
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrocatalytic NAD+ reduction via hydrogen atom-coupled electron transfer†
Fengyuan
Liu‡
ab,
Chunmei
Ding‡
bc,
Shujie
Tian
bc,
Sheng-Mei
Lu
bc,
Chengcheng
Feng
bd,
Dandan
Tu
bc,
Yan
Liu
 bc,
Wangyin
Wang
bc and
Can
Li
*abc
bc,
Wangyin
Wang
bc and
Can
Li
*abc
aZhang Dayu School of Chemistry, Dalian University of Technology, Dalian 116024, Liaoning, China
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian National Laboratory for Clean Energy, Dalian 116023, China. E-mail: canli@dicp.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dSchool of Chemistry and Materials Science, University of Science and Technology of China, Hefei 230026, China
First published on 24th October 2022
Abstract
Nicotinamide adenine dinucleotide cofactor (NAD(P)H) is regarded as an important energy carrier and charge transfer mediator. Enzyme-catalyzed NADPH production in natural photosynthesis proceeds via a hydride transfer mechanism. Selective and effective regeneration of NAD(P)H from its oxidized form by artificial catalysts remains challenging due to the formation of byproducts. Herein, electrocatalytic NADH regeneration and the reaction mechanism on metal and carbon electrodes are studied. We find that the selectivity of bioactive 1,4-NADH is relatively high on Cu, Fe, and Co electrodes without forming commonly reported NAD2 byproducts. In contrast, more NAD2 side product is formed with the carbon electrode. ADP-ribose is confirmed to be a side product caused by the fragmentation reaction of NAD+. Based on H/D isotope effects and electron paramagnetic resonance analysis, it is proposed that the formation of NADH on these metal electrodes proceeds via a hydrogen atom-coupled electron transfer (HadCET) mechanism, in contrast to the direct electron-transfer and NAD˙ radical pathway on carbon electrodes, which leads to more by-product, NAD2. This work sheds light on the mechanism of electrocatalytic NADH regeneration, which is different from biocatalysis.
Introduction
Most biocatalytic pathways depend on either of the two important cofactors, nicotinamide adenine dinucleotide (NAD+) or nicotinamide adenine dinucleotide phosphate (NADP+), which exist as redox couples with their corresponding reduced forms, NADH and NADPH (collectively abbreviated hereafter as NAD(P)H). NADH and NADPH differ in a phosphate group and function in different biological reactions in organisms. In natural photosynthesis, solar energy is harvested by photosystems, and the electrons and protons from the water oxidation reaction are utilized for NADPH production, which is then used as reducing equivalent for CO2 fixation dark reactions in the Calvin cycle.1–3 Nicotinamide adenine dinucleotide cofactor is a critical charge transfer mediator and energy carrier in photosynthesis and is involved in biocatalysis by oxidoreductases. NAD(P)H turns into its oxidized form (NAD(P)+) after donating two electrons and one proton and is difficult to use stoichiometrically for practical large-scale biocatalytic reactions.4 Therefore, efficient reduction of NAD+ to NADH with artificial catalysts is of significance for biocatalysis, understanding natural photosynthesis and constructing artificial photosynthetic systems for solar energy utilization.5–14Previously, NAD+ has been reduced to NADH through biocatalysis with sacrificial reagents, such as formic acid,15–18 and photo(electro)catalysis with noble metal complexes such as [Cp*Rh(bpy)(H2O)]Cl2,19–25 which is coupled with enzyme-catalyzed reduction reactions.19,24,26–31 However, there are product separation difficulties and serious problems of mutual inactivation of molecular and enzymatic catalysts due to their interaction.32–34 Heterogeneous NAD+ hydrogenation to NADH with H2 as the reducing agent can avoid the above issues, but the selectivity for 1,4-NADH is relatively low.35 Electrocatalysis by metal catalysts with H2O providing protons and electrons for NAD+ reduction is another important way for artificial NADH production,36–40 but the formation of 1,6-NADH and NAD2 side products leads to a low selectivity for 1,4-NADH.35–38,40–42 The intrinsic reasons behind this issue are not well understood. It has been reported that NADPH production in natural photosynthesis proceeds through direct hydride transfer from the flavin adenine dinucleotide hydride (FADH−) to the nicotinamide controlled by ferredoxin–NADP+ reductase (FNR).43 For NAD+ reduction to NADH by artificial catalysts, it has been found that the transfer of two electrons results in the formation of 1,4-NADH and its 1,6-isomer, whereas the uptake of one electron leads to the formation of the NAD˙ radical that undergoes irreversible dimerization. Nevertheless, there is an ongoing debate on the mechanism of NAD+ reduction that the reaction may proceed through a direct electron transfer pathway with the NAD˙ radical as an intermediate or a hydride transfer mechanism.21,23 In addition, product analysis which has long been a thorny issue was only based on the grounds of preliminary evidence with carbon, Hg, and several metal catalysts without detailed product quantification and mechanism analysis. Therefore, it is of importance to study the mechanisms of the electrocatalytic NAD+ reduction reaction with different catalysts and to understand their differences from the pathway of selective 1,4-NAD(P)H production in nature, which is of significance for the exploration of more efficient artificial catalysts for NAD+ reduction to 1,4-NADH.
Herein, taking various metal and carbon materials as catalysts, the electrocatalytic NAD+ reduction reaction and the mechanisms are studied with systematic quantification of products. We find that the selectivity of 1,4-NADH is relatively high on Cu, Fe, and Co electrodes without forming NAD2 side products, different from the literature.17 In contrast, obvious formation of NAD2 is observed on the carbon electrode with low activity and selectivity for 1,4-NADH. And ADP-ribose is confirmed to be a fragmentation side product. With the reaction kinetics, H/D isotope effect, and electron paramagnetic resonance (EPR) analysis, we propose a hydrogen atom-coupled electron transfer (HadCET) mechanism for the NAD+ reduction reaction on these metal electrodes in contrast to the NAD˙ radical process on the carbon electrode.
Results and discussion
Electrocatalytic NAD+ reduction to NADH was studied with Cu, Fe, Co, and carbon electrodes (see the ESI† for the preparation and morphology details, Fig. S1†). NAD+ is fully consumed after the reaction judging from the disappearance of the 1H nuclear magnetic resonance (1HNMR) signals of NAD+ (Fig. S2†). Typical signals of 1,4-NADH with diagnostic chemical shifts at 2.6 ppm, 5.8 ppm, and 6.8 ppm appear after the reaction, suggesting that 1,4-NADH is produced (Fig. 1a, S2 and S3†). The change in UV-visible absorption spectra as a function of time indicates the consumption of NAD+ and generation of NADH (Fig. S4†). NAD2 was commonly considered as an important side product on metal electrodes such as Au and Hg.44–46 However, in our results, only 1,4-NADH and 1,6-isomer are produced without the observation of NAD2 with Cu, Fe, and Co electrodes (Fig. 1a, S2 and S3†), indicating a distinct reaction mechanism. In sharp contrast, with carbon electrodes, there are NAD2 signals in 1HNMR spectra after the reaction, consistent with previous reports.34 It is noteworthy that there is some ADP-ribose, which may be caused by the fragmentation of NAD+ and has rarely been reported before for NAD+ reduction.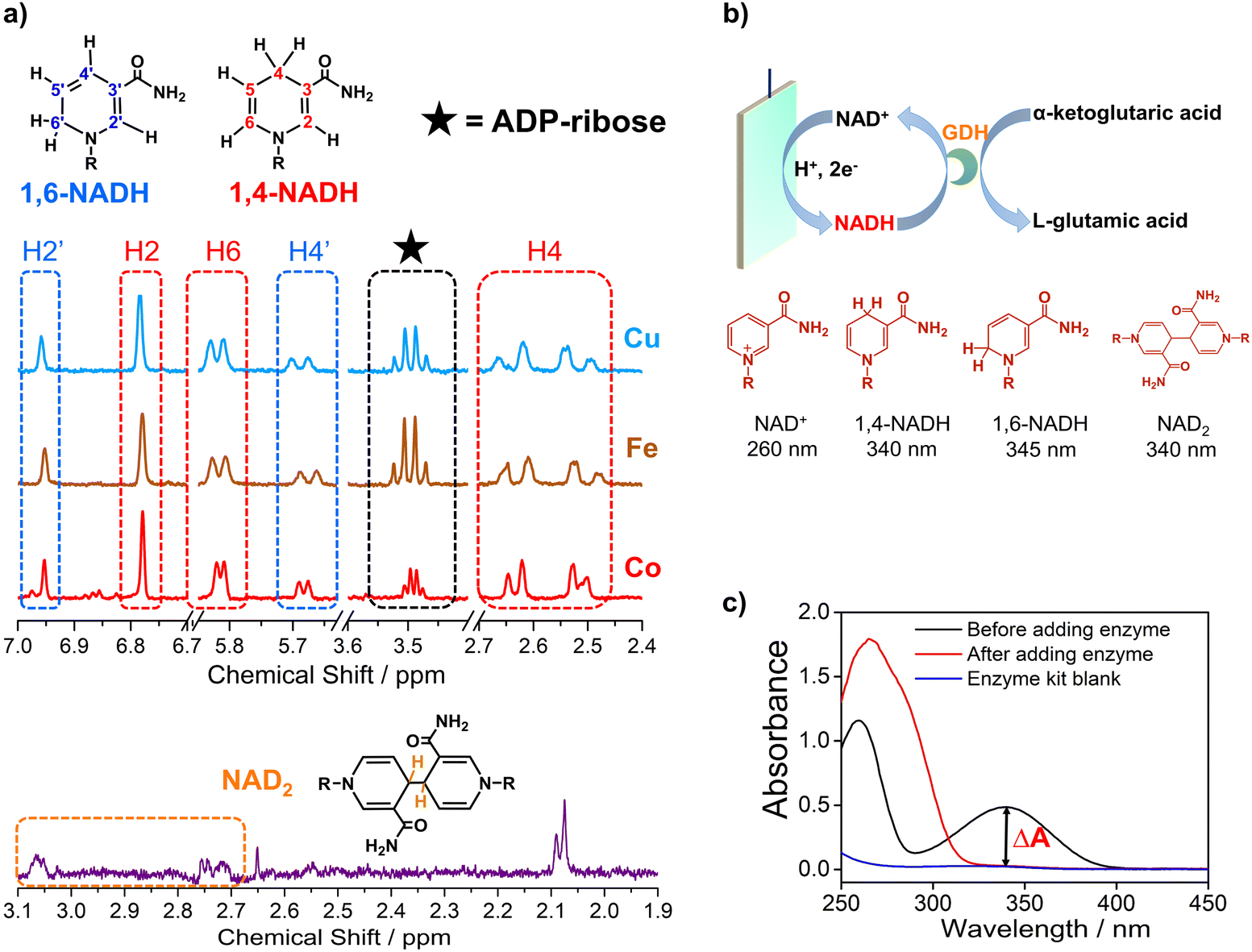 | ||
| Fig. 1 (a) The signals of 1,4-NADH (H2, H4, and H6), 1,6-NADH (H2′ and H4′), ADP-ribose, and NAD2 in the 1HNMR spectra of the products after electrocatalytic NAD+ reduction on Cu foam, Co foam, Fe foam, and carbon felt. Herein, the results of Cu, Fe, and carbon are measured with 400 MHz 1HNMR, while the result of Co is obtained with 700 MHz 1HNMR. Comparison of 400 MHz and 700 MHz 1HNMR is shown in Fig. S3.† (b, c) Schematic description of electrocatalytic NAD+ reduction and the determination of different products. | ||
More importantly, systematic quantitative analysis of products that has always been a tough issue was conducted combining UV-visible absorption spectra and NMR measurements (see the ESI† for details about the quantification of all products). The typical light absorption band of NAD+, 1,4-NADH, 1,6-NADH, and NAD2 is located at 260 nm, 340 nm, 345 nm, and 340 nm, respectively.47 Glutamate dehydrogenase (GDH) was used as an enzymatic catalyst for 1,4-NADH determination. The amount and selectivity of 1,4-NADH can be calculated from the change of absorbance (ΔA) before and after the enzyme catalyzed reaction, as shown in Fig. 1b and c. For the case of metal electrodes, the amount of 1,6-NADH is quantified from the residual absorbance after adding GDH. Then, the amount of ADP-ribose can be obtained from the difference of the initial NAD+ amount and NADH produced. And NAD2 can be quantified from the absorbance and the amount of NADH according to their molar extinction coefficient.
Fig. 2 displays the NAD+ conversion to different products when NAD+ is totally consumed. The highest yield of enzymatically active 1,4-NADH with Cu, Fe, and Co electrodes is about 57.9%, 64.1%, and 48.81% at −0.4 V vs. RHE (denoted as VRHE), respectively. In sharp contrast, the regeneration of 1,4-NADH is only 7.9% at −0.4 VRHE on the carbon electrode with a high percentage of NAD2 (above 40%).
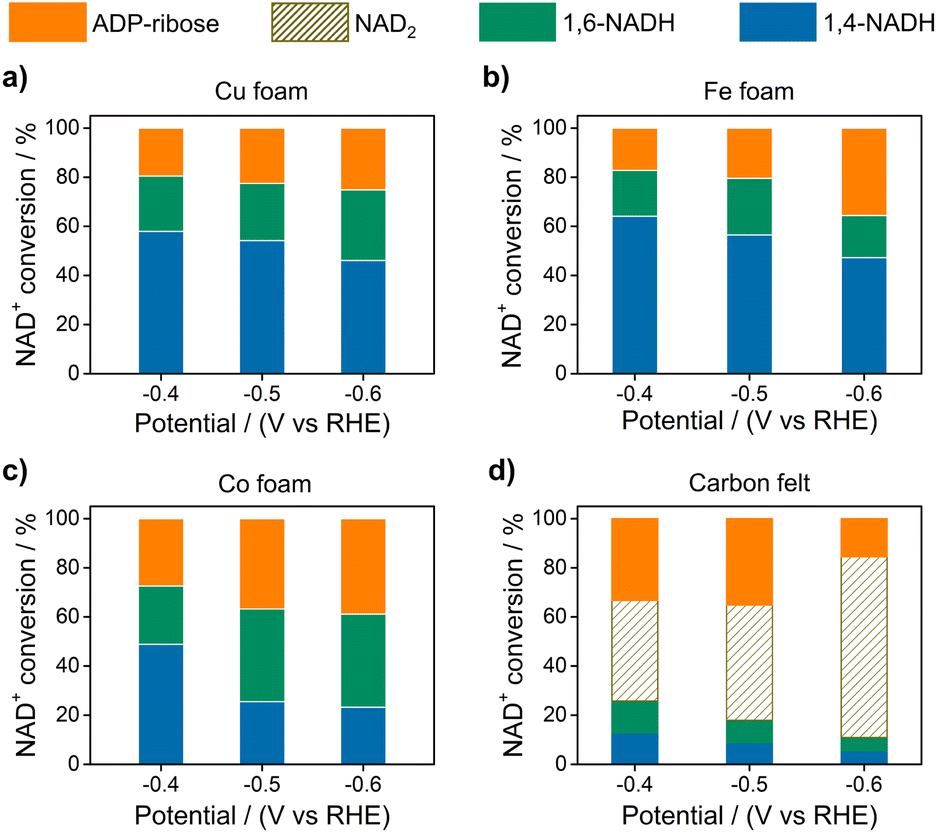 | ||
| Fig. 2 The conversion of NAD+ to different products on (a) Cu, (b) Fe, (c) Co foams, and (d) carbon felt at different potentials. Electrolyte: 0.1 M phosphate buffer (pH 7); catholyte: 12 mL electrolyte with an initial NAD+ concentration of 1 mM. | ||
Fig. 3a and S5† show the 1,4-NADH concentration trends during the NAD+ reduction reaction on the Cu, Fe, Co, and carbon electrodes at fixed potentials. The 1,4-NADH formation rate on the Cu electrode is highest among all of the tested electrodes. And the 1,4-NADH formation rate increases with the applied potential from −0.4 VRHE to −0.5 VRHE and then decreases at −0.6 VRHE (Fig. S5†). As shown in Fig. 3a and b, the concentration of the aimed 1,4-NADH product and the total concentration of NADH (1,4-NADH + 1,6-NADH) increase with the reaction time, and the production rate decreases with the consumption of NAD+. However, the percentage of 1,4-NADH in NADH remains at around 71% with negligible decline as a function of time, that's to say the selectivity for 1,4-NADH does not change, indicating that the reaction mechanism is independent of the reaction degree. More interestingly, ADP-ribose is observed with both kinds of materials. To confirm the existence of ADP-ribose derivatives and NAD2 products, the electrolyte after the reaction was analyzed with electrospray ionization mass spectrometry (ESI-MS). Typical signals of ADP-ribose derivatives (m/z 558.1 and 540.1) and NAD2 (m/z 1327.2) can be detected, as shown in Fig. 3c, d and S6.†
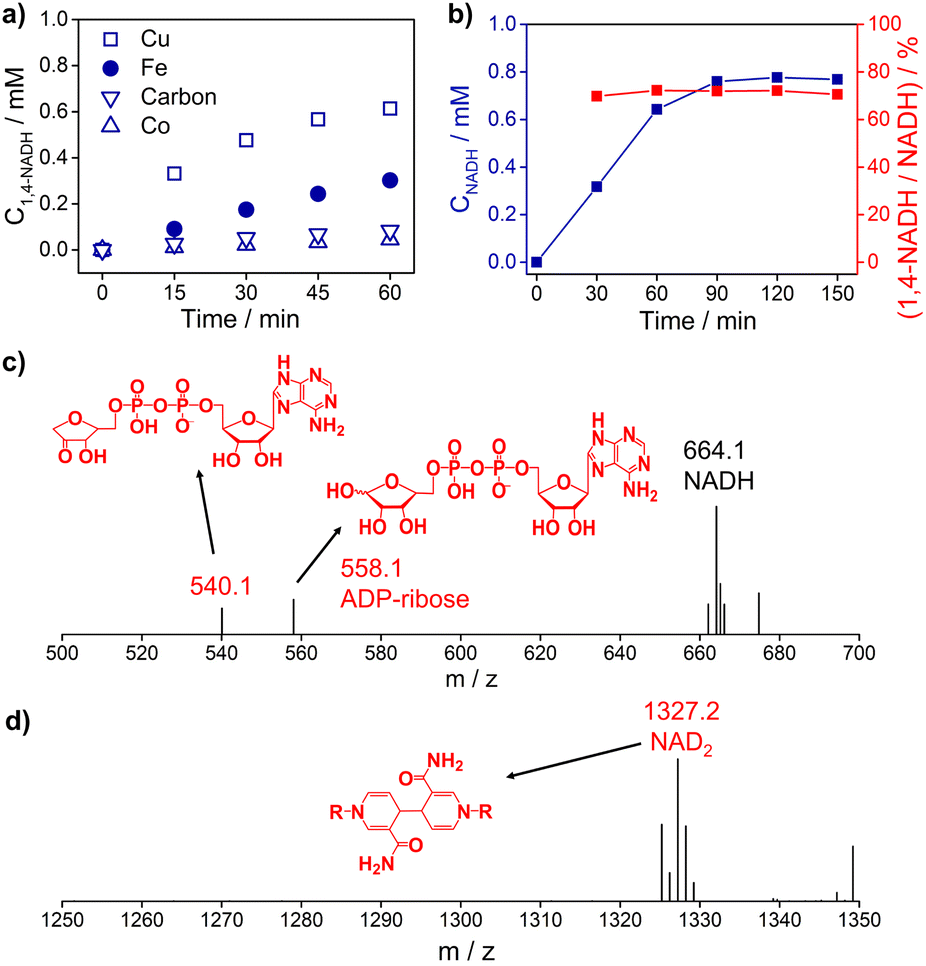 | ||
| Fig. 3 (a) The 1,4-NADH concentration trends on Cu foam, Fe foam, Co foam, and carbon felt under −0.5 VRHE. (b) The concentration of NADH and selectivity of 1,4-NADH versus total NADH (1,4-NADH + 1,6-NADH) concentration as a function of time. The electrospray ionization mass spectrometry (ESI-MS) of electrocatalytic NAD+ reduction products on (c) Cu foam and (d) carbon felt. Electrolyte: 0.1 M phosphate buffer (pH 7); catholyte: 12 mL electrolyte with an initial NAD+ concentration of 1 mM. ESI-MS full spectra of electrocatalytic NAD+ reduction products on Cu foam and carbon felt are shown in Fig. S6.† | ||
Encouraged by the above results, we propose a possible reaction mechanism for NAD+ reduction, as illustrated in Fig. 4. Considering that the nicotinamide moiety of NAD+ is positively charged, the H+ transfer as the initial step is difficult. For NAD+ reduction with organometallic homogenous catalysts, such as Rh complexes, it is well acknowledged that Rh–H is first formed, and then, NADH is produced through one-step hydride transfer between the reduced Rh–H and NAD+ by forming a special ring transition state.21 In previous reports, for (photo)electrocatalysis without a charge transfer mediator, the reaction mechanism was inconsistent. A hydride transfer mechanism has been proposed for NADH regeneration with the Pt/p-GaAs photocathode.39 However other studies performed on a variety of metal electrodes such as Au and Hg have led to a commonly established reduction mechanism that the first electron transfer is the initial step forming the NAD˙ radical, which tends to dimerize quickly and may also accept another electron and one proton forming 1,4-NADH and side products such as 1,6-NADH.48 Yet for the case of our tested metal catalysts, based on the above product analysis results, no NAD2 is produced in the reaction. Such being the case, we propose that no NAD˙ is produced in the reaction because the rate of NAD˙ dimerization is very fast with a rate constant of 3 × 106 M s−1,46 much faster than forming NADH by accepting a proton and one electron.48 This means that the NAD+ reduction mechanism on the tested metal electrodes is quite different from the NAD˙ pathway.
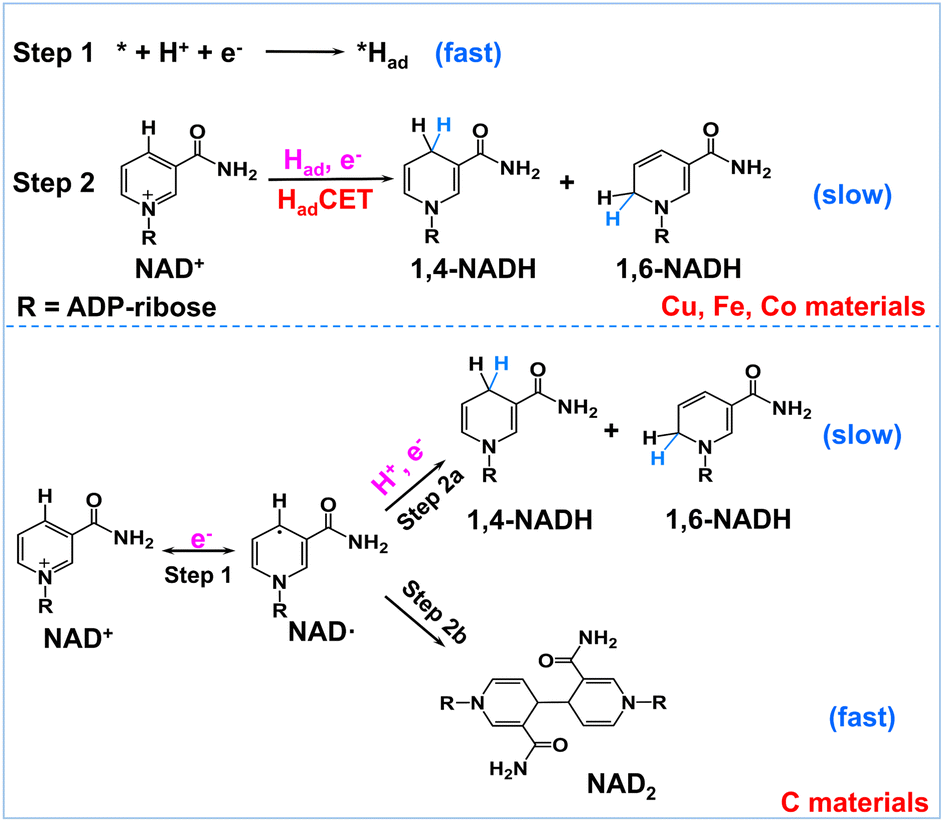 | ||
| Fig. 4 Schematic description of the possible reaction mechanism of NAD+ reduction on Had-rich metal electrodes and Had-poor carbon electrodes. | ||
In natural photosynthesis, NADPH is produced through the hydride transfer from FADH− to NADP+ controlled by the FNR enzyme.43 On metal electrodes, hydride species are usually difficult to form and the hydride transfer mechanism can be excluded. Nevertheless, many organic, organometallic, and biological systems involving (C, N, O, S)–H and metal–H bonds, such as Ru-based water oxidation complexes, artificial and natural water oxidation clusters, hydrogenases, flavin cofactors and heme proteins, are related to the proton-coupled electron transfer (PCET) process.49–51 Natural organisms are masterful in managing PCET to drive the photosynthesis and nitrogen fixation reactions. It is worth noting that the surface of metal electrodes may be covered with many surface-adsorbed hydrogen atoms (Had) as they have high proton activation ability, considering that they are widely used for hydrogen evolution or hydrogenation reactions. With these points in mind, we deduce that Had is primarily involved in the NAD+ reduction reaction on Had-rich metal catalysts. To be specific, Had atoms are first formed via the Volmer process (* + H+ + e− → *Had, * is the active site). Second, NADH is produced via the reaction of Had and NAD+ coupled with an electron transfer (NAD+ + *Had + e− → * + NADH). This Had-coupled electron transfer (HadCET) mechanism is similar and coincides with the commonly observed PCET process in natural biology except that the Had atom on the electrode surface is the hydrogen source for HadCET here. It may occur via a stepwise electron–Had or Had–electron transfer process, or through a concerted Had–electron transfer process, which needs more research. Interestingly, we also evaluated the performance of Pt, Ni, Ru, and Ag for comparison and found that no or trace NADH is produced with these electrodes (Fig. S7†). It is inferred that the Had atoms formed on Pt, Ni, and Ru surfaces tend to form H2 molecules and the formation of Had is relatively difficult on the Ag surface, judging from their ΔGH and exchange current density for hydrogen evolution in the literature (Table S1†).52–54 This means that a high coverage of Had on metal electrodes is favorable for NADH formation. In the case of the carbon electrode, it has poor proton activation ability and its surface is likely to have a low coverage of Had. Therefore, the NAD+ reduction may follow the direct one-electron transfer and NAD˙ radical mechanism mentioned in the literature.55
To obtain further insights into the reaction mechanism of NAD+ reduction, we used the EPR technique to detect the hydrogen species on the electrode surface. The Had atoms may diffuse into the nearby electrolyte forming H˙ radicals, which can be captured by the radical trapping reagent 5,5-dimethyl-1-pyrroline-N-oxide (DMPO, Fig. S8†). Fig. 5a displays the EPR results when adding DMPO to the NAD+-free electrolyte after electrolysis with Cu and carbon as working electrodes at −0.4 VRHE. The signals with Cu show an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2:1:2:1:2:1:1 and their hyperfine coupling constants are A(N) = 16.43 G and A(H) = 22.68 G, which are consistent with the characteristic signals of DMPO–H.56 The DMPO–H signals can be obviously observed with Cu, while the EPR signals are negligible with the carbon electrode, which confirms that the surface of Cu is rich in Had in sharp contrast to carbon which is poor in Had. And obvious DMPO–D signals can be observed in the EPR spectrum (Fig. S9†) of the solution after electrolysis with the Cu electrode in deuterated buffer, which consolidates our conclusion that there are many Had atoms on the Cu electrode surface. In addition, the probability of NADH production via the reaction between H˙ and NAD+ with the participation of another electron from the electrode is low. Therefore, the EPR results further rationalize that NADH on metal electrodes such as Cu, Fe, and Co is produced via the HadCET process proposed above.
:1:2:1:2:1:2:1:1 and their hyperfine coupling constants are A(N) = 16.43 G and A(H) = 22.68 G, which are consistent with the characteristic signals of DMPO–H.56 The DMPO–H signals can be obviously observed with Cu, while the EPR signals are negligible with the carbon electrode, which confirms that the surface of Cu is rich in Had in sharp contrast to carbon which is poor in Had. And obvious DMPO–D signals can be observed in the EPR spectrum (Fig. S9†) of the solution after electrolysis with the Cu electrode in deuterated buffer, which consolidates our conclusion that there are many Had atoms on the Cu electrode surface. In addition, the probability of NADH production via the reaction between H˙ and NAD+ with the participation of another electron from the electrode is low. Therefore, the EPR results further rationalize that NADH on metal electrodes such as Cu, Fe, and Co is produced via the HadCET process proposed above.
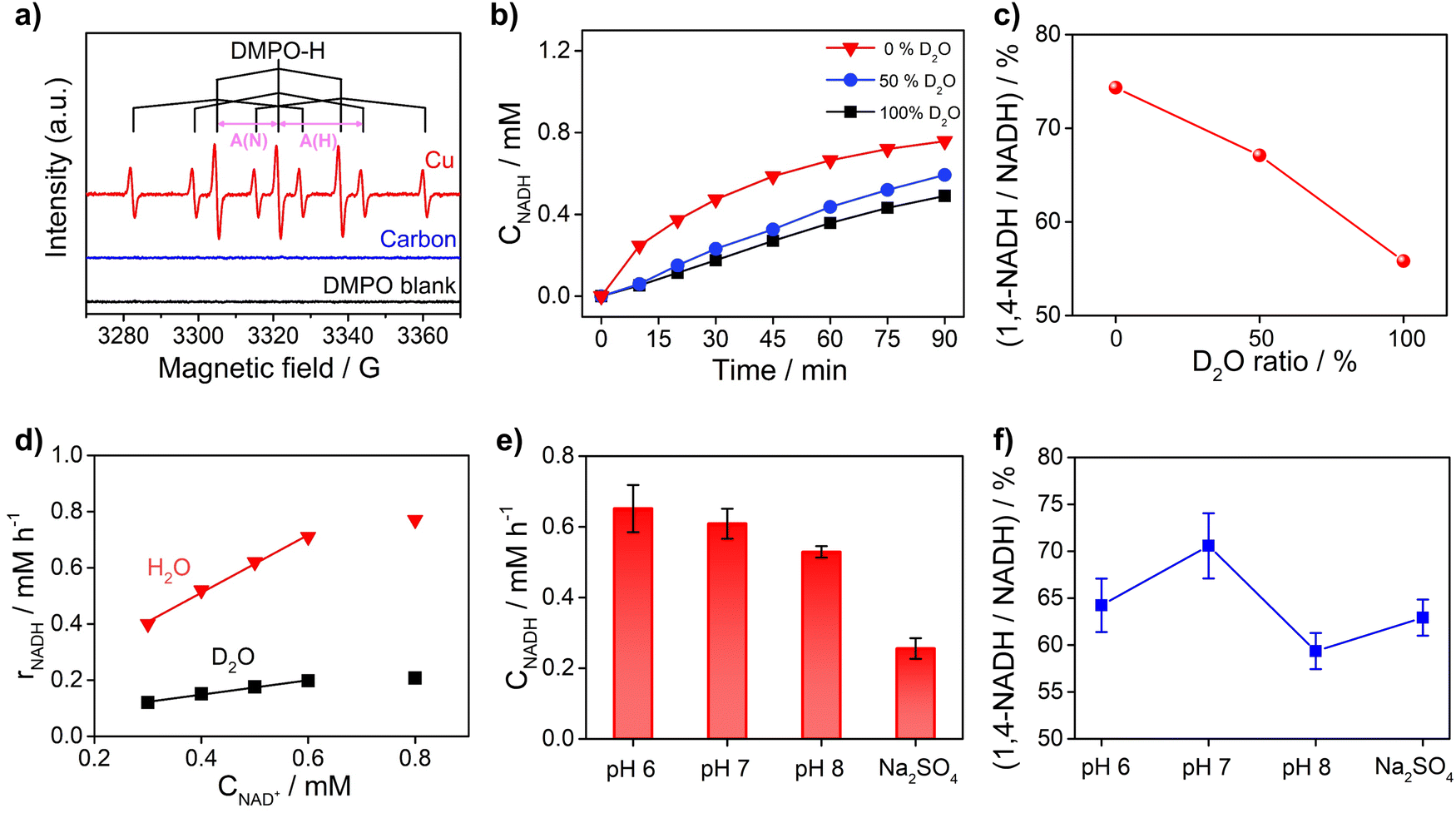 | ||
| Fig. 5 (a) EPR spectra in the X-band of the solutions obtained after electrolysis with Cu foam and carbon felt in 0.1 M phosphate buffer (pH 7) under argon using DMPO as the H˙ trapping reagent. The blank experiment is the EPR result of electrolyte without electrolysis after adding DMPO. (b) Activity and (c) selectivity of the NAD+ reduction reaction on Cu foam in 0.1 M phosphate buffer with different contents of D2O solvent. (d) Kinetic isotope effect of the NAD+ reduction reaction on Cu foam. (e) The activity and (f) selectivity of NADH regeneration at different pH and electrolytes. Electrolyte: 0.1 M phosphate buffer (pH 6, 7 and 8) and unbuffered 0.1 M Na2SO4 aqueous solution; catholyte: 12 mL electrolyte with an initial NAD+ concentration of 1 mM. | ||
Furthermore, the critical role of Had for the NAD+ reduction reaction on metal electrodes was evidenced by the H/D isotope effect and reaction kinetic analysis. As shown in Fig. 5b, the activity of NADH production in H2O electrolyte is 2–4 times of that in electrolyte with D2O as the solvent, suggesting that hydrogen species play a key role in the rate-determining step (RDS) of NAD+ reduction to NADH. More interestingly, the selectivity for 1,4-NADH declines obviously with the increase of the D2O ratio in the electrolyte (Fig. 5c), which can be explained by the different coverage of Had and Dad atoms due to their different production and consumption rates. In addition, as shown in Fig. 5d, the NADH formation rate (r) increases linearly with the initial NAD+ concentration from 0.3 mM to 0.6 mM (Fig. S10 and S11†), indicating a first-order reaction kinetics (r = kCNAD+, k = 1.03 h−1). Then, we explored the kinetic isotope effect (KIE, KIE = kH/kD) of the NAD+ reduction reaction in phosphate buffer (pH 7) with D2O as the solvent. A KIE value of 3.96 is calculated from the reaction rate constants in H2O and D2O electrolytes, suggesting a primary isotope effect. If the C–H formation is the only RDS, KIE should be around 7. A lower KIE means there is not only one RDS. The solvent isotope effect which results from isotopic differences in the medium can be excluded because H2O cannot directly participate in the NAD+ reduction reaction. The NADH formation by electrocatalytic NAD+ reduction involves the breaking of metal–Had and the electron transfer process, as shown in Fig. S12.† We speculate that the rates of metal–Had breaking and C–H formation of NADH may be similar. The RDSs of NAD+ reduction to NADH involve the breakage of the metal–Had bond and formation of the C–H bond during the hydrogenation of the nicotinamide group (Fig. S12†). The above results of the H/D isotope effect and KIE analysis all point to the HadCET mechanism for NADH production mentioned above.
Moreover, the pH effect on the activity of NAD+ reduction with the Cu electrode was also studied (Fig. 5e and f). The total NADH (1,4- and 1,6-NADH) production follows the trend of pH 6 > pH 7 > pH 8. That's because the formation of Had is difficult under alkaline conditions, and thus, the coverage of Had is low and the reaction between NAD+ and Had is also slow. Interestingly, the selectivity of 1,4-NADH at pH 6 is lower than that of pH 7, indicating that the relatively high Had coverage at pH 6 may result in more random attacking of Had on NAD+.57 In addition, 1,4-NADH tends to decompose through acid-catalyzed protonation of the nicotinamide ring.58 For the control experiment with unbuffered Na2SO4 electrolyte, the activity and selectivity are obviously lower than those of neutral buffered electrolyte. This can be explained that there is an increase in local pH during the NAD+ reduction reaction as it is a proton-consuming reaction. So, careful pH control with buffered electrolyte is important for the NAD+ reduction reaction, and neutral phosphate buffer (pH 7) electrolyte delivers the highest activity for 1,4-NADH production. Therefore, these results of KIE and pH effects on the NAD+ reduction reaction, similar to the features for the PCET process,49 further supports the proposed HadCET mechanism. In addition to the distinct coverage of Had on metal and carbon electrodes, their surface structure and the adsorption of NAD+ may be different in orientation and conformation, which needs more studies.
As for the formation of ADP-ribose, it was previously observed in the photocatalytic NADH oxidation reaction,59 but was rarely reported for the NAD+ reduction reaction. The possibility of the destructive hydrogenation reaction by H2 molecules can be excluded because there is no change of the NMR spectra after bubbling H2 through NAD+ solution for 3 h (Fig. S13†). Here, the formation of ADP-ribose may be caused by the fragmentation of NAD+ or the NAD˙ radical.59,60 For metal electrodes, there are many Had atoms on electrode surfaces. And three different sites in NAD+ may be attacked by Had, as shown in Fig. 6. The hydrogenation of C4 (path 1) and C2 (path 2) sites leads to the formation of 1,4-NADH and 1,6-NADH. The positively charged N site in the nicotinamide group tends to suffer nucleophilic attack (path 3) by Had, resulting in the breaking of the N–C bond and ADP-ribose fragments. For the carbon electrode, ADP-ribose comes from the fragmentation of the NAD˙ radical,59 and the amount of ADP-ribose is lower at more negative potential (Fig. 2d). However, the amount of ADP-ribose increases as the applied potential is more negative on metal electrodes (Fig. 2a–c), which shows little difference for Cu, Fe, and Co electrodes possibly as the adsorption configurations of NAD+ on these species are similar. Further work on suppressing ADP-ribose formation and promoting 1,4-NADH production by controlling the adsorption configuration of NAD+ and increasing the coverage of Had with rationally designed catalysts is still under research.
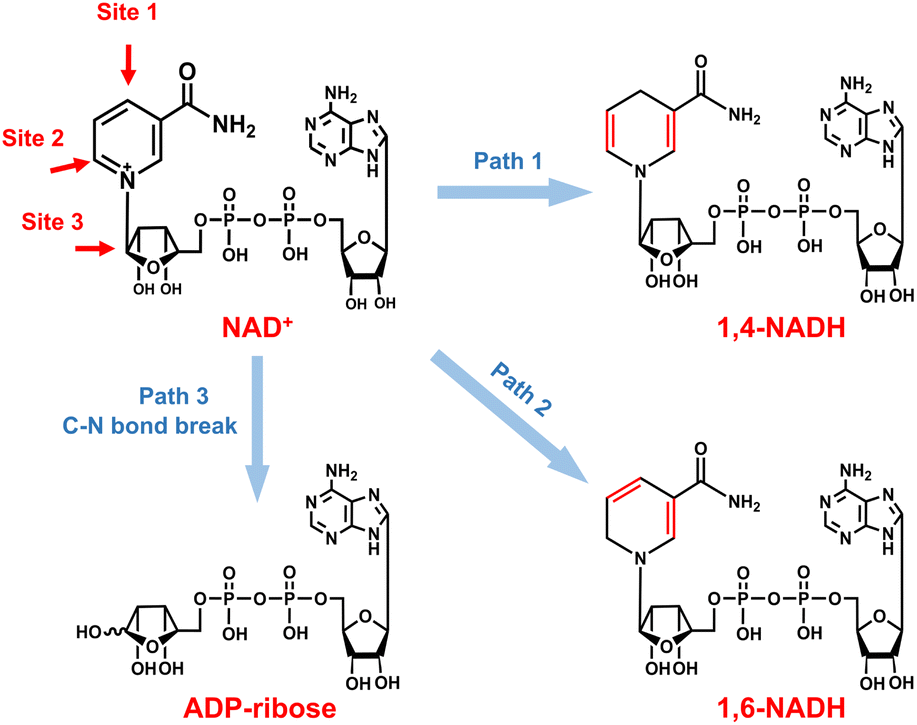 | ||
| Fig. 6 Possible reaction pathways of NAD+ with different attacking sites of Had and the corresponding products. | ||
Conclusions
Electrocatalytic NADH regeneration with various metal electrodes and carbon catalysts was investigated with quantification of all products, among which ADP-ribose is confirmed to be a side product caused by the fragmentation reaction. We find that the selectivity of bioactive 1,4-NADH is relatively high on Cu, Fe, and Co electrodes without forming side product NAD2, suggesting a mechanism involving Had on the surface rather than the previously reported NAD˙ intermediate. In contrast, there is obvious formation of NAD2 for the carbon electrode with low selectivity for 1,4-NADH. We propose that the formation of NADH on these metal electrodes proceeds via a HadCET mechanism, in contrast to the direct electron transfer and NAD˙ radical pathway on carbon electrodes.Author contributions
Fengyuan Liu and Chunmei Ding conceived the project and wrote the manuscript under the supervision of C. Li. All authors discussed the results and contributed to manuscript editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFC2103500). This work was also conducted by the Fundamental Research Center of Artificial Photosynthesis (FReCAP), financially supported by the National Natural Science Foundation of China (grant nos. 21872141 and 22088102), Dalian National Laboratory for Clean Energy (DNL201911), and Dalian Institute of Chemical Physics (DICP ZZBS201710). C. Ding acknowledges the support from Liaoning Revitalization Talents Program (XLYC2007002), Dalian Youth Science and Technology Program (2019RQ022), and Youth Innovation Promotion Association CAS (2022178). We thank Dr Guang Zeng for his help in the EPR test and data analysis.Notes and references
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- M. Medina, FEBS J., 2009, 276, 3942–3958 CrossRef CAS PubMed.
- A. Aliverti, V. Pandini, A. Pennati, M. de Rosa and G. Zanetti, Arch. Biochem. Biophys., 2008, 474, 283–291 CrossRef CAS PubMed.
- W. Jones, J. W. H. Burnett, J. Shi, R. F. Howe and X. Wang, Joule, 2020, 4, 2055–2059 CrossRef.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Z. Li, W. Luo, M. Zhang, J. Feng and Z. Zou, Energy Environ. Sci., 2013, 6, 347–370 RSC.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- J. H. Kim and J. S. Lee, Adv. Mater., 2019, 31, 1806938 CrossRef PubMed.
- X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 CrossRef CAS PubMed.
- W. Li, D. He, S. W. Sheehan, Y. He, J. E. Thorne, X. Yao, G. W. Brudvig and D. Wang, Energy Environ. Sci., 2016, 9, 1794–1802 RSC.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- S. Wang, G. Liu and L. Wang, Chem. Rev., 2019, 119, 5192–5247 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- F. Hollmann, I. W. C. E. Arends and K. Buehler, ChemCatChem, 2010, 2, 762–782 CrossRef CAS.
- T. W. Johannes, R. D. Woodyer and H. Zhao, Biotechnol. Bioeng., 2007, 96, 18–26 CrossRef CAS PubMed.
- T. Quinto, V. Köhler and T. R. Ward, Top. Catal., 2014, 57, 321–331 CrossRef CAS.
- H. Wu, C. Tian, X. Song, C. Liu, D. Yang and Z. Jiang, Green Chem., 2013, 15, 1773–1789 RSC.
- R. K. Yadav, G. H. Oh, N.-J. Park, A. Kumar, K.-j. Kong and J.-O. Baeg, J. Am. Chem. Soc., 2014, 136, 16728–16731 CrossRef CAS PubMed.
- V. Ganesan, D. Sivanesan and S. Yoon, Inorg. Chem., 2017, 56, 1366–1374 CrossRef CAS PubMed.
- H. C. Lo, O. Buriez, J. B. Kerr and R. H. Fish, Angew. Chem., Int. Ed., 1999, 38, 1429–1432 CrossRef CAS PubMed.
- B. Zhang, S. Xu, D. He, R. Chen, Y. He, W. Fa, G. Li and D. Wang, J. Chem. Phys., 2020, 153, 064703 CrossRef CAS PubMed.
- Q. Pan, H. Liu, Y. Zhao, S. Chen, B. Xue, X. Kan, X. Huang, J. Liu and Z. Li, ACS Appl. Mater. Interfaces, 2019, 11, 2740–2744 CrossRef CAS PubMed.
- S. Kim, S. Lee, T. F. Anjong, H. Y. Jang, J.-Y. Kim, C. Lee, S. Park, H. J. Lee, J. Yoon and J. Kim, J. Phys. Chem. C, 2016, 120, 28407–28414 CrossRef CAS.
- S. Y. Lee, S. Y. Lim, D. Seo, J.-Y. Lee and T. D. Chung, Adv. Energy Mater., 2016, 6, 1502207 CrossRef.
- S. K. Kuk, R. K. Singh, D. H. Nam, R. Singh, J.-K. Lee and C. B. Park, Angew. Chem., Int. Ed., 2017, 56, 3827–3832 CrossRef CAS PubMed.
- E. J. Son, J. W. Ko, S. K. Kuk, H. Choe, S. Lee, J. H. Kim, D. H. Nam, G. M. Ryu, Y. H. Kim and C. B. Park, Chem. Commun., 2016, 52, 9723–9726 RSC.
- S. H. Lee, G. M. Ryu, D. H. Nam, J. H. Kim and C. B. Park, ChemSusChem, 2014, 7, 3007–3011 CrossRef CAS PubMed.
- S. Choudhury, J.-O. Baeg, N.-J. Park and R. K. Yadav, Green Chem., 2014, 16, 4389–4400 RSC.
- Y. Cheng, J. Shi, Y. Wu, X. Wang, Y. Sun, Z. Cai, Y. Chen and Z. Jiang, Research, 2021, 2021, 8175709 CrossRef CAS PubMed.
- Y. Sun, J. Shi, Z. Wang, H. Wang, S. Zhang, Y. Wu, H. Wang, S. Li and Z. Jiang, J. Am. Chem. Soc., 2022, 144, 4168–4177 CrossRef CAS PubMed.
- J. Lutz, F. Hollmann, T. V. Ho, A. Schnyder, R. H. Fish and A. Schmid, J. Organomet. Chem., 2004, 689, 4783–4790 CrossRef CAS.
- M. Poizat, I. W. C. E. Arends and F. Hollmann, J. Mol. Catal. B: Enzym., 2010, 63, 149–156 CrossRef CAS.
- F. Hildebrand and S. Lütz, Chem.–Eur. J., 2009, 15, 4998–5001 CrossRef CAS PubMed.
- X. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. Shi, Chem, 2017, 2, 621–654 CAS.
- R. Barin, S. Rashid-Nadimi, D. Biria and M. A. Asadollahi, Electrochim. Acta, 2017, 247, 1095–1102 CrossRef CAS.
- H.-K. Song, S. H. Lee, K. Won, J. H. Park, J. K. Kim, H. Lee, S.-J. Moon, D. K. Kim and C. B. Park, Angew. Chem., Int. Ed., 2008, 47, 1749–1752 CrossRef CAS PubMed.
- A. Azem, F. Man and S. Omanovic, J. Mol. Catal. A: Chem., 2004, 219, 283–299 CrossRef CAS.
- P. Stufano, A. R. Paris and A. Bocarsly, ChemElectroChem, 2017, 4, 1066–1073 CrossRef CAS.
- I. Ali, T. Khan and S. Omanovic, J. Mol. Catal. A: Chem., 2014, 387, 86–91 CrossRef.
- I. Ali, N. Ullah, M. A. McArthur, S. Coulombe and S. Omanovic, Can. J. Chem. Eng., 2018, 96, 68–73 CrossRef CAS.
- A. Damian and S. Omanovic, J. Mol. Catal. A: Chem., 2006, 253, 222–233 CrossRef CAS.
- I. Lans, M. Medina, E. Rosta, G. Hummer, M. Garcia-Viloca, J. M. Lluch and À. González-Lafont, J. Am. Chem. Soc., 2012, 134, 20544–20553 CrossRef CAS PubMed.
- K. Cheikhou and T. Tzédakis, AIChE J., 2008, 54, 1365–1376 CrossRef CAS.
- S. K. Yoon, E. R. Choban, C. Kane, T. Tzedakis and P. J. A. Kenis, J. Am. Chem. Soc., 2005, 127, 10466–10467 CrossRef CAS PubMed.
- W. T. Bresnahan and P. J. Elving, J. Am. Chem. Soc., 1981, 103, 2379–2386 CrossRef CAS.
- B. A. Beaupre, M. R. Hoag, J. Roman, F. H. Foersterling and G. R. Moran, Biochemistry, 2015, 54, 795–806 CrossRef CAS PubMed.
- A. Damian, K. Maloo and S. Omanovic, Chem. Biochem. Eng. Q., 2007, 21, 21–32 CAS.
- D. G. Nocera, J. Am. Chem. Soc., 2022, 144, 1069–1081 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- S. L. J. Tan, J. M. Kan and R. D. Webster, J. Phys. Chem. B, 2013, 117, 13755–13766 CrossRef CAS PubMed.
- J. K. Noerskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed.
- H. Jaegfeldt, Bioelectrochem. Bioenerg., 1981, 8, 355–370 CrossRef CAS.
- Y. Wang, D. Zhao, H. Ji, G. Liu, C. Chen, W. Ma, H. Zhu and J. Zhao, J. Phys. Chem. C, 2010, 114, 17728–17733 CrossRef CAS.
- S. H. Lee, J. H. Kim and C. B. Park, Chem.–Eur. J., 2013, 19, 4392–4406 CrossRef CAS PubMed.
- P. L. Hentall, N. Flowers and T. D. H. Bugg, Chem. Commun., 2001, 2098–2099 RSC.
- S. Zhang, J. Shi, Y. Chen, Q. Huo, W. Li, Y. Wu, Y. Sun, Y. Zhang, X. Wang and Z. Jiang, ACS Catal., 2020, 10, 4967–4972 CrossRef CAS.
- U. Vitinius, K. Schaffner, M. Demuth, M. Heibel and H. Selbach, Chem. Biodiversity, 2004, 1, 1487–1497 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc02691k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |