
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of chromanone lactones via vinylogous conjugate addition of butenolide to 2-ester chromones†
Yi
Li
,
Shuang
Xin
,
Rui
Weng
,
Xiaohua
Liu
 * and
Xiaoming
Feng
*
* and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn
First published on 5th July 2022
Abstract
Chiral chromanone lactones are a class of natural products with important biological activity. We report a direct diastereo- and enantioselective vinylogous conjugate addition of butenolide to 2-ester substituted chromones. The transformation proceeded well in the presence of as low as 1 mol% of a chiral N,N′-dioxide/ScIII complex, 3 Å MS and a catalytic amount of hexafluoroisopropanol (HFIP). The scope of Michael acceptors includes a variety of substituted chromones at different positions, and the desired chromanone lactones upon reduction are afforded in good yield and diastereoselectivity, and excellent enantioselectivity (up to 99% ee). The strategy could be used in the concise synthesis of blennolide C and gonytolide A, C and G.
Introduction
Chromanone lactones represent a family of natural products with important biological activity, and the structure characterized as 4-chromanone bearing a five-membered lactone at the 2-position could also give access to tetrahydroxanthone natural products (Scheme 1a).1 In view of their intriguing bioactivities and structural diversity, the total synthesis of the chromanone lactone series has drawn active interest.2 The vinylogous conjugate addition of butanolide or its synthetic equivalents to chromones provided a straightforward route for this purpose. Porco and coworkers initiated a benzopyrylium species involving Mukaiyama addition with siloxyfurans from 5-hydroxyl-2-carboxylate chromones activated by diisopropylsilyl ditriflate (Scheme 1b),3 and the transformations following the resolution enabled the asymmetric total synthesis of several mono- or dimeric natural products.4 The strategy was expanded to the vinylogous addition of siloxyfurans to chromones promoted with TMSI by Zhang’s and Li's groups, which was used for the synthesis of racemic natural products.5 A stereodivergent variety using a chiral copper(I)-complex catalyst and siloxyfurans was realized by Shibasaki very recently.6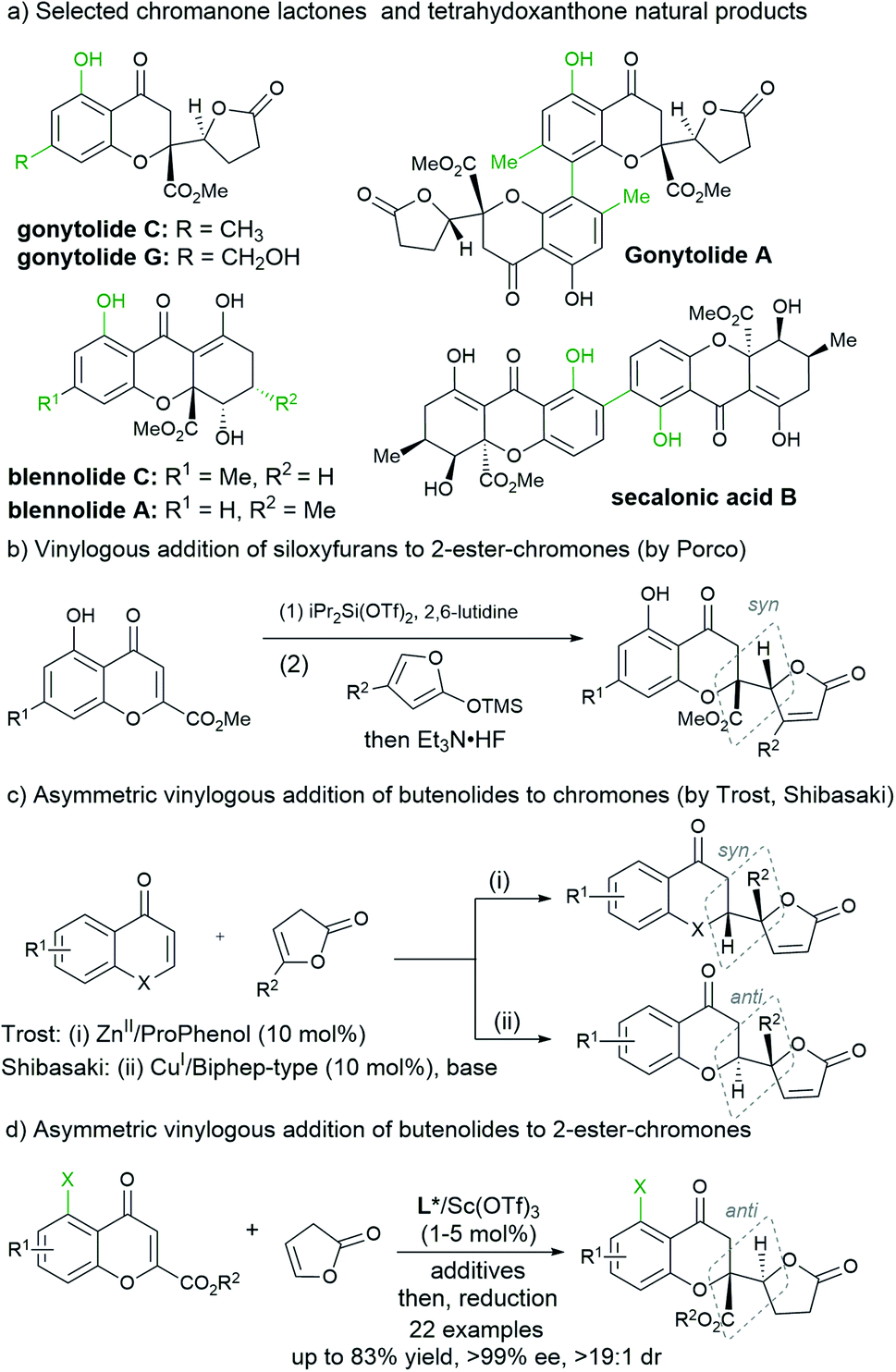 | ||
| Scheme 1 Background information about chromanone lactones and related compounds. | ||
On the other hand, the asymmetric catalytic preparation of butanolide-containing chromanones from butanolides seems to be more promising and challenging. Previously, Trost's7a and Shibasaki's groups7b reported asymmetric vinylogous addition of 2-chromones with unactivated 5-alkyl γ-butenolides as the pronucleophiles using Zn–ProPhenon and Cu–Biphep complex catalysts, respectively (Scheme 1c), providing an atom-economical method without preactivation of the reactants and desilylation.8 However, as evidenced by the structures presented in Scheme 1a, 2-ester and 5-hydroxyl substitutions are popular in these natural products, but the employment of 2-ester chromones as Michael acceptors6 overcomes the challenges of steric hindrance for the construction of a quaternary carbon center,9 the retro-oxo-Michael reaction and epimerization of the initial adducts for the achievement of high diastereo- and enantioselectivity.3,4 Our research group has realized vinylogous additions of both β,γ-unsaturated butenolides and their equivalents catalyzed by the chiral N,N′-dioxide/ScIII complex catalysts.10 In light of the performance of chiral Lewis acid catalysis using Feng N,N′-dioxides as ligands, we reasoned that it could be applied for the preparation of optically active chromanone lactones and tetrahydroxanthones (Scheme 1d). In fact, the direct vinylogous addition of butanolide to 2-ester chromones worked well promoted by as low as 1 mol% of the chiral scandium catalyst of N,N′-dioxide. The diastereoselectivity in the Mukaiyama addition of 2-ester chromones from Porco's study showed that the syn-diastereomer of the silylene adduct is kinetically favorable. Differently, the highly anti-selective and enantioselective chromanone lactone is more readily available in the presence of molecular sieves and alcohols in this study. Herein, we disclosed a highly diastereo- and enantioselective vinylogous conjugate addition for the synthesis of chromanone lactones, which could be used in the concise synthesis of (+)-blennolide C, (+)-gonytolide A and other natural product members.
Results and discussion
In the initial studies, the vinylogous addition reaction of 2-ester chromone 1a and 5-H-butenolide 2a was selected as the model reaction to optimize the reaction conditions (Table 1). The direct addition product 3aa was unstable during the purification with silica gel, and thus the target lactone-substituted chromanone 3a was isolated as the final product via the treatment with NaBH4/NiCl2·6H2O in one-pot after the catalytic reaction.3,4 The reaction in the presence of a chiral N,N′-dioxide L3-PrPr2/Sc(OTf)3 complex could deliver the desired product 3a with good enantioselectivity after 4 hours in THF at 30 °C (94% ee; entry 1), and although the yield increased gradually with a prolonged reaction time, both the diastereoselectivity and the ee value decreased during the course (entries 2 and 3), indicating that the epimerization of the adduct 3aa occurred during the process, similar to the observation from Porco’s work.3 To minimize the loss of stereoselectivity we further investigated different additives (see the ESI† for details). It was a delight to find that the medium yield (56% yield) with up to 99% ee and 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 anti:syn was obtained after 16 hours when 3 Å MS was added to the reaction system (entry 4). Remarkably, the addition of 20 mol% of hexafluoroisopropanol (HFIP) increased the yield of 3a to 82% (entry 5). With the established grounded system, we evaluated the performance of different N,N′-dioxide ligands, and the selected results are listed in entries 6–9. It was found that the L-proline-derived L3-PrPr2 gave better yield and diastereoselectivity than L-ramipril-derived L3-RaPr2 (entry 6), and higher outcome in terms of yield, and diastereo- and enantioselectivity than L-pipecolic acid-derived L3-PiPr2 (entry 7). Changing the ligand to 2,6-dimethylaniline-based L3-PrMe2 led to a dramatic drop in stereoselectivity (entry 8). Although the use of 2,6-diethyl aniline-based L3-PrEt2 resulted in decreased diastereoselectivity at 30 °C (entry 9), excellent results could be achieved by lowering the temperature to 20 °C (entry 10). Noteworthily, the loading of the catalyst L3-PrEt2/Sc(OTf)3 could be reduced to 1 mol%, and the lactone 3a was isolated in 82% yield, 99% ee and a 19:1 anti:syn ratio (entry 11).
:1 anti:syn was obtained after 16 hours when 3 Å MS was added to the reaction system (entry 4). Remarkably, the addition of 20 mol% of hexafluoroisopropanol (HFIP) increased the yield of 3a to 82% (entry 5). With the established grounded system, we evaluated the performance of different N,N′-dioxide ligands, and the selected results are listed in entries 6–9. It was found that the L-proline-derived L3-PrPr2 gave better yield and diastereoselectivity than L-ramipril-derived L3-RaPr2 (entry 6), and higher outcome in terms of yield, and diastereo- and enantioselectivity than L-pipecolic acid-derived L3-PiPr2 (entry 7). Changing the ligand to 2,6-dimethylaniline-based L3-PrMe2 led to a dramatic drop in stereoselectivity (entry 8). Although the use of 2,6-diethyl aniline-based L3-PrEt2 resulted in decreased diastereoselectivity at 30 °C (entry 9), excellent results could be achieved by lowering the temperature to 20 °C (entry 10). Noteworthily, the loading of the catalyst L3-PrEt2/Sc(OTf)3 could be reduced to 1 mol%, and the lactone 3a was isolated in 82% yield, 99% ee and a 19:1 anti:syn ratio (entry 11).
|
|
||||
|---|---|---|---|---|
| Entry | Conditions | Yield (%) | dr | ee (%) |
|
a Unless otherwise noted, all reactions were carried out with (1) Sc(OTf)3/L (1:1, 10 mol%), 1a (0.1 mmol), and 2a (3.0 equiv.) in THF (0.1 M) at 30 °C; 3 Å MS (0 or 20 mg); hexafluoroisopropanol (HFIP, 0 or 20 mol%). (2) NiCl2 6H2O (1.0 equiv.), NaBH4 (2.0 equiv.), THF/MeOH (3/1, 0.25 M), 0 °C, 10 min. The isolated yield of the product 3a is provided. The ee values were determined by UPC2 analysis and dr values were determined by 1H NMR.
b At 20 °C.
c 1 mol% catalyst loading.
|
||||
| 1 | L3-PrPr2, 4 h | 19 | 4:1 |
94 |
| 2 | L3-PrPr2, 8 h | 35 | 3.6:1 |
90 |
| 3 | L3-PrPr2, 24 h | 42 | 2.3:1 |
74 |
| 4 | L3-PrPr2, 3 Å MS, 16 h | 56 | >19:1 |
>99 |
| 5 | L3-PrPr2, 3 Å MS, HFIP, 16 h | 82 | >19:1 |
>99 |
| 6 | L3-RaPr2, 3 Å MS, HFIP, 16 h | 23 | 1:1 |
>99 |
| 7 | L3-PiPr2, 3 Å MS, HFIP, 16 h | 42 | 6:1 |
>99 |
| 8 | L3-PrMe2, 3 Å MS, HFIP, 16 h | 70 | 9:1 |
78 |
| 9 | L3-PrEt2, 3 Å MS, HFIP, 16 h | 79 | 12:1 |
>99 |
| 10b | L3-PrEt2, 3 Å MS, HFIP, 16 h | 83 | 19:1 |
>99 |
| 11b,c | L3-PrEt2, 3 Å MS, HFIP, 16 h | 82 | 19:1 |
>99 |
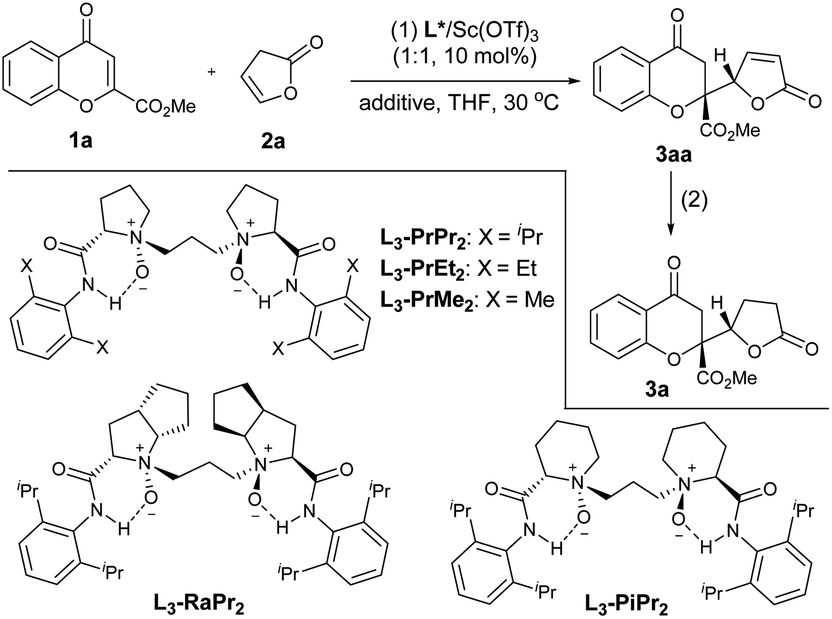
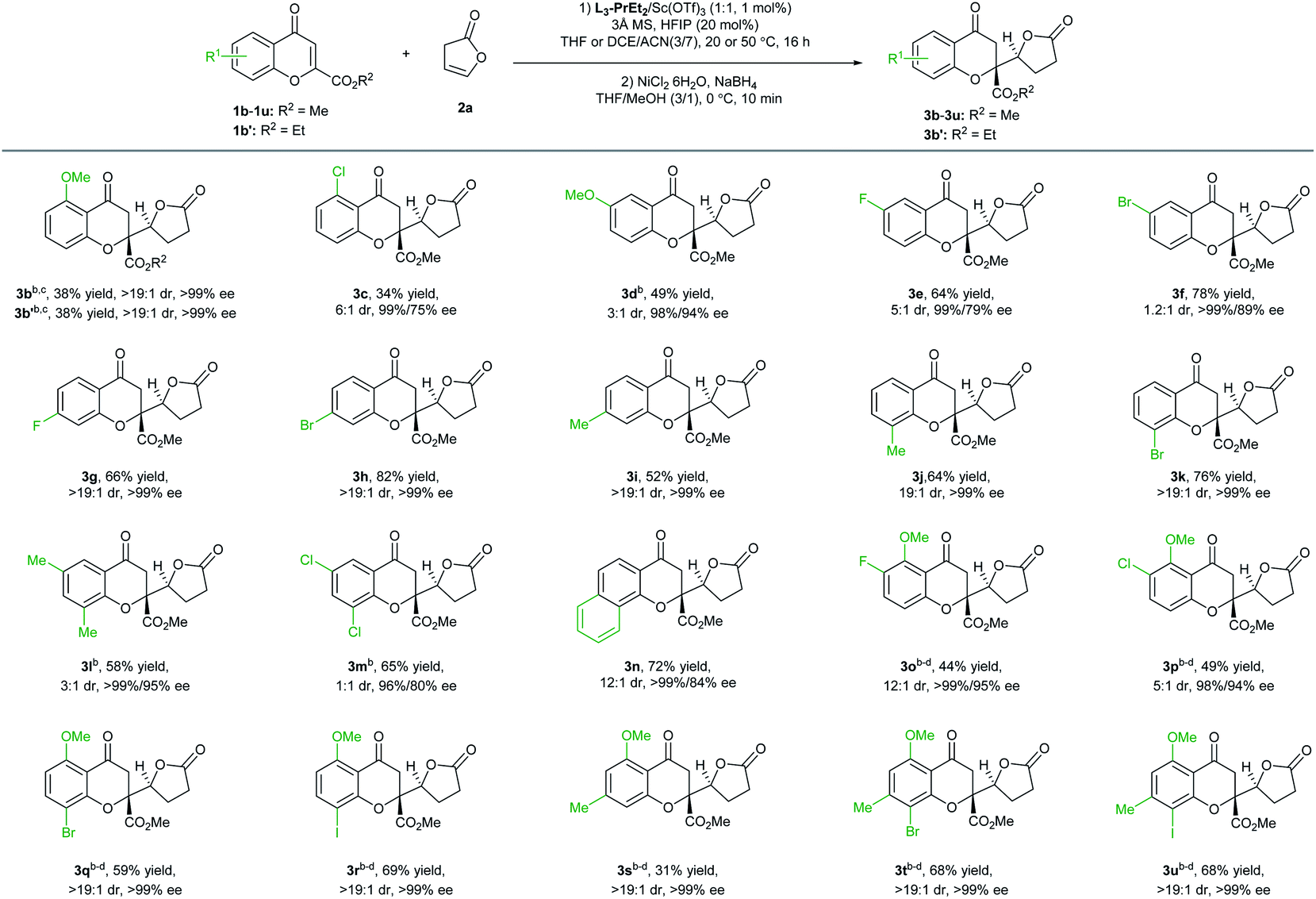
With the optimized reaction conditions in hand (Table 1, entry 11), the scope of chromones in the vinylogous addition reaction was evaluated (Table 2). Particularly, various substitutions at different positions of chromones have nearly no influence on the enantioselectivity, and all the tested reactions (22 examples) yielded the desired products with 96% to >99% ee. Generally, electron-withdrawing or donating substitution at 7- or 8-positions did not significantly affect the yield and diastereoselectivity (3g–3k; >19:1 dr), while 5- or 6-substituted ones led to varied reactivity and diastereoselectivity. A 5-MeO substituent did not impact the selectivity but reduced the yield (3b), and the ethyl ester substitution gave similar results (3b′), while a 5-Cl substituent resulted in a reduced dr value (3c, 9:1 dr). Moderate to good yield and excellent enantioselectivity were observed with 6-methoxylchromone (3d), 6-flurorochromone (3e), or 6-bromochromone (3f), but the diastereoselectivity was also obviously affected. Chromones with disubstitution at the 6- and 8-positions were also good electrophiles, and the adducts 3l and 3m were afforded in a moderate yield and dr value, with 99% and 96% ee, respectively in the presence of 5 mol% catalyst. With a fused-ring substrate, chromanone lactone 3n was formed in 72% yield, 12:1 dr, and 99% ee.
|
a Unless otherwise noted, the reactions were carried out with 1 (0.1 mmol), 2a (0.3 mmol), and L3-PrEt2/Sc(OTf)3 (1:1, 1 mol%) in THF (0.1 M) at 20 °C for 16 hours and then with NiCl2·6H2O (1.0 equiv.) and NaBH4 (2.0 equiv.) in THF/MeOH (3/1, 0.25 M) at 0 °C for 10 min. The isolated yield of 3 is provided. The ee values were determined by UPC2 and the dr values were determined by 1H NMR.
b
L3-PrEt2/Sc(OTf)3 (5 mol%).
c At 50 °C.
d DCE/MeCN (3:7, 0.1 M) as the solvent.
|
|---|
|
Owing to the fact that chromanone lactones and the related tetrahydroxanthone natural products contain a hydroxyl group at the 5-position, we then focused on the reactions of 5-MeO-chromones. The disubstituted and tri-substituted chromones were tolerable with excellent enantioselectivity (98–99% ee) and moderate yield with 5 mol% of the scandium catalyst. The 7-, 8-substituted chromanone lactones 3q–3u could be isolated as nearly single enantiomers (>19:1 dr and >99% ee), but 6-substitution resulted in decreased diastereoselectivity (3o and 3p). Halogen-substitution at the 6- or 8-position allowed further coupling reactions to prepare the corresponding dimeric natural products. Due to poor solubility of these polysubstituted chromones in tetrahydrofuran, the reaction solvent needed to be changed to a mixture of 1,1,2,2-tetrachloroethane and acetonitrile with increased reaction temperature (50 °C).
To evaluate the synthetic potential of the protocol, chromanone lactone 3a was prepared on a gram-scale with 1 mol% of the L3-PrEt2/Sc(OTf)3 complex with no deterioration in yield or stereoselectivity (Scheme 2a). Then, the synthetic utility of the chromanone lactones in the related natural products was probed. Treatment of 5-methoxyl-7-methyl substituted chromanone lactone 3s with BBr3 solution afforded 5-OH based gonytolide C, the prevalent backbone of chromones in natural products. The absolute configuration of 3s and its precursor 3sa was assigned as (2R,5′S) according to the structure of gonytolide C (Scheme 2b).2d Following Porco's work,3,4 gonytolide C can be smoothly transformed into the tetrahydroxanthone-type natural product blennolide C via NaH promoted Dieckmann cyclization. As an important intermediate, gonytolide C can also be converted to gonytolide A4d and G2d (Scheme 1a).
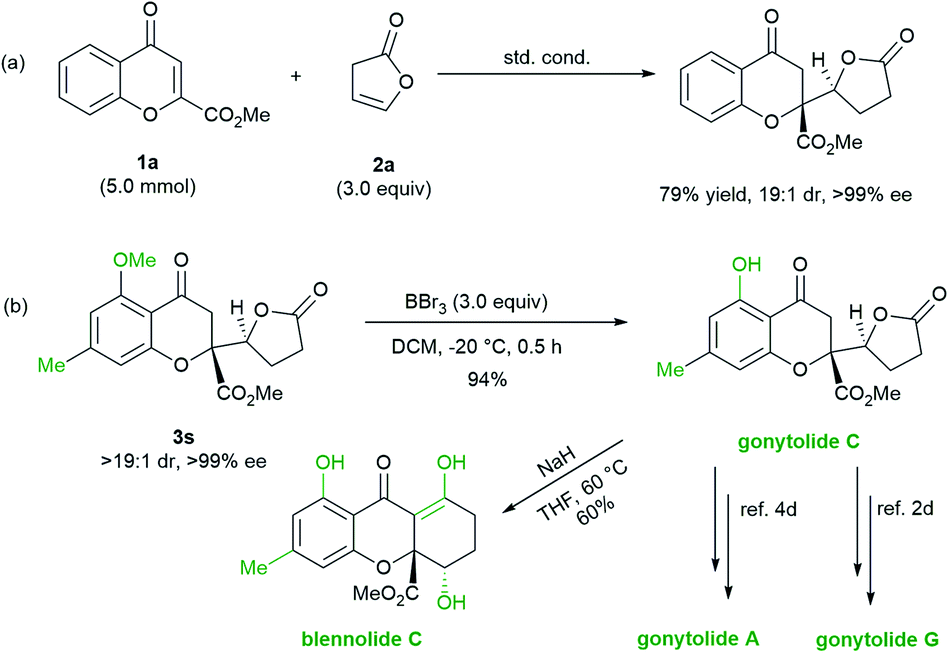 | ||
| Scheme 2 The gram scale synthesis of 3a and transformations into natural products. | ||
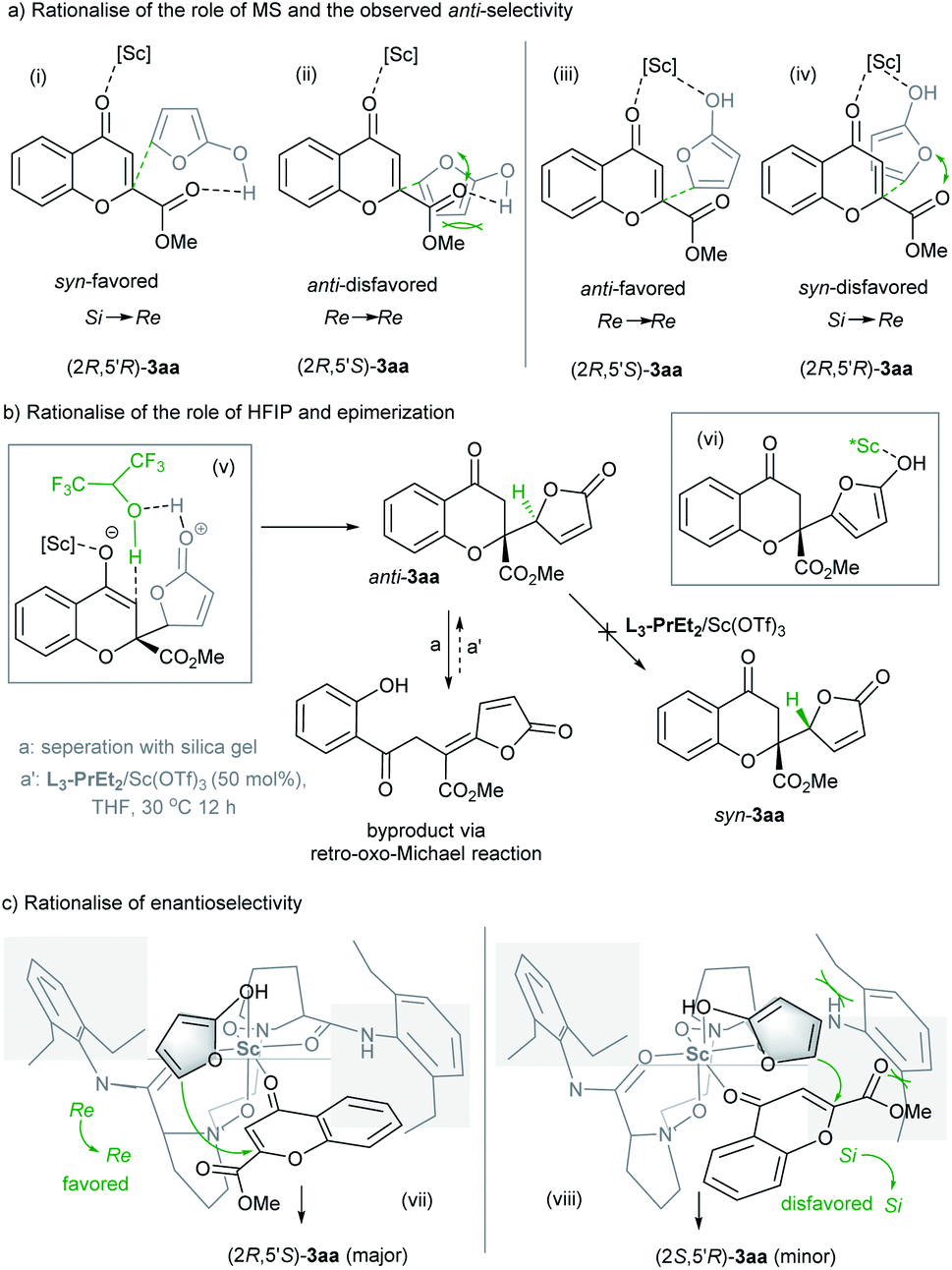
The major diastereomers obtained in our study differed from those obtained in the vinylogous addition of siloxyfuran to benzopyrylium in Porco's study, which is proposed to be due to the swing favored transition state.3 As shown in Table 1 and S6b in the ESI,† the addition of a desiccating agent dramatically increased the anti-selectivity. The main function of molecular sieves would be protection of the active catalyst from adventitious water. X-ray crystal structures of the Sc3+ complexes of N,N′-dioxide ligands11 manifest octahedral geometry in which one cis-position is occupied by two ancillary ligands (usually H2O and the TfO− ion). If water takes over one coordination to the scandium center prior to the enolized butanolide intermediate, the reaction could take place via transition state (i) as shown in Scheme 3a where the chromone directly biunds to the metal center rather than enolate to undergo addition. The epi-transition state (ii) is relatively disfavored due to the steric hindrance and long pair repulsion of the oxygens. As a result, the generation of a syn-selective product would be a competitive pathway if a trace amount of water existed. However, if the amount of water is minimized in the reaction system, the catalyst would enable the bonding of both chromone and the enolized butanolide simultaneously. As displayed in Scheme 3a, transition state (iii) to generate an anti-selective isomer (Re → Re approach or its antipodal) would be preferred over the generation of an opposite syn-isomer via transition state (iv) (Si → Re approach or its antipodal) where there exists long pair repulsion of oxygens. In addition, the basic condition in the presence of 3 Å MS might accelerate the enolization of butanolide 2a to improve the reactivity.
 | ||
| Scheme 3 Proposed explanation for the role of additives and selectivity. | ||
The positive role of HFIP in the reactivity is rationalized in Scheme 3b, and as shown in transition state (v), this weakly coordinated alcohol could act as a proton transfer agent to accelerate the isomerization of the enol intermediate after vinylogous addition, giving the adduct 3aa and regenerating the catalyst for another catalytic cycle. Epimerization of the adduct is common as observed in the previous study,3 which is proposed via an isomerization process due to the enolization of the lactone unit (vi). But under the optimized reaction conditions, the epimerization of 3aa was not observed (see the ESI† for details). In addition, the retro-oxo-Michael reaction should be avoided,4d and the byproduct was detected during the column chromatography with silica gel, which could not undergo addition to regenerate the adduct 3aa under the catalyst system (Scheme 3b).
To explain the observed high enantioselectivity, we propose a model based on the crystal structure of the N,N′-dioxide/Sc(OTf)3 complexes11 and previous DFT calculations.12 The scandium biunds the chromone 1a at the horizontal position, and the enolate of the butanolide substitutes the solvent or counterion at the top. As depicted in transition state (vii) of Scheme 3c, the reaction occurs via the Re → Re approach in the open front left quadrant of the catalyst, which leads to the major (2R,5′S) adduct. In contrast, the Si → Si approach (viii) to yield the (2S,5′R) adduct is disfavored due to a steric clash of the ester group of chromone and enolized butanolide with the left amide subunit of the catalyst.
Conclusions
In summary, we developed an efficient asymmetric vinylogous addition of butanolide to 2-ester chromones. The reaction proceeds well under mild reaction conditions with good to excellent diastereo- and enantioselectivity (up to 99% ee) and the yields are generally satisfactory. As low as 1 mol% of the chiral N,N′-dioxide/ScIII complex catalyst enables the generation of a variety of tolerable substituted chromones at different positions. The 5-MeO substituted ones provide access to chromanone lactones, which could be rapidly transformed into optically enriched natural products, including blennolide C, gonytolide C and other related natural products.Data availability
Further details of experimental procedure, 1 H, 13 C{1 H} and 19F{1 H} NMR, UPCC spectra are available in the ESI.†Author contributions
Y. L. performed the experiments. R. W. repeated data. X. M. F. and X. H. L. supervised the project. X. M. F., X. H. L., S. X. and Y. L. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the National Natural Science Foundation of China (21890723 and 21921002) for financial support.Notes and references
-
(a) F. McPhee, P. S. Caldera, G. W. Bemis, A. F. McDonagh, I. D. Kuntz and C. S. Craik, Biochem. J., 1996, 320, 681 CrossRef CAS PubMed
; (b) S. Bräse, A. Encinas, J. Keck and C. F. Nising, Chem. Rev., 2009, 109, 3903 CrossRef PubMed
-
(a) M. C. Bröhmer, E. Bourcet, M. Nieger and S. Bräse, Chem.–Eur. J., 2011, 17, 13706 CrossRef PubMed
- T. Qin, R. P. Johnson and J. A. Porco Jr, J. Am. Chem. Soc., 2011, 133, 1714 CrossRef CAS PubMed
-
(a) T. Qin and J. A. Porco Jr, Angew. Chem., 2014, 126, 3171 (
Angew. Chem., Int. Ed.
, 2014
, 53
, 3107
) CrossRef
-
(a) L. A. Stubbing, F. F. Li, D. P. Furkert, V. E. Caprio and M. A. Brimble, Tetrahedron, 2012, 68, 6948 CrossRef CAS
- During the preparation of this manuscript, an enantioselective version was reported, see J. Cui, R. Oriez, H. Noda, T. Watanabe and M. Shibasaki, Angew. Chem., Int. Ed., 2022, 61, e202203128 CAS
-
(a) B. M. Trost, E. Gnanamani, C. A. Kalnmals, C. I. Hung and J. S. Tracy, J. Am. Chem. Soc., 2019, 141, 1489 CrossRef CAS PubMed
- The review of butenolides:
(a) B. Mao, M. Fañanás-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502 CrossRef CAS PubMed
-
(a) D. Baek, H. Ryu, J. Y. Ryu, J. Lee, B. M. Stoltz and S. Hong, Chem. Sci., 2020, 11, 4602 RSC
-
(a) L. Zhou, L. L. Lin, J. Ji, M. S. Xie, X. H. Liu and X. M. Feng, Org. Lett., 2011, 13, 3056 CrossRef CAS PubMed
-
(a) Y. L. Liu, D. J. Shang, X. Zhou, X. H. Liu and X. M. Feng, Chem.–Eur. J., 2009, 15, 2055 CrossRef CAS PubMed
-
(a) F. Tan, M. P. Pu, J. He, J. Z. Li, J. Yang, S. X. Dong, X. H. Liu, Y. D. Wu and X. M. Feng, J. Am. Chem. Soc., 2021, 143, 2394 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC [1957788]. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02541h |
| This journal is © The Royal Society of Chemistry 2022 |