
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNavigating chemical reaction space – application to DNA-encoded chemistry†
Silvia
Chines
a,
Christiane
Ehrt
b,
Marco
Potowski‡
a,
Felix
Biesenkamp
a,
Lars
Grützbach
a,
Susanne
Brunner
c,
Frederik
van den Broek
 d,
Shilpa
Bali
d,
Katja
Ickstadt
c and
Andreas
Brunschweiger
*a
d,
Shilpa
Bali
d,
Katja
Ickstadt
c and
Andreas
Brunschweiger
*a
aTU Dortmund University, Department of Chemistry and Chemical Biology, Otto-Hahn-Str. 6, 44227, Dortmund, Germany. E-mail: andreas.brunschweiger@tu-dortmund.de
bUniversität Hamburg, Bundesstr. 43, 20146, Hamburg, Germany
cTU Dortmund University, Department of Statistics, Vogelpothsweg 87, 44227, Dortmund, Germany
dElsevier B.V., Radarweg 29, 1043 NX Amsterdam, The Netherlands
First published on 1st September 2022
Abstract
Databases contain millions of reactions for compound synthesis, rendering selection of reactions for forward synthetic design of small molecule screening libraries, such as DNA-encoded libraries (DELs), a big data challenge. To support reaction space navigation, we developed the computational workflow Reaction Navigator. Reaction files from a large chemistry database were processed using the open-source KNIME Analytics Platform. Initial processing steps included a customizable filtering cascade that removed reactions with a high probability to be incompatible with DEL, as they would e.g. damage the genetic barcode, to arrive at a comprehensive list of transformations for DEL design with applicability potential. These reactions were displayed and clustered by user-defined molecular reaction descriptors which are independent of reaction core substitution patterns. Thanks to clustering, these can be searched manually to identify reactions for DEL synthesis according to desired reaction criteria, such as ring formation or sp3 content. The workflow was initially applied for mapping chemical reaction space for aromatic aldehydes as an exemplary functional group often used in DEL synthesis. Exemplary reactions have been successfully translated to DNA-tagged substrates and can be applied to library synthesis. The versatility of the Reaction Navigator was then shown by mapping reaction space for different reaction conditions, for amines as a second set of starting materials, and for data from a second database.
Introduction
DNA-encoded library technologies, conceptualized by Brenner and Lerner 30 years ago, are today a rapidly growing research field.1 Positioned at the interface between chemistry and biology, the technology is based on tracking chemical reaction steps with DNA-tags. It allows for accessing ultra-large numbers of compounds for screening on biological targets by combining the power of combinatorial synthesis with DNA barcoding.2DELs have proven to be productive for hit identification for many target classes such as hydrolases, kinases and epigenetic targets.3–7
Despite its substantial success, this technology still poses many challenges. One particular challenge is the accessible chemical space of “DEL-compatible” chemistry, defined by the limits of DNA chemical stability. Extreme pH values are certainly prohibited, as well as strong oxidants and many metal catalysts, especially in combination with forcing reaction conditions, and mutagens.8 To date, validated DEL chemical reaction space was quite narrow, as stated in the review article by Franzini and Randolph in 20169 and then further supported by Fitzgerald and Paegel in 2021.10 Both concluded that DEL design depended on “a few good reactions”,10 namely robust transformations which use a vast scope of building blocks. However, the limited toolbox of validated DEL chemistry has been criticized in the literature as it biases chemical space coverage.9 Encoded chemical space coverage might have contributed to the lower productivity of DELs in terms of identification of clinical development candidates as compared to other hit-finding technologies.11
As chemical reactions are a key source of scaffold diversity, expanding the chemical reactions space for DNA-encoded libraries is an attractive strategy to diversify encoded library design. Accordingly, in recent years, large efforts have been dedicated towards implementing diverse reactions into DEL design.12 They included application of modern methods in catalysis, such as photocatalysis13 and micelles,14 and use of resins for DNA-immobilization,15 as well as development of barcoding strategies, such as solid-support-initiated encoded chemistry,16 and PNA-encoded chemistry (peptide nucleic acids).17 Despite the technological advances that enable a larger DEL reaction scope, the main modus operandi for reaction selection remains to screen scientific journals according to combinatorial chemistry awareness, to the medicinal chemist's toolbox, or to concepts such as DOS (diversity-oriented synthesis).18,19 An alternative to individual researchers' knowledge of chemical reactions would be a data science-based approach allowing for navigation of huge chemistry databases such as USPTO,20 Reaxys21 and CAS.22 Nowadays, data science tools have been applied successfully to support chemists23,24 and remarkable work has been conducted, mainly covering three aspects: predicting synthetic strategies (Fig. 1A), classifying reactions, and designing libraries (Fig. 1B). MEGAN (Molecule Edit Graph Attention Network) from the Jastrzębski group25 and DeepReact + by Gong et al.26 stand out for reaction conditions optimization, while the landmark research works of Segler et al.27 and Chematica from the Grzybowski group, as well as MEGAN, support retro-synthetic analysis.25,28 For reaction classification and mapping, some examples are: the attention based neural network developed by Schwaller et al.;29 the ReactionCode by Delannèe et al., which classifies, searches and balances reactions;30 the Reaction Recommender by Ghiandoni et al., which assists with reaction selection via a multi-label classification algorithm;31 and the reaction difference fingerprints developed by Andronov et al.32 Lastly, in the area of library design, two tools shall be mentioned: eDESIGNER, developed in 2020 by Martìn et al.,33 and Synthl, presented by Zabolotna et al.34 The former uses established reactions in the DEL field, and the latter, though being useful for building block-based library design, is not tailored to DEL chemistry. Thus, there is a need for computational tools that aid in identification of suitable reactions for DEL synthesis from large chemistry databases. However, chemists, who lack the technical knowledge of programming, may find it challenging to adapt existing computational workflows to a task such as DEL chemistry design, because they are based on complex algorithms such as machine learning, deep learning or attention networks.
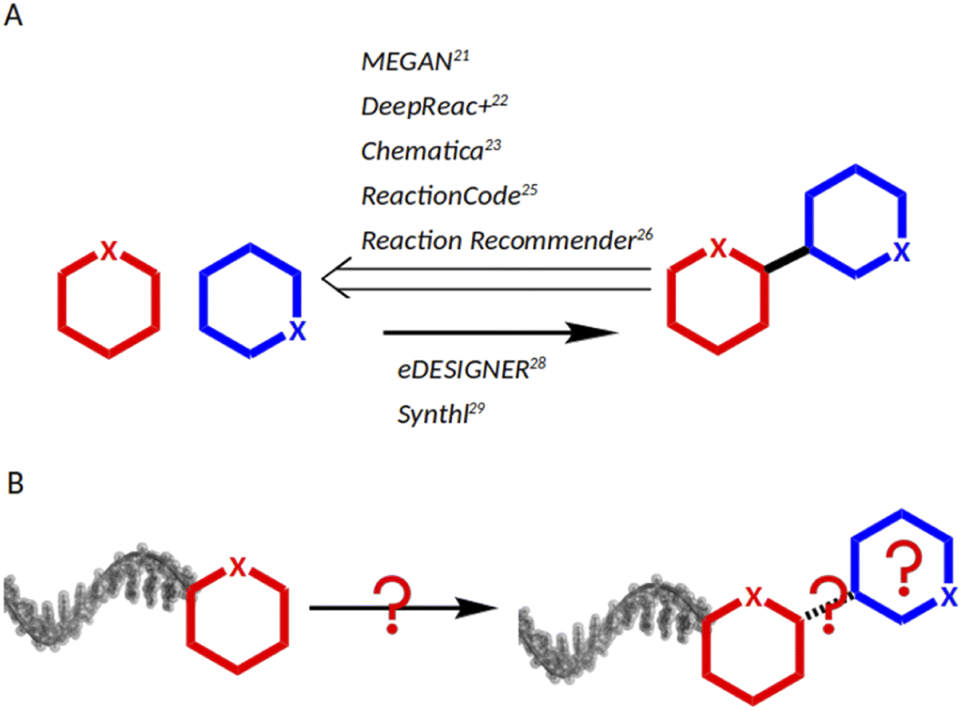 | ||
| Fig. 1 (A) Background work: existing algorithms for retro-synthetis analysis , for chemical reaction space mapping, and for library design. (B) This work: a computational workflow that considers the idiosyncratic challenges of reaction identification for DNA-encoded chemistry. | ||
With our computational workflow called “Reaction Navigator”, we aim to efficiently identify a comprehensive suite of reactions for DNA-encoded library synthesis from building blocks sharing a common functional group. This tool charts the chemical reactions space to facilitate the process of reaction identification and, by consequence, of DELs design. The algorithm is user-friendly, as it is based on pre-programmed nodes within the KNIME Analytics Platform interface, to process, analyze and visualize large data.35 This software has already proven its utility in the chemical sciences36 and it is supported by a large community via the KNIME Forum and the KNIME Hub.37
Results and discussion
The Reaction Navigator workflow was developed using a sample of reactions on aldehydes from the Reaxys® database.21§ Aldehydes were selected for their known chemical versatility38 (Fig. 2A). This algorithm utilized the chemistry-oriented extensions, such as RDKit and Indigo,39,40 implemented in KNIME for the purpose of filtering conditions, describing the reactions and plotting them according to their reaction cores. It consists of five modules which are described in upcoming paragraphs and are depicted in Fig. 2B. The workflow is publicly accessible in the KNIME Hub at the following link: https://kni.me/s/R8gFmu9rgDDqVxtp.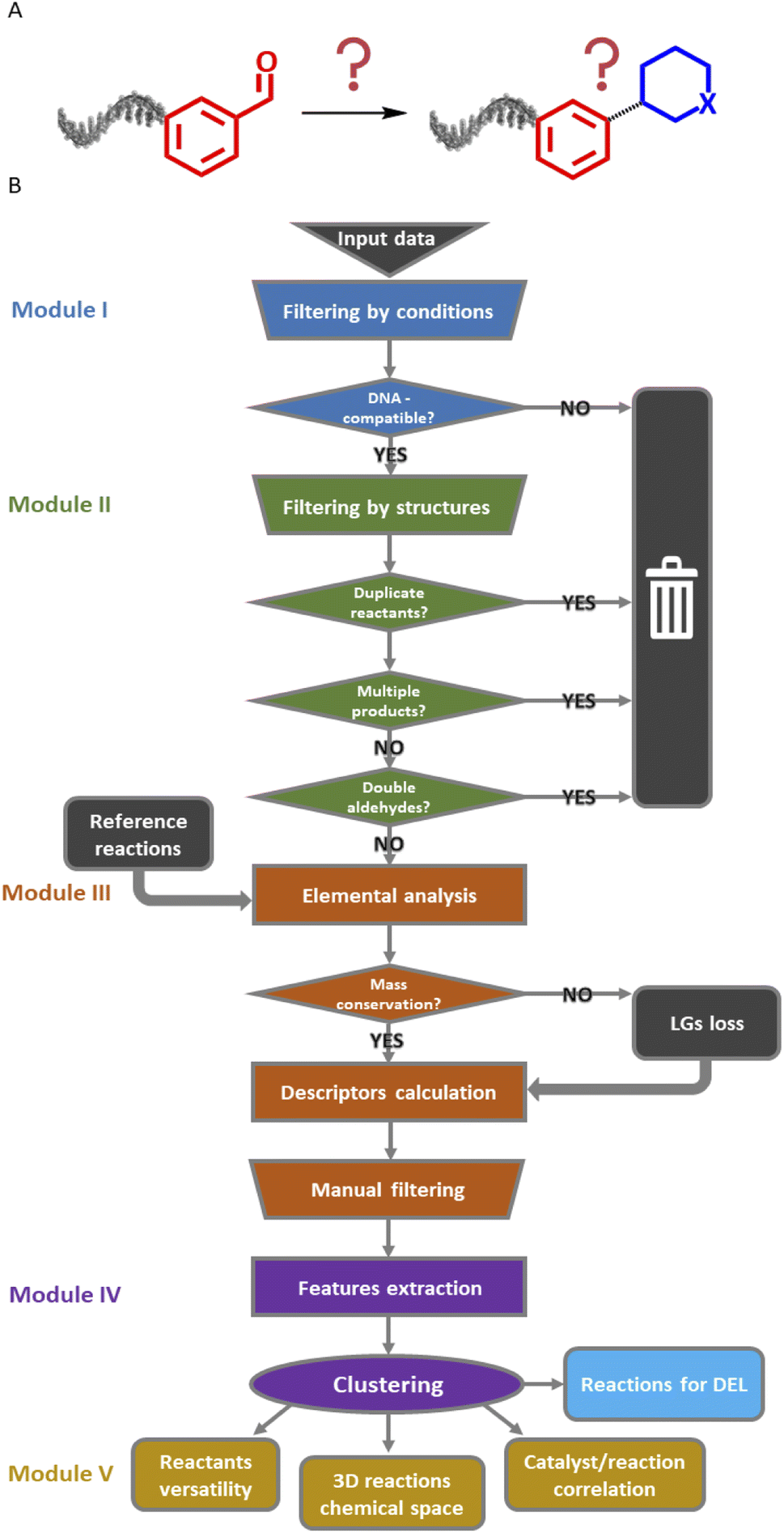 | ||
| Fig. 2 (A) Input and rationale: reactions starting with the carboxaldehyde building block with unknown products. (B) Complete scheme of the KNIME workflow with the five modules. LGs: leaving groups. | ||
Filter modules of the Reaction Navigator
The input data consisted of a list of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 reactions of aldehydes. As a first step, the data was curated in module I. This curation step removed reactions that were incompletely described or that used not commercially available reactants, reagents, or solvents. Additionally, the first module excluded reactions involving well-known DNA-incompatible reagents or forming metal complexes.8,41 At this point, we did not limit the dataset to reactions performed in aqueous solvents as solid-support strategies have been developed to allow for the use of organic solvents in DEL synthesis.16,17 The temperature threshold was set to 200 °C, which is in general high for DNA, but some reactions may be feasible on DNA at lower temperatures by simply prolonging reaction times, increasing reagents excess or catalysts loadings.42
000 reactions of aldehydes. As a first step, the data was curated in module I. This curation step removed reactions that were incompletely described or that used not commercially available reactants, reagents, or solvents. Additionally, the first module excluded reactions involving well-known DNA-incompatible reagents or forming metal complexes.8,41 At this point, we did not limit the dataset to reactions performed in aqueous solvents as solid-support strategies have been developed to allow for the use of organic solvents in DEL synthesis.16,17 The temperature threshold was set to 200 °C, which is in general high for DNA, but some reactions may be feasible on DNA at lower temperatures by simply prolonging reaction times, increasing reagents excess or catalysts loadings.42
The remaining app. 44000 reactions following the filtering step in module I were subsequently scored according to the reagents/catalysts. As in the provided dataset the distinction between reagents and catalysts was not clear, we merged them under the name mediators. The compatibility for DNA-encoded chemistry was based on the potential of a reagent for mutagenic lesions, and especially the redox potential of a given reagent, and the practical feasibility. The first two criteria affect the DNA stability and, by consequence, its reliability as a barcode at later stages and the latter influences the applicability of the reaction conditions in a DEL environment, for example working under inert atmosphere with 96-well plates could be challenging. KNIME report I describes the scoring system that we devised. A “0” means that the catalyst has higher potential to damage DNA: these are for instance many transition metal-based reagents. A “1” was given to reagents that need to be tested, and a “2” was given to reagents with high probability for or proven DNA compatibility. These are for instance proline-based organocatalysts. Finally, the score “3” was assigned to catalysts that proved to be DNA-compatible. Furthermore, solvents with higher boiling point were given preference, as they are more suitable for microliter-scale reactions. When choosing a cluster of interesting reactions, the reaction conditions can be sorted by the score, reaction temperature, and solvent boiling point and the top-ranking conditions can be selected for testing on DNA-encoded substrates. The scored reactions were processed in the second module. In this step, reactions yielding unstable products such as acetals, side products or involving duplicate identical reactants were excluded.43,44 The last part of this module deleted salts and additives from the reaction schemes, as they would have affected the following descriptors calculation (KNIME report II in the ESI†). This module removed roughly 11000 reactions.
With 33000 reactions in hand, we manually added 33 published DEL reactions, although some were already present in the provided dataset, to function as landmarks in the final map that facilitate the navigation of the chemical reactions space. The complete list is depicted in Fig. S6 in the ESI.† Some reference examples are the Ugi four-component reaction,45 the Biginelli three-component reaction,46 or the SnAP reaction.47
Module III – reaction description
The third module was initiated with an elemental analysis, namely the count of all elements (excluding hydrogen) in the products and the reactants to identify unbalanced reactions (mass conservation). In the following subtraction, a loss of one or two oxygen atoms was tolerated, due to possible condensation steps. A positive difference in count hinted at an incorrect list of reactants or at a mistake in reaction presentation. Such reactions were excluded from further analysis (ca. 2000 entries). Reactions with a negative difference were treated separately, as a loss of leaving groups was plausible and needed to be addressed (ca. 6000 entries, see below). Finally, the descriptors were calculated for the over 24000 reactions with a null difference as they were balanced. Those descriptors were inspired by the work of Feher and Schmidt48 and included structural properties such as number of rings or bond types (e.g. heteroatom–carbon bonds, carbon–carbon bonds) for a total of 21 features (Fig. 3). Similar to the elemental analysis, reaction descriptors were calculated as difference between reactants and products, to ensure that the varying substituents would not influence the counts.20 Notably, the reactions descriptors were characterized by a positive value when the considered substructure was formed and by a negative value when it was converted during the reaction. Unbalanced reactions due to leaving groups were addressed with a parallel workflow in which the descriptors of respective leaving group were subtracted from the reactant descriptors before calculating the reaction descriptors. This approach is an alternative to existing protocols, in which the leaving groups are identified and added to the product side.27 With this strategy, ca. 2000 reactions could be reinserted in the workflow for a total number of approximately 26000 reactions.
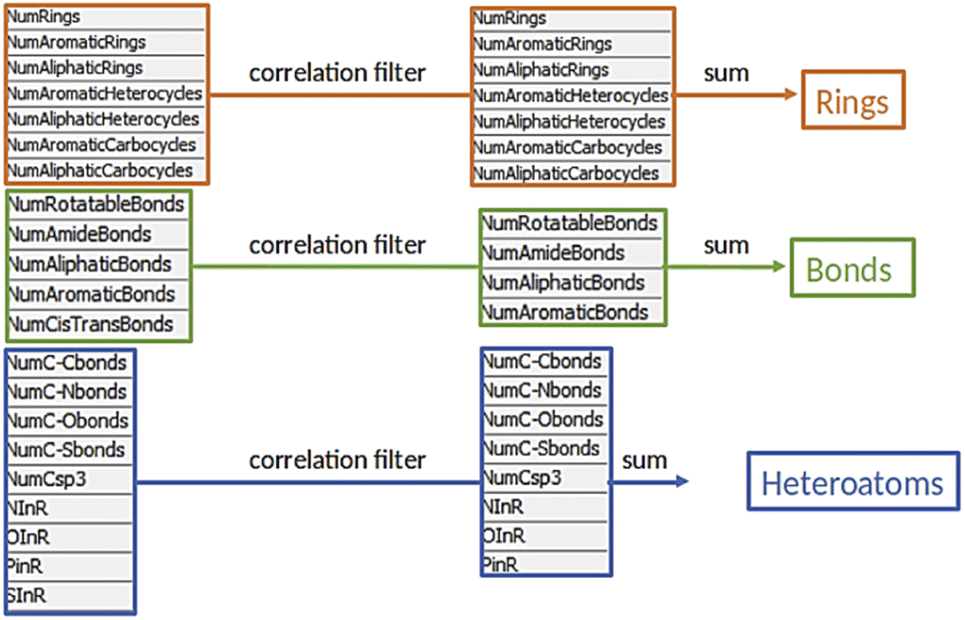 | ||
| Fig. 3 Composition of the three factors starting from the 21 descriptors, after correlation filter. NInR = number of nitrogen atoms in rings, OInR = number of oxygen atoms in rings, PInR = number of phosphorus atoms in rings, SInR = number of sulphur atoms in rings. | ||
As a result of the descriptors calculation two reactions featured prominently in the data set: reductive amination accounted for app. 7500 reactions, while aldol condensation of aldehydes and ketones corresponded to roughly 800 entries. We decided to filter them by similarity search, as they represented the equivalent of the hay in the haystack for our purpose. This additional filter left us with around 18000 transformations. A noteworthy observation is that only one sixth of the initial data set was kept at this point.
Module IV – charting the chemical reaction space
Before we charted the chemical reaction space of the descriptors, we grouped the 21 reaction features into three blocks that we deemed most important for DEL reaction selection: ring formation/opening, bond formation, and reactions involving heteroatoms. The correlation matrix, depicted in Fig. S13 in the ESI,† indicated high correlation between some reaction features. Thus, within each block, we performed a correlation filter in order to avoid redundancy. We summed up the remaining features in each of the three blocks to obtain a new set of three variables, not strongly correlated and easily representable in a three-dimensional plot (Fig. 3). The three new variables were used in the clustering protocol via the fuzzy c-means algorithm. This algorithm allows for a looser clustering, and it performs better than the hierarchical or the k-means methods, especially with sparse databases.49 The optimization of the clustering method was based on the silhouette coefficient as validation parameter50 and it is described in detail in the ESI (Fig. S14†). Fig. 4A shows the scatter plot of the clustered reactions, in which three major geometric planes are visible. This characteristic is explained by the integer nature of the variables, yet the fuzzy c-means algorithm split each plane in distinct clusters. A validation of the clustering and the silhouette coefficient was represented, for example, by the three reference reactions highlighted in Fig. 4A: the two highlighted purple dots are the Ugi and the Cushman reactions, sharing similar starting materials (imines and carbonyls), and consequently appearing in two adjacent clusters, while the green dot positioned further away is the Petasis reaction. The output of the clustering gave 59 clusters and 45 unclustered reactions. It is notable that the output contained five densely occupied clusters and all of them were characterized by reactions such as aldol reactions, aminoalkylation/amidoalkylation and cyanation. Those clusters were also spangled with Grignard reactions, which belong to the class of alkylation reactions. This finding highlights the predominance of this class of reactions in the context of the aldehyde functional group. The rest of the data was divided into the remaining 54 clusters ranging from 9 to 902 entries.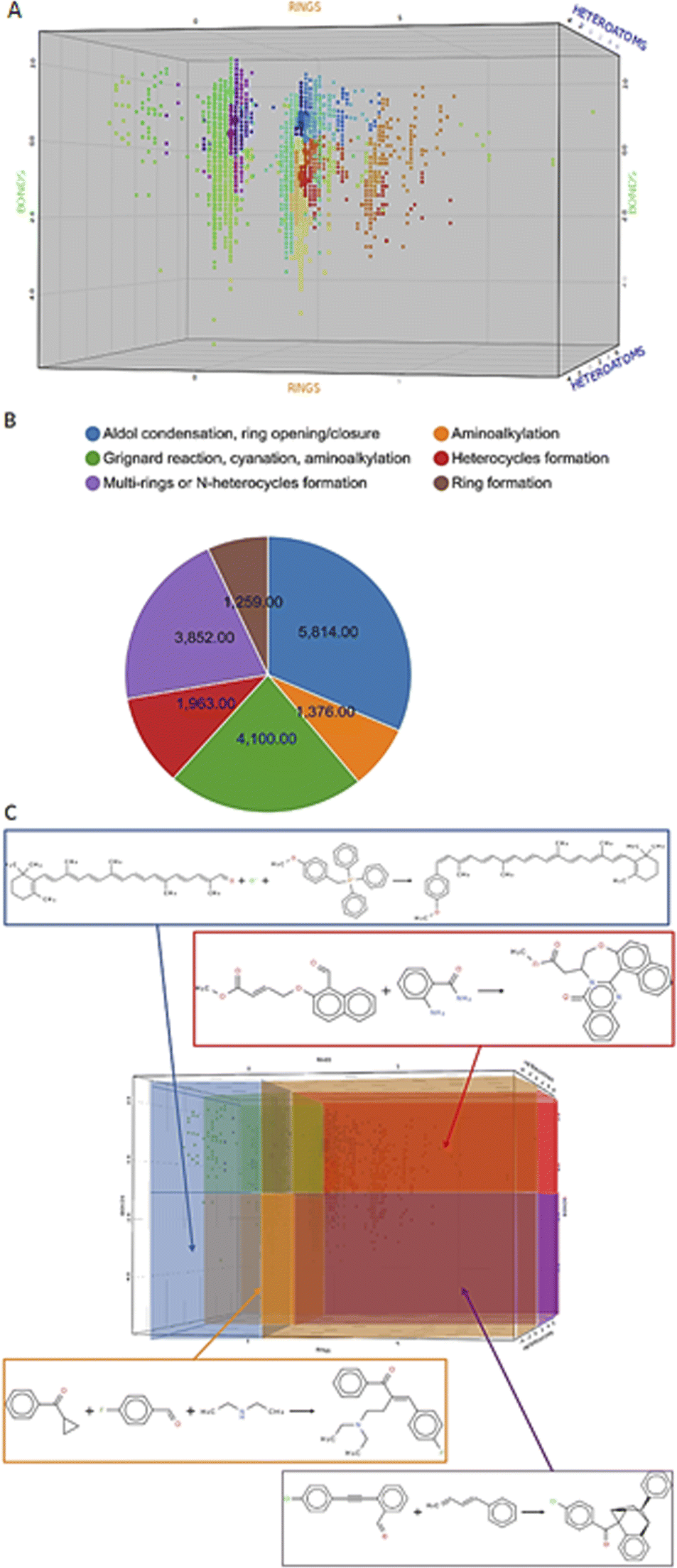 | ||
| Fig. 4 Charting and classification. (A) Scatter plot of the clustered reactions in the 3D chemical space. The dimensions are the three variables RINGS, BONDS and HETEROATOMS and the colors of the dots depend on the cluster affiliation. The small dots belong to the Reaxys data set, while the big dots represent the reference reactions. The cluster affiliation confirms the chemical similarity as emphasized by the reference reactions circled in yellow: the two closer purple dots are the Ugi and the Cushman reactions, while the green dot is the Petasis reaction. (B) Pie chart featuring the classes proportion in the data set. (C) Highlighted areas in the scatter plot, identified by the rule-based classes, and examples for each area.64–67 Enlarged in the ESI.† | ||
Module V – analysis of chemical reaction space
According to the values of three new variables, we could classify the reactions and define six areas in the final plot (Fig. 4C), according to predefined rules. The rules were manually assigned according to sampled reactions presenting certain combinations of “forming” or “breaking” bonds/substructures (see Table S5 in the ESI†). In fact, each area of the 3D scatter plot is characterized by a combination of variables, which can either be “forming” or “breaking” bonds/substructures, depending on their sign. For example, ring-opening reactions will show a “breaking” RINGS variable as seen for the linear transformations populating the bottom-left area of the plot (in orange in Figure 4C),51 while annulation reactions occupied the top-right area (in purple in Fig. 4C) which featured all three “forming” variables.52In this last module, we investigated three aspects of the chemical reaction space: versatile starting materials, common vs. rare catalysts and the accessible scaffolds. Firstly, by counting the number of clusters in which reactants were found, we could identify versatile starting materials, which form a variety of diverse scaffolds. Ten of these are shown in the ESI (Fig. S17†). The most prominent ones were malononitrile, found in 42 clusters, and dimethylcyclohexane-1,3-dione, found in 35 clusters.
Secondly, we aimed to identify versatile metal-catalysts and reagents with the intent of testing their DNA-compatibility as they potentially gave access to many different reactions for DEL design. To do so, we extracted the metal centers (metal ions in salts or complexes as well as the elements) from the database and analyzed their shares (Table S6 in the ESI†). In the pie chart in Fig. 5 it is noticeable that 70% of reactions did not contain any reagents in the provided data set, and the metal centers we defined covered slightly more than half of the remaining portion. The pie chart on the right-hand side in Fig. 5 shows that among the defined metal centers, the most common are zinc in the form of zinc powder and diethylzinc with 365 reactions, and titanium as titanium tetrachloride with 280 reactions. Copper as copper(II) bis(trifluoromethanesulfonate) and silver mainly in the form of silver acetate followed with 202 and 197 reactions respectively. Molybdenum and hafnium, with 3 and 4 reactions each, were examples of rare metal centers in this database.
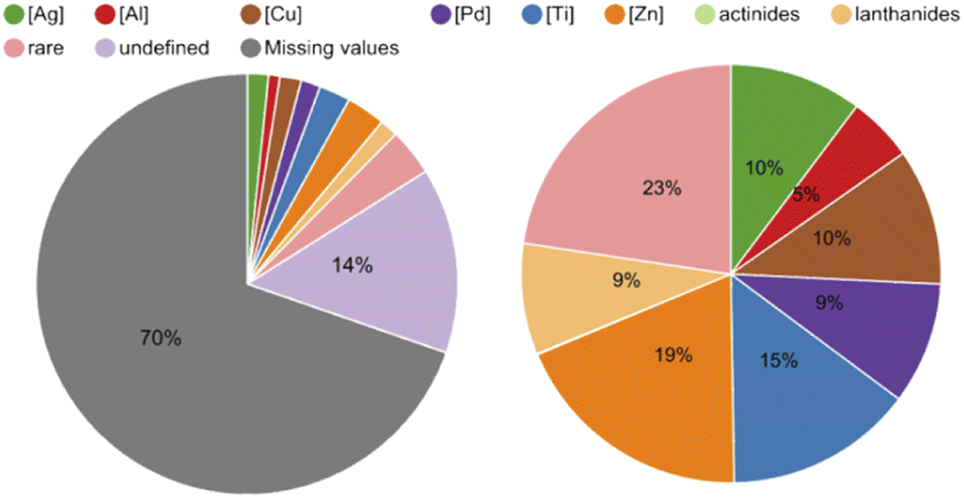 | ||
| Fig. 5 Pie chart on the left-hand side: 70% of the reaction data did not contain a mediator in the provided table and 14% showed mediators that fell outside the defined mediators (e.g. organocatalysts). Pie chart on the right-hand side: shares of the defined metal centers in the provided data set. Zinc accounted for 19%, titanium for 15%, silver and copper for 10% and palladium for 9%. The rest of the metal centers were grouped as rare (see ESI for the full detailed Table S6†). | ||
Additionally, we were curious to learn about a possible correlation between metal centers and reaction clusters. This information would guide the choice of catalysts when optimizing reaction conditions. A note must be made as a premise: on average, roughly 70% entries per each cluster featured an undefined mediator, for example organocatalysts. It is striking, that four out of the five clusters mentioned above, containing aldol additions or condensations, aminoalkylations or amidoalkylations and cyanation reactions, show a similar pattern in terms of catalysts, as visible in the heat map in Fig. S18 in the ESI,† plotting clusters vs. metal centers. Indeed, cluster 12, 48 and 52 each comprise 40% zinc-catalyzed and 30% titanium-catalyzed reactions. A clear correlation is visible for zinc and cluster 48, as depicted in the bar chart and respective pie chart in Fig. 6. Fig. 6 also illustrates one exemplary reaction from this cluster.53
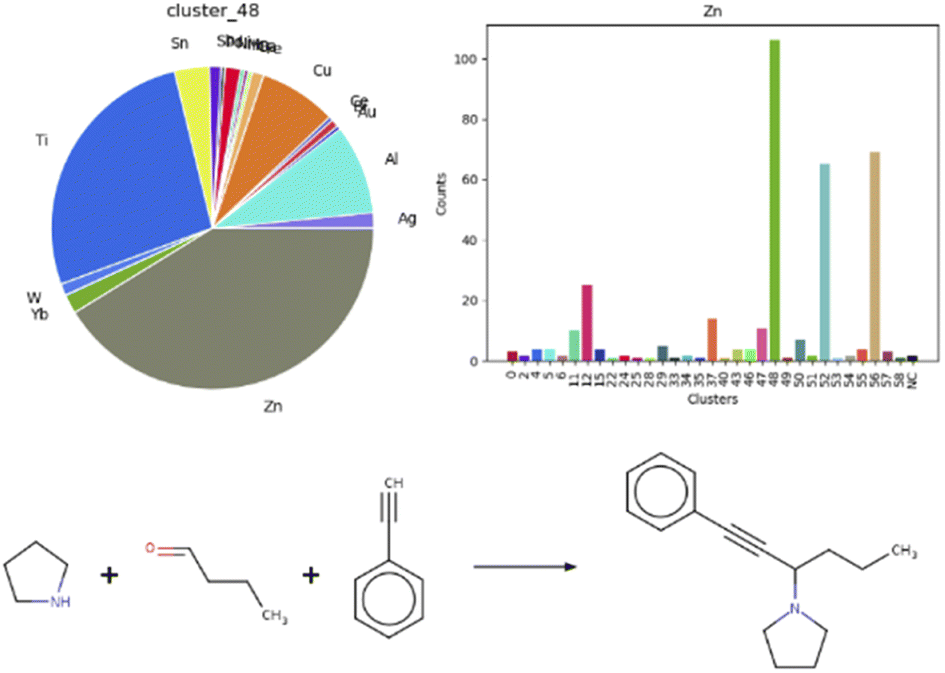 | ||
| Fig. 6 Correlation between cluster 48 and the zinc metal center, with an example of a zinc-catalyzed reaction in cluster 48.68 | ||
A similar trend, but less marked, is detectable for titanium, which promoted the same reaction types. This observation may signify that these two metals are often employed to catalyze the same class of reactions, in some cases even as a combination. Another correlation could be noticed for palladium and cluster 50, which consist of acylations. The comprehensive heat map and the results of the whole analysis are described extensively in the ESI (Fig. S21A–C†).
Finally, we aimed to analyze the accessibility of frequently used or “privileged” scaffolds in medicinal chemistry from the aldehyde functional group. For this, we referred to the work of Taylor et al., who enumerated the most common scaffolds in approved drugs.54 We searched for the first 15 entries from that publication in our data set and could determine tetrahydropyran to be the most commonly formed scaffold in 35 reactions, followed by pyridine with 19 reactions (Fig. S22 in the ESI†). In contrast, three out of 15 scaffolds were not detected in this dataset: cephem, indole and penam.
Additionally, our analysis uncovered four reactions that are likely not suitable for DEL synthesis as they employ nucleosides as starting materials. We would call them anti-reactions, as they may compromise DNA integrity. They involve malononitrile, epoxides and acrylic aldehyde as shown in Fig. S23 in the ESI.†55–58 Such reactions may be feasible on nucleobase-protected DNA barcodes.
Experimental validation of selected reactions
The ultimate application of the workflow was to extract reactions for expanding the chemistry toolbox for DEL design. At first, we skimmed the data set, extracting five central and five peripheral reactions from each cluster to pinpoint the clusters that were worth to be considered (Table S7†). Heterocycle forming multi-component reactions (MCRs) were in the focus of our search because heterocycles are abundantly represented in drug space. Furthermore, MCR strategies provide side-chain diversity and exit vectors for further library synthesis in one step. Hence, we inspected the suitable clusters and selected three reactions according to the produced scaffold.The first reaction extracted was a metal-catalyzed quinoline synthesis. The synthesis of this scaffold was reported in several publications under the usage of diverse metals that should be compatible to an on-DNA approach.59 Therefore, we started to invest the transfer of the quinoline synthesis onto a CPG-bound 10mer pyrimidine–aldehyde conjugate 1 with aniline 2a and N-Boc propargylamine 3a in the presence of different metal salts in a mixture of dimethylacetamide and triethyl orthoformate at 50 °C (Scheme 1 and Table S8,† entries 1–5). In absence of any metal salt no product formation was detectable (Table S8,† entry 1). Also in case of FeCl3, Yb(OTf)3 or Sc(OTf)3 as promoter no desired product 4 was formed (Table S4,† entries 2–4). To our delight in the presence of Cu(OTf)2 the desired DNA-quinoline 4 was formed with 59% conversion (Table S8,† entry 5), a finding which is consistent with the literature that Cu(OTf)2 is a suitable catalyst for the quinoline synthesis.59 Encouraged by these results we further improved the product formation by varying the concentrations of aniline 2a (Table S8,† entries 5–7), N-Boc propargylamine 3a (Table S8,† entries 5, 8–10) and of the promotor Cu(OTf)2 (Table S8,† entries 9 and 11), and investigating of the impact of the solvent (Table S8,† entries 11 and 17) as well as the reaction temperature (Table S8,† entries 12 and 18). As optimal reaction conditions we found the treatment of CPG-bound TC–aldehyde conjugate 1 with aniline 2a (200 mM), N-Boc propargylamine 3a (400 mM) and Cu(OTf)2 (20 mM) in dimethylacetamide at 80 °C overnight (Table S8,† entry 17). The DNA-quinoline conjugate 4 was formed with a conversion of 75% and 16% of undefined by-products, that could be easily removed by semipreparative HPLC.
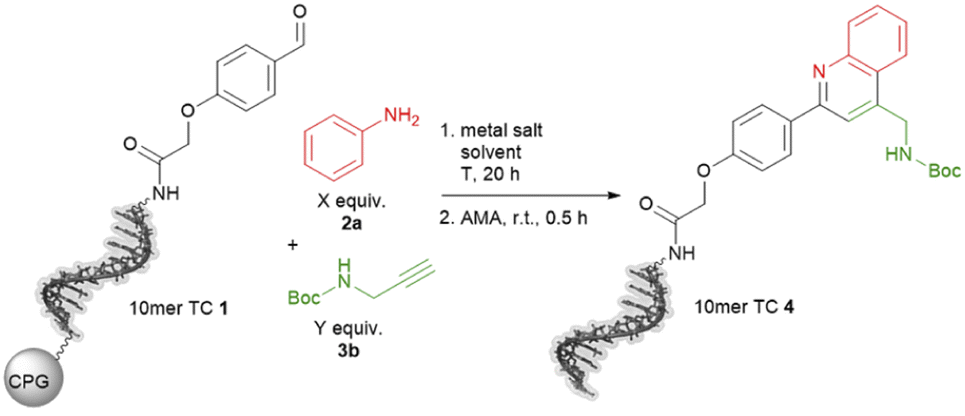 | ||
| Scheme 1 On-DNA reaction to form the quinoline scaffold. 10mer TC = 5′-TTC CTC TCC T-3′-CPG. AMA = 30% aqueous ammonia/40% aqueous methylamine, 1:1 (vol/vol). | ||
With the optimized reaction conditions in hand, we started to transfer the reaction onto a CPG-coupled 10mer ATGC–aldehyde conjugate 5. However, beside the product formation with 60% conversion we observed 33% DNA degradation (Table S9,† entry 1). To reduce the DNA damage we fine-tuned the reaction conditions further by using lower amounts of the promoter Cu(OTf)2 or reducing the reaction time (Table S9,† entries 2–4). To our delight the reaction proceeded with 5 mM concentration of the promotor with a conversion of 67% and an acceptable degree of DNA damage of 14% (Table S9,† entry 3). Based on these findings we investigated the scope of the quinoline synthesis on CPG-bound ATGC oligonucleotide–aldehyde conjugate 5 using diverse anilines 2 (Scheme 2). Independent of the electronic properties of the substituents, reactions with fluorine, bromine, ethyl, tert-butyl as well as methoxy groups proceeded smoothly leading to the desired products (6b–q). However, the position of the substituents had an impact on the reactivity of the anilines. While substituents in para or meta position resulted in moderate to excellent conversions of 61 to 87%, substituents in ortho position decreased the conversions to lower degrees of 38%. In case of bulkier substituents like a bromine or tert-butyl in ortho position the conversion dropped to below 5% (6h and 6m). For meta substituents the formation of regioisomers is possible, however the formation was not detected by the analytical methods. Cyano-substituted aniline led to the desired product 6r with a lower conversion of 19%. Nevertheless, hydroxy and ester functions were well tolerated with conversions up to 69% (6s–6w). The methyl ester is not stable under cleavage conditions and reacts in the presence of AMA to the corresponding methyl amide (6u). Also, anilines containing two different substituents were competent substrates for the quinoline synthesis with conversions of 75 and 83% (6x and 6y).
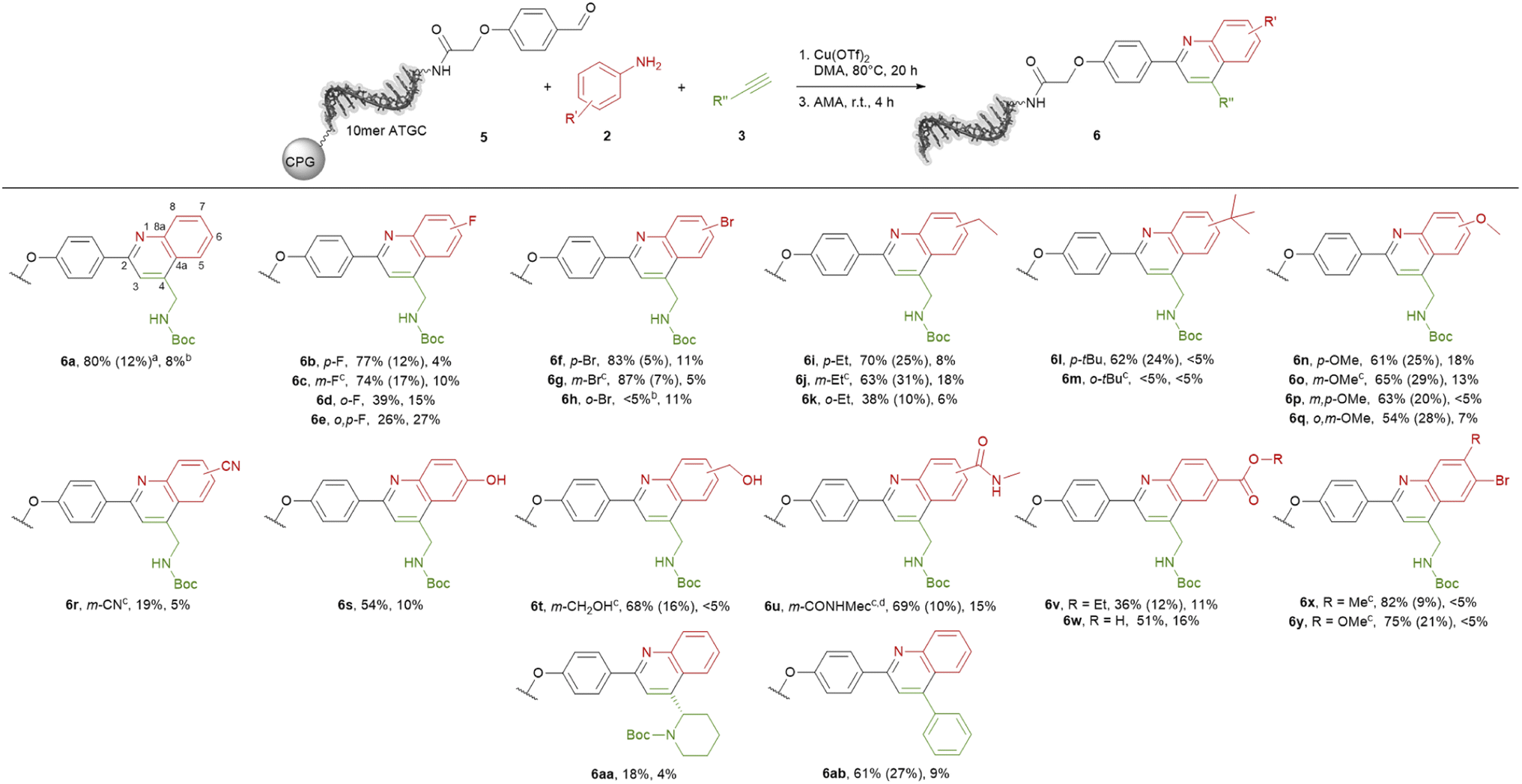 | ||
| Scheme 2 Scope of quinoline synthesis on CPG-coupled DNA aldehyde conjugate 5 with diverse anilines 6 and alkynes 7. Reaction conditions: Cu(OTf)2 (5 mM), aniline 6 (200 mM), alkyne 7 (400 mM) and DNA conjugate 9 (20 nmol) in DMA, 80 °C, 20 h. DNA cleavage: AMA, rt, 4 h. a Conversion was determined by RP-HPLC analysis based on the ratios of 5 to 6; the fraction of by-products is given in brackets. b DNA damage was determined by RP-HPLC analysis relative to the purity of 5. c Formation of a mixture of 5- and 7-regioisomers. d The corresponding methyl ester was used for the reaction, methyl amide formation occured during AMA cleavage. 10mer ATGC = 5′-NH2-C6-GTCATGATCT-3′-CPG. DMA = dimethylacetamide, AMA = 30% aqueous ammonia/40% aqueous methylamine, 1:1 (vol/vol). | ||
Last we tested further alkynes 3. Tert-butyl (S)-2-ethynylpiperidine-1-carboxylate can be used in the quinoline synthesis. The conversion to product 6aa proceeded with a low value of 18%. Phenylacetylene reacted smoothly to the desired product 6ab with 61% conversion, even if the product is quite aromatic and therefore a less attractive scaffold. In most cases the formation of undefined by-products as well as an acceptable degree of DNA damage were observed. However, the target DNA-quinoline conjugates 6 could be isolated by semi-preparative HPLC purification in all cases.
The second interesting scaffold-forming reaction was promoted by iron(III) or nickel(II) salts and produced a substituted pyrrole scaffold from an aldehyde, an amine and β-ketocarbonyls with nitromethane as solvent.60 Only few conditions were screened and good product formation was achieved at 80 °C for 6 hours (Table S13 in the ESI†). Notably, the temperature was lower than the original publication as well as the reaction time. The reaction of five different amines 8 was explored on the aldehyde-substrate 7: benzylamine, p-methoxyaniline, p-nitroaniline, 2-aminopyridine and cyclopentylamine (Scheme 3). We could clearly detect product formation for the electron-rich benzylamine and p-methoxyaniline, while the electron-poor p-nitroaniline, 2-aminopyridine, and cyclopentylamine were not competent substrates (Table S14 in the ESI†). Currently, we are investigating a broader scope of starting materials for this reaction.
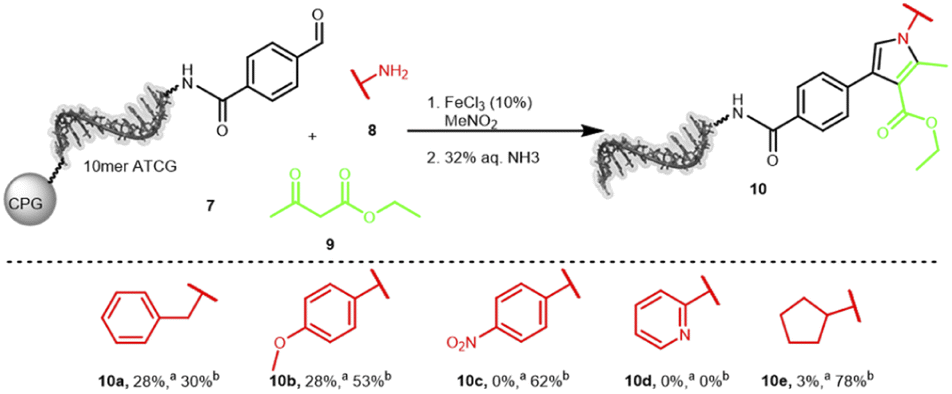 | ||
| Scheme 3 On-DNA amine scope for the synthesis of the pyrrole scaffold. Reaction conditions: FeCl3 (25 mM), amine 8 (250 mM), ethylacetoacetate 9 (250 mM) and DNA conjugate 7 (20 nmol) in MeNO2, 80 °C, 6 h. DNA cleavage: 23% aq. ammonia, 4 h. a Conversion was determined by RP-HPLC analysis based on the ratios of 7 to 10. b DNA damage was determined by RP-HPLC analysis relative to the purity of conjugate 7. 10mer ATGC = 5′-NH2-C6-GTCATGATCT-3′-CPG, MeNO2 = nitromethane. | ||
The third transformation was a retro-aza-Michael addition involving a ring expansion reaction, promoted by ethyl aluminium iodide (Et2AlI). The reaction between a cyclopropane-based thioester 12 and an imine was expected to form a diversely substituted pyrrolidine scaffold.61 When applied to the protected DNA substrate 11 on controlled pore glass (CPG), the reaction did not yield any product 14 but we observed significant DNA degradation. Therefore, we decided to test MgI2 as a suggested alternative mediator, but this Lewis acid surprisingly cleaved the C6-amino linker at the 5′-terminus of the oligonucleotide (Scheme 4 and Table S16 in the ESI†).
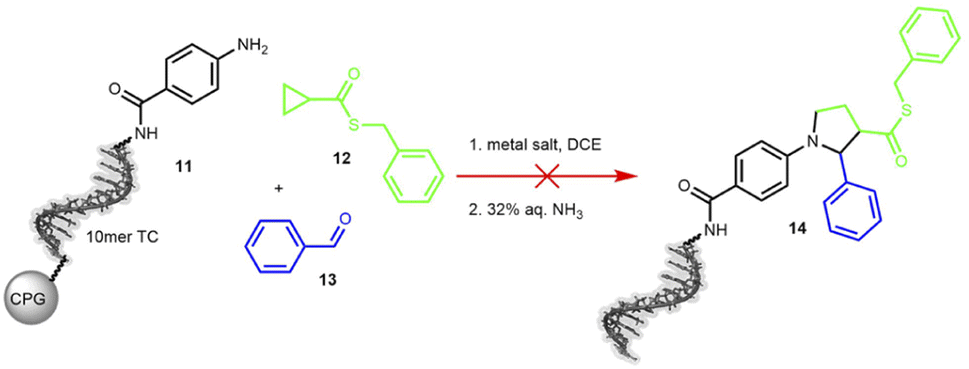 | ||
| Scheme 4 On-DNA reaction to form the pyrrolidine scaffold. Product formation was detected by MALDI-TOF. 10mer TC = 5′-TTC CTC TCC T-3′-CPG, DCE = dichloroethane. | ||
Experimental data from unsuccessful reactions are still very valuable as they allow us to improve the filtering cascade.
Adaptability of the Reaction Navigator
In order to ensure the adaptability of the algorithm to different applications, we changed the filtering cascade to select reactions that are tolerating water, applied the workflow to amines as functional group, and applied the workflow to the USPTO dataset.000 reactions survived this filtering step (Fig. 7). Further filtering of these 4000 reactions gave a final dataset of only 1900 reactions that were clustered and projected in the chemical reaction space depicted in Fig. S24.† Reactions tollerant to water are rare in the organic chemistry landscape, and this finding justifies the effort in searching for potentially compatible reactions and in adapting them to DNA-tagged substrates.
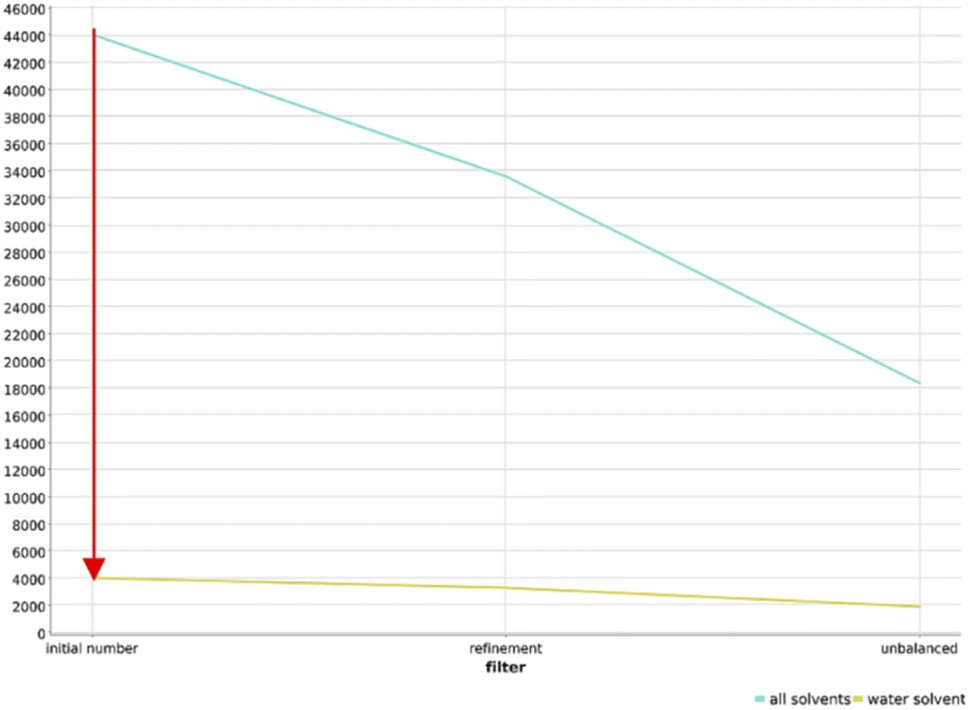 | ||
| Fig. 7 Main reductions in the number of reactions over the course of the workflow for the two data sets: reactions with all solvents (blue) and reactions with water (yellow). Notably, the biggest drop occurs when filtering reactions involving water as solvent, highlighted by the red arrow. | ||
000 reactions, approximately 1000 were excluded because the nitrogen was represented by an amide or amidine group. Additionally, the workflow excluded 63% reactions due to duplicate starting material, multiple side products, and especially due to unknown leaving groups. The clustering produced a good silhouette coefficient of 0.766, although 451 reactions could not be clustered and were assigned to the noise (red data points in Fig. S25†).
In the scatter plot in Fig. S25,† where some examples are highlighted with the respective reaction cores, it is noticeable that reactions clustered together either form similar scaffolds or involve similar transformations.
000 reactions starting with aldehydes extracted from the USPTO database. The reactions could not be filtered or ranked by compatibility as such data set do not provide well-organized information about the conditions. Furthermore, in many entries, reactants were represented as “above the arrow” species, not figuring among the reactants. Nevertheless, the same workflow was applied also on reactions starting with primary amines with the only difference that reactions with unreacted amines were excluded, instead of unreacted aldehydes. The clustering was as effective as for the initial data set and the respective chemical reaction space scatter plots with examples are depicted in Fig. S26 and S27,† respectively. The silhouette coefficient, which was proved to correlate with the clustering quality, showed good results: 0.736 and 0.732 respectively.
In both cases a striking reduction in the number of reactions was observed during the treatment of reactions with leaving groups. From 10000 reactions, only ca. 1400 aldehyde-starting and 1000 amine-starting reactions were clustered, respectively. The leaving groups treatment is still to be improved by machine learning paving the way for fruitful collaborations, using for example the rxnmapper to identify the missing substructures in the products.69
Conclusions
As chemical reaction space is vast, it is essential to support forward design of screening libraries by efficient navigation strategies. This process can be facilitated with data science tools as an effective alternative to the tedious screening of large numbers of publications in the search for attractive, yet under-exploited, reactions. Considerable effort has been put into the development of advanced computational tools to support synthesis of target molecules.29–34 However, a computer-based tool for chemical reaction space navigation that can be operated and tailored without expert knowledge in information technologies was lacking to date. We employed the open-access KNIME interface that allows for data analysis without the need for scripting to set up simple mathematical operations for chemical reaction filtering, mapping, clustering and statistical analysis. A dataset of 100000 reactions utilizing arylaldehydes was initially filtered to remove reactions that use less accessible reactants, reaction conditions with higher probability of DNA damage, and products with low appeal for encoded library design, e.g., due to expected instability in aqueous solutions. Following reduction of the dataset, we described and clustered chemical transformations according to three predefined variables. The feature extraction and reactions classification ensured minimal loss of information and improved interpretability of the very large dataset. In parallel, we developed the nucleus of a computational workflow for treating reactions involving leaving groups. It is worth mentioning that the KNIME interface allows for adapting the workflow to different research foci at different stages such us the filtering cascade, the descriptors selection, or the clustering method.
Despite its simplicity, the Reaction Navigator conferred us deep insight into the potential DEL-compatible chemical reactions space (Fig. 4) and precious information to guide future research, such as correlations between metal catalysts and certain reaction types, reactant versatility and scaffold accessibility. Above all, the charted reactions suggested some examples that were successfully tested on DNA-tagged substrates and could potentially be implemented in library synthesis.
However, limitations remain which are due to incomplete knowledge about DNA chemical stability under given reaction conditions. These are exemplified by the identification of clusters of reactions that have a low probability to be translatable to DEL, such as Grignard reactions which need to be performed under strictly dry conditions, or titanium tetrachloride-promoted reactions which will set free large amounts of hydrogen chloride. Data quality is a second limiter. In fact, a standardized method for presenting reaction schemes and reaction conditions data is indispensable for further development of cheminformatics or machine learning techniques. Promising advancements in this direction have been proposed, on the one hand, with ELNs (Electronic Laboratory Notebooks)62 and, on the other hand, with the creation of the ORD (Open Reaction Database) in 2021.63 Furthermore, manual steps such as the scoring process and the leaving groups treatments might be improved with machine learning techniques to reduce the human intervention to a minimum. Finally, the chemical stability of DNA needs to be investigated under a multitude of reaction conditions to improve the filter cascade, as indicated by the failed reaction.
Obvious applications and extensions of the Reaction Navigator are experimental testing of more examples from the charted reactions on DNA-tagged substrates, and an application of the workflow to further functional groups commonly used in DEL synthesis such as amines, aryl/alkyl halides, and carboxylic acids. Additionally, the Reaction Navigator could be tailored to navigate the plausible reaction space for other classes of (bio-)molecules such as proteins, peptides, or natural products that shall be diversified by chemical methods. In summary, the Reaction Navigator is a data science tool with a low barrier to use for the non-expert which can aid the chemist in decision making. With this rational, we expect to see a surge of approaches directed toward the expansion of the chemical and biological reaction space in library design.
Data availability
The ESI† contains experimental data describing the synthesis and characterization of all DNA conjugates. It contains a detailled description of the computational workflows, too. The workflow is publicly accessible in the KNIME Hub at the following link: https://kni.me/s/R8gFmu9rgDDqVxtp.Author contributions
SC developed the computational workflow, performed on-DNA reactions, and wrote the manuscript (data curation, formal analysis, investigation, methodology, validation, visualization and writing – original draft). CE developed part of the computational workflow, and edited the manuscript (conceptualization, writing – review and editing). MP performed on-DNA reactions, and wrote the manuscript (investigation, methodology, validation, writing – original draft). FB and LG analysed the catalyst use (data curation and investigation). S. B. and K. I. supported the use of statistics tools, and edited the manuscript (supervision, writing – review and editing). FvdB and S. B. provided the resources, curated data, supervised the project, and edited the manuscript (resources, data curation, writing – review and editing). A. B. conceptualized and supervised the project and wrote the manuscript (conceptualization, project administration, supervision and writing – review and editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the support of the DFG grant No. BR 5049/3-1.Notes and references
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 1992, 89(12), 5381–5383 CrossRef CAS PubMed.
- Y. K. Sunkari, Y. Krishna, V. Kumar Siripuram, T. Nguyen and M. Flajolet, Trends Pharmacol. Sci., 2022, 43(1), 4–15 CrossRef CAS PubMed.
- (a) C. A. Reiher, D. P. Schuman, N. Simmons and S. Wolkenberg, ACS Med. Chem. Lett., 2021, 12(3), 343–350 CrossRef CAS PubMed; (b) M. Song and G. T. Hwang, J. Med. Chem., 2020, 63, 6578–6599 CrossRef CAS PubMed.
- Y. Ding, H. O'Keefe, J. L. DeLorey, D. I. Israel, J. A. Messer, C. H. Chiu, S. R. Skinner, R. E. Matico, M. F. Murray-Thompson, F. Li, M. A. Clark, J. W. Cuozzo, C. Arico-Muendel and B. A. Morgan, ACS Med. Chem. Lett., 2015, 6(8), 888–893 CrossRef CAS.
- H. Deng, J. Zhou, F. Sundersingh, J. A. Messer, D. O. Somers, M. Ajakane, C. Arico-Muendel, A. Beljean, S. L. Belyanskaya, R. Bingham, E. Blazensky, A.-B. Boullay, E. Boursier, J. Chai, P. Carter, C.-W. Chung, A. Daugan, Y. Ding, K. Herry, C. Hobbs, E. Humphries, C. Kollmann, V. L. Nguyen, E. Nicodeme, S. E. Smith, N. Dodic and N. Ancellin, ACS Med. Chem. Lett., 2016, 7(4), 379–384 CrossRef CAS PubMed.
- C. R. Wellaway, D. Amans, P. Bamborough, H. Barnett, R. A. Bit, J. A. Brown, N. R. Carlson, C. Chung, A. W. J. Cooper, P. D. Craggs, R. P. Davis, T. W. Dean, J. P. Evans, L. Gordon, I. L. Harada, D. J Hirst, P. G. Humphreys, K. L. Jones, A. J. Lewis, M. J. Lindon, D. Lugo, M. Mahmood, S. McCleary, P. Medeiros, D. J. Mitchell, M. O'Sullivan, A. Le Gall, V. K. Patel, C. Patten, D. L. Poole, R. R. Shah, J. E. Smith, K. A. J Stafford, P. J. Thomas, M. Vimal, I. D. Wall, R. J. Watson, N. Wellaway, G. Yao and R. K. Prinjha, J. Med. Chem., 2020, 63(2), 714–746 CrossRef CAS PubMed.
- H. Richter, A. L. Satz, M. Bedoucha, B. Buettelmann, A. C. Petersen, A. Harmeier, R. Hermosilla, R. Hochstrasser, D. Burger, B. Gsell, R. Gasser, S. Huber, M. N. Hug, B. Kocer, B. Kuhn, M. Ritter, M. G. Rudolph, F. Weibel, J. Molina-David, J.-J. Kim, J. V. Santos, M. Stihle, G. J. Georges, R. D. Bonfil, R. Fridman, S. Uhles, S. Moll, C. Faul, A. Fornoni and M. Prunotto, ACS Chem. Biol., 2019, 14(1), 37–49 CrossRef CAS PubMed.
- M. Potowski, F. Losch, E. Wünnemann, J. K. Dahmen, S. Chines and A. Brunschweiger, Chem. Sci., 2019, 10, 10481–10492 RSC.
- R. M. Franzini and C. Randolph, J. Med. Chem., 2016, 59(14), 6629–6644 CrossRef CAS PubMed.
- P. R. Fitzgerald and B. M. Paegel, Chem. Rev., 2021, 121, 7155–7177 CrossRef CAS PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2018, 61, 9442–9468 CrossRef CAS PubMed.
- (a) A. L. Satz, A. Brunschweiger, M. E. Flanagan, A. Gloger, N. J. V. Hansen, L. Kuai, V. B. K. Kunig, X. Lu, D. Madsen, L. A. Marcaurelle, C. Mulrooney, G. O'Donovan, S. Sakata and J. Scheuermann, Nat. Rev. Methods Primers, 2022, 2, 3 CrossRef CAS; (b) K. Götte, S. Chines and A. Brunschweiger, Tetrahedron Lett., 2020, 61, 151889 CrossRef.
- J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723–3732 CrossRef CAS PubMed.
- M. Klika Škopić, K. Götte, C. Gramse, M. Dieter, S. Pospich, S. Raunser, R. Weberskirch and A. Brunschweiger, J. Am. Chem. Soc., 2019, 141, 10546–10555 CrossRef PubMed.
- D. T. Flood, S. Asai, X. Zhang, J. Wang, L. Yoon, Z. C. Adams, B. C. Dillingham, B. B. Sanchez, J. C. Vantourout, M. E. Flanagan, D. W. Piotrowski, P. Richardson, S. A. Green, R. A. Shenvi, J. S. Chen, P. S. Baran and P. E. Dawson, J. Am. Chem. Soc., 2019, 141, 9998–10006 CrossRef CAS PubMed.
- (a) V. B. K. Kunig, C. Ehrt, A. Dömling and A. Brunschweiger, Org. Lett., 2019, 21, 7238 CrossRef CAS PubMed; (b) M. Potowski, B. B. K. Kunig, F. Losch and A. Brunschweiger, MedChemComm, 2019, 10(7), 1082–1093 RSC.
- D. Chouikhi, M. Ciobanu, C. Zambaldo, V. Duplan, S. Barluenga and N. Winssinger, Chem.–Eur. J., 2012, 18, 12698–12704 CrossRef CAS PubMed.
- R. J. Fair, R. T. Walsh and C. D. Hupp, Bioorg. Med. Chem. Lett., 2021, 51, 128339 CrossRef CAS.
- C. J. Gerry, M. J. Wawer, P. A. Clemons and S. L. Schreiber, J. Am. Chem. Soc., 2019, 141, 10225–10235 CrossRef CAS PubMed.
- N. Schneider, D. M. Lowe, R. A. Sayle, M. A. Tarselli and G. A. Landrum, J. Med. Chem., 2016, 59, 4385–4402 CrossRef CAS PubMed.
- https://www.reaxys.com .
- https://www.cas.org/ .
- C. W. Coley, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2018, 51, 1281–1289 CrossRef CAS.
- H. J. Kulik and M. S. Sigman, Acc. Chem. Res., 2021, 54, 2335–2336 CrossRef CAS PubMed.
- M. Sacha, M. Błaż, P. Byrski, P. Dąbrowski-Tumański, M. Chromiński, R. Loska, P. Włodarczyk-Pruszyński and S. Jastrzębski, J. Chem. Inf. Model., 2021, 61, 3273–3284 CrossRef CAS PubMed.
- Y. Gong, D. Xue, G. Chuai, J. Yu and Q. Liu, Chem. Sci., 2021, 12, 14459–14472 RSC.
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. J. Trice and B. A. Grzybowski, Chem, 2018, 4, 522–532 CAS.
- P. Schwaller, D. Probst, A. C. Vaucher, V. H. Nair, D. Kreutter, T. Laino and J.-L. Reymond, Nat. Mach. Intell., 2021, 3, 144–152 CrossRef.
- V. Delannée and M. C. Nicklaus, J. Cheminf., 2020, 12, 72 Search PubMed.
- G. M. Ghiandoni, M. J. Bodkin, B. Chen, D. Hristozov, J. E. A. Wallace, J. Webster and V. J. Gillet, J. Comput.-Aided Mol. Des., 2020, 34, 783–803 CrossRef CAS PubMed.
- M. Andronov, M. V. Fedorov and S. Sosnin, ACS Omega, 2021, 6, 30743–30751 CrossRef CAS PubMed.
- A. Martín, C. A. Nicolaou and M. A. Toledo, Commun. Chem., 2020, 3, 127 CrossRef.
- Y. Zabolotna, D. M. Volochnyuk, S. V. Ryabukhin, K. Gavrylenko, D. Horvath, O. Klimchuk, O. Oksiuta, G. Marcou and A. Varnek, J. Chem. Inf. Model., 2021, 41, 2100289 Search PubMed.
- M. R. Berthold, N. Cebron, F. Dill, T. R. Gabriel, T. Kötter, T. Meinl, P. Ohl, C. Sieb, K. Thiel and B. Wiswedel, in Studies in Classification, Data Analysis, and Knowledge Organization, Springer, 2007 Search PubMed.
- A. J. Kooistra, M. Vass, R. McGuire, R. Leurs, I. J. P. de Esch, G. Vriend, S. Verhoeven and C. de Graaf, ChemMedChem, 2018, 13, 614–626 CrossRef CAS PubMed.
- (a) https://hub.knime.com/ ; (b) https://forum.knime.com/. .
- N. G. Paciaroni, J. M. Ndungu and T. Kodadek, Chem. Commun., 2020, 56, 4656–4659 RSC.
- RDKit, Open-source cheminformatics. https://www.rdkit.org/ Search PubMed.
- Indigo toolkit, GGA Software Services, https://ggasoftware.com/ Search PubMed.
- M. L. Malone and B. M. Paegel, ACS Comb. Sci., 2016, 18, 182–187 CrossRef CAS PubMed.
- M. Potowski, R. Lüttig, A. Vakalopoulos and A. Brunschweiger, Org. Lett., 2021, 23, 5480–5484 CrossRef CAS PubMed.
- H. Park, J. Choi, M. Kim, S. Choi, M. Park, J. Lee, Y.-G. Suh, H. Cho, U. Oh, H.-D. Kim, Y. H. Joo, S. S. Shin, J. K. Kim, Y. S. Jeong, H.-J. Koh, Y.-H. Park and S. Jew, Bioorg. Med. Chem. Lett., 2005, 15, 631–634 CrossRef CAS PubMed.
- N. Edayadulla and P. Ramesh, Eur. J. Med. Chem., 2015, 106, 44–49 CrossRef CAS PubMed.
- I. Ugi, R. Meyr, U. Fetzer and C. Steinbrückner, Angew. Chem., 1959, 71, 373–388 Search PubMed.
- P. Biginelli, Ber. Dtsch. Chem. Ges., 1891, 24, 1317–1319 CrossRef.
- C.-V. T. Vo, G. Mikutis and J. W. Bode, Angew. Chem., Int. Ed., 2013, 52, 1705–1708 CrossRef CAS PubMed.
- M. Feher and J. M. Schmidt, J. Chem. Inf. Comput. Sci., 2003, 43, 218–227 CrossRef CAS PubMed.
- J. C. Bezdek, J. Cybern., 1973, 3, 58–73 CrossRef.
- P. J. Rousseeuw, J. Comput. Appl. Math., 1987, 20, 53–65 CrossRef.
- E. S. Hand, K. A. Belmore and L. D. Kispert, Helv. Chim. Acta, 1993, 76, 1928–1938 CrossRef CAS.
- Z. Cao, H. Zhu, X. Meng, L. Tian, G. Chen, X. Sun and J. You, J. Org. Chem., 2016, 81, 12401–12407 CrossRef CAS.
- A. Franche, C. Imbs, A. Fayeulle, F. Merlier, M. Billamboz and E. Léonard, Chin. Chem. Lett., 2020, 31, 706–710 CrossRef CAS.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- M. Shekouhy and A. Khalafi-Nezhad, Green Chem., 2015, 17, 4815–4829 RSC.
- R. Kordnezhadian, M. Shekouhy, S. Karimian and A. Khalafi-Nezhad, J. Catal., 2019, 380, 91–107 CrossRef CAS.
- B. T. Golding, P. K. Slaich, G. Kennedy, C. Bleasdale and W. P. Watson, Chem. Res. Toxicol., 1996, 9, 147–157 Search PubMed.
- M. Sako and I. Yaekura, Tetrahedron, 2002, 58, 8413–8416 CrossRef CAS.
- (a) S. Yu, J. Wu, H. Lan, H. Xu, X. Shi, X. Zhu and Z. Yin, RSC Adv., 2018, 8, 33968–33971 RSC; (b) J. Tang, L. Wang, D. Mao, W. Wang, L. Zhang, S. Wu and Y. Xie, Tetrahedron, 2011, 67, 8465–8469 CrossRef CAS; (c) X. Zhang, B. Liu, X. Shu, Y. Gao, H. Lv and J. Zhu, J. Org. Chem., 2012, 77, 501–510 CrossRef CAS PubMed; (d) P. B. Sarode, S. P. Bahekar and H. S. Chandak, Tetrahedron Lett., 2016, 57, 5753–5756 CrossRef CAS; (e) C. E. Meyet and C. H. Larsen, J. Org. Chem., 2014, 79, 9835–9841 CrossRef CAS PubMed.
- (a) A. T. Khan, M. Lal, P. Ray Bagdi, R. Sidick Basha, P. Saravanan and S. Patra, Tetrahedron Lett., 2012, 53, 4145–4150 CrossRef CAS; (b) S. Maiti, S. Biswas and U. Jana, J. Org. Chem., 2010, 75, 1674–1683 CrossRef CAS PubMed.
- W. Huang, J. Chin, L. Karpinski, G. Gustafson, C. M. Baldino and L. Yu, Tetrahedron Lett., 2006, 47(28), 4911–4915 CrossRef CAS.
- G. M. Ghiandoni, M. J. Bodkin, B. Chen, D. Hristozov, J. E. A. Wallace, J. Webster and V. J. Gillet, J. Chem. Inf. Model., 2019, 59, 4167–4187 CrossRef CAS PubMed.
- S. M. Kearnes, M. R. Maser, M. Wleklinski, A. Kast, A. G. Doyle, S. D. Dreher, J. M. Hawkins, K. F. Jensen and C. W. Coley, J. Am. Chem. Soc., 2021, 143, 18820–18826 CrossRef CAS.
- E. S. Hand, K. A. Belmore and L. D. Kispert, Helv. Chim. Acta, 1993, 76, 1928–1938 CrossRef CAS.
- V. Srinivasulu, I. Shehadeh, M. A. Khanfar, O. G. Malik, H. Tarazi, I. A. Abu-Yousef, A. Sebastian, N. Baniowda, M. J. O'Connor and T. H. Al-Tel, J. Org. Chem., 2019, 84, 934–948 CrossRef CAS PubMed.
- F. Bertozzi, M. Gustafsson and R. Olsson, Org. Lett., 2002, 4, 4333–4336 CrossRef CAS PubMed.
- S. Zhu, Z. Guo, Z. Huang and H. Jiang, Chem.–Eur. J., 2014, 20, 2425–2430 CrossRef CAS PubMed.
- J. Rosales, J. M. Garcia, E. Ávila, T. González, D. S. Coll and E. Ocando-Mavárez, Inorg. Chim. Acta, 2017, 467, 155–162 CrossRef CAS.
- P. Schwaller, B. Hoover, J.-L. Reymond, H. Strobelt and T. Laino, Sci. Adv., 2021, 7, eabe4166 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02474h |
| ‡ Current address: Serengen GmbH, Emil-Figge-Str. 76a, 44227, Dortmund, Germany. |
| § Copyright© 2022 Elsevier Limited except certain content provided by third parties. Reaxys® is a trademark of Elsevier Limited. Reaxys data were made accessible to our research project via the Elsevier R&D Collaboration Network. |
| This journal is © The Royal Society of Chemistry 2022 |