diketiminate (BDI) ancillary ligand (right), and relative energy and symmetry considerations for the combination of the [M(NR)2]+ fragment and a monoanionic, π-donor ligand (center). Relative energies of molecular orbitals are not drawn to scale. | ||
The nonbonding LUMO (2a1), antibonding LUMO+1 (2b2), and antibonding LUMO+2 (3a1) are oriented along a plane bisecting the two imido ligands (the yz plane). These unoccupied orbitals lack any π-bonding component in the xz plane. As such, incorporation of a π-donor ligand at these positions would cause competition between the π-donor ligand and the occupied metal-based π-symmetry MOs (1b1 and 1b2), causing the energy of the HOMO and HOMO-1 (1b1 and 1b2) to increase.29 On the right side of Fig. 2, this phenomenon is depicted for two commonly employed monoanionic π-donor ligands in the early transition metal literature: cyclopentadienyl and β-diketiminate.
Computational investigations into three different group 5 bis(imido) systems provide support for the qualitative molecular orbital diagram shown in Fig. 2. First, the C2v-symmetric, cationic vanadium bis(imido) complexes [V(PEt3)2(NtBu)2]+ and [V(η1-CO)(PEt3)2(NtBu)2]+ were optimized at the B3LYP level of density functional theory (DFT), with the goal of gaining insight into the nature of V → CO π-back bonding in this system.40 The Nimido–V–Nimido bond angle in the optimized CO-adducted complex was found to be 118.4°. In line with this small Nimido–V–Nimido angle, the HOMO is suggested to be b1-symmetric for both complexes, while the HOMO-1 is b2-symmetric. Calculations suggest the π* orbitals of CO interact with the b1-symmetric HOMO of the base-free complex [V(PEt3)2(NtBu)2]+, generating a lower-energy bonding combination (the HOMO of [V(η1-CO)(PEt3)2(NtBu)2]+) and a higher-energy antibonding combination. The b2-symmetric orbitals of [V(PEt3)2(NtBu)2]+ are also involved in π-back bonding to CO in this system. Thus, π-back bonding interactions in this system serve to lower the energy of the imido-based HOMO and HOMO-1, decreasing the effects of π-loading.
Second, the complex (BDI′)Nb(NMe)2 (BDI′ = HC[C(Me)N(2,6-Me2-C6H3)]2) was optimized at the BP86 level of DFT.29 The optimized structure was C2v symmetric, with a Nimido–Nb–Nimido bond angle of 110.8°. The HOMO-1 was suggested to be b2-symmetric and polarized toward the imido nitrogen atoms (11.5% metal character, 53.3% imido nitrogen p-character, 6.8% BDI-ligand p-character). The HOMO of the optimized complex was suggested to be the antibonding combination of the b1-symmetric [Nb(NMe)2]+ fragment and the BDI ligand (5.5% metal character, 47.4% imido nitrogen p-character, 33.1% BDI-ligand p-character). The lower energy bonding combination of the b1-symmetric orbitals was suggested to be primarily composed of BDI-ligand p-character (9.5% metal character, 19.6% imido nitrogen p-character, 52.2% BDI-ligand p-character). The composition of these orbitals and their energies relative to related orbitals in the [Nb(NMe)2]+ fragment suggest that π-loading does indeed increase the energy and polarization of the imido-based HOMO and HOMO−1 in this system.
Third, a computational study was carried out to investigate the bonding in the dimeric bis(imido) complex [(CCC)Ta(NtBu2)·LiI]2 (CCC = 2,6-(BuIm)2-C6H3, Im = imidazole) at the PBEPBE level of DFT.32 The HOMO and HOMO−1 of the optimized complex were suggested to both be π-symmetric imido N-centered (ca. 45%) orbitals; the Nimido–Ta–Nimido bond angle in the optimized complex was not given. Thus, in all three cases presented above, the HOMO is localized on the bis(imido) groups, consistent with imido-based reactivity patterns displayed by these types of complexes.
In addition to computational studies, the electronic structure of vanadium bis(imido) complexes has been investigated via X-ray absorption near-edge structure (XANES) spectroscopy and 13C and 51V nuclear magnetic resonance (NMR) spectroscopy. A systematic study of complexes V(NtBu)2ClL2 (L = PMe3, PEt3, PMe2Ph) and V(NtBu)2(PMe3)2X (X = Cl, I, Br) by La Pierre et al. revealed the electronic structure in these complexes to be dominated by the imido ligands.41 For the family of complexes that varies by identity of the phosphine donor ligand, all three spectroscopic studies suggested that donating ability and steric profile of the variable ligand influences electronic structure: larger cone angles and more electron-rich phosphines led to increased localization of electron density at the imido groups. However, the overall trends in the spectroscopic data of this family of complexes were quite subtle. Furthermore, for the halide family, no discernable trends in the data could be observed. These data stand in contrast to reported 51V NMR trends displayed by the well-studied family of complexes CpV(NR)X2 (X = Me, Cl, Br, I), where the identity of the X ligand has dramatic consequences on the electronic structure of the complex.42–45 Thus, the data reported by La Pierre et al. suggest that the electronic structure in these complexes is dominated by π-bonding interactions with the imido ligands, with little influence from the other ligands present.
Finally, the effects of π-loading manifest in increased bond lengths for M-imido groups in bis(imido) complexes, relative to their mono(imido) congeners. In Fig. 3, we present eight pairs of closely related group 5 bis(imido) and mono(imido) complexes, as well as an oxo-imido complex and a closely related mono(imido) complex. Pairs of complexes were selected such that the oxidation state at the metal center and net charge on the complex are identical in all cases, while the identity of supporting ligand(s) and imido substituent group(s) are as similar as possible. In all cases, the crystallographically determined M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond lengths displayed by π-loaded bis(imido) or oxo-imido complexes are lengthened relative to their mono(imido) congeners, suggesting that π-loading serves to weaken the M-imido interaction and therefore increase reactivity of M-imido groups in these complexes. Indeed, in all but one of the cases presented below, the bis(imido) complex reacts across one of the two imido groups either via 1,2-addition or cycloaddition (no reactivity was reported for Tsai and coworker's (BDI)V(NAd)2 complex46). Of particular note is the Nb oxo-imido complex (BDI)NbO(NAr)DMAP, which reacts via 1,2-addition across the π-loaded oxo group.30 In contrast, none of the mono(imido) complexes presented in Fig. 3 display imido-based reactivity.29,47,49,50,52,54 These opposing reactivity trends serve to drive home our point: the imido moieties in group 5 mono(imido) complexes tend to serve solely as supporting ligands, whereas π-loaded group 5 bis(imido) complexes display reactive imido groups. These reactivity patterns are detailed in the following section.
N bond lengths displayed by π-loaded bis(imido) or oxo-imido complexes are lengthened relative to their mono(imido) congeners, suggesting that π-loading serves to weaken the M-imido interaction and therefore increase reactivity of M-imido groups in these complexes. Indeed, in all but one of the cases presented below, the bis(imido) complex reacts across one of the two imido groups either via 1,2-addition or cycloaddition (no reactivity was reported for Tsai and coworker's (BDI)V(NAd)2 complex46). Of particular note is the Nb oxo-imido complex (BDI)NbO(NAr)DMAP, which reacts via 1,2-addition across the π-loaded oxo group.30 In contrast, none of the mono(imido) complexes presented in Fig. 3 display imido-based reactivity.29,47,49,50,52,54 These opposing reactivity trends serve to drive home our point: the imido moieties in group 5 mono(imido) complexes tend to serve solely as supporting ligands, whereas π-loaded group 5 bis(imido) complexes display reactive imido groups. These reactivity patterns are detailed in the following section.
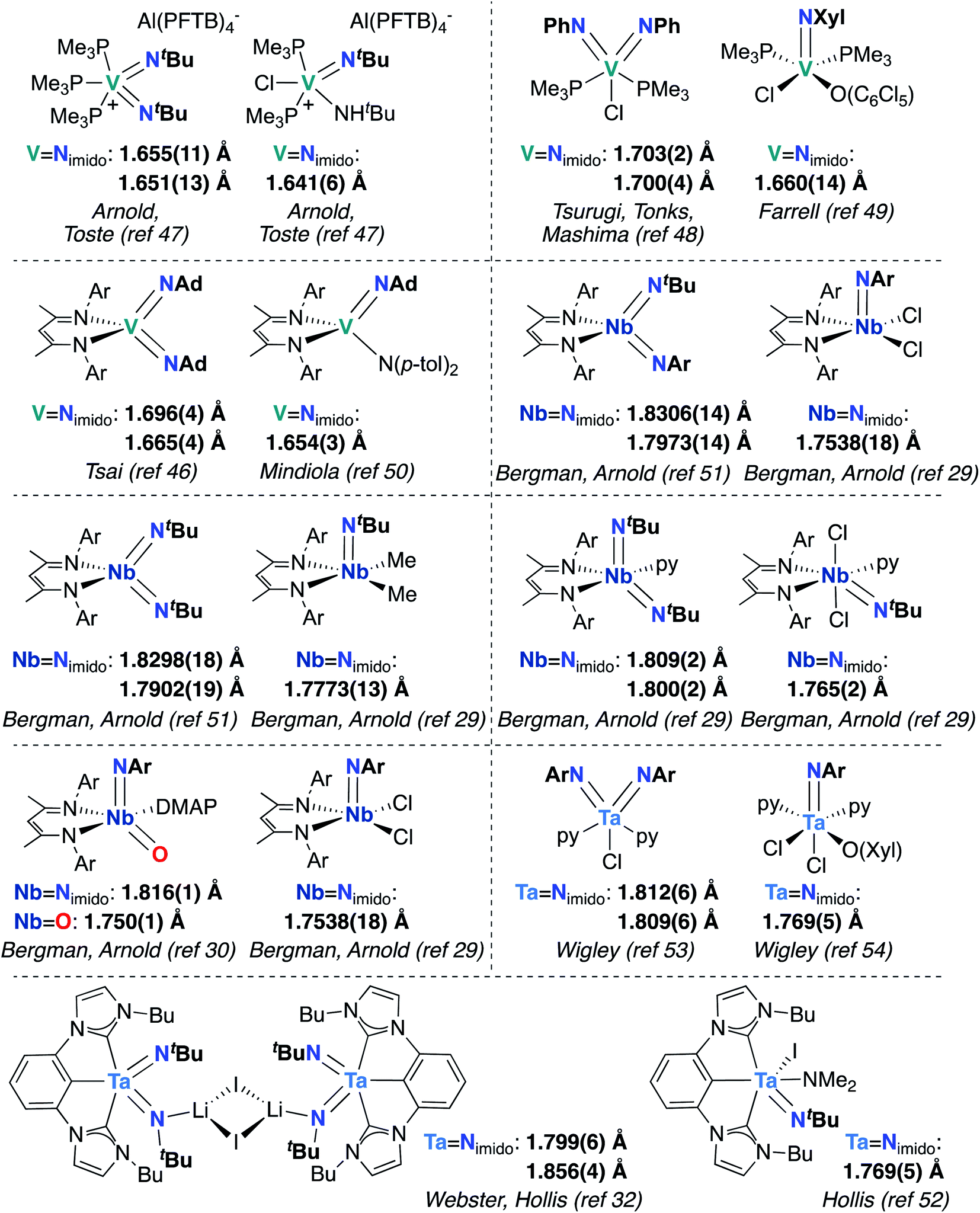 | ||
| Fig. 3 Crystallographically characterized pairs of closely related group 5 bis(imido) (or oxo-imido) and mono(imido) complexes, with MNimido (or MO) bond lengths in Å. Ar = 2,6-diisopropylphenyl. | ||
3. Synthetic routes to group 5 bis(imido) complexes
Bis(imido) complexes are typically accessed from mono(imido) precursors via one of several routes, as outlined in Fig. 4: (1) oxidation of a d2 imido precursor;46,48,55–58 (2) base-induced α-elimination from an imido-amido precursor;39,41,59 and (3) α-hydrogen elimination from an imido-amino-halide precursor.29,32,53,60 The first route necessitates generating a low-valent imido precursor; d2 group 5 transition metals are known to be highly reactive, so if the precursor is to be isolated, bulky supporting ligands are required to enforce kinetic stability.61 Masking of low valence via π-back bonding with arene or CO ligands can also help in isolating stable low valent precursors: for example, Bergman and Arnold have demonstrated facile Nb bis(imido) synthesis via reaction of the Nb(III) complex (BDI)Nb(NtBu)(η6-C6H6) with various organic azides.57 In other cases, a low valent intermediate can be generated in situ: Tsurugi, Tonks, and Mashima were able to generate V(NPh)2Cl(PMe3)2 upon reduction of V(NPh)Cl3 in the presence of azobenzene, followed by addition of PMe3.48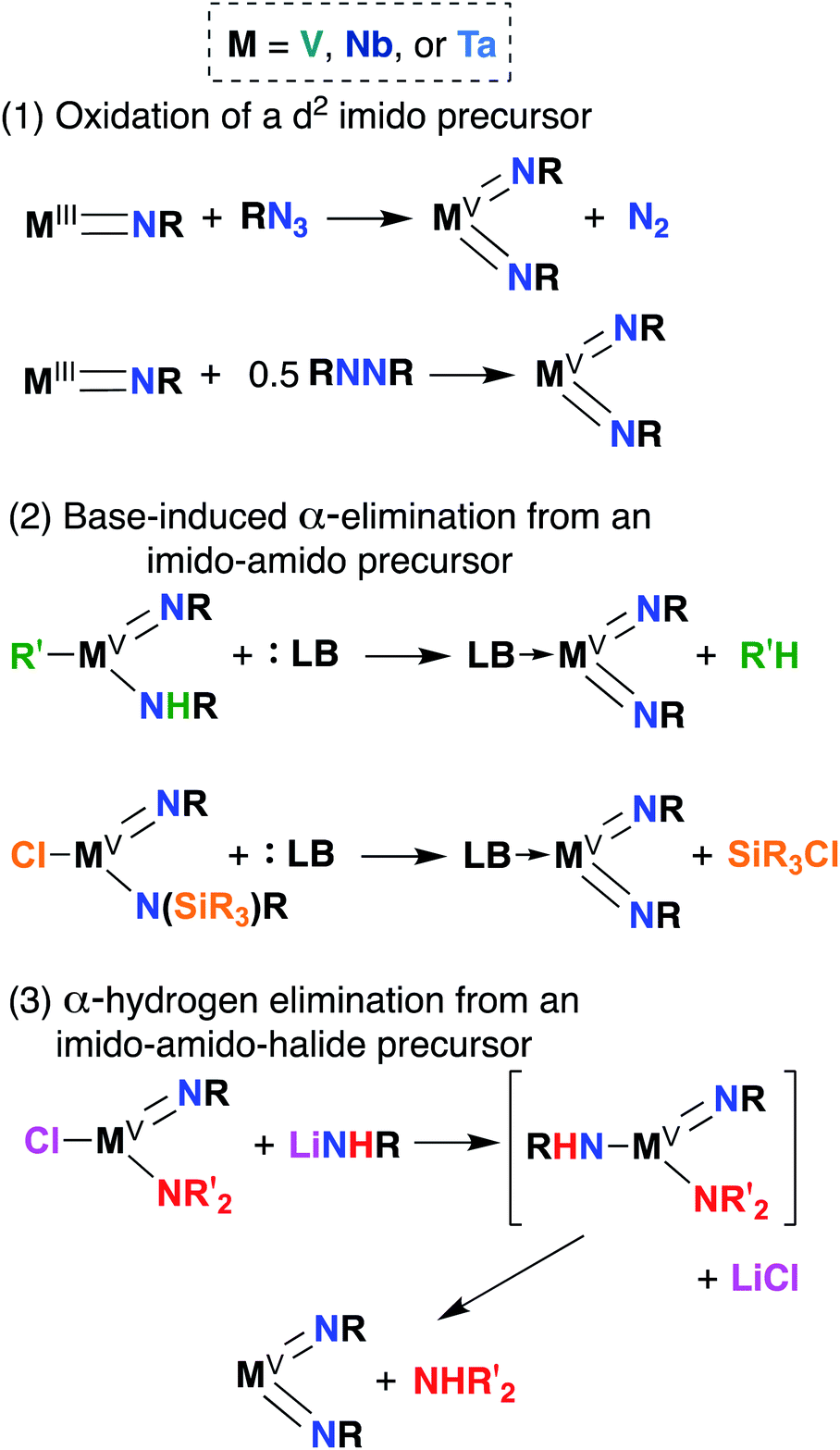 | ||
| Fig. 4 Synthetic routes to group 5 bis(imido) complexes. | ||
The second route depicted in Fig. 4 is commonly employed to access sterically unencumbered bis(imido) complexes supported by labile ligands. For example, Wolczanski found that heating the mono(imido) complex Ta(NHSitBu3)(NSitBu3)Me2 in pyridine or THF led to generation of the bis(imido) complex Ta(NSitBu3)2MeL2 (L = py, THF) and methane.39 Elimination of silyl chlorides can also provide impetus for formation of bis(imido) complexes: La Pierre et al. reported an imido-silylamido precursor V(NtBu)(N(TMS)tBu)Cl2 that liberated TMSCl upon addition of a Lewis base and heat to yield complexes V(NtBu)2ClL2 (L = PMe3, PEt3, PMe2Ph, py).41
Finally, the third route shown in Fig. 4 stands alone in that it can be employed to synthesize bis(imido) complexes directly from amido-halide precursors in a one-pot, multistep synthesis (although this route is also commonly employed starting from imido-amido-halide precursors, as depicted). This route was pioneered by Wigley, who reported synthesis of M(NDipp)Cl(py)2 (M = Nb, Ta) via reaction of two equivalents of LiNH(Dipp) with either Ta(NEt2)2Cl3(OEt2) or [Nb(NEt2)2Cl3]2.53 In the same work, Wigley showed that the Ta bis(imido) product could be also isolated upon reaction of Ta(NDipp)(NEt)2Cl2(py)2 with one equivalent of LiNH(Dipp). As with route 2, route 3 can provide access to sterically unencumbered bis(imido) complexes supported by labile ligands.
4. Reactivity of π-loaded bis(imido) complexes
4.1. Catalytic reactions mediated by group 5 bis(imido) complexes
To date, there have been four reports of catalytic transformations mediated by group 5 metal bis(imido) complexes; all reports suggest key elementary steps that occur across a metal–imido bond. In 2011, La Pierre et al. reported cationic V(V) bis(imido) 1 (Scheme 1), supported by labile trimethyl phosphine ligands, that was capable of mediating Z-selective semihydrogenation of alkynes.47 V bis(imido) complexes are relatively rare, and there are only a handful of other cationic V bis(imido) complex reported to date.40,41 When a solution of 1 was placed under an atmosphere of H2, the authors observed spectroscopic evidence of equilibrium between 1 and hydrido-imido-amido intermediate IM1 (Scheme 1), the product of 1,2-addition of H2 across the V–imido bond. In the presence of alkynes and H2, 1 readily and selectively converted alkynes to Z-alkenes, with 44–100% conversion rates observed for internal aryl, alkyl, and silyl alkynes. Significantly lower yields (10–52%) were observed with terminal aryl and alkyl alkynes, presumably due to competitive addition of the terminal C–H bond across the π-loaded V-imido group.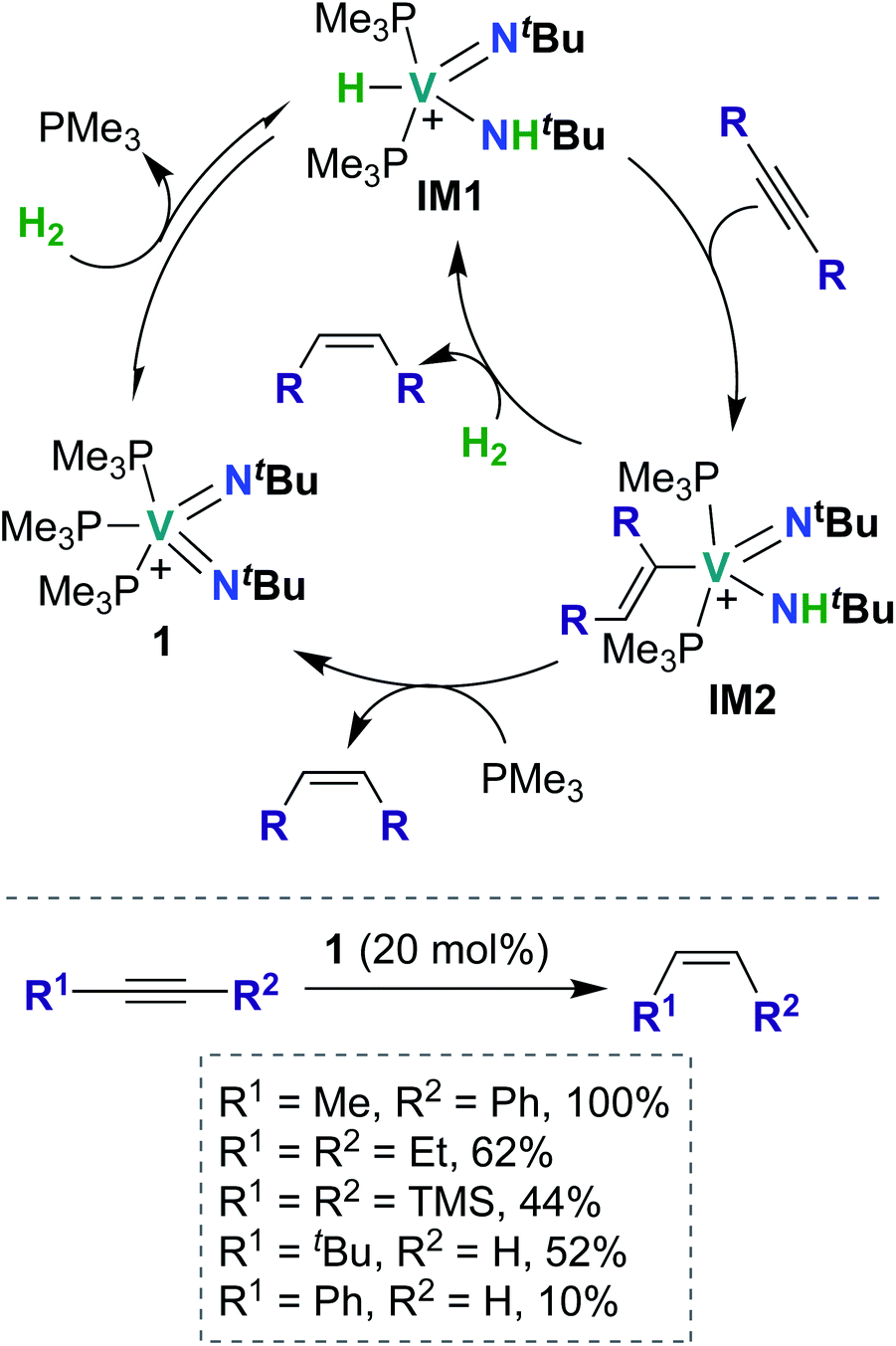 | ||
| Scheme 1 V bis(imido)-catalyzed semihydrogenation of alkynes to Z-alkenes. | ||
Using a combination of synthetic and computational studies, the authors proposed two likely catalytic mechanisms, as outlined in Scheme 1. First, H2 undergoes 1,2-addition across one of the V–imido bonds to generate IM1. One equivalent of alkyne then inserts into the V–hydrido bond to yield imido-amido-alkenyl IM2. Intermediate IM2 can then undergo σ-bond metathesis with another equivalent of H2 to give IM1 (center route) or undergo 1,2-α-NH elimination to yield 1 (left route). A combination of H2/D2 crossover experiments and parahydrogen-induced polarization NMR experiments provided strong support for the path involving 1,2-α-NH elimination (left route): both sets of experiments suggested that a single molecule of H2 is involved in reduction of the alkyne substrate. Three features of catalyst 1 likely contribute to its unique catalytic ability: π-loaded imido groups, positive charge (the neutral analogue to 1 was not found to be catalytically active), and labile supporting ligands.
In 2019, Tonks, Mashima, and co-workers found that a combination of VCl3(THF)3 and N,N-bis(trimethylsilyl)aniline is an efficient catalyst for the [2 + 2 + 1] coupling reaction of alkynes and azobenzenes to yield substituted pyrroles; they obtained strong evidence supporting V bis(imido) intermediate IM4 (Scheme 2) as the catalytically active species.48 Preliminary catalyst screening experiments indicated that formation of bis(imido) V species is important to catalysis: under catalytic conditions, a combination of mono(imido) complex VCl3(NPh) and N,N-bis(trimethylsilyl)aniline produced the desired pyrrole product in quantitative yield, while in the absence of azobenzene, VCl3(NPh) resulted in poor yield (6%) of the desired product. Substrate scope was investigated, as detailed in the bottom of Scheme 2: highest yields were obtained upon combining azobenzene with symmetrical aliphatic alkenes, such as 3-hexyne (74%) and 4-octyne (92%).
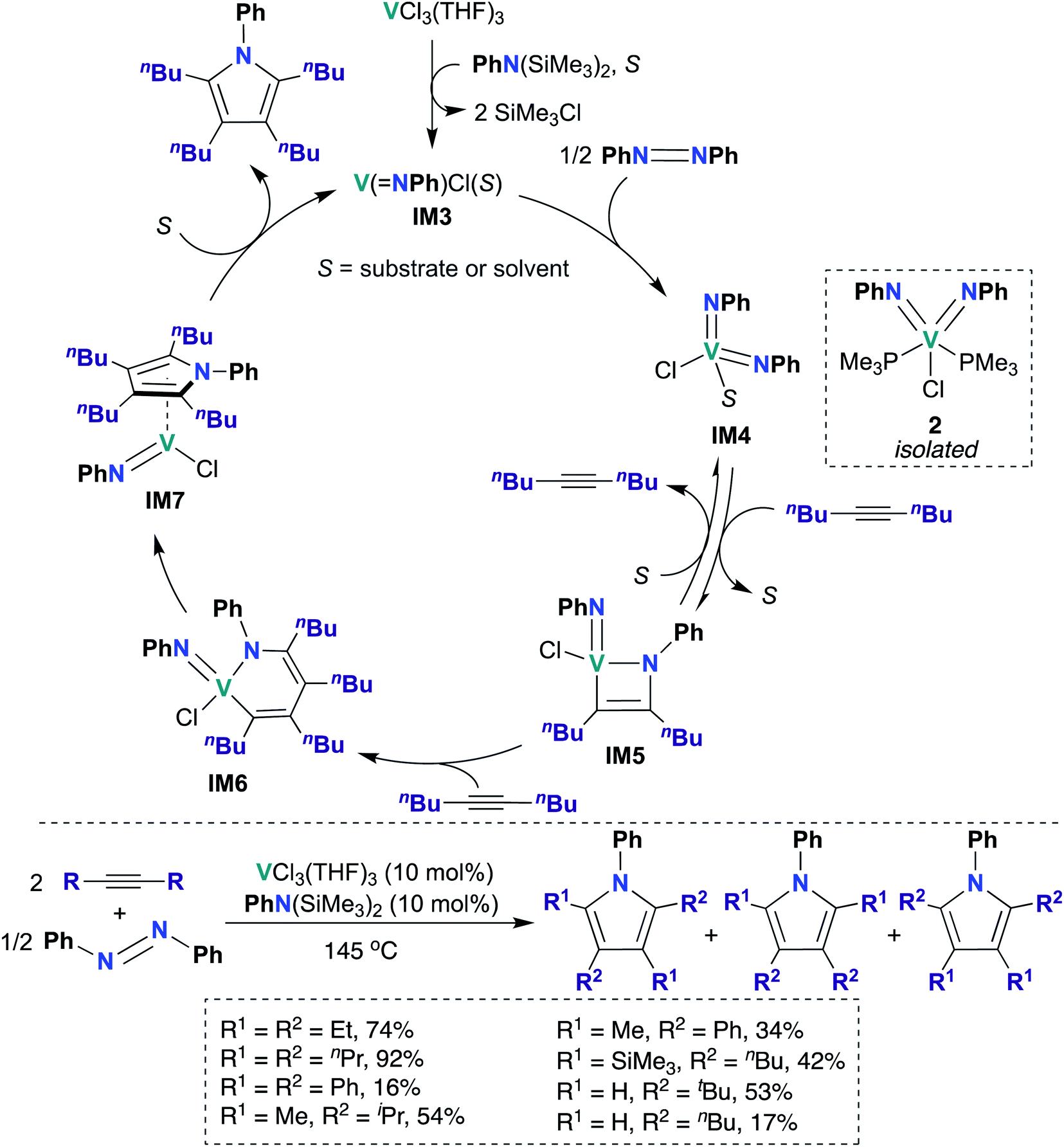 | ||
| Scheme 2 V bis(imido)-catalyzed [2 + 2 + 1] coupling of alkynes and azobenzenes to give multisubstituted pyrroles. | ||
Kinetic studies revealed a second-order rate dependence on VCl3(THF)3, suggesting bimetallic activation of azobenzene by a V species; it was found to be first-order with respect to 5-decyne and 0.8-order with respect to azobenzene. The authors suggested that a V(III) mono(imido) species was responsible for azobenzene cleavage, yielding a V(V) bis(imido) species. To test this hypothesis, V(NPh)Cl3 was treated with an organosilicon reducing reagent62 in the presence of azobenzene and 5-decyne to yield “V(NPh)Cl”, as evidenced by observation of two equivalents of SiMe3Cl by 1H NMR spectroscopy. Upon heating this reaction mixture, 91% conversion to the desired pyrrole product was observed, indicating that similar mono(imido) V(III) and bis(imido) V(V) species may be generated along the catalytic cycle. Additionally, the authors were able to trap a V(V) bis(imido) species by generating V(NPh)Cl in situ as described above, followed by subsequent addition of PMe3 to yield bis(imido) V(V) 2 (Scheme 2). Complex 2 was subsequently demonstrated to be a competent catalyst for the [2 + 2 + 1] coupling reaction; this experiment clearly indicates that a V(V) bis(imido) species is catalytically relevant.
Based on these kinetic and other experimental studies, the authors proposed a mechanism for the coupling of azobenzene and 5-decyne, as outlined in Scheme 2: first, reaction of VCl3(THF)3 with N,N-bis(trimethylsilyl)aniline yields the on-cycle mono(imido) V(III) species IM3, with concomitant generation of two equivalents of SiMe3Cl. Then, bimetallic reductive cleavage of azobenzene furnishes bis(imido) V(V) intermediate IM4, which subsequently reacts with alkyne via [2 + 2] cycloaddition to form azavanadacyclobutene IM5. Next, a second equivalent of alkyne inserts into the V–C bond to form azavanadacyclohexadiene IM6; reductive elimination from IM6 yields the substituted pyrrole product and a mono(imido) V(III) intermediate IM7, which may be stabilized by back-bonding to pyrrole product. Finally, dissociation of pyrrole regenerates mono(imido) V(III) species IM3, thus completing the catalytic cycle.
A 2015 report by Bergman, Arnold, and co-workers detailed a series of BDI-supported Nb bis(imido) complexes capable of mediating stoichiometric nitrene transfer reactions, as well as catalytic nitrene transfer from organic azides and isocyanides to yield asymmetric dialkylcarbodiimines.57 Four-coordinate bis(imido) Nb complex 3 (Scheme 3, top) reacts with isocyanide to give five-coordinate bis(imido) 4-Xyl. When this complex was subjected to heat, either in solution or in the solid state, clean conversion to five-coordinate bis(imido) 5-Xyl was observed, in which the nitrene fragments from one tert-butyl imido group and the coordinated isocyanide were exchanged to generate a complex containing a thermodynamically favored aryl(imido) ligand and coordinated tert-butyl isocyanide. Complexes 5-R (R = 2,6-dimethylphenyl, 4-methoxy-2,6-dimethylphenyl, or 4-methoxyphenyl) could also be generated from 3 upon heating in the presence of the appropriate aryl isocyanide. The authors hypothesized that these stoichiometric transformations occurred via an η2-carbodiimide mono(imido) intermediate, akin to IM8 in the bottom of Scheme 3. This work represents the only example of exchange of nitrene groups to generate a new isocyanide and imido group, which could potentially be leveraged as a new method for incorporation of imido moieties into metal complexes and integration of various substituents into isocyanides.
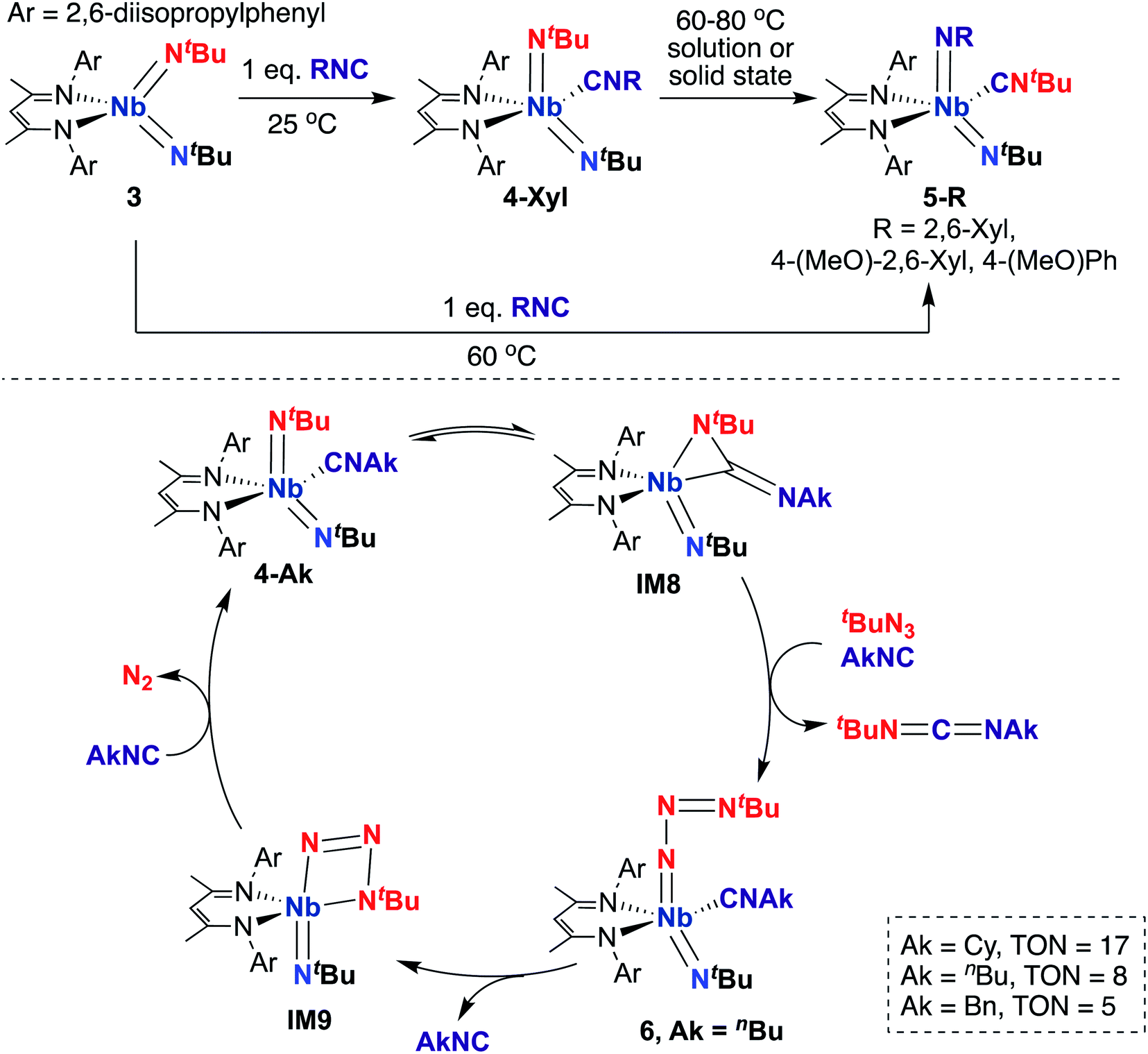 | ||
| Scheme 3 Nb bis(imido)-promoted stoichiometric nitrene metathesis (top) and catalytic nitrene transfer from organic azides and isocyanides to give dialkylcarbodiimides (bottom). | ||
Inspired by these results, the authors considered whether these complexes could carry out catalytic nitrene transfer. They found that addition of excess tert-butyl azide and alkyl isocyanide to 4-Ak (Scheme 3, bottom; Ak = n-butyl, cyclohexyl, or benzyl) resulted in catalytic generation of asymmetric carbodiimides upon heating, as observed by 1H NMR spectroscopy. Notably, they observed build-up of an intermediate by 1H NMR spectroscopy under catalytic conditions. Upon leaving a mixture of 4-nBu, excess tert-butyl azide, and n-butyl isocyanide undisturbed at room temperature for several hours, single crystals of terminal tert-butylazido mono(imido) 6 (Scheme 3, bottom) were obtained; isolated 6 was found to be a competent catalyst for carbodiimide formation. Based on these experimental results, the authors proposed the mechanism outlined in the bottom of Scheme 3. Upon heating, 4-Ak reaches dynamic equilibrium with η2-carbodiimide mono(imido) intermediate IM8, in which coordinated isocyanide has inserted into one of the two Nb-imido groups. Then, IM8 eliminates asymmetric alkylcarbodiimide, coordinates isocyanide, and reductively binds organic azide to generate terminal azido 6. Next, coordinated isocyanide is released to open a binding site at the metal center, allowing generation of azaniobacycle intermediate IM9, which subsequently liberates N2 and binds isocyanide to regenerate 4-Ak and complete the catalytic cycle.
In 2016, Hollis, Webster, and co-workers reported Ta bis(imido) 7 (Scheme 4), supported by a tridentate, monoanionic pincer ligand with two N-heterocyclic carbene donor groups, that is capable of catalytically mediating intermolecular transfer hydrogenation of α,ω-aminoalkenes to yield cyclic imines, cyclic amines, and hydrogenated substrate.32 Upon heating 7 in the presence of various α,ω-aminoalkenes (Scheme 4, bottom), complete conversion to a distribution of three products was observed: a cyclic imine, the product of oxidative amination; a monosubstituted amine, the product of reductive hydrogenation; and a smaller amount of a cyclic amine, the product of intramolecular hydroamination.
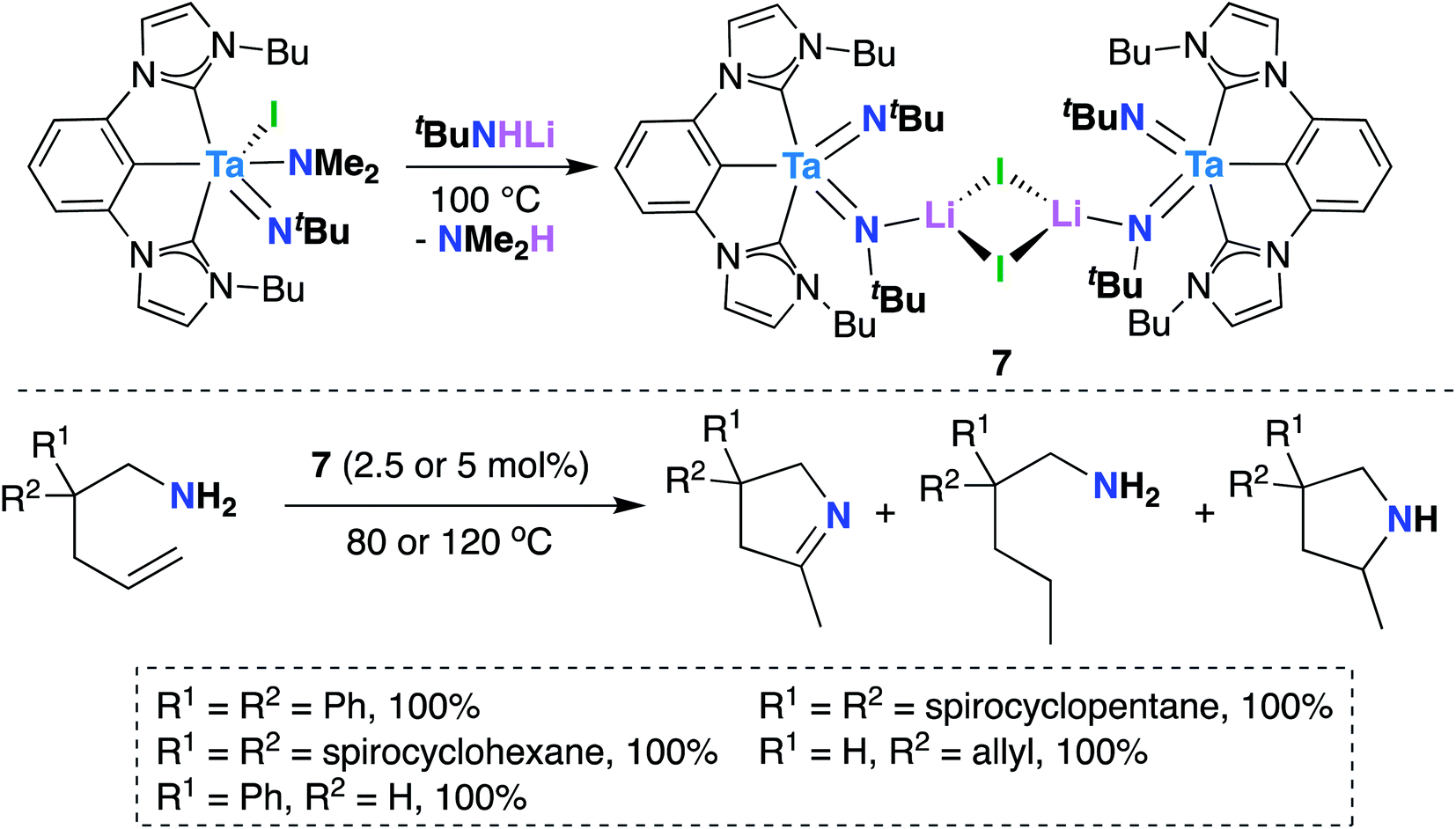 | ||
| Scheme 4 Ta bis(imido)-catalyzed oxidative amination of alkenes to yield cyclic imines, cyclic amines, and hydrogenated substrate. | ||
Varying reaction conditions led to different product distributions, with shorter reaction times favoring the oxidative amination product. In line with this experimental observation, a computational study suggested that the oxidative amination product is favored kinetically, while hydrogenated substrate is the thermodynamic product.63 Secondary amines did not react under catalytic conditions, suggesting that 7 initially reacts with the substrate via imido ligand exchange to form a Ta-imidoalkene species. Based on these experimental and computational investigations, the authors propose a possible, albeit complex, mechanistic pathway. A key elementary step in the transformation involves intramolecular [2 + 2] cycloaddition across Ta-imido groups to give an azatantallacyclobutane intermediate.
4.2. Stoichiometric reactivity of group 5 bis(imido) complexes
While reports of group 5 bis(imido)-catalyzed processes are relatively rare and have only been reported within the last decade, examples of stoichiometric reactivity across a π-loaded group 5 metal–imido bond are more numerous, particularly for Nb, with reports dating back to the early 1990s. Reactivity patterns include 1,2-additions, reductive eliminations, and overall [2 + 2], [2 + 4], and [3 + 2] cycloadditions and retro-cycloadditions. Below, we outline these examples of stoichiometric reactivity, starting with V and moving to Nb and then to Ta.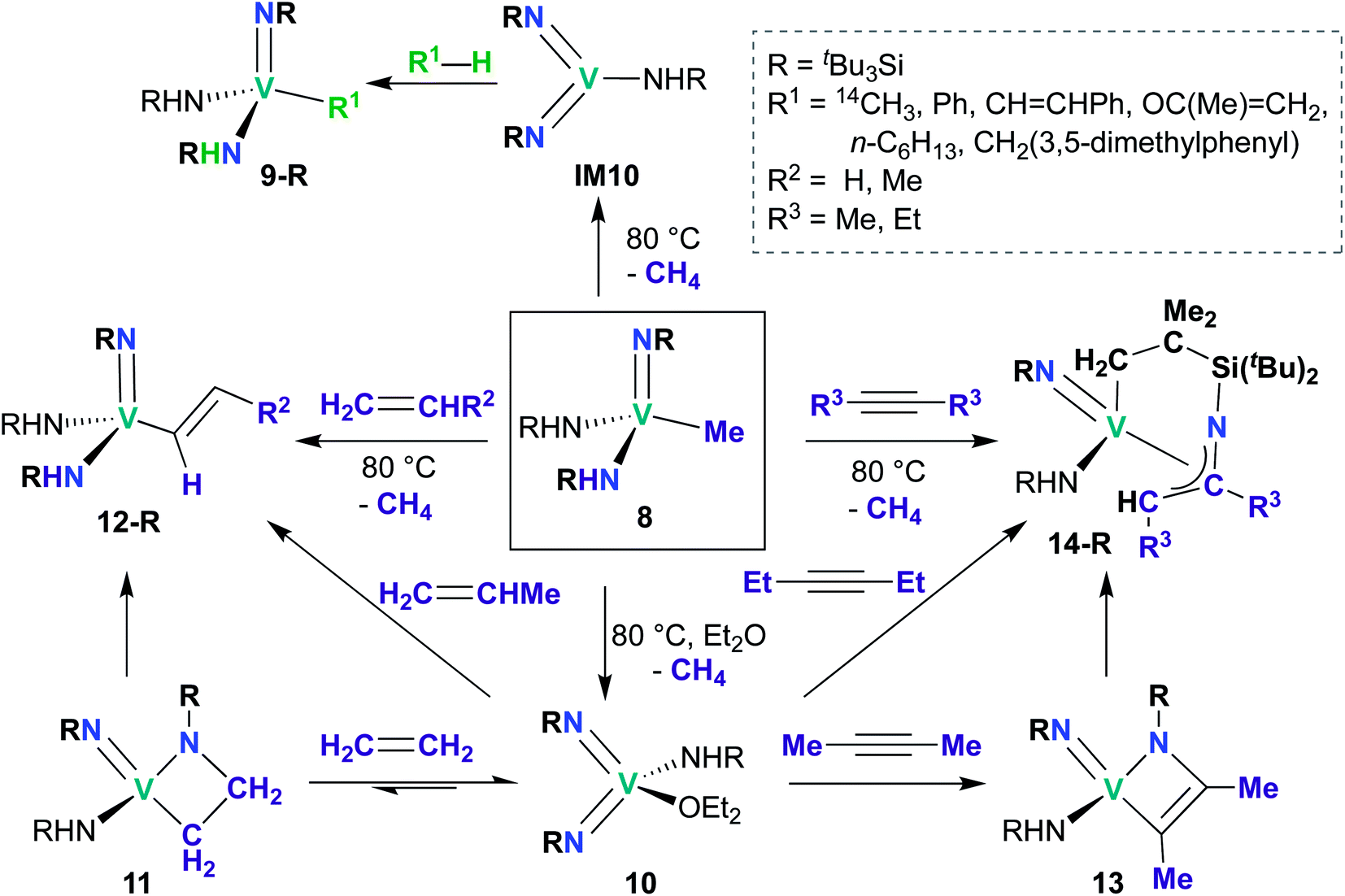 | ||
| Scheme 5 1,2-Addition and [2 + 2] cycloaddition reactivity of V bis(imido) complexes. | ||
A subsequent report detailed [2 + 2] cycloadditions of imido-amido 8 and bis(imido) 10 (Scheme 5).59 Mono(imido) 8 reacts with alkenes to yield alkenyl complexes 12-R (R = H, Me), and with alkynes to yield η3-azallyl complexes 14-R (R = Me, Et). These reactions presumably proceed through bis(imido) intermediate IM10, which subsequently undergoes [2 + 2] cycloaddition of alkene or alkyne across an Nb-imido bond, followed by isomerization to yield the final product. Products 12-R and 14-R could also be synthesized via addition of alkene or alkyne to base-stabilized bis(imido) 10; vanadaazacyclobutane 11 and vanadaazacyclobutene 13 could be isolated from these reactions.
The third and final example of stoichiometric V bis(imido) reactivity was published in 2010 by Tsai and co-workers, who synthesized a series of V bis(imido) complexes, (BDI)V(NR)2 (R = p-tolyl, 1-adamantyl, SiMe3) and observed that trimethylsilyl analogue 15 (Scheme 6) underwent isomerization to the mixed 2,6-diisopropylphenylimido-trimethylsilylimido 16 upon heating.46 Kinetic studies suggested that this transformation proceeds intramolecularly via the proposed transition state depicted in Scheme 6.
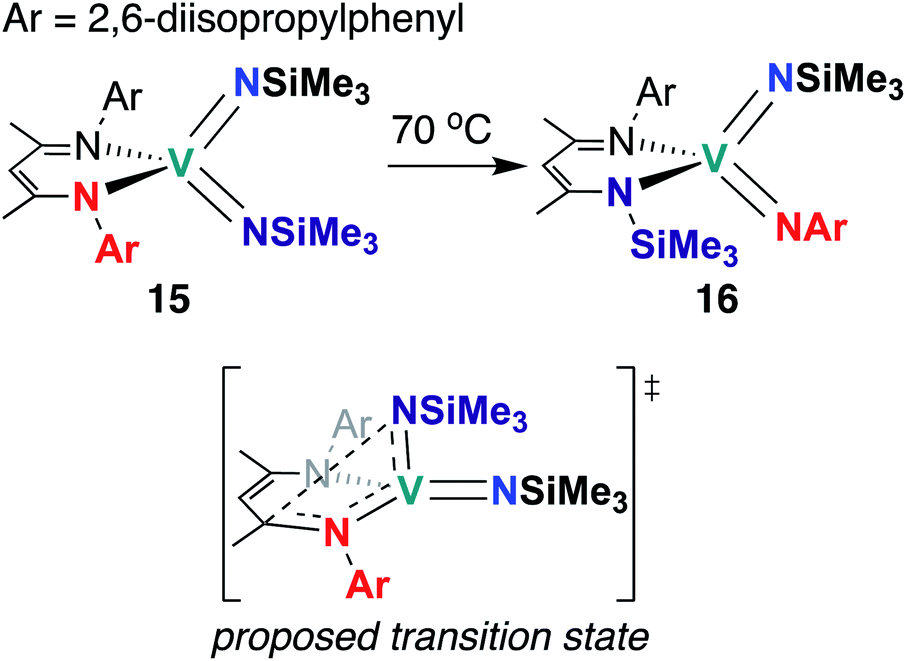 | ||
| Scheme 6 Intramolecular ligand insertion reactivity of a V bis(imido) complex. | ||
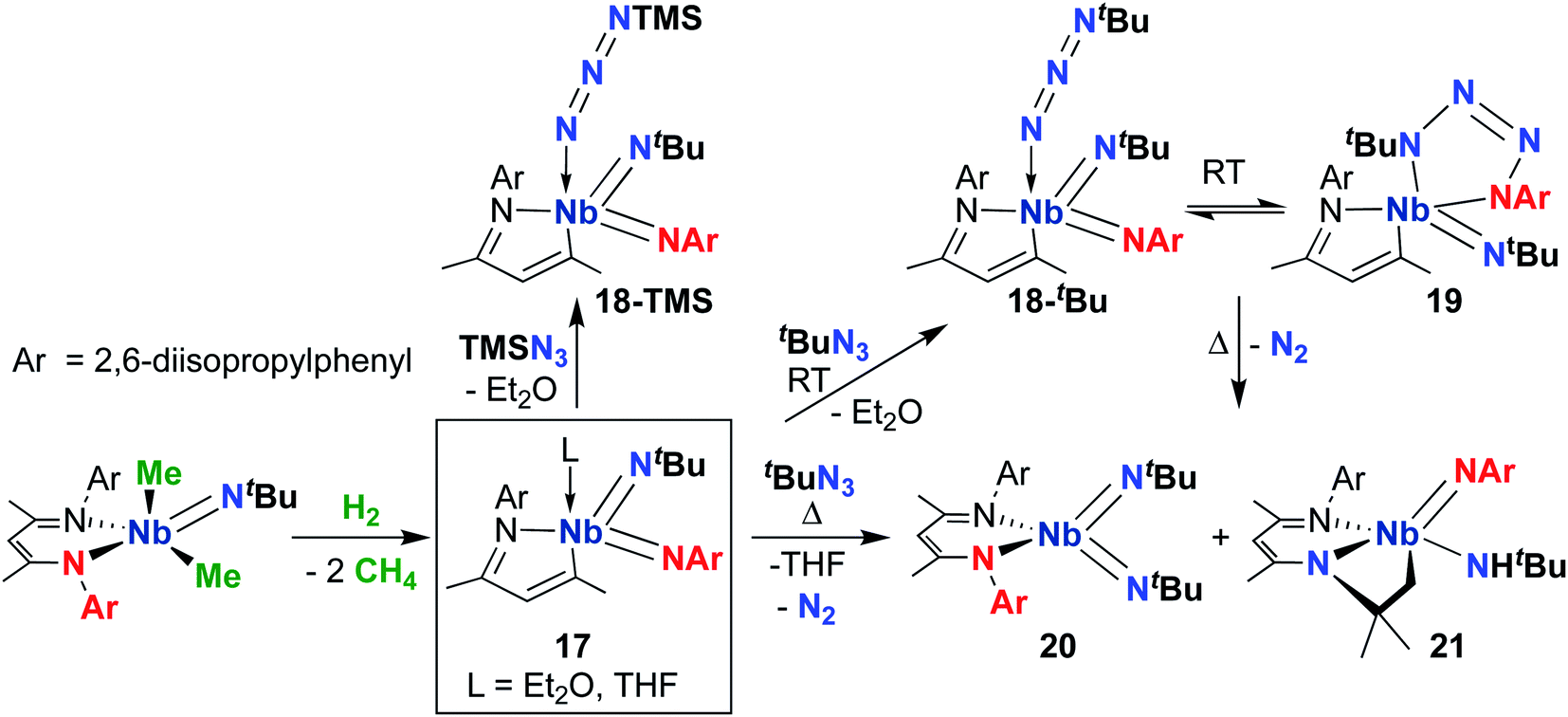 | ||
| Scheme 7 Reactivity of a MAD-supported Nb bis(imido) complex with organic azides. | ||
Focusing on the MAD system first, a 2014 report detailed synthesis of MAD-supported Nb bis(imido) 17 (Scheme 7) from reaction of a BDI-supported mono(imido) dimethyl complex with H2.51 The presumed low-valent intermediate, (BDI)Nb(NtBu), is unstable and reductively cleaves the BDI ligand to form a high-valent bis(imido) complex. Intriguingly, 17 reacted with organic azides to give bis(imido) complexes 18-R (R = trimethylsilyl, tert-butyl), which featured a rare example of a terminally-bound, unreduced azide ligand. A variable temperature NMR study revealed that tert-butyl derivative 18-tBu exists in dynamic equilibrium with niobatetrazene 19, the product of [3 + 2] cycloaddition of an organic azide across a π-loaded Nb-imido group; such cycloadditions are rare in the early transition metal literature.65–67 Remarkably, heating this equilibrating mixture to temperatures above 35 °C resulted in unprecedented reformation of the BDI ligand, yielding complexes 20 and 21 in an 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 ratio. Complex 21 presumably arose from a four-coordinate bis(imido) complex, which undergoes C–H activation across the Nb-(tert-butyl)imido group. The remarkable reactivity of bis(imido) 20 is reported in subsequent publications (see below).
:15 ratio. Complex 21 presumably arose from a four-coordinate bis(imido) complex, which undergoes C–H activation across the Nb-(tert-butyl)imido group. The remarkable reactivity of bis(imido) 20 is reported in subsequent publications (see below).
A 2015 publication detailed cycloaddition reactivity of MAD-supported bis(imido) 17 with unsaturated substrates (Scheme 8).68 Addition of acetylene or hex-3-yne to 17 resulted in formation of complexes 22 and 23, each bearing an eight-membered metallacycle composed of the MAD ligand, an imido group, and a substrate C–C multiple bond. DFT calculations suggested this transformation involves initial [2 + 2] cycloaddition of alkyne across one of the Nb-imido moieties to yield an azaniobacyclobutene intermediate, followed by conjugative addition of the newly formed Nb–C bond to the MAD ligand. The authors suggested that the observed regioselectivity (i.e., reactivity occurs at either the (tert-butyl)imido or (2,6-diisopropylphenyl)imido group) arises due to different activation energies for the conjugative addition step. Similarly, 17 reacted with norbornene to give 24, presumably via [2 + 2] cycloaddition to yield an azaniobacyclobutane intermediate. Complex 17 was also observed to react with oxygen-containing unsaturated substrates: addition of (E)-4-chloro-chalcone resulted in stereoselective [4 + 2] cycloaddition across the Nb-(tert-butyl)imido group to give 25. Reaction of 17 with para-bromo-benzaldehyde resulted in formation of dimeric bis(µ-oxo)-imido 26, which likely forms via a [2 + 2] cycloaddition–cycloreversion sequence.
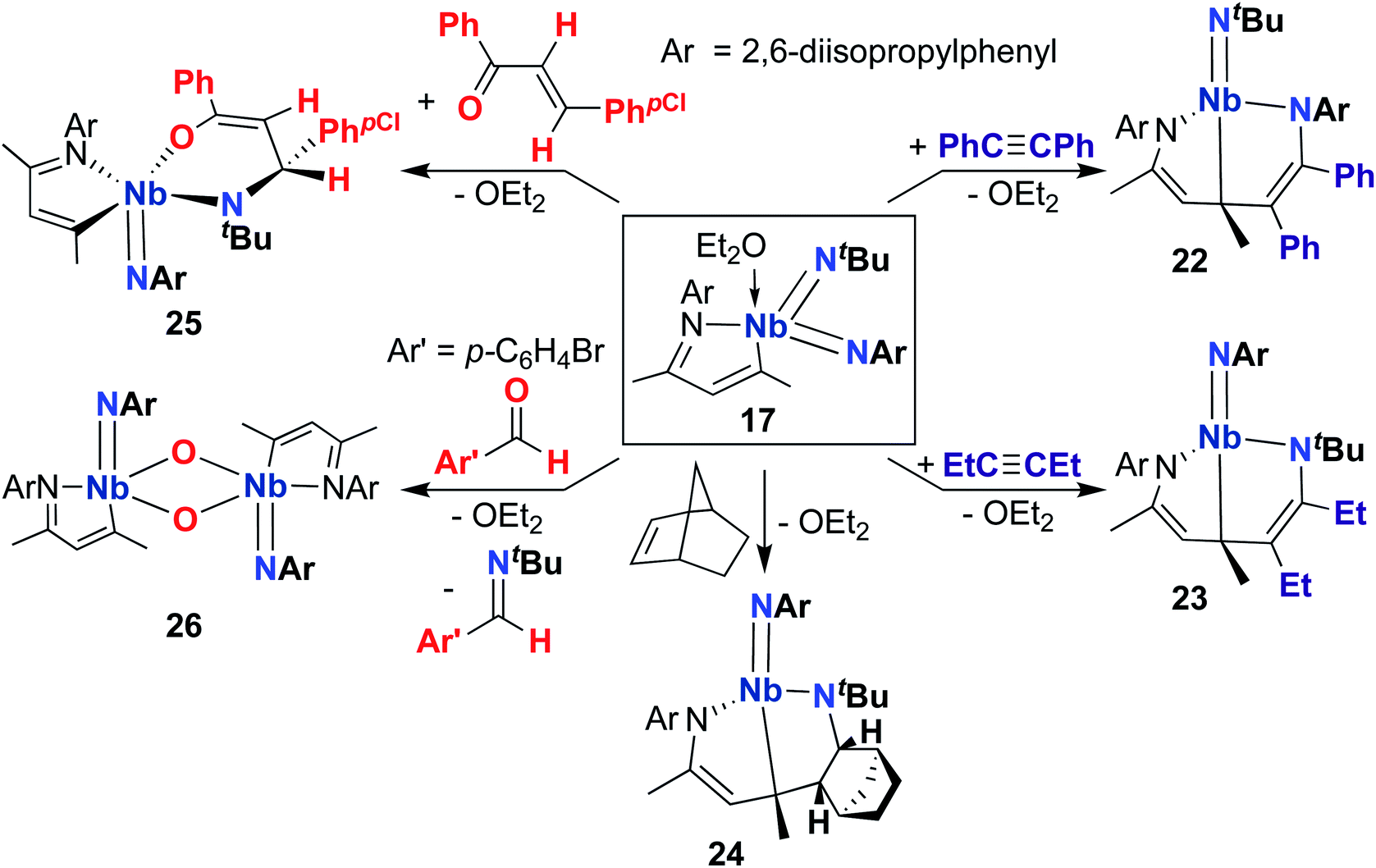 | ||
| Scheme 8 [4 + 2] and [2 + 2] cycloaddition reactivity of a MAD-supported Nb bis(imido) complex. | ||
From 2010–2022, reactivity of BDI-supported Nb bis((tert-butyl)imido) 20 was investigated and several synthetic routes to 20 were discovered, as outlined in Scheme 9. Complex 20 can be accessed via pyridine abstraction from five-coordinate Nb bis(imido) 27,29 or via addition of organic azide to MAD-supported bis(imido) 17,51 as described above. In 2016, Kriegel et al. reported an efficient synthesis for bis(imido) complexes (including 20, as well as two asymmetric bis(imido) complexes) involving addition of organic azides to low-valent Nb precursor 28.57,69 This route proved to be the highest-yielding and cleanest. With an abundant supply of 20 on hand, Arnold, Bergman, and several co-workers proceeded to investigate the remarkable reactivity of this complex.
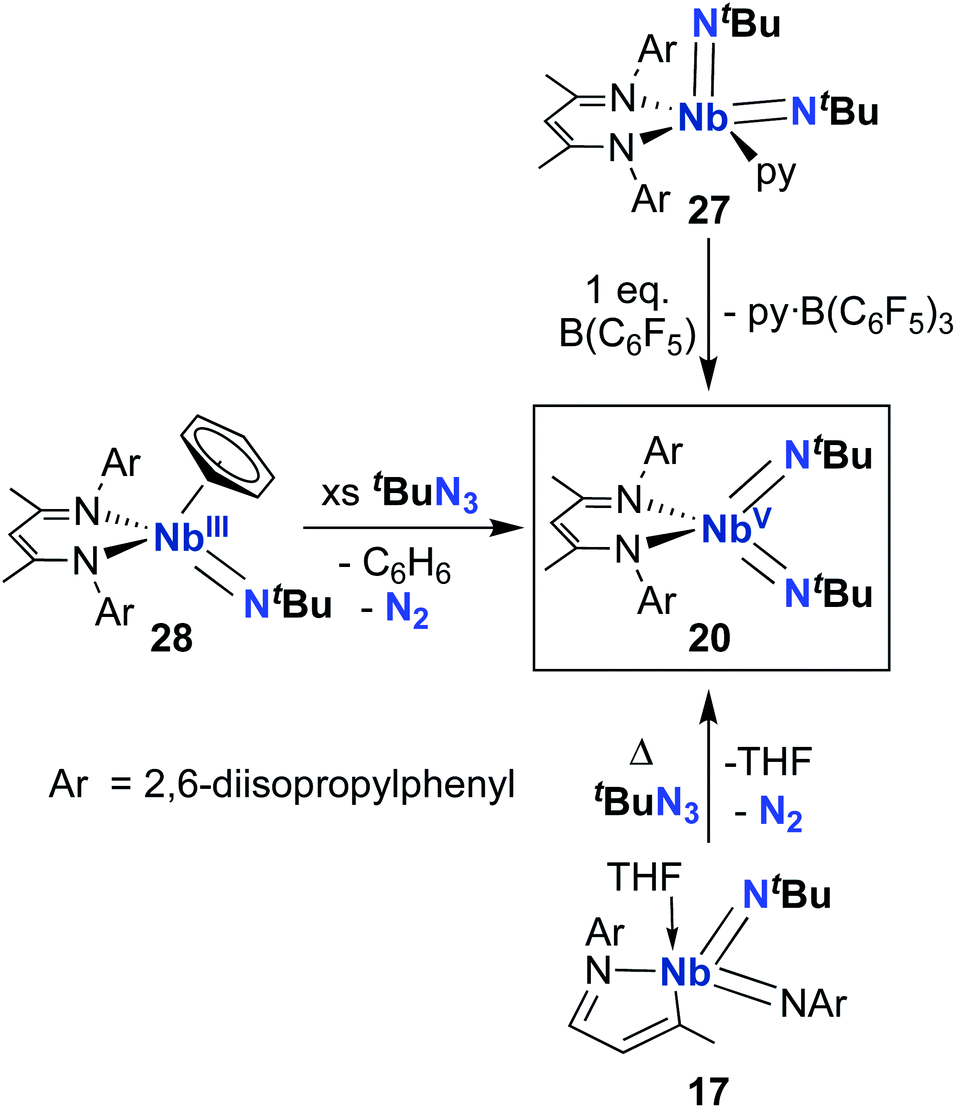 | ||
| Scheme 9 Synthetic routes to the symmetric Nb bis(imido) complex 20. | ||
Complex 20 was observed to mediate catalytic nitrene transfer of alkyl-substituted isocyanides and stoichiometric nitrene metathesis with aryl-substituted isocyanides, as outlined above in Scheme 3, Section 4.1.57 Complex 20 was also observed to react with three equivalents of benzyl isocyanide to yield 29 (Scheme 10), the product of benzyl group transfer from one isocyanide to another to yield a terminally-bound cyanide ligand and an η2-iminoacyl group.57 Remarkably, 20 was observed to react with six equivalents of n-butyl isocyanide to yield 30 (Scheme 10); this transformation involves formation of four new C–C bonds and a C–N bond to generate a heavily substituted κ2-N,N-pyrrolediamido ligand. To date, this represents the only reported example of such reactivity across the entire transition metal block, highlighting the unique chemistry that can be accessed with group 5 bis(imido) complexes.
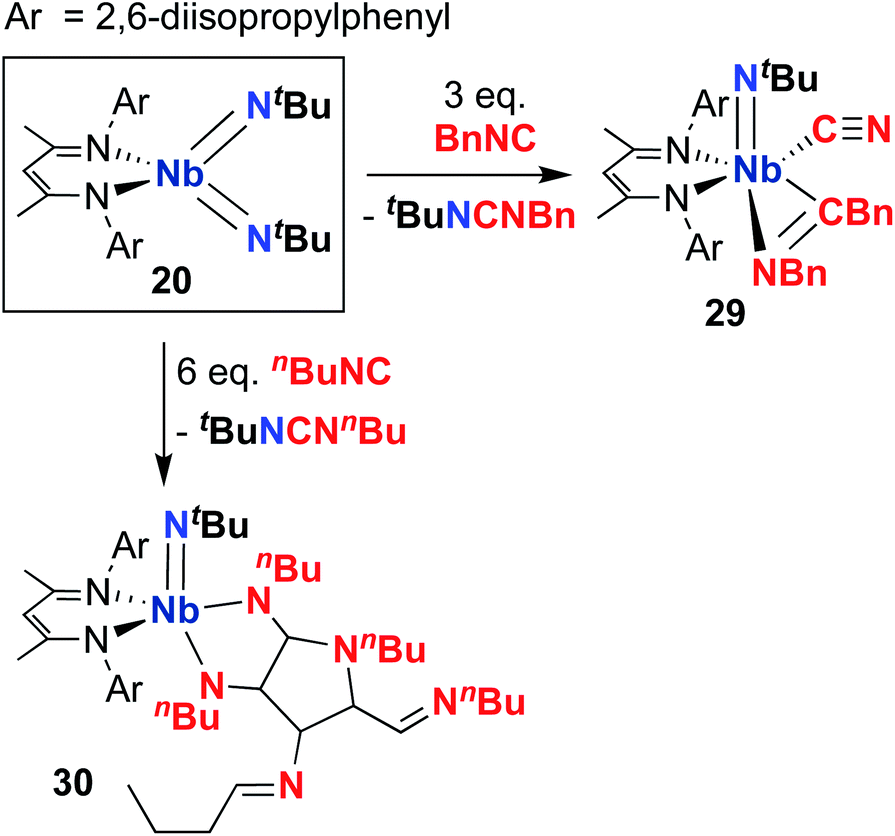 | ||
| Scheme 10 Reactivity of Nb bis(imido) complex 20 with excess alkyl-substituted isocyanide. | ||
Complex 20 was also reported to be active toward 1,2-addition across one of the two Nb-imido double bonds.30 Exposing a solution of 20 to an H2 atmosphere established a dynamic equilibrium between 20 and amido-imido-hydrido 31 (Scheme 11). The ratio of 31 to 20 varied based on H2 pressure, with Keq ≈ 8.0 at 20 °C under 1 atmosphere of H2. Placing crystals of 31 under vacuum or exposing a solution of 31 to an N2 atmosphere resulted in liberation of H2 and regeneration of bis(imido) 20. Complex 20 was also observed to react with primary silanes, pinacolborane, tert-butylacetylene, and triphenylmethanethiol via 1,2-addition across an Nb-imido double bond, resulting in amido-imido products 32-R, 33, 34, and 35, respectively (Scheme 11). In all cases, the more electronegative substrate fragment was observed to bind to the electropositive Nb center: silanes and boranes resulted in Nb-hydrides, while more acidic substrates led to imido protonation. In the case of amido-imido-thiolate 35, the BDI ligand binds in a κ1-fashion, presumably to relieve steric clash with the triphenylmethanethiolato ligand. The related mono(imido) complexes (BDI)Nb(NtBu)Cl2 and (BDI)Nb(NtBu)F2 were not observed to react with H2, silanes, boranes, acetylene, or thiols,29 suggesting that π-loading enhances reactivity at imido groups in this system.
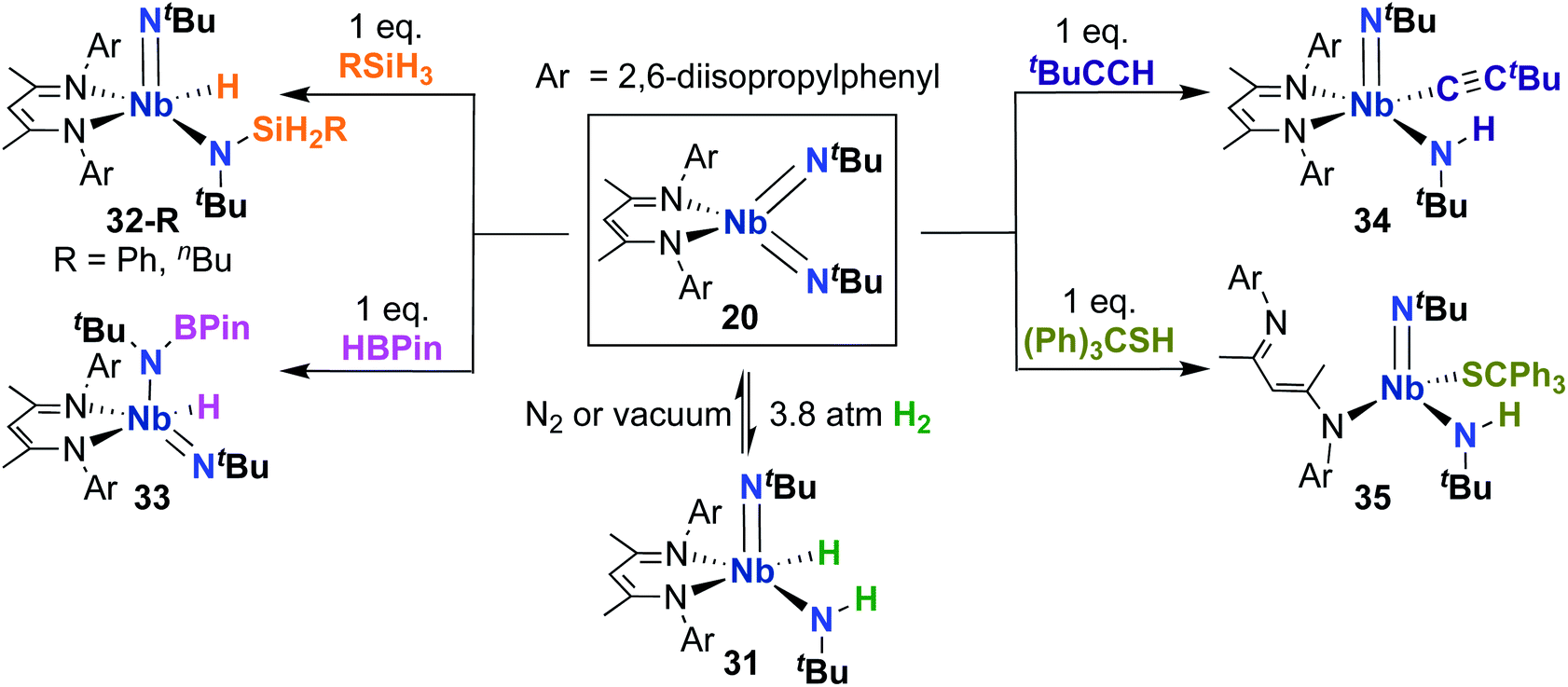 | ||
| Scheme 11 1,2-Addition reactivity of Nb bis(imido) complex 20 toward element-hydrogen bonds. | ||
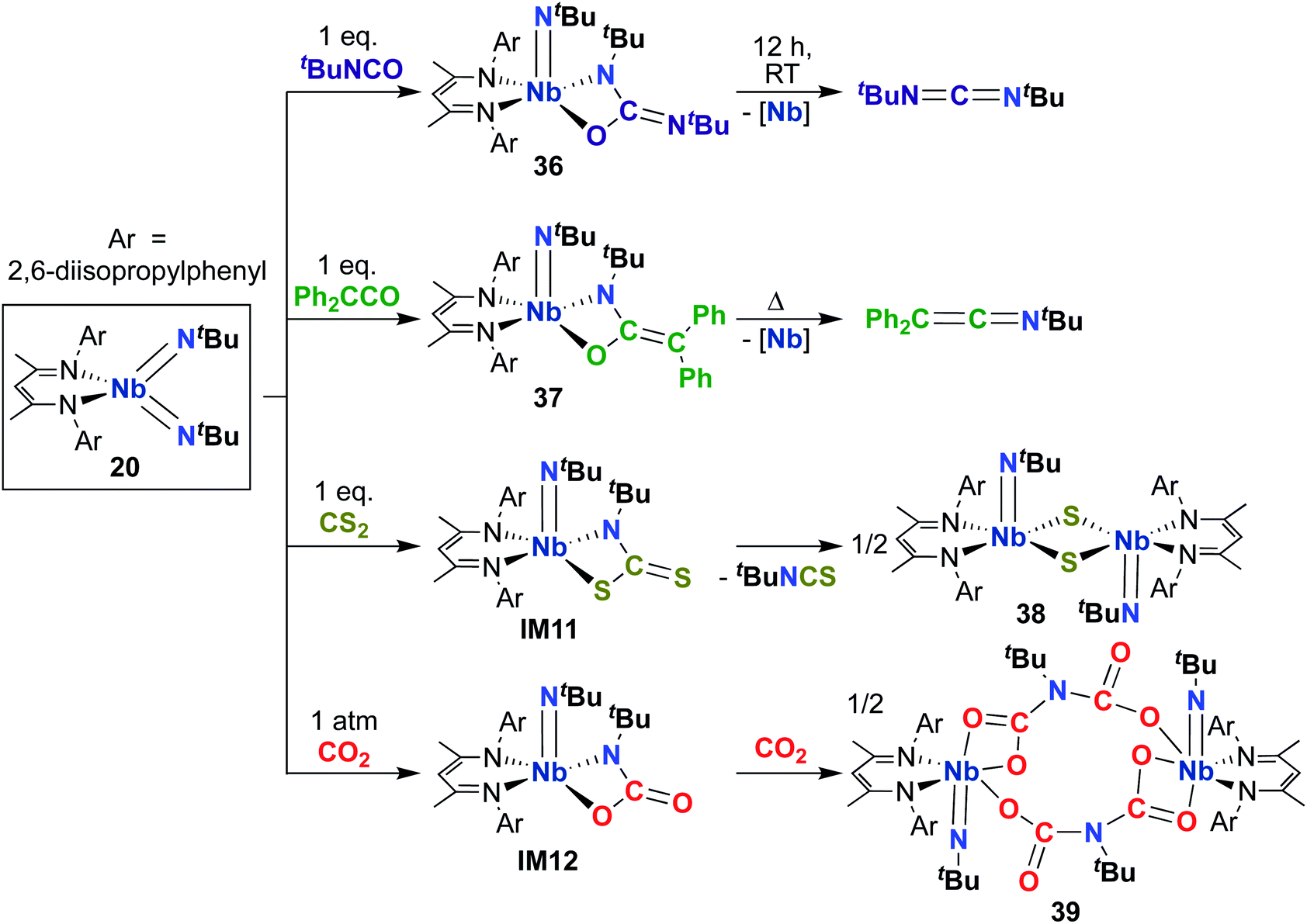
Building on the 1,2-addition and metathesis reactivity described above, 20 was also found to be capable of mediating [2 + 2] cycloadditions of heteroallenes across one of the two Nb-imido bonds.29,30 Complex 20 was observed to undergo [2 + 2] cycloaddition with tert-butyl isocyanate29 and diphenylketene30 to yield oxaazaniobacyclobutane products 36 and 37, respectively (Scheme 12). Complex 36 was found to decompose readily in solution to liberate carbodiimide, while 37 was stable in solution at room temperature and liberated diphenylethenimine upon heating. The related reactions of 20 with CS2 or CO2 led instead to dimeric products 38 and 39, respectively (Scheme 12).30 Reaction with CS2 presumably occurs via initial [2 + 2] cycloaddition to generate sulfaazaniobacyclobutane intermediate IM11, which proceeds to liberate isothiocyanate via a retro-[2 + 2] process and generate a transient imido–sulfido species, which dimerizes to give the bis(µ-sulfido)-imido product. Similarly, reaction with CO2 also likely forms oxaazaniobacyclobutane intermediate IM12 via [2 + 2] cycloaddition; this species then inserts another equivalent of CO2 into the metallacycle before dimerizing, suggesting that insertion of CO2 occurs more rapidly than retrocycloaddition in this case. These reactions showcase the highly reactive nature of π-loaded bis(imido) complexes and highlight the driving force provided by formation of strong Nb-oxo bonds.
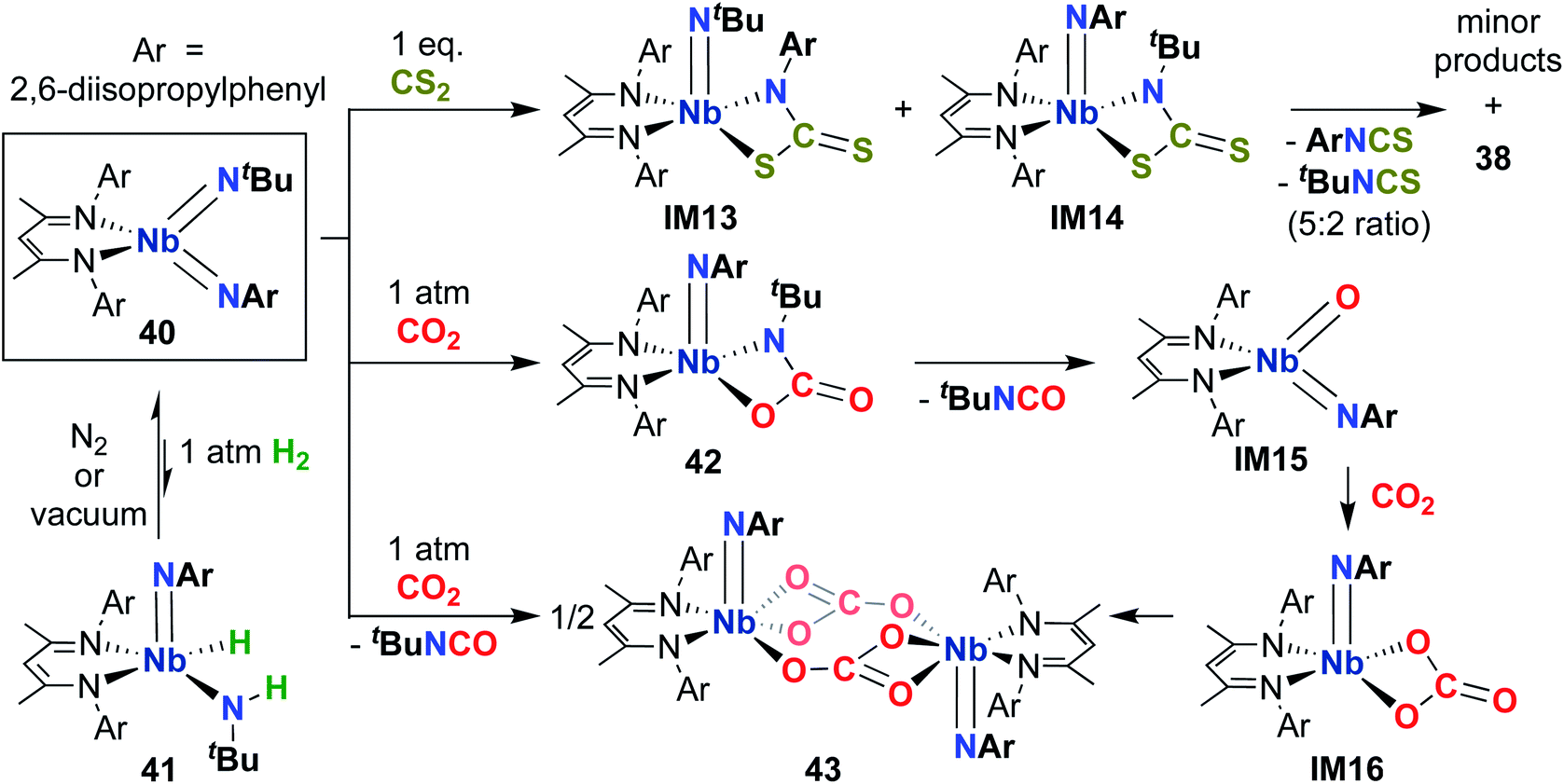 | ||
| Scheme 12 [2 + 2] Cycloaddition reactivity of Nb bis(imido) complex 20 toward heteroallenes. | ||
What factors, aside from π-loading, contribute to the significant reactivity of symmetric bis(imido) 20? Comparison of reactivity patterns between 20 and asymmetric bis(imido) 40 provides answers (Scheme 13).70 First, 40 was observed to be far less reactive toward 1,2-addition: addition of H2 resulted in low conversion to amido-imido-hydrido product 41, with Keq ≈ 2.5 at 20 °C under 1 atmosphere of H2 (Keq ≈ 8.0 for symmetric bis(imido) 20). 1,2-Addition of H2 occurred solely across the Nb-(tert-butyl)imido bond. Complex 40 reacted sluggishly with other 1,2-addition substrates (silanes, boranes, thiols); these reactions were not pursued further. The authors suggested that the weakly electron withdrawing nature of the 2,6-diisopropylphenyl group serves to stabilize the NbNAr bonding interaction, while the weakly electron donating tert-butyl moiety increases electron density at the imido group, leading to heightened reactivity at the NbNtBu bond. These subtle electronic factors also come into play when comparing reactivity patterns of bis((tert-butyl))imido) 20 with (tert-butyl)imido-(aryl)imido 40. As such, π-loading can be fine-tuned by choice of imido group substituent, with electron donating alkyl groups leading to more reactive bis(imido) complexes.
 | ||
| Scheme 13 1,2-Addition and [2 + 2] cycloaddition reactivity of asymmetric Nb bis(imido) complex 40. | ||
Complex 40 was also observed to react with heteroallenes across one of the two Nb-imido bonds: addition of CO2 led to oxaazaniobacycle 42 (Scheme 13), the product of [2 + 2] cycloaddition across the Nb-(tert-butyl)imido bond.30 Complex 42 was observed to undergo retro-cycloaddition in solution under an atmosphere of CO2 to release isocyanate, yielding the four-coordinate oxo-imido intermediate IM15. This intermediate can insert another equivalent of CO2 to yield carbonate intermediate IM16, which dimerizes to yield the observed carbonate-bridged product 43. Surprisingly, 40 was found to react with CS2 across both the (tert-butyl)imido and (aryl)imido groups, yielding bis(µ-sulfido)-imido dimer 38, a 5:2 mixture of aryl- and (tert-butyl)-isothiocyanate, and a mixture of minor side products. This reaction was proposed to proceed through sulfaazaniobacyclobutane intermediates IM13 and IM14; the observed reactivity across the (aryl)imido group was attributed to increased steric bulk, which prevents dimerization to form a stable bis(µ-sulfido)-(aryl)imido product akin to 38.
Indeed, observed reactivity of the (BDI) Nb bis(imido) system appears to be quite sensitive to imido group steric profile. As mentioned previously, several bis(imido) complexes can be accessed via reaction of low-valent Nb mono(imido) precursor 28 with organic azides (Scheme 9), including bis((tert-butyl))imido) (20), (tert-butyl)imido-(2,6-diisopropylphenyl)imido (40), and (tert-butyl)imido-(trimethylsilyl)imido.57 However, when less sterically demanding substituents are employed, such as cyclohexyl or benzyl, mono(imido) tetrazene complexes are instead isolated (44 and 45, respectively; Scheme 14, top).67 These complexes were hypothesized to form from initial nitrene transfer to yield unstable four-coordinate bis(imido) intermediates IM17 and IM19, followed by [3 + 2] cycloaddition of a second equivalent of azide. Cyclohexyl derivative 44 was observed to undergo azide metathesis to form benzyl derivative 45; experimental and computational investigations support a mechanism in which bis(imido) intermediates IM17 and IM19 are formed via retro-[3 + 2]-cycloaddition from tetrazene complex 44 and spectroscopically observed tetrazene intermediate IM18, respectively. Notably, bis(imido) intermediate IM17 could be trapped upon addition of a bulky Lewis base to yield stable five-coordinate bis(imido) complexes 46-R (R = tert-butyl, cyclohexyl; Scheme 14, bottom).
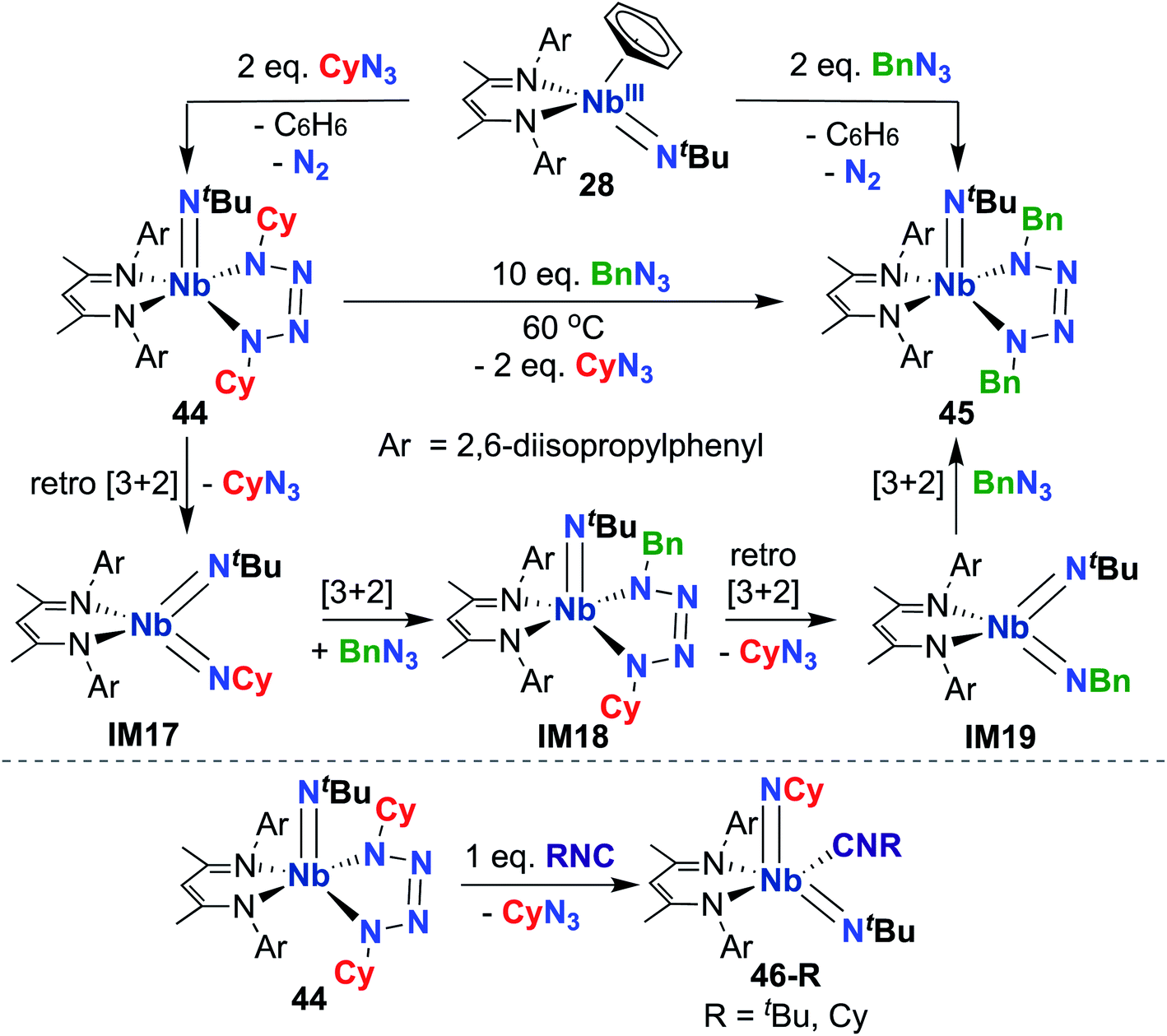 | ||
| Scheme 14 [3 + 2] Cycloaddition and retro-cycloaddition reactivity of Nb tetrazene complexes. | ||
The chemistry of Bergman and Arnold's (BDI)Nb bis(imido) system has been explored extensively, leading to the emergence of several themes. First, π-loading via addition of a second imido group makes bis(imido) complexes reactive toward nitrene transfer, 1,2-addition, and overall [2 + 2], [2 + 4], and [3 + 2] cycloadditions; in contrast, related mono(imido) systems do not display these reactivity patterns. Second, reactivity of bis(imido) complexes can be tuned by choice of imido substituent, with electron-donating groups leading to weaker Nb–Nimido bonding interactions and enhanced reactivity. Finally, bulky R-groups at BDI and imido ligands serve to kinetically stabilize reactive four-coordinate bis(imido) complexes. These guiding principles can be used to inform the design of bis(imido) complexes of vanadium, tantalum, and of other metals of the early transition metal block.
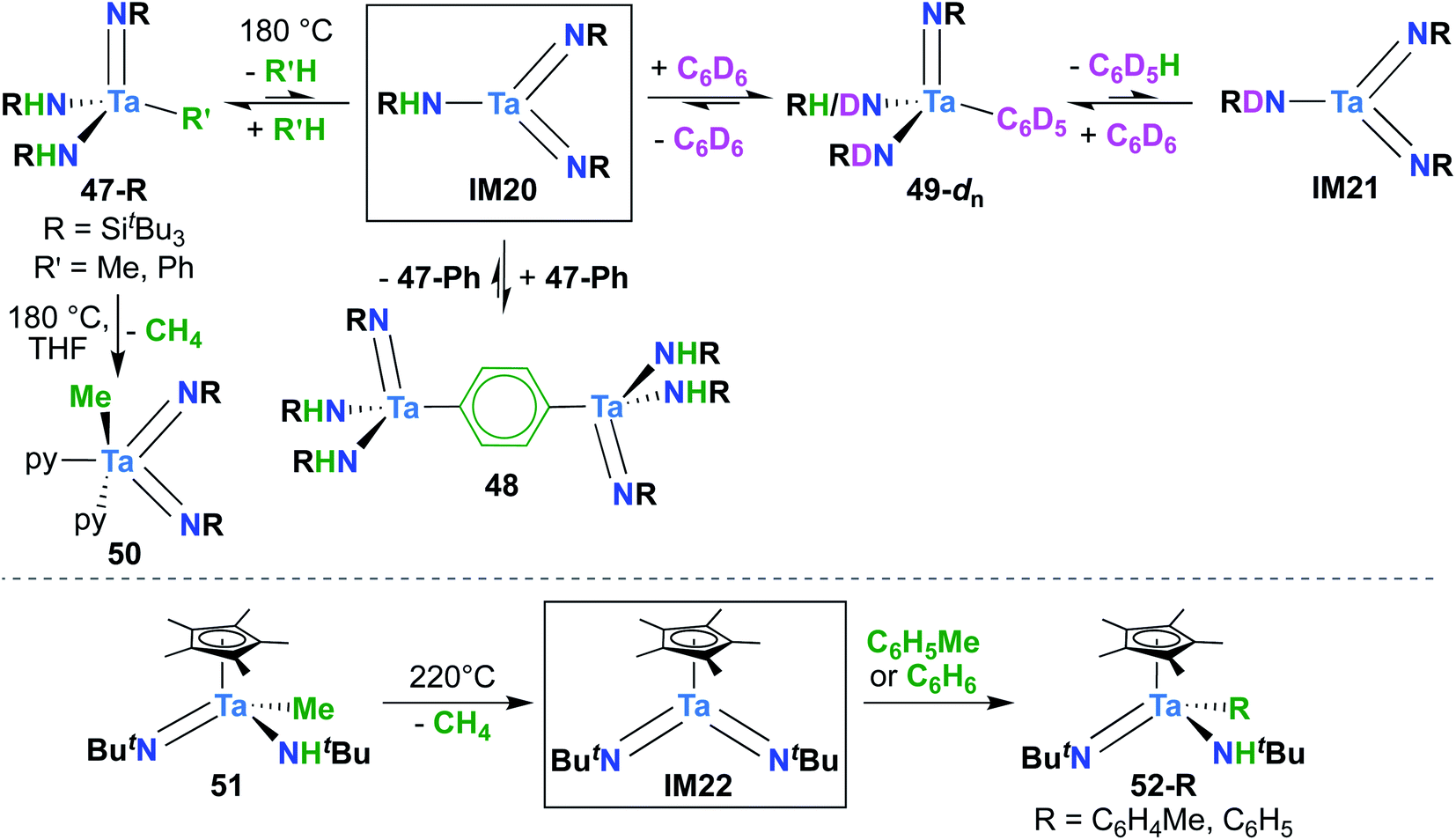 | ||
| Scheme 15 C–H activation reactivity of transient Ta bis(imido) complexes. | ||
A report from Royo and Sánchez-Nieves in 2000 represents the second and final example of a reactive of Ta bis(imido) complex.27 Mono(imido) 51 (Scheme 15, bottom) was observed to yield mono(imido) complexes 52-R (R = C6H5, C6H4Me) upon thermolysis in benzene or toluene solvent. Based on literature precedent,39 it seems likely that this transformation proceeds via reductive elimination of methane to yield π-loaded bis(imido) intermediate IM22, which can activate aromatic C–H bonds across one of the two Ta-imido groups. However, since no isotope labelling studies were carried out and IM22 could not be trapped, a simple σ-bond metathesis mechanism cannot be ruled out.64
5. Synthesis and reactivity of π-loaded chalcogenido complexes
The examples of catalytic and stoichiometric bis(imido) reactivity given above provide support for the claim that π-loading leads to increased reactivity at group 5 metal–imido linkages. A few researchers have begun to extend this strategy toward d0 group 5 chalcogenido–imido and bis(chalcogenido) complexes, which are isolobal to the bis(imido) complexes discussed above. This emerging field of research has already led to new types of complexes and new modes of reactivity for the early transition metals.In 2009, Mindiola and co-workers reported V bis(tellurido) 53 (Scheme 16), the first example of such a complex in the group 5 transition metal literature.71 A crystal structure of 53 supported assignment a V(V) metal center, as the intramolecular Te–Te distance was observed to be too long for an η2-Te22−-type ligand bound to a V(III) metal center. However, the V–Te bond lengths in 53 were found to be shorter than a corresponding mono(tellurido) complex;71 these bond metrics are inconsistent with the π-loading hypothesis. Synthesis of 53 also resulted in formation of ditelluretane 54 and tritellurolane 55. Bis(tellurido) 53 was found to react with one or two equivalents of organic azide to yield tellurido-imido 56 and bis(imido) 57, respectively. Unfortunately, a crystal structure of tellurido-imido 56 was not obtained, nor was reactivity reported, so the presence or extent of π-loading in this complex cannot be evaluated. The authors hypothesized that reaction of 53 with organic azides proceeds via elimination of Te0 to form a transient V(III) complex, as opposed to mechanism involving [2 + 2] cycloaddition. Thus, it appears that V bis(tellurido) complexes are not strictly analogous to π-loaded V bis(imido) complexes (although the nature of tellurido–imido complexes remains an open question).
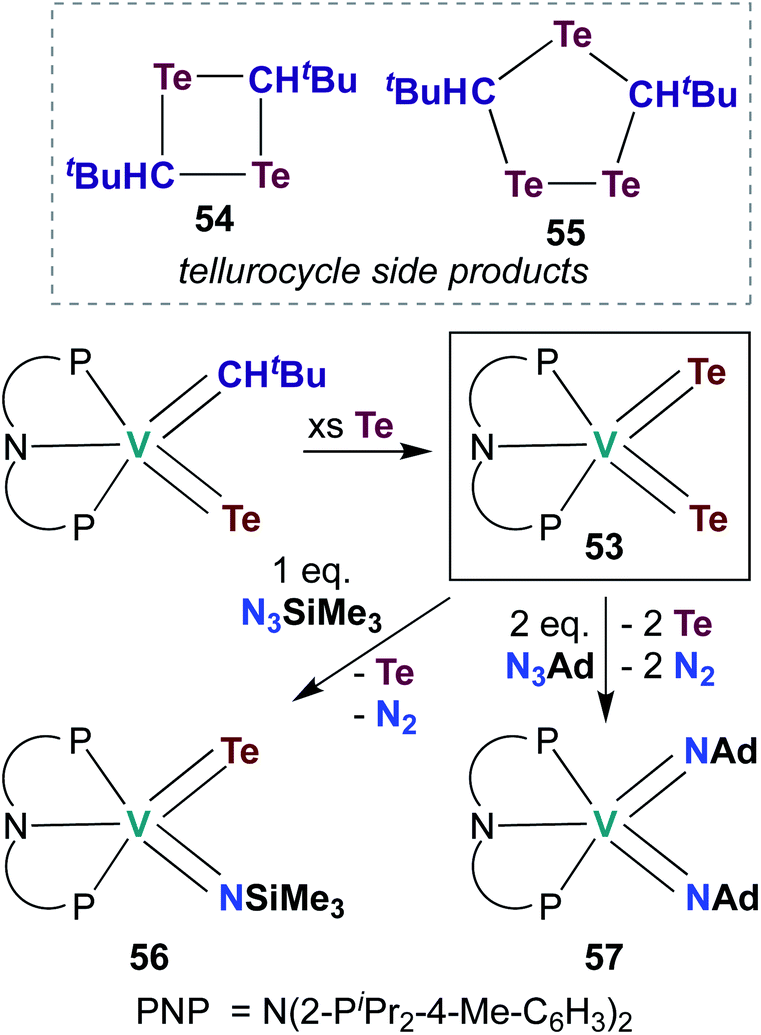 | ||
| Scheme 16 Synthesis and reactivity of V bis(tellurido) complex 53. | ||
Chirik and co-workers reported two more examples of V bis(chalcogenido) complexes in 2012: bis(imido), bis(oxo), and bis(sulfido) complexes 58-E (E = NPh, O, S; Scheme 17) were synthesized from a dinitrogen-bridged V(III) precursor.56 Bond metrics in complexes 58-O and 58-S cannot be compared to analogous mono(chalcogenido) complexes to evaluate the extent of π-loading, as such complexes have not been reported. Similarly, reactivity of complexes 58-O and 58-S has not yet been reported.
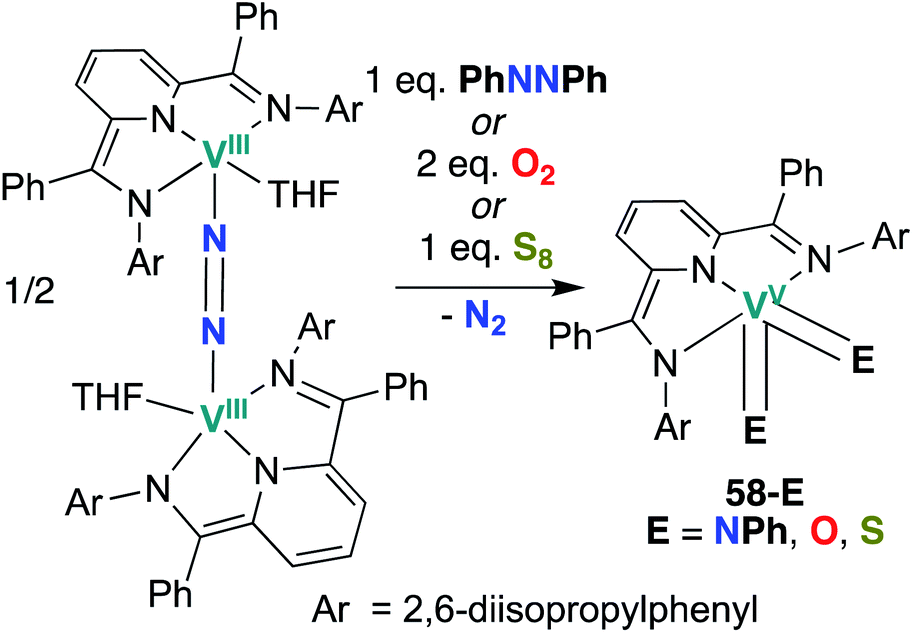 | ||
| Scheme 17 Syntheses of V bis(imido), bis(oxo), and bis(sulfido) complexes 58-E. | ||
Finally, in 2020, Bergman, Arnold, and co-workers reported the synthesis of a reactive, π-loaded oxo-imido complex.30 As described earlier, imido-oxaazaniobacycle 42 can be accessed from bis(imido) 40 (Scheme 13); addition of Lewis bases to 42 resulted in expulsion of isocyanate to yield five-coordinate oxo–imido complexes 59 and 61 (Scheme 18). The pyridine derivative 59 was found to be unstable in solution, liberating half an equivalent of pyridine and dimerizing to give bis(µ-oxo)-imido dimer 60, which was observed to be quite unreactive. However, the 4-dimethylaminopyridne derivative 61 was more stable than 59, enabling a crystal structure of this complex to be obtained; this represents the only structurally characterized group 5 terminal oxo-imido complex reported to date. The bond metrics in this structure were found to be consistent with a π-loaded complex, as detailed in Section 2. Complex 61 was found to react with primary, secondary, and tertiary silanes across the Nb-oxo bond via 1,2-addition to yield complexes 62a–c. 1,2-Addition reactions across group 5-oxo complexes are rare,72 and to date, no other examples of such reactivity have been reported for Nb-oxo complexes.73 In the case of 61, it's clear that π-loading contributes to polarizing the Nb-oxo group, engendering reactivity at a group that usually spectates. This result suggests that the π-loading strategy can be extended to chalcogenido–imido complexes, enabling new modes of reactivity for these complexes.
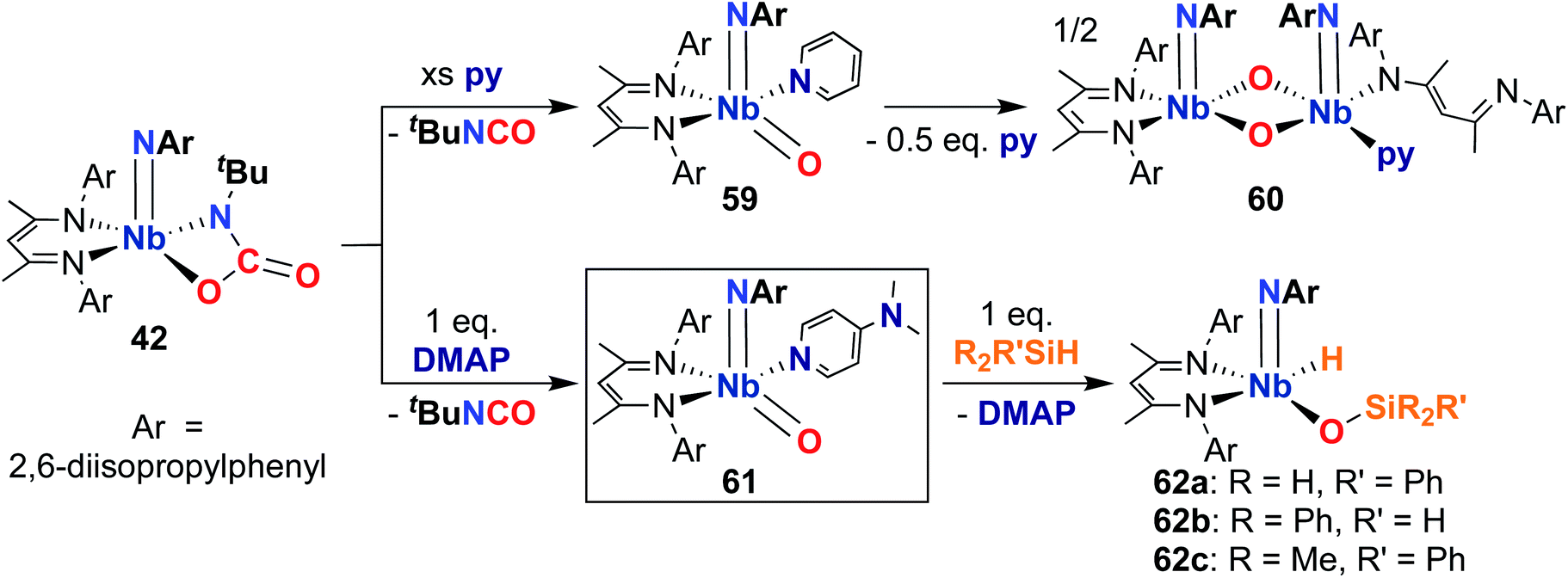 | ||
| Scheme 18 Synthesis and 1,2-addition reactivity of π-loaded Nb oxo-imido complex 61. | ||
6. Conclusions and future outlook
The above examples illustrate instances in which imido-based reactivity is observed in π-loaded bis(imido) complexes of the group 5 triad. While there are numerous examples of such reactivity reported for bis(imido) complexes, there are far fewer examples of mono(imido)-mediated reactivity, suggesting that π-loading activates group 5-imidos towards reactivity. Of the examples presented above, catalysis is dominated by V bis(imido) complexes supported by labile and relatively unbulky ligands, while Nb bis(imido) complexes supported by bulky bidentate ligands represent the most stoichiometrically active species. In general, the stoichiometric reactivity of V and Ta bis(imido) complexes is underexplored, as is the chemistry of Nb bis(imido) systems supported by ligands other than BDI and MAD.In terms of catalysis, it seems likely that labile ligands (i.e., PR3, THF, py), less steric bulk at ancillary ligand(s), and cationic charge could lead to more examples of V- and Nb-mediated catalytic processes. Ta systems are particularly underexplored: to date, only three structurally characterized examples of Ta bis(imido) complexes have been reported. It seems likely that bulky ancillary ligands will be needed to stabilize Ta bis(imido) species, as the few examples of such complexes reported to date are highly reactive and often transient. Notably, verified examples of cycloadditions across imido groups of Ta bis(imido) complexes are absent from the literature (although it seems likely that [2 + 2] cycloaddition occurs along Hollis' catalytic cycle32).
Chalcogenido–imido and reactive bis(chalcogenido) species are underexplored for all metals of the group 5 triad. Promising results with a reactive Nb oxo-imido species suggest that the π-loading strategy can be extended towards this class of complexes. The synthesis and study of group 5 terminal oxo-imido, sulfido-imido, and bis(chalcogenido) species is an open field, and one that will surely lead to interesting chemistry.
The rational design of highly reactive, π-loaded group 5 complexes will lead to systems capable of mediating challenging transformations with implications for a sustainable chemical future. Desirable transformations include N–H bond activation of ammonia, C–H bond activation of methane, conversion of CO2 into larger Cn products (such as isocyanates and carbamates, as described above for niobium bis(imido) complexes), and synthesis of higher-value cyclic and linear hydrocarbons from plentiful feedstock molecules. Employing relatively abundant and affordable early transition metals to carry out these transformations will help ameliorate the overuse of expensive precious metal catalysts, and ultimately contribute to a greener chemical industry.
Author contributions
J. I. F. conceived and wrote the manuscript. J. M., S. L.-F., and D. A. contributed to schemes and figures. R. G. B. and J. A. conceived and contributed to all portions of the manuscript.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This version replaces the manuscript published on 29th June 2022. The images of Schemes 4–7 had been associated with the incorrect captions and were out of order. The RSC apologises for any confusion.Acknowledgements
This work was funded by the NSF (Grant No. CHE-1465188 and 1954612 to J. A. and R. G. B.). J. I. F. acknowledges support from an NSF graduate research fellowship (Grant No. DGE 1752814). We thank Dr Michael A. Boreen, Erik T. Ouellette, I. Joseph Brackbill, Sheridon N. Kelly, and Anukta Jain for helpful discussions.Notes and references
- D. E. Wigley, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc., 1994, pp. 239–482 Search PubMed.
- K. Kawakita, B. F. Parker, Y. Kakiuchi, H. Tsurugi, K. Mashima, J. Arnold and I. A. Tonks, Coord. Chem. Rev., 2020, 407, 213118–213141 CrossRef CAS PubMed.
- J. R. Webb, S. A. Burgess, T. R. Cundari and T. B. Gunnoe, Dalton Trans., 2013, 42, 16646–16665 RSC.
- P. T. Wolczanski, Organometallics, 2018, 37, 505–516 CrossRef CAS.
- J. Chu, E. Lu, Y. Chen and X. Leng, Organometallics, 2013, 32, 1137–1140 CrossRef CAS.
- A. P. Duncan and R. G. Bergman, Chem. Rec., 2002, 2, 431–445 CrossRef CAS PubMed.
- J. Chu, X. Han, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 10894–10897 CrossRef CAS PubMed.
- A. F. Heyduk, R. A. Zarkesh and A. I. Nguyen, Inorg. Chem., 2011, 50, 9849–9863 CrossRef CAS PubMed.
- P. J. Walsh, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1988, 110, 8729–8731 CrossRef CAS.
- C. C. Cummins, S. M. Baxter and P. T. Wolczanski, J. Am. Chem. Soc., 1988, 110, 8731–8733 CrossRef CAS.
- J. Chu, E. Lu, Z. Liu, Y. Chen, X. Leng and H. Song, Angew. Chem., Int. Ed., 2011, 50, 7677–7680 CrossRef CAS PubMed.
- N. Hazari and P. Mountford, Acc. Chem. Res., 2005, 38, 839–849 CrossRef CAS PubMed.
- S. W. Krska, R. L. Zuckerman and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 11828–11829 CrossRef CAS.
- K. E. Meyer, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1994, 116, 2669–2670 CrossRef CAS.
- Y. Li, Y. Shi and A. L. Odom, J. Am. Chem. Soc., 2004, 126, 1794–1803 CrossRef CAS PubMed.
- A. L. Odom and T. J. McDaniel, Acc. Chem. Res., 2015, 48, 2822–2833 CrossRef CAS PubMed.
- R. T. Ruck, R. L. Zuckerman, S. W. Krska and R. G. Bergman, Angew. Chem., Int. Ed., 2004, 43, 5372–5374 CrossRef CAS PubMed.
- F. Basuli, H. Aneetha, J. C. Huffman and D. J. Mindiola, J. Am. Chem. Soc., 2005, 127, 17992–17993 CrossRef CAS PubMed.
- Z. W. Davis-Gilbert, L. J. Yao and I. A. Tonks, J. Am. Chem. Soc., 2016, 138, 14570–14573 CrossRef CAS PubMed.
- Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2016, 8, 63–68 CrossRef CAS PubMed.
- E. P. Beaumier, M. E. McGreal, A. R. Pancoast, R. H. Wilson, J. T. Moore, B. J. Graziano, J. D. Goodpaster and I. A. Tonks, ACS Catal., 2019, 9, 11753–11762 CrossRef CAS PubMed.
- A. J. Pearce, R. P. Harkins, B. R. Reiner, A. C. Wotal, R. J. Dunscomb and I. A. Tonks, J. Am. Chem. Soc., 2020, 142, 4390–4399 CrossRef CAS PubMed.
- A. I. Nguyen, R. A. Zarkesh, D. C. Lacy, M. K. Thorson and A. F. Heyduk, Chem. Sci., 2011, 2, 166–189 RSC.
- M. C. Burland, T. W. Pontz and T. Y. Meyer, Organometallics, 2002, 21, 1933–1941 CrossRef CAS.
- R. E. Blake, D. M. Antonelli, L. M. Henling, W. P. Schaefer, K. I. Hardcastle and J. E. Bercaw, Organometallics, 1998, 17, 718–725 CrossRef CAS.
- S. M. Rocklage and R. R. Schrock, J. Am. Chem. Soc., 1980, 102, 7809–7811 CrossRef.
- P. Royo and J. Sánchez-Nieves, J. Organomet. Chem., 2000, 597, 61–68 CrossRef CAS.
- J. W. Bruno and X. J. Li, Organometallics, 2000, 19, 4672–4674 CrossRef CAS.
- N. C. Tomson, J. Arnold and R. G. Bergman, Organometallics, 2010, 29, 2926–2942 CrossRef CAS PubMed.
- J. I. Fostvedt, L. N. Grant, B. M. Kriegel, A. H. Obenhuber, T. D. Lohrey, R. G. Bergman and J. Arnold, Chem. Sci., 2020, 11, 11613–11632 RSC.
- D. P. Smith, K. D. Allen, M. D. Carducci and D. E. Wigley, Inorg. Chem., 1992, 31, 1319–1320 CrossRef CAS.
- T. R. Helgert, X. Zhang, H. K. Box, J. A. Denny, H. U. Valle, A. G. Oliver, G. Akurathi, C. E. Webster and T. K. Hollis, Organometallics, 2016, 35, 3452–3460 CrossRef CAS.
- A. M. Baranger, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 2753–2763 CrossRef CAS.
- G. Parkin, L. R. Whinnery, A. van Asselt, D. J. Leahy, N. G. Hua, R. W. Quan, L. M. Henling, W. P. Schaefer, B. D. Santarsiero and J. E. Bercaw, Inorg. Chem., 1992, 31, 82–85 CrossRef CAS.
- D. N. Huh, Y. Cheng, C. W. Frye, D. T. Egger and I. A. Tonks, Chem. Sci., 2021, 12, 9574–9590 RSC.
- P. D. Bolton and P. Mountford, Adv. Synth. Catal., 2005, 347, 355–366 CrossRef CAS.
- P. A. Zhizhko, N. S. Bushkov, A. V. Pichugov and D. N. Zarubin, Coord. Chem. Rev., 2021, 448, 214112 CrossRef CAS.
- D. S. Williams, M. H. Schofield and R. R. Schrock, Organometallics, 1993, 12, 4560–4571 CrossRef CAS.
- C. P. Schaller and P. T. Wolczanski, Inorg. Chem., 1993, 32, 131–144 CrossRef CAS.
- H. S. La Pierre, J. Arnold, R. G. Bergman and F. D. Toste, Inorg. Chem., 2012, 51, 13334–13344 CrossRef CAS PubMed.
- H. S. La Pierre, S. G. Minasian, M. Abubekerov, S. A. Kozimor, D. K. Shuh, T. Tyliszczak, J. Arnold, R. G. Bergman and F. D. Toste, Inorg. Chem., 2013, 52, 11650–11660 CrossRef CAS PubMed.
- D. Rehder, T. Polenova and M. Bühl, Annu. Rep. NMR Spectrosc., 2007, 62, 49–114 CrossRef CAS.
- D. Rehder, Coord. Chem. Rev., 2008, 252, 2209–2223 CrossRef CAS.
- F. Preuss, T. Wieland and B. Günther, Z. Anorg. Allg. Chem., 1992, 609, 45–50 CrossRef CAS.
- F. Preuss, H. Becker and T. Weiland, Z. Naturforsch., 1990, 45b, 191–198 CrossRef.
- K. M. Lin, P. Y. Wang, Y. J. Shieh, H. Z. Chen, T. S. Kuo and Y. C. Tsai, New J. Chem., 2010, 34, 1737–1745 RSC.
- H. S. La Pierre, J. Arnold and F. D. Toste, Angew. Chem., Int. Ed., 2011, 50, 3900–3903 CrossRef CAS PubMed.
- K. Kawakita, E. P. Beaumier, Y. Kakiuchi, H. Tsurugi, I. A. Tonks and K. Mashima, J. Am. Chem. Soc., 2019, 141, 4194–4198 CrossRef CAS PubMed.
- W. S. Farrell, C. Greene, P. Ghosh, T. H. Warren and P. Y. Zavalij, Organometallics, 2020, 39, 3906–3917 CrossRef CAS.
- B. L. Tran, M. Singhal, H. Park, O. P. Lam, M. Pink, J. Krzystek, A. Ozarowski, J. Telser, K. Meyer and D. J. Mindiola, Angew. Chem., Int. Ed., 2010, 49, 9871–9875 CrossRef CAS PubMed.
- A. H. Obenhuber, T. L. Gianetti, X. Berrebi, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2014, 136, 2994–2997 CrossRef CAS PubMed.
- T. R. Helgert, T. K. Hollis, A. G. Oliver, H. U. Valle, Y. Wu and C. E. Webster, Organometallics, 2014, 33, 952–958 CrossRef CAS.
- Y.-W. Chao, P. A. Wexler and D. E. Wigley, Inorg. Chem., 1990, 29, 4594–4595 CrossRef.
- Y.-W. Chao, P. A. Wexler and D. E. Wigley, Inorg. Chem., 1989, 28, 3860–3868 CrossRef CAS.
- Y. Tsai, P. Wang, K. Lin, S. Chen and J. Chen, Chem. Commun., 2008, 2008, 205–207 RSC.
- C. Milsmann, Z. R. Turner, S. P. Semproni and P. J. Chirik, Angew. Chem., Int. Ed., 2012, 51, 5386–5390 CrossRef CAS PubMed.
- B. M. Kriegel, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2016, 138, 52–55 CrossRef CAS PubMed.
- N. C. Tomson, J. Arnold and R. G. Bergman, Organometallics, 2010, 29, 5010–5025 CrossRef CAS PubMed.
- J. de With, A. D. Horton and A. G. Orpen, Organometallics, 1993, 12, 1493–1496 CrossRef CAS.
- S. G. Bott, D. M. Hoffman and S. P. Rangarajan, Inorg. Chem., 1995, 34, 4305–4310 CrossRef CAS.
- E. P. Beaumier, A. J. Pearce, X. Y. See and I. A. Tonks, Nat. Rev. Chem, 2019, 3, 15–34 CrossRef PubMed.
- T. Saito, H. Nishiyama, H. Tanahashi, K. Kawakita, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2014, 136, 5161–5170 CrossRef CAS PubMed.
- G. Liang, T. K. Hollis and C. E. Webster, Organometallics, 2018, 37, 1671–1681 CrossRef CAS.
- J. de With and A. D. Horton, Angew. Chem., Int. Ed., 1993, 32, 903–905 CrossRef.
- T. Gehrmann, J. Lloret Fillol, H. Wadepohl and L. H. Gade, Organometallics, 2012, 31, 4504–4515 CrossRef CAS.
- K. E. Meyer, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 974–985 CrossRef CAS.
- A. Jain, J. I. Fostvedt, B. M. Kriegel, D. W. Small, L. N. Grant, R. G. Bergman and J. Arnold, Inorg. Chem., 2022, 61, 6574–6583 CrossRef CAS PubMed.
- A. H. Obenhuber, T. L. Gianetti, R. G. Bergman and J. Arnold, Chem. Commun., 2015, 51, 1278–1281 RSC.
- T. L. Gianetti, G. Nocton, S. G. Minasian, N. C. Tomson, A. L. D. Kilcoyne, S. A. Kozimor, D. K. Shuh, T. Tyliszczak, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2013, 135, 3224–3236 CrossRef CAS PubMed.
- B. M. Kriegel, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2016, 138, 52–55 CrossRef CAS PubMed.
- U. J. Kilgore, J. A. Karty, M. Pink, X. Gao and D. J. Mindiola, Angew. Chem., Int. Ed., 2009, 48, 2394–2397 CrossRef CAS PubMed.
- J. I. Fostvedt, M. A. Boreen, R. G. Bergman and J. Arnold, Inorg. Chem., 2021, 60, 9912–9931 CrossRef CAS PubMed.
- G. Parkin, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc., 1998, pp. 1–165 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |