
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmino group induced structural diversity and near-infrared emission of yttrium-tetracarboxylate frameworks†
Hai-Lun
Xia
a,
Kang
Zhou
a,
Jiandong
Guo
a,
Jian
Zhang
ab,
Xiaoxi
Huang
 a,
Dawei
Luo
b,
Xiao-Yuan
Liu
*a and
Jing
Li
*ac
a,
Dawei
Luo
b,
Xiao-Yuan
Liu
*a and
Jing
Li
*ac
aHoffmann Institute of Advanced Materials, Shenzhen Polytechnic, 7098 Liuxian Blvd, Nanshan District, Shenzhen, 518055, P. R. China. E-mail: liuxiaoyuan1989@szpt.edu.cn
bSchool of Materials and Environmental Engineering, Shenzhen Polytechnic, 7098 Liuxian Blvd, Nanshan District, Shenzhen, 518055, P. R. China
cDepartment of Chemistry and Chemical Biology, Rutgers University, 123 Bevier Road, Piscataway, New Jersey 08854, USA. E-mail: jingli@rutgers.edu
First published on 21st July 2022
Abstract
Near-infrared (NIR)-emitting materials have been extensively studied due to their important applications in biosensing and bioimaging. Luminescent metal–organic frameworks (LMOFs) are a new class of highly emissive materials with strong potential for utilization in biomedical related fields because of their nearly unlimited structural and compositional tunability. However, very little work has been reported on organic linker-based NIR-MOFs and their emission properties. In the present work, a series of yttrium-tetracarboxylate-based LMOFs (HIAM-390X) are prepared via judicious linker design to achieve NIR emission with diverse structures. The introduction of an amino group not only offers the remarkable emission bathochromic shift from 521 nm, 665 nm to 689 nm for the resultant MOFs, but also influences the linker conformations, leading to the topology evolution from (4,12)-c ftw, (4,8)-c scu, which is rarely reported in rare earth element-based MOFs, to an unprecedented topology hlx for HIAM-3901 (without an amino group), HIAM-3905 (with one amino group) and HIAM-3906 (with two amino groups). Among these MOFs, HIAM-3907 shows an emission maximum at ∼790 nm, with the emission tail close to 1000 nm. The NIR emission may be attributed to the combination of the strongly electron-donating amino group and the strongly electron-withdrawing acceptor naphtho[2,3-c][1,2,5]selenadiazole. This work sheds light on the rational design of organic linker-based LMOFs with controlled structures and NIR emission, and inspires future interest in biosensing and bioimaging related applications of NIR-MOFs.
Introduction
Luminescent metal–organic frameworks (LMOFs) built on emissive organic linkers exhibit strong promise in various applications, including, but not limited to, solid-state-light,1–6 sensing7–13 and bioimaging,14,15 owing to their easily tunable linker structures and highly accessible functional sites.16 However, most LMOFs reported to date emit light within the visible light spectrum except those using a rare-earth-element as the metal node, particularly Yb, Nd and Er, which usually need organic chromophores as sensitizers and exhibit limited emission tunability due to the specific electronic structures of rare earth elements.17–19 There is a lack of study on near-infrared emissive MOFs (NIR-MOFs) based on organic linkers that emit light in the NIR region.20 The major challenge is to design and synthesize organic linkers that are suitable for the synthesis of MOFs with NIR emission.In recent years, different kinds of NIR emissive materials, such as carbon nanotubes,21 quantum dots,22 rare earth element-based nanoparticles23,24 and small molecules,25,26 have been gradually developed to explore their bio-related applications. To enhance the performances, it is important to extend the emission to a longer wavelength range, to ensure negligible tissue absorption and minimum auto-fluorescence and reduced scattering, particularly for materials that emit in the NIR-II region. Among them, molecular probes with the emission wavelengths within the 700–1700 nm range, such as benzo[1,2-c:4,5-c′]bis([1,2,5]thiadiazole) (BBTD) and its derivative-based compounds, have been extensively studied, particularly for bioimaging and biosensing related research,25,27–29 due to their high spatiotemporal resolution, good penetration depth and non-invasiveness. However, to achieve NIR emission, most of the organic compounds are designed with large conjugated backbones, which usually suffer from low quantum efficiency due to strong intermolecular interactions and the aggregation-caused quenching (ACQ) effect.29–31 Recent studies have shown that ACQ could be suppressed by confining or immobilizing organic linkers or emissive guests in the MOF matrix.32,33 In addition, the unique properties of MOFs, such as high porosity and pore surface tunability, will also endow NIR-MOFs with more potential for practical applications, e.g. drug delivery and light-triggered treatment of diseases. Therefore, developing organic linker-based NIR-MOFs is an important and much desired research area due to their applications in bioimaging and sensing.
The key to success is to design suitable linker structures to achieve broad light absorption and emission, and to use them to prepare NIR-MOFs. It has been reported that introduction of electron-donating amino groups into BBTD-based molecules can lead to a significant emission red-shift.34–36 We have also discovered a 229 nm red-shift when carboxylic acid groups were substituted by amino groups, from 4,4′-(benzo[c][1,2,5]thiadiazole-4,7-diyl)dibenzoic acid (H2BTBA, 480 nm) to 4,4′-(benzo[c][1,2,5]thiadiazole-4,7-diyl)dianiline (BTDA, 709 nm) (Fig. 1a and b). Furthermore, in principle, NIR emissive organic linkers can be designed and synthesized via increasing the energy of the highest occupied molecular orbitals (HOMO) or decreasing the energy of the lowest unoccupied molecular orbitals (LUMO). Particularly, for donor–acceptor–donor (D–A–D) type compounds, three different strategies can be employed to achieve a bathochromic shift in emission: (i) decreasing the electron density of acceptors, (ii) increasing the electron density of donor groups and (iii) elongating the conjugated chain between the donor and acceptor.26,37,38 For example, when the acceptor group was changed from benzo[c][1,2,5]thiadiazole in H2BTBA to naphtho[2,3-c][1,2,5]selenadiazole to form H2NSB, a 154 nm bathochromic shift was observed (Fig. 1b).39
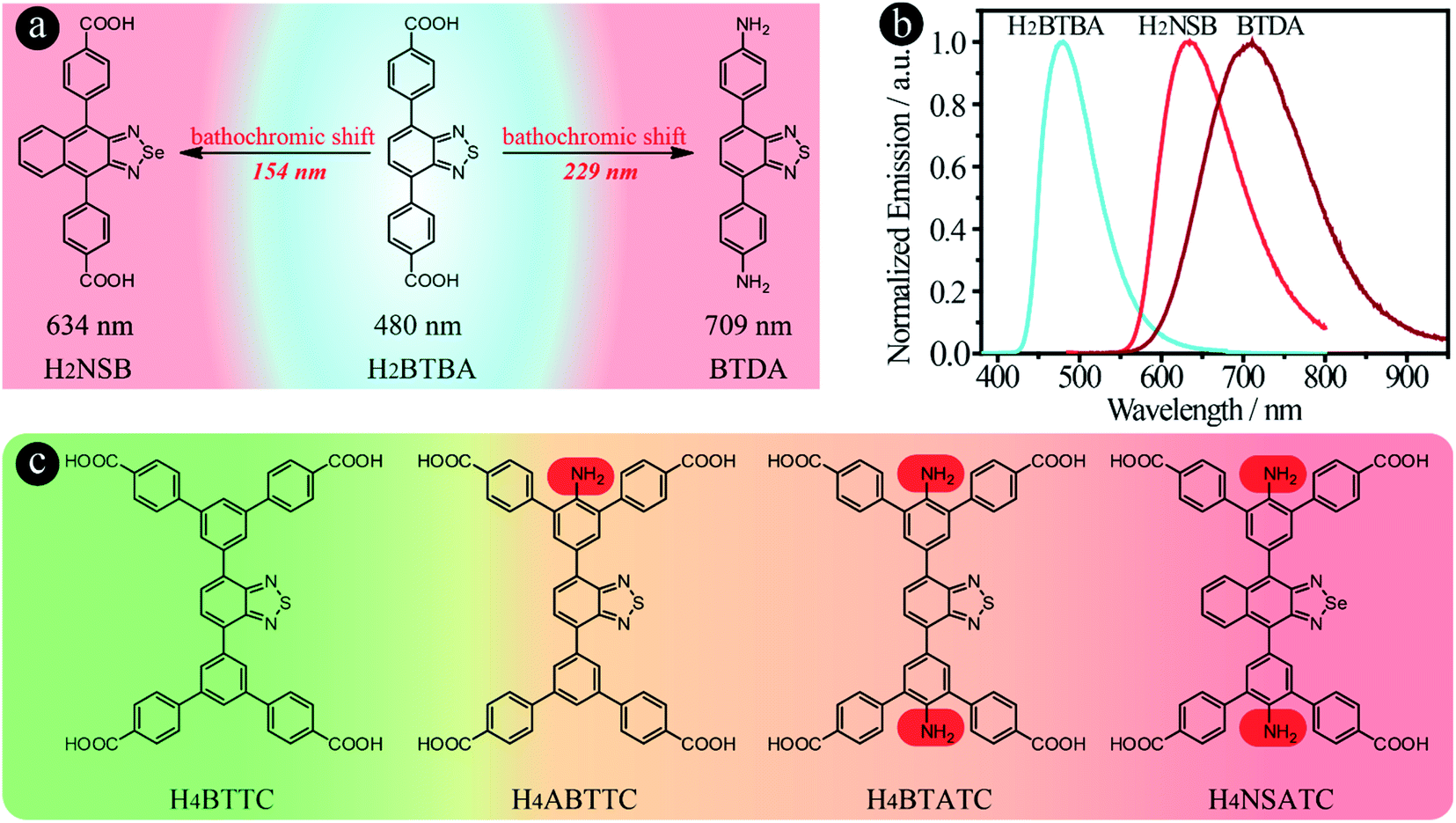 | ||
| Fig. 1 The molecular structures of H2NSB, H2BTBA and BTDA with the corresponding emission wavelengths (a); the normalized emission spectra of H2NSB, H2BTBA and BTDA in DMF under 365 nm excitation (b); the molecular structures of H4BTTC, H4ABTTC, H4BTATC and H4NSATC used in this work to prepare LMOFs with controllable structures and emission toward the NIR range (c). | ||
One useful approach to introduce amino groups into organic linkers for synthesizing MOFs is to use tetracarboxylate-based compounds as the skeleton.40 On the other hand, tetratopic carboxylate linkers used to prepare MOFs usually lead to various topologies, thereby enriching the structural diversity which leads to enhancement in the performance or extension of the applications.40–42 In the case of benzo[c][1,2,5]thiadiazole and its derivative-based tetratopic carboxylate linkers, i.e. 5′,5′′′′-(5,6-dimethylbenzo[c][1,2,5]thiadiazole-4,7-diyl)bis([1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid) (H4DTTC), 5′,5′′′′-(benzo[c][1,2,5]thiadiazole-4,7-diyl)bis(([1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid)) (H4BTTC), 5′,5′′′′-(benzo[c][1,2,5]selenadiazole-4,7-diyl)bis(([1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid)) (H4BSTC), 5′,5′′′′-(naphtho[2,3-c][1,2,5]thiadiazole-4,9-diyl)bis(([1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid)) (H4NTTC), and 5′,5′′′′-(naphtho[2,3-c][1,2,5]selenadiazole-4,9-diyl)bis(([1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid)) (H4NSTC),43 three topologies (scu, scp and csq) have been reported with the Zr6 cluster as the secondary building unit (Fig. 2).44 However, apart from the fact that the ftw topology of Zr-based MOFs has strict requirement regarding the linker size (i.e., aspect ratio), we find that it is very difficult to obtain the ftw structure even when using linkers that have a suitable size. Attempts to form Zr6-based ftw structures have failed even after using numerous combinations of three common organic solvents and six acids under different reaction conditions. However, the Y6 cluster shows high flexibility to the aspect ratio of linkers in forming the ftw topology.45,46 Therefore, yttrium was chosen as the metal in the present work to investigate the structural diversity and achieve NIR-MOFs. In addition, as it is well known that addition of substituents, such as amino, hydroxyl and methyl groups, on the molecular skeleton will introduce steric hindrance, which would dictate the resulting MOFs’ structures,40,47,48 we thus speculate that the introduction of amino groups may not only induce the emission red-shift, but also lead to diverse structures of the resultant MOFs due to steric hindrance, which is rarely studied in Y-MOFs.
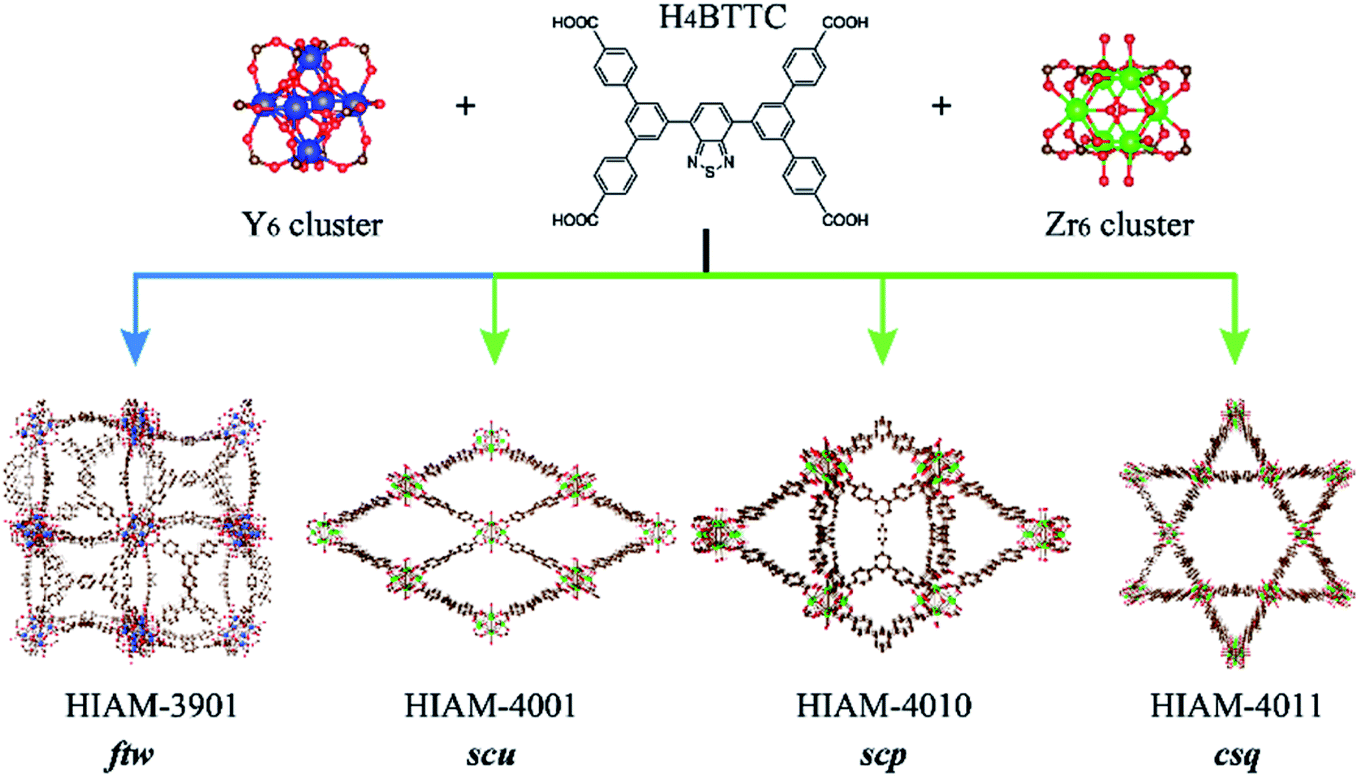 | ||
| Fig. 2 The topology evolution of LMOFs prepared using Zr6/Y6 clusters and H4BTTC as the organic linker (color scheme: brown, C; green, Zr; dark blue, Y; red, O; yellow, S; blue, N). | ||
Results and discussion
ftw-type Y-MOFs with tunable emission toward the NIR region
Bearing the aforementioned discussions in mind, we first attempted to construct Y-MOFs with the ftw topology using H4DTTC, H4BTTC, H4BSTC, H4NTTC and H4NSTC as the organic linkers (Fig. 3a). Single crystals of five Y-MOFs, HIAM-390X (HIAM = Hoffmann Institute of Advanced Materials; 39 = yttrium), were obtained according to the reported procedures with some modifications.45 A typical synthesis is schematically shown in Fig. 3a: the reaction was carried out in a 5 mL vial containing 2.5 mL DMF, Y(NO3)3·6H2O (13.0 mg, 0.034 mmol), organic linker and varying amounts of 2-fluorobenzoic acid in a preheated oven at 120 °C for 24 hours. The synthetic procedures for each MOF are described in detail in the Experimental section. Depending on the properties of the organic linker, colorless (HIAM-3900), light yellow (HIAM-3901), yellow (HIAM-3902), light orange (HIAM-3903) and red (HIAM-3904) cube-shaped single crystals were obtained with blue, green, yellow, orange and red emission (Fig. 3b). Although we were able to obtain single crystals of appropriate sizes for all five MOFs, it was still very difficult to solve their crystal structures due to the weak diffraction. However, we found that the powder X-ray diffraction (PXRD) patterns of these MOFs match well with those of simulated Y-ftw-MOF-2 (Fig. 3c),45 where a similar linker, 5′,5′′′-bis(4-carboxyphenyl)-[1,1′:3′,1′′:4′′,1′′′:3′′′,1′′′′-quinque-phenyl]-4,4′′′′-dicarboxylic acid, was used as the organic linker, indicating that they are isoreticular structures. Therefore, the structural characterization of the HIAM-390X (X = 0, 1, 2, 3, 4) series was based on Y-ftw-MOF-2, which crystallizes in the cubic system with the Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group. Each Y6 cluster is coordinated by twelve linkers and each linker is connected to four Y6 clusters, leading to a (4,12)-c ftw topology.
space group. Each Y6 cluster is coordinated by twelve linkers and each linker is connected to four Y6 clusters, leading to a (4,12)-c ftw topology.
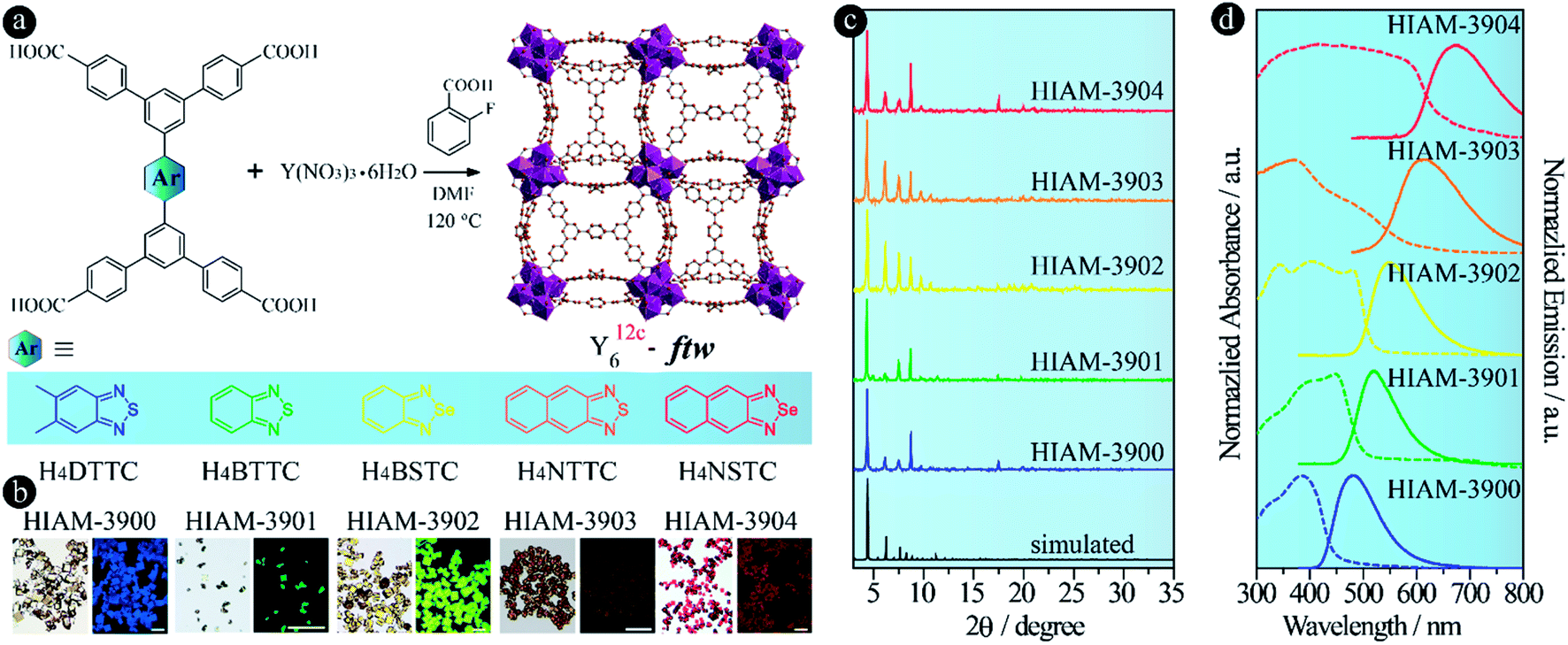 | ||
| Fig. 3 The molecular structures of the linkers, synthetic conditions and single crystal structure of HIAM-390X (a) (color scheme: brown, C; dark blue, Y; red, O); crystal images under the daylight and irradiation (excitation under 365 nm for HIAM-3900, HIAM-3901 and HIAM-3902, and excitation under 450 nm for HIAM-3903 and HIAM-3904, scale bar: 200 μm) (b); the PXRD patterns (c) and the normalized solid-state emission spectra under 365 nm excitation and UV-vis absorption spectra (d) of HIAM-3900, HIAM-3901, HIAM-3902, HIAM-3903 and HIAM-3904. | ||
The optical absorption and emission spectra of HIAM-390X were measured at room temperature. As shown in Fig. 3d, HIAM-3900, HIAM-3901, HIAM-3902, HIAM-3903 and HIAM-3904 emit at different energies with the peak maxima at 480 nm, 521 nm, 550 nm, 615 nm and 675 nm, respectively, consistent with the emission color of the single crystals (Fig. 3b). The corresponding solid-state photoluminescence quantum yields (PLQY) are 28.9%, 31.7%, 26.1%, 5.30% and 6.20% for HIAM-3900, HIAM-3901, HIAM-3902, HIAM-3903 and HIAM-3904, respectively, under 365 nm excitation, and are much higher than the 6.5%, 1.3%, 0.7%, 0.7% and 0.8% for the corresponding linkers H4DTTC, H4BTTC, H4BSTC, H4NTTC and H4NSTC, respectively. These data further confirm that immobilization and confinement of linkers in the MOF matrix is an efficient strategy to prevent them from aggregation, leading to increased PLQY.1,49 It should be noted that HIAM-3901, HIAM-3902, HIAM-3903 and HIAM-3904 can also be excited under blue light (450 nm) with PLQY of 27.0%, 20.0%, 4.10% and 4.70%, respectively. In addition, the emission tails of HIAM-3903 and HIAM-3904 extend into the energy region (>700 nm) of near-infrared light (NIR-I, 700–900 nm), which might be useful for bio-related applications if they have aqueous stability under specific conditions. A monotonic bathochromic shift is also observed in the absorption spectra from HIAM-3900 to HIAM-3904 (Fig. 3d). These results demonstrate that using acceptor groups with decreased electron density is an effective approach to achieve NIR-MOFs, and it has no effect on the underlying net of the resultant LMOFs. When acceptors with electron density much lower than that of naphtho[2,3-c][1,2,5]selenadiazole, such as benzo[1,2-c:4,5-c′]bis([1,2,5]thiadiazole), can be incorporated to the linker skeleton, we believe that LMOFs with NIR-I, and even second near-infrared region (NIR-II) emission can be achieved, which is an ongoing study in our group.
Structural analysis and description of amino group induced topology evolution of Y-MOFs
In addition to the use of acceptors with lower electron density to red-shift emission as discussed above, pre-synthesis linker modifications, such as the introduction of amino groups, have also been utilized to prepare MOFs with emission/absorption red-shift behaviour, i.e. MIL-125-NH2 and MIL-125-(NH2)2.50 To introduce amino groups into organic linkers and confirm their effect on the emission properties of the resultant LMOFs, three benzo[c][1,2,5]thiadiazole-based tetratopic carboxylate linkers, H4BTTC without an amino group, 2′-amino-5′-(7-(4,4′′-dicarboxy-[1,1′:3′,1′′-terphenyl]-5′-yl)benzo[c][1,2,5]thiadiazol-4-yl)-[1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid (H4ABTTC) with one amino group and 5′,5′′′′-(benzo[c][1,2,5]thiadiazole-4,7-diyl)bis(2′-amino-[1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid) (H4BTATC) with two amino groups, were designed and synthesized as shown in Fig. 1c. To further extend the light absorption into the lower energy region to realize NIR emission, the acceptor was changed from benzo[c][1,2,5]thiadiazole to naphtho[2,3-c][1,2,5]selenadiazole to produce 5′,5′′′′-(naphtho[2,3-c][1,2,5]selenadiazole-4,9-diyl)bis(2′-amino-[1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid) (H4NSATC). Since either adding amino groups or using electron-deficient acceptors can red-shift the emission wavelength of benzo[c][1,2,5]thiadiazole-based linkers,32,43 we envision that such a combination would lead to a much larger bathochromic shift. In addition, introduction of amino groups will offer more options for post-synthesis alteration via modification of the amino groups, which can provide multiple possibilities for tuning the emission and facilitate related applications, such as biosensing.51–54 As expected, a remarkable emission bathochromic shift is observed for H4ABTTC (λem = 717 nm) with one amino group and H4BTATC (λem = 697 nm) with two amino groups compared with the one without an amino group, H4BTTC (λem = 498 nm), in DMF solution (Fig. S1†). This reveals that introducing an amino group into the linker skeleton indeed is a powerful approach to realize emission red shift.Subsequently, HIAM-3905 and HIAM-3906 were grown as orange and red single crystals following a similar method to that for HIAM-3901 except using H4ABTTC and H4BTATC as the organic linkers (Fig. 4a1–a3 and b1–b3). However, single crystal X-ray diffraction (sc-XRD) analysis revealed that HIAM-3905 and HIAM-3906 possess totally different structures compared with HIAM-3901. As shown in Fig. 4a4 and a5 and Table S1,† HIAM-3905 crystallized in the orthorhombic crystal system with the Cmmm space group. Each Y6 cluster is coordinated by eight ABTTC linkers and eight terminal H2O/OH− groups. Each ABTTC is connected to four Y6 clusters, resulting in a (4,8)-c scu net for HIAM-3905, which is rarely reported in rare-earth element-based MOFs.55–57 Four types of one-dimensional open channels co-exist in HIAM-3905, i.e. rhombic channels and hexagonal channels along the a-axis, and two different rhombic channels along the b- and c-axis (Fig. S2†). However, in the structure of HIAM-3906 (Fig. 4b4, b5, S3 and Table S2†), each Y6 cluster is coordinated with ten BTATC and four terminal H2O/OH− groups. Each deprotonated BTATC is connected to four Y6 clusters. Further analysis demonstrates that HAIM-3906 is a 3-nodal (4,4,10)-c net with the point symbol of {424·620·8}2{44·62}5, which is an unprecedented topology in MOFs and denoted as hlx in the present work.
 | ||
| Fig. 4 The molecular structure of 4-connected H4ABTTC in HIAM-3905 (a1) and H4BTATC in HIAM-3906 (b1); the 8-connected Y6 cluster in HIAM-3905 (a2) and the 10-connected Y6 cluster in HIAM-3906 (b2); the bright-field (above) and photoluminescence (below) single-crystal images of HIAM-3905 (a3) and HIAM-3906 (b3) under 450 nm excitation; the scu net topology (a4) and single-crystal structure (a5) of HIAM-3905; the hlx net topology (b4) and single-crystal structure (b5) of HIAM-3906; conformations of ABTTC in HIAM-3905 (a6) and BTATC in HIAM-3906 (b6) with the dihedral angle between the inner phenyl ring and outer phenyl ring (color scheme: brown, C; green, F; dark blue, Y; red, O; yellow, S; blue, N). | ||
The amino group induced structural diversity can be considered as the most interesting structural feature of HIAM-3901, HIAM-3905 and HIAM-3906, where three types of linker conformations co-exist in HIAM-3906 (Fig. 4b6), while only one and two types of conformations are found in HIAM-3905 and HIAM-3901, respectively (Fig. 4a6 and S4†). In fact, the dihedral angles between the outer phenyl ring (with carboxylate groups) and the inner phenyl ring (with amino groups) in HIAM-3906 are 53.22°, 58.66° and 90.00° (Fig. 4b6), respectively. The three types of linkers all adopt the C2v symmetry, in which the pairs of phenyl rings rotate toward each other. The dihedral angles are 15.19° and 37.45° for HIAM-3901 with both linker types adopting the C2v symmetry (Fig. S4†), and it is 45.29° for HIAM-3905 with the C2h symmetry (Fig. 4a6). Regarding the structural effect, substituting the hydrogen atoms in H4BTTC with a bulkier amino group would introduce steric hindrance, which will change the rotation of the phenyl rings during the MOF synthesis and dictate the structures of the resultant MOFs.40,47,48 The experimental PXRD patterns of the as-synthesized HIAM-3906 match well with the simulated ones, indicative of phase purity (Fig. 5a). However, for HIAM-3905, due to the structural flexibility of the (4,8)-c scu net, notable peak shifts were observed between the simulated and experimental patterns. It should also be noted that the secondary building units (SBUs) in HIAM-3905 and HIAM-3906 are fluoro-bridged clusters, Y6(F8)(COO)8 and Y6(F8)(COO)10 (Fig. 4a2 and b2), which is consistent with the recent work which reported that using μ3-F rather than μ3-O will offer a much better crystallographic fit when 2-fluorobenzoic acid is employed as the modulator.58 The fluorine presence in HIAM-3905 and HIAM-3906 was confirmed by SEM-EDS analysis (Fig. S5 and S6†).
 | ||
| Fig. 5 The PXRD patterns of simulated and synthesized HIAM-3905, HIAM-3906 and HIAM-3907 (a); the normalized solid-state emission (365 nm excitation for HIAM-3901, and 450 nm excitation for HIAM-3905, HIAM-3906 and HIAM-3907) (b) and UV-vis absorption spectra (c) of HIAM-3901, HIAM-3905, HIAM-3906 and HIAM-3907; the calculated HOMO–LUMO energy levels of H4BTTC, H4ABTTC, H4BTATC and H4NSATC (d). | ||
Characterization of amino group induced NIR emissive Y-MOFs
The single crystals of HIAM-3905 and HIAM-3906 show red and dark red emissions with peak maxima at 665 nm and 689 nm, which are bathochromically shifted by 144 nm and 168 nm compared with that of HIAM-3901 (Fig. 5b), confirming that the amino group has the capacity to red-shift the emission energy/wavelength. To further extend the emission wavelength to a lower energy region, we used the acceptor group naphtho[2,3-c][1,2,5]selenadiazole, with even lower electron density than benzo[c][1,2,5]thiadiazole in H4BTATC to form H4NSATC, which was then utilized as the organic linker to prepare HIAM-3907. The PXRD pattern of HIAM-3907 shows excellent agreement with that of simulated HIAM-3906 (Fig. 5a), indicating its isoreticular nature and phase purity. Not surprisingly, HIAM-3907 emits at 790 nm, with a red-shift of 115 nm and 101 nm compared with that of HIAM-3904 (the one without the amino group) and HIAM-3906, respectively, which brings HIAM-3907 to the NIR-I range. Together, a 269 nm bathochromic shift has been achieved by the combination of amino and naphtho[2,3-c][1,2,5]selenadiazole groups in the linker skeleton of H4BTTC to form HIAM-3907.HIAM-3907 is one of the few organic-linker-based LMOFs with the emission maximum beyond 750 nm, covering the entire NIR-I range with the emission tail towards the NIR-II window.20 These results further demonstrate that the use of acceptors with lower electron density is an effective method to achieve low energy emissions. The corresponding PLQY are 9.0%, 5.0% and 2.0% for HIAM-3905, HIAM-3906 and HIAM-3907, respectively, under 450 nm excitation, which are also much higher than the 3.1%, 0.40% and 0.10% for the corresponding linkers H4ABTTC, H4BTATC and H4NSATC, respectively. In addition, the absorption edges of HIAM-3906 and 3907 reach 650 and 750 nm (Fig. 5c). The broad light harvesting energy range makes these MOFs promising for application in photocatalysis. To further investigate the amino group effect on the emission behavior of the designed linkers, density functional theory (DFT) calculation was utilized to study the electronic structures of these linkers. As depicted in Fig. 5d, after addition of one or two amino groups on H4BTTC to form H4ABTTC and H4BTATC, the LUMO energies gradually increased from −2.71 eV to −2.57 eV and to −2.34 eV, while a much larger increase was observed for the HOMO energies, from −6.13 eV to −5.53 eV and to 5.17 eV. As a result, the energy gaps between the HOMO and LUMO are significantly decreased from 3.42 eV for H4BTTC to 2.96 eV for H4ABTTC, and 2.83 eV for H4BTATC, which are consistent with the experimental results. These results demonstrate that the amino group exhibits a much more significant effect on the HOMO than on the LUMO. When an acceptor group possessing much lower electron density than benzo[c][1,2,5]thiadiazole was used, such as naphtho[2,3-c][1,2,5]selenadiazole, to form H4NSATC, a 0.40 eV lower LUMO energy and a 0.16 eV higher HOMO energy were achieved, which leads to a further decreased energy gap of 2.27 eV for H4NSATC.
As summarized in Table 1, although a series of Y-MOFs were prepared with emission toward the near-infrared range and good water stability, for example HIAM-3907 (Fig. S7†), it is still difficult to apply them in bioimaging and biosensing in the present work due to the inability to achieve nanocrystallization. Attempts to achieve nano-sized NIR-MOFs with excellent aqueous compatibility, such as stability and dispersibility, for bio-related applications are ongoing.
| Linker | MOFs | Connectivity of the Y6 cluster | Topology | Emission wavelength (nm) | PLQY (%, excitation wavelength/nm) |
|---|---|---|---|---|---|
| H4DTTC | HIAM-3900 | 12 | ftw | 480 | 28.9 (365 nm) |
| H4BTTC | HIAM-3901 | 12 | ftw | 521 | 31.7 (365 nm) |
| 27.0 (450 nm) | |||||
| H4BSTC | HIAM-3902 | 12 | ftw | 550 | 26.1 (365 nm) |
| 20.0 (450 nm) | |||||
| H4NTTC | HIAM-3903 | 12 | ftw | 615 | 5.30 (365 nm) |
| 4.10 (450 nm) | |||||
| H4NSTC | HIAM-3904 | 12 | ftw | 675 | 6.20 (365 nm) |
| 4.70 (450 nm) | |||||
| H4ABTTC | HIAM-3905 | 8 | scu | 665 | 9.0 (450 nm) |
| H4BTATC | HIAM-3906 | 10 | hlx | 689 | 5.0 (450 nm) |
| H4NSATC | HIAM-3907 | 10 | hlx | 790 | 2.0 (450 nm) |
Conclusion
In conclusion, a series of luminescent Y-MOFs with the ftw topology, HIAM-3900 to HIAM-3904, have been prepared using 2,1,3-benzothiadiazole and its derivative-based tetratopic carboxylate linkers. The emission tails of HIAM-3903 and HIAM-3904 fall in the NIR-I region. In addition, introducing an amino group and acceptor with lower electron density into the linker skeleton will not only redshift the emission of the resultant LMOFs to cover the entire NIR-I window (i.e. HIAM-3907, λem = 790 nm), but also induce topology evolution, from (4,12)-c ftw, (4,8)-c scu, which is rarely reported in rare-earth element-based MOFs, to an unprecedented topology (4,10)-c hlx, as a result of steric hindrance caused by the amino group. The present work sheds light on the rational design of emissive organic linkers to obtain NIR-MOFs with controllable structures, which no doubt will facilitate their applications in biosensing and bioimaging.Data availability
All the data have been included in the ESI.†Author contributions
X.-Y. L. conceived the idea and designed the experiment; H.-L. Xia and J. Zhang worked on the synthesis and characterization of all materials. K. Zhou carried out and analyzed the single crystal structures. J. Guo did the density functional theory calculations. X. Huang and D. Luo provided detailed structural analysis and discussion. X.-Y. L. and J. Li wrote the paper with the help from all authors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
X.-Y. Liu acknowledges the financial support from start-up funding for Shenzhen High-Caliber Personnel of Shenzhen Polytechnic (6022310053K), Guangdong Basic and Applied Basic Research Foundation (2020A1515110420), and Shenzhen Science and Technology Program (RCBS20200714114941230). D. Luo acknowledges the Special Funds for the Science and Technology Innovation Project of Guangdong Provincial Department of Education (2021KTSCX278). H.-L. Xia gratefully acknowledges the support from Shenzhen Polytechnic (6021330018K).References
- Q. Gong, Z. Hu, B. J. Deibert, T. J. Emge, S. J. Teat, D. Banerjee, B. Mussman, N. D. Rudd and J. Li, J. Am. Chem. Soc., 2014, 136, 16724 CrossRef CAS PubMed.
- J. Cornelio, T. Y. Zhou, A. Alkas and S. G. Telfer, J. Am. Chem. Soc., 2018, 142, 15470 CrossRef PubMed.
- W. P. Lustig and J. Li, Coord. Chem. Rev., 2018, 373, 116 CrossRef CAS.
- W. J. Newsome, S. Ayad, J. Cordova, E. W. Reinheimer, A. D. Campiglia, J. K. Harper, K. Hanson and F. J. Uribe-Romo, J. Am. Chem. Soc., 2019, 11298 CrossRef CAS PubMed.
- W. P. Lustig, Z. Shen, S. J. Teat, N. Javed, E. Velasco, D. M. O'Carroll and J. Li, Chem. Sci., 2020, 11, 1814 RSC.
- W. J. Newsome, A. Chakraborty, R. T. Ly, G. S. Pour, D. C. Fairchild, A. J. Morris and F. Uribe-Romo, J. Chem. Sci., 2020, 11, 4391 CAS.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- Z. Hu, W. P. Lustig, J. Zhang, C. Zheng, H. Wang, S. J. Teat, Q. Gong, N. D. Rudd and J. Li, J. Am. Chem. Soc., 2015, 137, 16209 CrossRef CAS PubMed.
- A. Mallick, A. M. El-Zohry, O. Shekhah, J. Yin, J. Jia, H. Aggarwal, A. H. Emwas, O. F. Mohammed and M. Eddaoudi, J. Am. Chem. Soc., 2019, 141, 7245 CrossRef CAS.
- J. Li, S. Yuan, J.-S. Qin, J. Pang, P. Zhang, Y. Zhang, Y. Huang, H. F. Drake, W. R. Liu and H.-C. Zhou, Angew. Chem., Int. Ed., 2020, 59, 9319 CrossRef CAS PubMed.
- C. R. Martin, P. Kittikhunnatham, G. A. Leith, A. A. Berseneva, K. C. Park, A. B. Greytak and N. B. Shustova, Nano Res., 2020, 14, 338 CrossRef.
- Y. Zhao, H. Zeng, X.-W. Zhu, W. Lu and D. Li, Chem. Soc. Rev., 2021, 50, 4484 RSC.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242 RSC.
- J. Park, Q. Jiang, D. Feng, L. Mao and H. C. Zhou, J. Am. Chem. Soc., 2016, 138, 3518 CrossRef CAS PubMed.
- J. Park, M. Xu, F. Li and H. C. Zhou, J. Am. Chem. Soc., 2018, 140, 5493 CrossRef CAS PubMed.
- S. Ali Akbar Razavi and A. Morsali, Coord. Chem. Rev., 2019, 399, 213023 CrossRef CAS.
- D. Zou, J. Zhang, Y. Cui and G. Qian, Dalton Trans., 2019, 48, 6669 RSC.
- T.-Y. Luo, P. Das, D. L. White, C. Liu, A. Star and N. L. Rosi, J. Am. Chem. Soc., 2020, 142, 2897 CrossRef CAS PubMed.
- P. F. Muldoon, G. Collet, S. V. Eliseeva, T.-Y. Luo, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2020, 142, 8776 CrossRef.
- F. Wang, J. Wang, S. F. Maehrlein, Y. Ma, F. Liu and X. Y. Zhu, J. Phys. Chem. Lett., 2020, 11, 762 CrossRef CAS PubMed.
- K. Welsher, Z. Liu, S. P. Sherlock, J. T. Robinson, Z. Chen, D. Daranciang and H. Dai, Nat. Nanotechnol., 2009, 4, 773 CrossRef CAS PubMed.
- O. T. Bruns, T. S. Bischof, D. K. Harris, D. Franke, Y. Shi, L. Riedemann, A. Bartelt, F. B. Jaworski, J. A. Carr, C. J. Rowlands, M. W. B. Wilson, O. Chen, H. Wei, G. W. Hwang, D. M. Montana, I. Coropceanu, O. B. Achorn, J. Kloepper, J. Heeren, P. T. C. So, D. Fukumura, K. F. Jensen, R. K. Jain and M. G. Bawendi, Nat. Biomed. Eng., 2017, 1, 0056 CrossRef CAS PubMed.
- D. J. Naczynski, M. C. Tan, M. Zevon, B. Wall, J. Kohl, A. Kulesa, S. Chen, C. M. Roth, R. E. Riman and P. V. Moghe, Nat. Commun., 2013, 4, 2199 CrossRef CAS.
- P. Pei, Y. Chen, C. Sun, Y. Fan, Y. Yang, X. Liu, L. Lu, M. Zhao, H. Zhang, D. Zhao, X. Liu and F. Zhang, Nat. Nanotechnol., 2021, 16, 1011 CrossRef CAS PubMed.
- A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. Hong, C. Qu, S. Diao, Z. Deng, X. Hu, B. Zhang, X. Zhang, O. K. Yaghi, Z. R. Alamparambil, X. Hong, Z. Cheng and H. Dai, Nat. Mater., 2016, 15, 235 CrossRef CAS.
- Z. Lei and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16294 CrossRef CAS.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38 RSC.
- S. Zhu, R. Tian, A. L. Antaris, X. Chen and H. Dai, Adv. Mater., 2019, 31, e1900321 CrossRef.
- S. Liu, Y. Li, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2020, 12, 3427 RSC.
- W. Xu, D. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 7476 CrossRef CAS.
- S. Xu, Y. Duan and B. Liu, Adv. Mater., 2020, 32, e1903530 CrossRef.
- S. Wu, D. Ren, K. Zhou, H.-L. Xia, X.-Y. Liu, X. Wang and J. Li, J. Am. Chem. Soc., 2021, 143, 10547 CrossRef CAS PubMed.
- X.-Y. Liu, K. Xing, Y. Li, C.-K. Tsung and J. Li, J. Am. Chem. Soc., 2019, 141, 14807 CrossRef CAS PubMed.
- Y. Sun, M. Ding, X. Zeng, Y. Xiao, H. Wu, H. Zhou, B. Ding, C. Qu, W. Hou, A. G. A. Er-bu, Y. Zhang, Z. Cheng and X. Hong, Chem. Sci., 2017, 8, 3489 RSC.
- L. e. Zhang, C. Liu, S. Zhou, R. Wang, Q. Fan, D. Liu, W. Wu and X. Jiang, Adv. Healthcare Mater., 2020, 9, 1901470 CrossRef CAS.
- Y. Fang, J. Shang, D. Liu, W. Shi, X. Li and H. Ma, J. Am. Chem. Soc., 2020, 142, 15271 CrossRef CAS PubMed.
- B. Li, M. Zhao and F. Zhang, ACS Mater. Lett., 2020, 2, 905 CrossRef CAS.
- H.-L. Xia, K. Zhou, S. Wu, D. Ren, K. Xing, J. Guo, X. Wang, X.-Y. Liu and J. Li, Chem. Sci., 2022, 13, 8036 RSC.
- G. Han, S. Wu, K. Zhou, H.-L. Xia, X.-Y. Liu and J. Li, Inorg. Chem., 2022, 61, 3363 CrossRef CAS PubMed.
- J. Pang, S. Yuan, J. Qin, C. Liu, C. Lollar, M. Wu, D. Yuan, H.-C. Zhou and M. Hong, J. Am. Chem. Soc., 2017, 139, 16939 CrossRef CAS PubMed.
- Z. Chen, S. L. Hanna, L. R. Redfern, D. Alezi, T. Islamoglu and O. K. Farha, Coord. Chem. Rev., 2019, 386, 32 CrossRef CAS.
- Y. Chen, X. Zhang, M. R. Mian, F. A. Son, K. Zhang, R. Cao, Z. Chen, S. J. Lee, K. B. Idrees, T. A. Goetjen, J. Lyu, P. Li, Q. Xia, Z. Li, J. T. Hupp, T. Islamoglu, A. Napolitano, G. W. Peterson and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 21428 CrossRef CAS PubMed.
- D. Ren, H.-L. Xia, K. Zhou, S. Wu, X.-Y. Liu, X. Wang and J. Li, Angew. Chem., Int. Ed., 2021, 60, 25048 CrossRef CAS PubMed.
- H.-L. Xia, K. Zhou, L. Yu, H. Wang, X.-Y. Liu, D. M. Proserpio and J. Li, Inorg. Chem., 2022, 61, 7980 CrossRef CAS PubMed.
- R. Luebke, Y. Belmabkhout, L. J. Weselinski, A. J. Cairns, M. Alkordi, G. Norton, L. Wojtas, K. Adil and M. Eddaoudi, Chem. Sci., 2015, 6, 4095 RSC.
- H. Wang, X. Dong, V. Colombo, Q. Wang, Y. Liu, W. Liu, X.-L. Wang, X.-Y. Huang, D. M. Proserpio, A. Sironi, Y. Han and J. Li, Adv. Mater., 2018, 30, 1805088 CrossRef PubMed.
- B. Wang, X. L. Lv, D. Feng, L. H. Xie, J. Zhang, M. Li, Y. Xie, J. R. Li and H. C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204 CrossRef CAS PubMed.
- X. L. Lv, S. Yuan, L. H. Xie, H. F. Darke, Y. Chen, T. He, C. Dong, B. Wang, Y. Z. Zhang, J. R. Li and H. C. Zhou, J. Am. Chem. Soc., 2019, 141, 10283 CrossRef CAS PubMed.
- Z. Hu, G. Huang, W. P. Lustig, F. Wang, H. Wang, S. J. Teat, D. Banerjee, D. Zhang and J. Li, Chem. Commun., 2015, 51, 3045 RSC.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D'arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942 CrossRef CAS PubMed.
- Z. Wang and S. M. Cohen, Angew. Chem., Int. Ed., 2008, 47, 4699 CrossRef CAS PubMed.
- Y. Zhang, X. Feng, H. Li, Y. Chen, J. Zhao, S. Wang, L. Wang and B. Wang, Angew. Chem., Int. Ed., 2015, 54, 4259 CrossRef CAS PubMed.
- A. M. Fracaroli, P. Siman, D. A. Nagib, M. Suzuki, H. Furukawa, F. D. Toste and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 8352 CrossRef CAS PubMed.
- Y. Wen, J. Zhang, Q. Xu, X.-T. Wu and Q.-L. Zhu, Coord. Chem. Rev., 2018, 376, 248 CrossRef CAS.
- X.-L. Lv, L. Feng, L.-H. Xie, T. He, W. Wu, K.-Y. Wang, G. Si, B. Wang, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2021, 143, 2784 CrossRef CAS PubMed.
- L. Feng, J. Pang, P. She, J.-L. Li, J.-S. Qin, D.-Y. Du and H.-C. Zhou, Adv. Mater., 2020, 32, 2004414 CrossRef PubMed.
- F. Saraci, V. Quezada-Novoa, P. R. Donnarumma and A. J. Howarth, Chem. Soc. Rev., 2020, 49, 7949 RSC.
- J. P. Vizuet, M. L. Mortensen, A. L. Lewis, M. A. Wunch, H. R. Firouzi, G. T. McCandless and K. J. Balkus, J. Am. Chem. Soc., 2021, 143, 17995 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2165296 and 2151211. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02683j |
| This journal is © The Royal Society of Chemistry 2022 |