
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanical interlocking of SWNTs with N-rich macrocycles for efficient ORR electrocatalysis†
Wanzheng
Zhang
 a,
Melanie
Guillén-Soler
b,
Sara
Moreno-Da Silva
a,
Alejandro
López-Moreno
a,
Luisa R.
González
c,
María del Carmen
Giménez-López
*b and
Emilio M.
Pérez
*a
a,
Melanie
Guillén-Soler
b,
Sara
Moreno-Da Silva
a,
Alejandro
López-Moreno
a,
Luisa R.
González
c,
María del Carmen
Giménez-López
*b and
Emilio M.
Pérez
*a
aIMDEA Nanociencia, C/ Faraday 9, Madrid, 28049, Spain. E-mail: emilio.perez@imdea.org
bCIQUS, Universidad de Santiago de Compostela, Rua Jenaro de la Fuente, Santiago de Compostela, 15782, Spain. E-mail: maria.gimenez.lopez@usc.es
cDepartamento de Química Inorgánica, Universidad Complutense de Madrid, Madrid, 28040, Spain
First published on 25th July 2022
Abstract
Substitutional N-doping of single-walled carbon nanotubes is a common strategy to enhance their electrocatalytic properties in the oxygen-reduction reaction (ORR). Here, we explore the encapsulation of SWNTs within N-rich macrocycles as an alternative strategy to display electroactive sites on the surface of SWNTs. We design and synthesize four types of mechanically interlocked derivatives of SWNTs (MINTs) by combining two types of macrocycles and two types of SWNT samples. Comprehensive electrochemical characterization of these MINTs and their reference SWNTs allows us to establish structure–activity relationships. First, we show that all MINT samples are superior electrocatalysts compared to pristine SWNTs, which serves as general validation of our strategy. Secondly, we show that macrocycles displaying both N atoms and carbonyl groups perform better than those with N atoms only. Finally, we demonstrate that a tighter fit between macrocycles and SWNTs results in enhanced catalytic activity and stability, most likely due to a more effective charge-transfer between the SWNTs and the macrocycles. These results, focusing on the ORR as a testbed, show the possibility of understanding electrocatalytic performance of SWNTs at the molecular level and thus enable the design of more active and more stable catalysts in the future.
Introduction
Functionalization of single-walled carbon nanotubes (SWNTs) with molecular fragments using covalent or noncovalent chemistry is a common strategy to alter their physical properties for their further application.1–4 Substitutional heteroatom doping of SWNTs is another strategy towards the same end.5–7 Among possible dopants (B, N, S, F, and P), the N dopant is perhaps the most frequently studied, since the doping process is typically facile, and the N-doped SWNTs maintain high electrical conductivity and are typically easier to disperse in organic solvents.8–11 As potentially negative outcomes, N-doping introduces defects into the SWNT structure, and the insertion of N groups produces changes in the energy band arrangements leading to creation of additional energy levels. Framed within the search for carbon-based metal-free catalysts12–17 for fuel cells18,19 and metal–air batteries20–23,24,25 N-doped carbon nanotubes have been reported as metal-free catalysts for the oxygen-reduction reaction (ORR) with much better electrocatalytic activity and operational stability than platinum.26,27 The intramolecular charge-transfer caused by heteroatom/defect doping modifies the charge/spin distribution of the sp2-conjugated carbon networks, yielding a more accessible chemisorption mode of O2 (and intermediates) and the subsequent electron transfer for the breaking and forming of chemical bonds.28,29 In addition, physically adsorbing polymers or conjugated molecules have also been demonstrated to promote intermolecular-charge transfer in carbon-based metal free electrocatalysts for enhancing ORR activity.30 Previous reports demonstrated that the adsorption of molecules such as anthraquinone (AQ) and fullerene (C60) on carbon nanotubes boosted ORR catalytic activities in alkaline and neutral solutions.31,32 Campidelli et al. used a nanotube as a template for the formation of a polymeric porphyrin layer which showed a higher ORR catalytic activity compared with the monomeric porphyrin systems.33 A few years ago, we developed a clipping strategy to thread SWNTs through macrocycles to form rotaxane-like mechanically interlocked nanotubes (MINTs).34 Since then, groups led by Kruss, Von Delius, and Ohe have described the encapsulation of SWNTs into peptide barrels,35 the use of disulfide bonds to connect curved π-extended tetrathiafulvalene moieties36 forming macrocycles around the SWNTs, and the direct threading of rigid cycloparaphenyleneacetylenes,37 respectively. We have demonstrated that a wide number of macrocycles based on different building blocks, including pyrene,38 π-extended tetrathiafulvalene,34,39 anthraquinone,40 naphthalenetetracarboxylic diimide41 or porphyrins,42,43 can be used for the encapsulation of SWNTs.Beyond the synthetic challenge, we have shown that the effect on the physical properties of SWNTs upon formation of MINTs is fundamentally different from other functionalization strategies.44,45 Based on this, we are investigating if/how the mechanical interlocking strategy is advantageous in fields where other chemistries did not succeed. For instance, we have already shown that the use of MINTs as fillers in polymers resulted in remarkable improvements in the mechanical properties of the composites.46 The intrinsic catalytic properties of SWNTs can be modulated by n or p doping upon encapsulation with electron-donating or withdrawing macrocycles, respectively.47 Of more relevance to the present work, we have shown that the formation of MINTs is a very promising immobilization strategy when the main objective is to graft functional molecules to the SWNT surface in a kinetically stable fashion, but without covalent modification of the SWNTs. For example, to attach catalytically active anthraquinones for the ORR,40 or to place and address molecular qubits for spintronic devices.43
In this work, we investigate the formation of MINTs using N-rich macrocycles as a novel strategy for preparation of SWNT derivatives displaying N atoms on their surface and demonstrate their ability as electrocatalysts in the ORR.
Results and discussion
The dibromo U-shape was prepared from bis(4-hydroxyphenyl)methane through two consecutive Williamson's etherifications with 11-bromoundecene and 1,4-dibromo-2,5-bis(bromomethyl)benzene, respectively. Then, the U-shape molecule containing two triphenylamine (TPA) units (U-TPA, ESI†) was prepared via Suzuki–Miyaura coupling in 72% yield in the presence of Pd(PPh3)4 from dibromo U-shape. Similarly, the U-shape containing two TPA and benzophenone units (U-CO-TPA, ESI†) was also prepared from the corresponding dibromo-U-shape. Two consecutive Williamson's etherifications from 4,4′- dihydroxybenzophenone and a later Suzuki–Miyaura coupling in 93% yield in the presence of Pd(PPh3)4 led to U-CO-TPA. Macrocycles Mac-TPA and Mac-CO-TPA were synthesized by ring-closing metathesis (RCM) of U-shape molecules. See Scheme 1 for a summary of the synthetic procedure and the chemical structures of the MINTs. Detailed procedures and full characterization are described in the ESI.† We expected the diphenylmethane or benzophenone spacer to provide sufficient flexibility to form Mac-TPA or Mac-CO-TPA around SWNTs by RCM of the terminal alkenes, which were facilitated through supramolecular interactions between the aromatic units (TPA, diphenylmethane, and benzophenone) and SWNTs.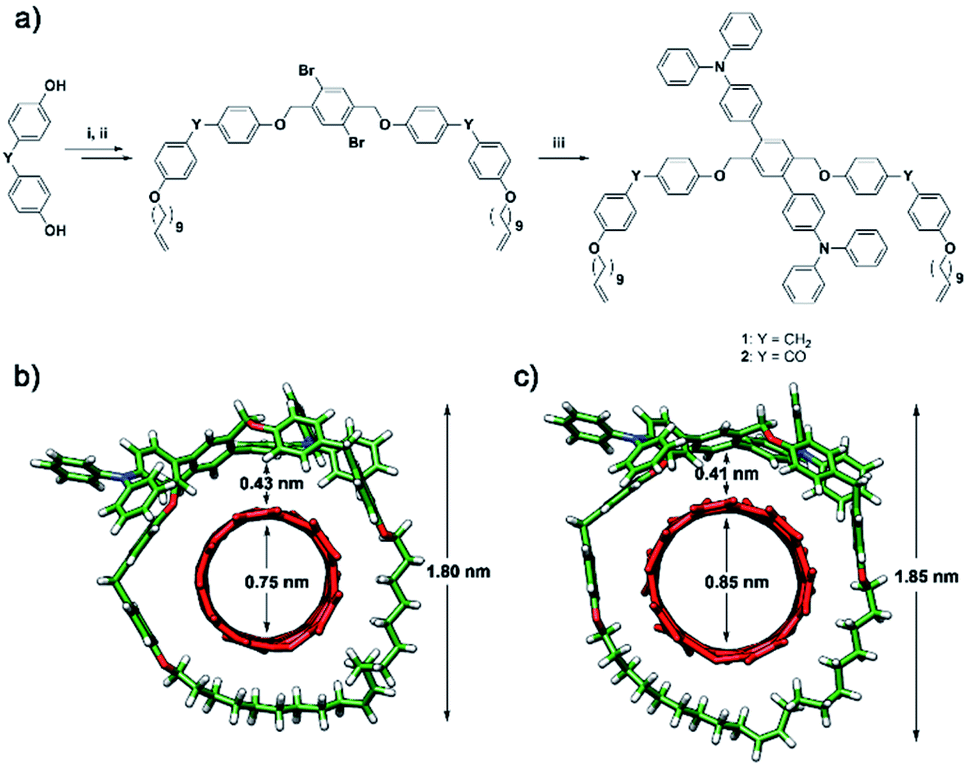 | ||
| Scheme 1 (a) Synthetic pathway to U-shapes 1 and 2: (i) 11-bromo-1-undecene (1 eq.), tetrabutylammonium bromide (0.5 eq.), NaOH (1 eq.), butanone/water, 85 °C, and 1 h; (ii) 1,4-bis-bromomethyl-benzene (0.4 eq.), K2CO3 (1 eq.), KI (cat), DMF, 80 °C, and 20 h; (iii) 4-(Diphenylamino)phenylboronic acid pinacol ester (2.5 eq.), Cs2CO3 (2.5 eq.), Pd(PPh3)4 (mol 10%), toluene/ethanol/water, 90 °C, and 15 h; an energy-minimized (MM94) model of (b) MINT(6,5)-1 and (c) MINT(7,6)-1. The MINT models are displayed with the alkene in E geometry arbitrarily, as the size of the cavity hardly changes in Z configuration. Note that, experimentally, MINTs are obtained as a mixture of E/Z isomers. | ||
We explored the formation of MINT-TPA and MINT-CO-TPA through templated RCM of U-TPA or U-CO-TPA in the presence of (6,5) and (7,6)-enriched SWNTs, which showed diameters of 0.78 nm and 0.88 nm, respectively, and are a good fit for our macrocycles. SWNTs (20 mg) were dispersed in 20 mL of tetrachloroethane (TCE) by sonication. Then, U-TPA or U-CO-TPA (0.01 mmol) and the 2nd generation Grubbs catalyst (0.01 mmol, 8.5 mg) were added, and the reaction was stirred at 60 °C under nitrogen for 96 hours.48 Afterwards, the suspension was filtered through a 0.2 μm pore-size polytetrafluoroethylene (PTFE) membrane and washed profusely with CH2Cl2 to remove any unreacted linear precursors, non-interlocked macrocycles, weakly adsorbed U-TPA/U-CO-TPA materials, remaining catalyst, etc. This washing procedure was repeated three times, after which the samples were dried under vacuum and subjected to thermogravimetric analysis (TGA) to quantify the degree of functionalization (Fig. S1 and S2 in the ESI†). The results are summarized in Table 1.
All samples showed a significant weight loss (21–32%) at around 400 °C corresponding to the macrocyclic material and showed no sign of significant participation of oligomeric species.39 The degree of functionalization is similar to the one found in previously reported MINTs (pyrene, exTTF, porphyrins, or NDI-based). The degree of functionalization of (6,5)-SWNT samples was slightly higher than that of (7,6)-SWNTs. This tendency was most likely due to the more suitable size of the cavity of the macrocycles for encapsulation of (6,5)-SWNTs. Although the changes are quantitatively small, for a given type of SWNT, the degree of functionalization of U-CO-TPA was higher than that of U-TPA; for instance, MINT(6,5)-2 shows a 32% loading compared to 29% for MINT(6,5)-1. This tendency was most likely due to a larger π-system of benzophenone than the spacer diphenylmethane, which results in more efficient U-shape-SWNT interaction. Control experiments with preformed linear U-shapes without Grubbs' catalyst lead to negligible functionalization (see the ESI†). By adjusting the type of SWNT or the U-shape molecules, the N-content according to TGA could be tuned from 0.47 to 0.68 wt%. This is a relatively low N-content compared to that of other N-doping methods such as catalytic CVD synthesis of carbon nanotubes using nitrogen precursors or ball milling using melamine, ranging 0–10% of N content, in which not only surface doping is produced but also bulk doping.49 Compared with methods in which only surface doping is produced, such as ball-milling using urea,50 the MINT approach results in a similar degree of N-doping.
Raman spectra were recorded using 532, 633, and 785 nm as excitation wavelengths. The analysis of the Raman spectra is summarized in Table 2 and illustrated in Fig. S3 and S4,† which show the Raman spectra of the (6,5)-SWNTs, (7,6)-SWNTs, MINT(6,5)-1-2, and MINT(7,6)-1-2 under 532 nm, 633 nm and 785 nm excitations, respectively. For (6,5)-SWNTs, the Raman spectra reveal changes for MINTs compared to pristine SWNTs in line with expectations for the noncovalent functionalization of SWNTs with TPA-based fragments. In particular, we observed no significant increase in the ID/IG ratio, which confirmed that there was no covalent modification of the SWNTs. With regard to doping effects, a shift from 1587 cm−1 to 1594 cm−1 of the G+ band is observed under 785 nm excitation for both the MINT(6,5) samples, which provides indirect evidence of n-doping of the SWNTs through encapsulation within the TPA macrocycles. A similar shift of the G+ band was observed under 532 nm excitation for MINT(7,6) samples, from 1587 cm−1 to 1592 cm−1.
| Sample | 532 nm | 633 nm | 785 nm | |||
|---|---|---|---|---|---|---|
| I D/IG | G+ | I D/IG | G+ | I D/IG | G+ | |
| a Average of at least 60 different Raman spectra. G+ Raman shifts in cm−1. | ||||||
| (6,5)-SWNTs | 0.07 | 1590 | 0.05 | 1593 | 0.20 | 1587 |
| MINT(6,5)-1 | 0.09 | 1592 | 0.05 | 1593 | 0.16 | 1594 |
| MINT(6,5)-2 | 0.08 | 1591 | 0.06 | 1593 | 0.17 | 1594 |
| (7,6)-SWNTs | 0.16 | 1587 | 0.13 | 1593 | 0.33 | 1594 |
| MINT(7,6)-1 | 0.20 | 1592 | 0.08 | 1594 | 0.11 | 1595 |
| MINT(7,6)-2 | 0.23 | 1592 | 0.11 | 1594 | 0.21 | 1598 |
In the absorption spectra (D2O, 1% sodium dodecyl sulphate, 298 K, Fig. S5†), the UV region is dominated by the nanotube absorption. The S22 and S11 transitions of the (6,5)-SWNTs were prominent in the vis-NIR region, appearing at λmax = 587 and 1023 nm for the pristine SWNTs. Both transitions are not shifted upon derivatization to form MINT(6,5)-1 or 2. The S22 and S11 transitions of the (7,6)-SWNTs were prominent in the vis-NIR region, appearing at λmax = 656 and 1137 nm for the pristine nanotubes. The transitions were red shifted to λmax = 661, and 1165 nm for MINT(7,6)-1. A red shift to 660 and 1175 nm upon derivatization to form MINT(7,6)-2 was observed. The photoluminescence excitation (PLE) maps (Fig. S6†) of the (6,5)-SWNTs showed an intense peak at λexc = 565 nm and λem = 975 nm, characteristic of the (6,5) chirality and residual peaks corresponding to (7,5) and (8,3) chiralities. In MINT(6,5)-1, the luminescence of the (6,5) nanotubes is quenched to approximately 25%, and this quenching is increased up to 40% for MINT(6,5)-2. The PLE map from (7,6)- SWNTs shows the presence of (6,5)-SWNTs and (7,6)-SWNTs and a minor amount of other chiralities. For MINT(7,6)-1 and MINT(7,6)-2, the luminescence from all chiralities is very efficiently quenched (90–95%) compared with that of (7,6)-SWNTs.
The investigation of a sample of MINT(6,5)-1 by atomic force microscopy (AFM) was also in agreement with the formation of the rotaxane-type species. Fig. S7a and b† show a topographic image of several SWNTs with a height of approximately 1 nm on the non-functionalized part. On the other hand, the densely functionalized parts of SWNTs show a height of around 1.5 nm which is perfectly consistent with the formation of 1 around (6,5)-SWNTs. Analysis by HR-TEM of the samples of MINT(6,5)-1 drop cast from a TCE suspension shows mostly bundled nanotubes with heavily functionalized sidewalls, in agreement with the TGA data. Fig. 1a and b show SWNTs (0.8 nm in diameter) densely covered with an organic material. Macrocycles of 1.9 nm and 2.0 nm in diameter are observed, in accordance with the expected size. To perform a more precise characterization, we also employed an aberration-corrected microscope. The microscope was operated at 60 kV to prevent damage to the nanotubes and macrocycles as much as possible. Fig. 1c and d show the aberration-corrected HR-TEM images of the three macrocycles of diameter 1.8–2.0 nm around SWNTs of 0.8 nm in diameter. Remarkably, the distances between macrocycles and the walls of the SWNTs correspond to very close (0.40 nm) van der Waals contacts.
 | ||
| Fig. 1 a) and (b) HR-TEM images of MINT(6,5)-1. Scale bars are 5 nm; (c) and (d) ac-HRTEM images of MINT(6,5)-1. Scale bars are 5 nm for (a) and (b) and 1 nm for (c) and (d). | ||
To investigate the electrochemical properties of the synthesized MINTs, cyclic voltammetry (CV) measurements using a three-electrode set-up in alternately a N2 or O2 saturated 0.1 M NaOH aqueous solution were performed, and the synthesized MINTs were compared with (6,5)-SWNTs and (7,6)-SWNTs measured under the same conditions. As shown in Fig. S12,† all the MINTs show similar redox features: two anodic peaks (around 0.7 and 0.9 V vs. RHE) and their corresponding cathodic peaks (at 0.65 V and 0.4 V vs. RHE), indicating good redox reversibility for the MINTs. These peaks were only observed for the MINTs, but not for the pristine SWNTs. When comparing the (6,5)-SWNT derivate MINTs, the MINT(6,5)-2 shows higher reversibility since its anodic peaks are 30 mV less positive than the ones observed for the MINT(6,5)-1, and its anodic-cathodic peak separation (ΔEP) is smaller (70 mV for MINT(6,5)-2 and 80 mV for MINT(6,5)-1). However, for the (7,6)-SWNT derivate MINTs, no changes in reversibility are observed, both MINTs (MINT(7,6)-1 and MINT(7,6)-2) exhibit identical redox features: an anodic peak at 0.66 V and a ΔEP of around 60 mV. It is also worth noting that for the (7,6)-SWNT derivate MINTs, the capacitive current values are higher than for the (6,5)-SWNT derivate MINTs (Fig. S8†), implying that the electron transfer is more favorable for the interlocked (7,6)-SWNTs. Additionally, electrochemical impedance spectroscopy (EIS) measurements (Fig. S13 and Table S2†) show that all the MINTs possess low interfacial resistance (Rr) (which is indicative of the fast charge transfer at the interface of the semiconductor and electrolyte) in comparison with the respective unfunctionalized SWNT counterpart. Besides, the (6,5)-SWNT and (7,6)-SWNT electrodes did not fit well with the proposed circuit model. All these measurements confirm that not only the presence of the rings, but also the presence of the carbonyl groups in U-CO-TPA favors charge transfer, which in turn induces a decrease in the resistance leading to faster ORR kinetics, as shown by CV and impedance measurements. When the CV measurements in N2 were compared with those carried out in an O2 saturated 0.1 M NaOH aqueous solution, the observance of more intense reduction current peaks suggests that the obtained MINTs could catalyze ORR effectively. As a consequence of the interlocking effect, the reduction potential peaks for the MINTs appear to be slightly shifted to more positive potentials regardless of the type of SWNT used for the MINTs.
The electrocatalytic performance of the obtained MINTs towards the ORR was carefully investigated and compared with that of the corresponding pristine SWNTs. Fig. 2a and b exhibit the typical linear sweep voltammetry (LSV) curves for the ORR obtained at a fixed rotation rate of 1600 rpm for each electrocatalyst together with the corresponding Tafel curves. Considering onset potential values, the oxygen reduction activities of these electrocatalysts follow the next trend (MINT(7,6)-2 (0.75 V) > MINT(6,5)-2 (0.74 V) > MINT(7,6)-1 (0.72 V) > MINT(6,5)-1 (0.70 V)). The observed improvement in the oxygen reduction activities for MINT-2 compared to the MINT-1 samples is in agreement with the observed ∼19 mV shift of the oxidation peak of 2 with respect to the oxidation peak of 1 giving rise to a higher EHOMO (eV) in absolute values (Fig. S14 and Table S3†). All the MINTs exhibit improved values with respect to the pristine (6,5) and (7,6) SWNT. To prove the importance of the MINT structure, we carried out the electrochemical analysis of the physical mixtures obtained by mixing 1 and 2, respectively, with the corresponding (6,5)-SWNT (Fig. S15–17 and Table S4†) under the same conditions showing a significantly poorer ORR activity. As can be seen in Table 3, a similar trend is also observed for the half-wave potential values demonstrating the suitability of the mechanical interlocking methodology for boosting OOR activities in a controllable manner in SWNTs through the introduction of the nitrogen species. To confirm that the enhancement in catalytic activity is indeed due to the introduction of nitrogen atoms, we synthesized a control sample with an identical structure to MINT(6,5)-1, but without the diphenylamine groups (see Fig. S8–11 and Table S1† for characterization). This sample presents electrocatalytic activity similar to that of naked SWNTs, confirming that the nitrogen atoms in TPA moieties act as active sites to enhance the ORR activity of the N-containing MINT electrodes. (Fig. S18 and Table S5†). Likewise, in the absence of the SWNTs, the macrocyclic species do not catalyze the reduction of oxygen as demonstrated in Fig. S19 and S20,† where the high resistance and poor electrical conductivity make the SWNT a requirement.
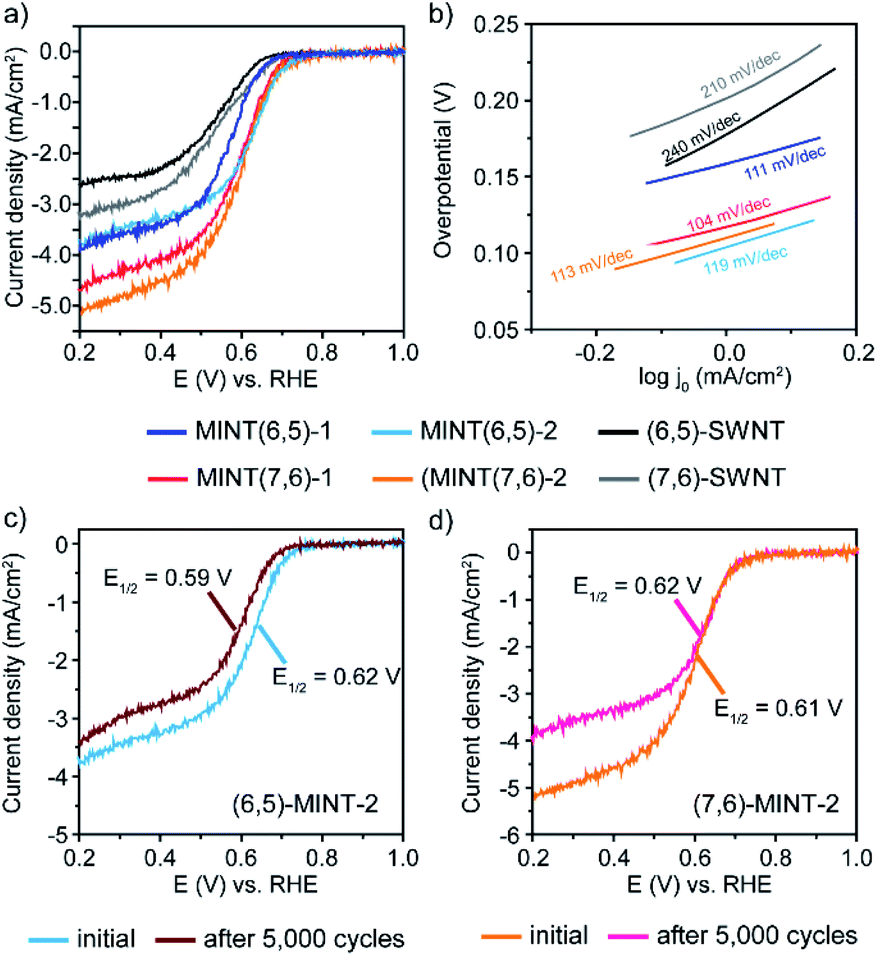 | ||
| Fig. 2 Electrochemical characterization of the as-prepared MINT electrocatalysts. (a) Comparison of the ohmic drop corrected ORR polarization curves and (b) Tafel plots for the MINT electrocatalysts with their respective pristine SWNT counterpart. iR-LSV polarization curves of (c) MINT(6,5)-2 and (d) MINT(7,6)-2 initially and after ORR stability tests (5000 cycles). | ||
| Catalyst | Half-wave potential (E1/2, V vs. RHE) | Onset potential (Eonset, V vs. RHE) | Current density jL at 0.2 V (mA cm−2) | Tafel (mV/dec) | ne− (at 0.4 V vs. RHE) | ECSAs (cm2) |
|---|---|---|---|---|---|---|
| MINT(6,5)-1 | 0.58 | 0.70 | 3.8 | 111 | 2.3 | 53.7 |
| MINT(6,5)-2 | 0.62 | 0.74 | 3.7 | 119 | 2.0 | 62.9 |
| MINT(6,5)-2 after 5,000 cycles | 0.59 | 0.70 | 3.4 | 120 | 2.1 | — |
| MINT(7,6)-1 | 0.60 | 0.72 | 4.7 | 104 | 2.4 | 70.2 |
| MINT(7,6)-2 | 0.62 | 0.75 | 5.3 | 113 | 2.5 | 74.2 |
| MINT(7,6)-2 after 5000 cycles | 0.62 | 0.75 | 4.0 | 111 | 2.6 | — |
| (6,5)-SWNTs | 0.54 | 0.65 | 2.6 | 240 | 1.8 | 46.3 |
| (7,6)-SWNTs | 0.56 | 0.68 | 3.2 | 210 | 1.8 | 51.2 |
As expected, the diffusion-limited current densities for all the studied electrocatalysts increase with increasing rotation rates (Fig. S21–S26†). All the as-prepared MINTs catalysts show an improved dispersion and wettability and exhibit higher diffusion-limited current densities than the (6,5) and (7,6) SWNTs. After comparing the limited-current density values at 0.2 V for each studied material (Table 3), it can be concluded that MINTs obtained from (7,6)-SWNTs achieve higher current values, MINT(7,6)-2 being the one with the highest value (5.3 mA cm−2) that may exhibit more accessible electrochemically active sites. No significant differences were found between MINT(6,5)-1 and MINT(6,5)-2, indicating that both systems possess similar charge transfer capacity.
The electro–kinetic parameters such as the onset and the half-wave potentials, the Tafel slopes, the limited current density at 0.2 V, and the electrochemical surface area (ECSA) of all synthesized electrocatalysts are summarized and compared with those of the pristine (6,5) and (7,6) SWNTs in Table 3. The combined observation of these parameters clearly demonstrates that the presence of the rings on the SWNT is linked to a significant increase of the electrocatalytic activity for the ORR.
The quantitative estimation of the electron-transfer number (n) was conducted using the Koutecky–Levich (K–L) plot (Fig. S21–S26†). The linear K–L plots demonstrated that the electron transfer number (n) was 2.0–2.5 in the range of 0.35–0.45 V for the MINTs (Table 3 and Fig. S21–24†), confirming that the oxygen reduction takes place mainly through a two-electron transfer process. For the pristine (6,5)-SWNTs and (7,6)-SWNTs, the n value obtained from the K–L plot was 1.8 in the range of 0.3–0.45 V, suggesting a pseudo 2 electron pathway. On the other hand, the H2O2 yield obtained by a rotating disk electrode (RRDE) was in the range of 10% and 15% for all the materials, giving the lowest yields for the MINT electrodes. Zheng et al.51 demonstrated that the ORR on pyridinic-N-doped graphene mainly occurs via the 2-electron process and not effectively via the 4-electron process. The calculated electron number and Tafel slope values are therefore in accordance with N-doped graphene electrocatalytic systems previously reported. To investigate the role of the different structural features in MINTs that might play an important role as active sites towards the ORR, we calculated the electrochemical surface areas (ECSAs) for each material by CV at different scan rates (ESI Fig. S19, S20 and Table S3†). Among all the catalysts, MINT(7,6)-2 possesses a considerably larger electrochemical surface area, which is 1.4 times greater than the observed value for (7,6)-SWNTs. As shown in Table 3, the following order was found: MINT(7,6)-2 > MINT(7,6)-1 > MINT(6,5)-2 > MINT(6,5)-1. In view of these results, along with the electrochemical parameters calculated for each material, we deeply analyze the different aspects contributing to the improvement of ORR kinetics. First, all MINTs present significantly larger ECSAs than the corresponding SWNTs. That is, the presence of the macrocycles enhances the electrocatalytic activity. Secondly, this increase in activity cannot be ascribed to the N atoms only. Instead, the presence of the carbonyl groups in U-CO-TPA units also seems relevant, as both MINTs MINT(7,6)-2 and MINT(6,5)-2 show larger ECSAs and much better onset and half-wave potentials than their counterparts MINT(7,6)-1 and MINT(6,5)-1.
The long-term stability of the MINT electrocatalysts showing the best performance for each type of SWNT (MINT(6,5)-2 and MINT(7,6)-2) was tested after 5000 potential cycles by continuously applying linear potential sweeps between 0.6 and 1.0 V vs. RHE at 100 mV s−1 scan rate. The half-wave and onset potentials of the MINT(6,5)-2 catalyst material slightly decrease after 5000 ORR cycles (from 0.62 V to 0.59 V and from 0.74 V to 0.69 V for the half-wave and the onset potentials, respectively) (Fig. 2c and Table 3). The limited-current density and Tafel slope also decrease after stability tests (Fig. S27† and Table 3). Even though the decrease is minimal compared to the stability observed for the physical mixture (2 + (6,5)-SWNT) (Fig. S30 and Table S6†), these results indicate that the material deteriorates upon cycling.
On the contrary, the MINT(7,6)-2 catalyst material revealed no significant difference neither in the half-wave potential nor in the onset potential parameters. However, the limited-current density decreased considerably after 5000 cycles of the ORR, as shown in Fig. 2d and Table 2, which may be due to the loss of material during stability tests upon rotation.
This material also maintains its Tafel slope value. It is worth noting the different stability behavior after 5000 potential cycles: while MINT(6,5)-2 deteriorates, the MINT(7,6)-2 remains stable. When calculating the number of electrons transferred from the linear K–L plots for both materials after the stability test (Fig. S28 and S29† and Table 3), we find that the electron transfer numbers for MINT(6,5)-2 and MINT(7,6)-2 were 2.0–2.2 and 2.5–2.7, respectively, which are slightly higher than the initial values.
Conclusions
We have synthesized MINTs containing TPA groups as a new strategy for displaying catalytically active N atoms on the surface of carbon nanotubes. The N-containing MINTS demonstrated an improved ORR performance which is ascribed to the introduction of nitrogen heteroatoms in the MINTs acting as effective active sites for the ORR, since a performance improvement is observed in comparison to the SWNT counterparts, and no improvement is observed in structurally related MINTs lacking the N atoms. Somewhat unexpectedly, the CO groups in the U-CO-TPA molecules also act as electroactive sites, resulting in a further improvement in ORR performance. An enhanced intermolecular charge-transfer effect due to a tighter interlocking fit for (7,6)-SWNT derivate MINTs than for the corresponding (6,5)-SWNT derivate translates in an increase for the ORR performance (activity & stability). These results indicate that understanding of SWNT electrocatalyst performance is possible at the molecular level, directly enabling design of more active and more stable catalysts.Data availability
All data available within the main text or ESI† or from the authors upon request.Author contributions
W. Z., M. G. S., S. M. D. S., A. L. M. and L. R. G. carried out and analysed experiments. M. C. G. L. and E. M. P. supervised and coordinated research. All authors contributed to analysing the data and writing the manuscript, and have given it final approval.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Ministry of Science of Spain (Projects No. RTI2018-101097-A-I00 and RyC-2016-20258 for M. G. L. and CTQ2017-86060-P; PID2020-116661RB-I00 for E. M. P.), Comunidad de Madrid (P2018/NMT-4367), the European Research Council (ERC) (Starting Investigator Grant (NANOCOMP-679124) for M. G. L.), the Xunta de Galicia (Centro singular de investigación de Galicia accreditation 2019–2022, ED431G 2019/03) and the European Union (European Regional Development Fund – ERDF). IMDEA Nanociencia received support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant CEX2020-001039-S).References
- Y.-P. Sun, K. Fu, Y. Lin and W. Huang, Acc. Chem. Res., 2002, 35, 1096–1104 CrossRef CAS PubMed.
- S. Mallakpour and S. Soltanian, RSC Adv., 2016, 6, 109916–109935 RSC.
- V. Georgakilas, K. Kordatos, M. Prato, D. M. Guldi, M. Holzinger and A. Hirsch, J. Am. Chem. Soc., 2002, 124, 760–761 CrossRef CAS PubMed.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105–1136 CrossRef CAS PubMed.
- C. Hu and L. Dai, Adv. Mater., 2019, 31, 1804672 CrossRef PubMed.
- J.-C. Li, P.-X. Hou and C. Liu, Small, 2017, 13, 1702002 CrossRef PubMed.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- R. Czerw, M. Terrones, J. C. Charlier, X. Blase, B. Foley, R. Kamalakaran, N. Grobert, H. Terrones, D. Tekleab, P. M. Ajayan, W. Blau, M. Rühle and D. L. Carroll, Nano Lett., 2001, 1, 457–460 CrossRef CAS.
- Y. Xia and R. Mokaya, Adv. Mater., 2004, 16, 1553–1558 CrossRef CAS.
- L. S. Panchakarla, A. Govindaraj and C. N. R. Rao, Inorg. Chim. Acta, 2010, 363, 4163–4174 CrossRef CAS.
- M. Steinmetz, D. Lima, R. R. L. Machado, U. Sundararaj, M. Arjmand, A. B. da Silva, J. P. Santos, C. A. Pessôa and K. Wohnrath, Diamond Relat. Mater., 2020, 110, 108093 CrossRef CAS.
- J. Zhang, Z. Xia and L. Dai, Sci. Adv., 2015, 1, e1500564 CrossRef PubMed.
- C. Hu, R. Paul, Q. Dai and L. Dai, Chem. Soc. Rev., 2021, 50, 11785–11843 RSC.
- X. Wang, A. Vasileff, Y. Jiao, Y. Zheng and S.-Z. Qiao, Adv. Mater., 2019, 31, 1803625 CrossRef PubMed.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 2230–2234 CrossRef CAS PubMed.
- J. Shui, M. Wang, F. Du and L. Dai, Sci. Adv., 2015, 1, e1400129 CrossRef PubMed.
- S. Wang, E. Iyyamperumal, A. Roy, Y. Xue, D. Yu and L. Dai, Angew. Chem., Int. Ed., 2011, 50, 11756–11760 CrossRef CAS PubMed.
- C. Hu, H. Cheng, Y. Zhao, Y. Hu, Y. Liu, L. Dai and L. Qu, Adv. Mater., 2012, 24, 5493–5498 CrossRef CAS PubMed.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- N. R. Sahraie, U. I. Kramm, J. Steinberg, Y. Zhang, A. Thomas, T. Reier, J.-P. Paraknowitsch and P. Strasser, Nat. Commun., 2015, 6, 8618 CrossRef CAS PubMed.
- L. Tao, Q. Wang, S. Dou, Z. Ma, J. Huo, S. Wang and L. Dai, Chem. Commun., 2016, 52, 2764–2767 RSC.
- Q. Liu, Y. Wang, L. Dai and J. Yao, Adv. Mater., 2016, 28, 3000–3006 CrossRef CAS PubMed.
- Q. Li, R. Cao, J. Cho and G. Wu, Adv. Energy Mater., 2014, 4, 1301415 CrossRef.
- J. Li, Z. Xi, Y.-T. Pan, J. S. Spendelow, P. N. Duchesne, D. Su, Q. Li, C. Yu, Z. Yin, B. Shen, Y. S. Kim, P. Zhang and S. Sun, J. Am. Chem. Soc., 2018, 140, 2926–2932 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- K. Gao, B. Wang, L. Tao, B. V. Cunning, Z. Zhang, S. Wang, R. S. Ruoff and L. Qu, Adv. Mater., 2019, 31, 1805121 CrossRef PubMed.
- C. Tang and Q. Zhang, Adv. Mater., 2017, 29, 1604103 CrossRef PubMed.
- R. Paul, Q. Dai, C. Hu and L. Dai, Carbon Energy, 2019, 1, 19–31 CrossRef.
- Z. Gong, G. Zhang and S. Wang, J. Chem., 2013, 2013, 756307 Search PubMed.
- R. Gao, Q. Dai, F. Du, D. Yan and L. Dai, J. Am. Chem. Soc., 2019, 141, 11658–11666 CrossRef CAS PubMed.
- I. Hijazi, T. Bourgeteau, R. Cornut, A. Morozan, A. Filoramo, J. Leroy, V. Derycke, B. Jousselme and S. Campidelli, J. Am. Chem. Soc., 2014, 136, 6348–6354 CrossRef CAS PubMed.
- A. de Juan, Y. Pouillon, L. Ruiz-González, A. Torres-Pardo, S. Casado, N. Martín, Á. Rubio and E. M. Pérez, Angew. Chem., Int. Ed., 2014, 53, 5394–5400 CrossRef CAS PubMed.
- F. A. Mann, J. Horlebein, N. F. Meyer, D. Meyer, F. Thomas and S. Kruss, Chem. – Eur. J., 2018, 24, 12241–12245 CrossRef CAS PubMed.
- B. Balakrishna, A. Menon, K. Cao, S. Gsänger, S. B. Beil, J. Villalva, O. Shyshov, O. Martin, A. Hirsch, B. Meyer, U. Kaiser, D. M. Guldi and M. von Delius, Angew. Chem., Int. Ed., 2020, 59, 18774–18785 CrossRef CAS PubMed.
- K. Miki, K. Saiki, T. Umeyama, J. Baek, T. Noda, H. Imahori, Y. Sato, K. Suenaga and K. Ohe, Small, 2018, 14, 1800720 CrossRef PubMed.
- A. López-Moreno and E. M. Pérez, Chem. Commun., 2015, 51, 5421–5424 RSC.
- A. de Juan, M. Mar Bernal and E. M. Pérez, ChemPlusChem, 2015, 80, 1153–1157 CrossRef CAS PubMed.
- D. Wielend, M. Vera-Hidalgo, H. Seelajaroen, N. S. Sariciftci, E. M. Pérez and D. R. Whang, ACS Appl. Mater. Interfaces, 2020, 12, 32615–32621 CrossRef CAS PubMed.
- S. Leret, Y. Pouillon, S. Casado, C. Navío, Á. Rubio and E. M. Pérez, Chem. Sci., 2017, 8, 1927–1935 RSC.
- L. de Juan-Fernández, P. W. Münich, A. Puthiyedath, B. Nieto-Ortega, S. Casado, L. Ruiz-González, E. M. Pérez and D. M. Guldi, Chem. Sci., 2018, 9, 6779–6784 RSC.
- S. Moreno-Da Silva, J. I. Martínez, A. Develioglu, B. Nieto-Ortega, L. de Juan-Fernández, L. Ruiz-Gonzalez, A. Picón, S. Oberli, P. J. Alonso, D. Moonshiram, E. M. Pérez and E. Burzurí, J. Am. Chem. Soc., 2021, 143, 21286–21293 CrossRef CAS PubMed.
- E. Martinez-Perinan, A. de Juan, Y. Pouillon, C. Schierl, V. Strauss, N. Martín, A. Rubio, D. M. Guldi, E. Lorenzo and E. M. Pérez, Nanoscale, 2016, 8, 9254–9264 RSC.
- J. Villalva, B. Nieto-Ortega, M. Melle-Franco and E. M. Pérez, J. Phys. Chem. C, 2020, 124, 15541–15546 CrossRef CAS.
- A. López-Moreno, B. Nieto-Ortega, M. Moffa, A. de Juan, M. M. Bernal, J. P. Fernández-Blázquez, J. J. Vilatela, D. Pisignano and E. M. Pérez, ACS Nano, 2016, 10, 8012–8018 CrossRef PubMed.
- M. Blanco, B. Nieto-Ortega, A. de Juan, M. Vera-Hidalgo, A. López-Moreno, S. Casado, L. R. González, H. Sawada, J. M. González-Calbet and E. M. Pérez, Nat. Commun., 2018, 9, 2671 CrossRef PubMed.
- S. Mena-Hernando and E. M. Pérez, Chem. Soc. Rev., 2019, 48, 5016–5032 RSC.
- M. Inagaki, M. Toyoda, Y. Soneda and T. Morishita, Carbon, 2018, 132, 104–140 CrossRef CAS.
- O. S. G. P. Soares, R. P. Rocha, A. G. Gonçalves, J. L. Figueiredo, J. J. M. Órfão and M. F. R. Pereira, Carbon, 2015, 91, 114–121 CrossRef CAS.
- B. Zheng, X.-L. Cai, Y. Zhou and X.-H. Xia, ChemElectroChem, 2016, 3, 2036–2042 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02346f |
| This journal is © The Royal Society of Chemistry 2022 |