
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePractical asymmetric amine nucleophilic approach for the modular construction of protected α-quaternary amino acids†
Teng
Liu
a,
Shaofei
Ni
b and
Wusheng
Guo
 *a
*a
aFrontier Institute of Science and Technology (FIST), Xi'an Jiaotong University, Yanxiang Road 99, Xi'an 710045, China. E-mail: wusheng.guo@mail.xjtu.edu.cn
bDepartment of Chemistry, Shantou University, Shantou 515063, China
First published on 5th June 2022
Abstract
We report the first amine nucleophilic approach for the modular construction of enantioenriched protected α-quaternary amino acids. The key to success is the use of an alcohol solvent, which makes a rationally designed COOMe-bonded Cu-allenylidene electrophilic intermediate stable enough to couple with amine nucleophiles before its decomposition. The reaction features wide functional group tolerance with high enantioselectivity, typically >90% ee, and is amenable to the modification of commercially available bioactive molecules. The resultant protected α-amino acids could be readily converted into a number of precious enantioenriched amines featuring α-hindered tertiary carbon centers, which are otherwise synthetically quite challenging, including those of α-amino aldehyde, peptides or α-vinyl amino ester with >92% ee in excellent yields. This protocol could be utilized for the synthesis of the protected bioactive α-ethylnorvaline in 3 steps, a significant advancement in comparison to an 11-step sequence reported previously.
Introduction
Nowadays, the pharmaceutical industry has witnessed a paradigm shift. The application of protein-, peptide- and monoclonal antibody-based biologics is experiencing a breakthrough in comparison with small molecules in the process of new drug development. Enantioenriched α-quaternary amino acids (α-QAAs) feature two modular non-hydrogen functionalities and are thus ideal candidates for the preparation of novel peptides with desired properties in addition to being important building blocks or intermediates in synthetic chemistry.1,2 Specifically, the two extra groups in chiral α-QAAs are projected along well-defined vectors, which is vital for the design of molecules that interact with biological acceptors. Though quite promising in biological applications, the synthesis of chiral α-QAAs is typically quite challenging and requires a multi-step process (exemplified in Scheme 1a)2 mainly because the construction of a hindered carbon stereocenter while keeping the reactive amino and carboxyl groups intact is not an easy task. As such, synthetic chemists have never stopped exploring new procedures for the efficient and versatile synthesis of chiral α-QAAs.1,2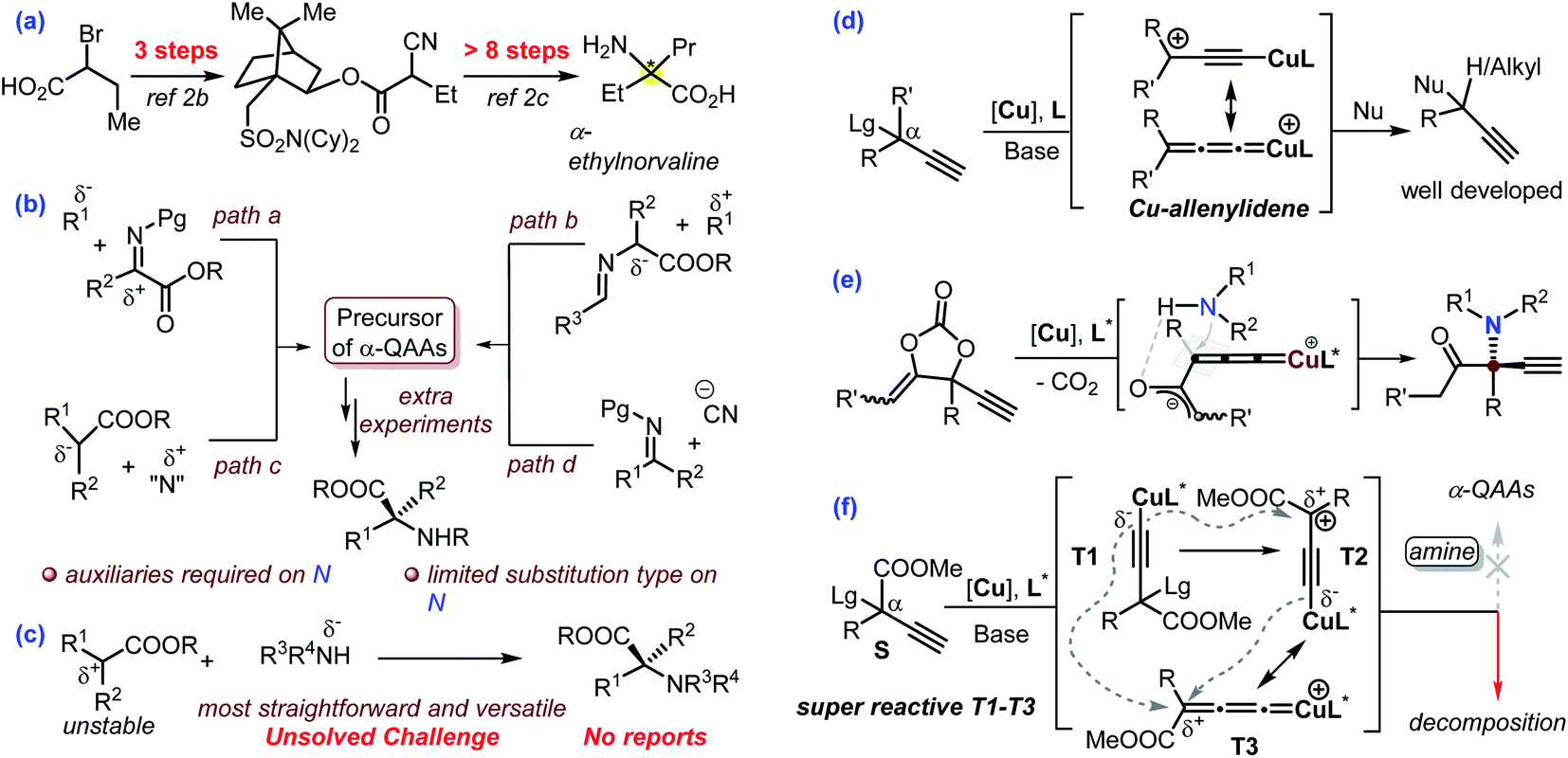 | ||
| Scheme 1 The asymmetric syntheses of (protected) α-quaternary amino acids: advances and challenges. | ||
Over the past 60 years, a number of methodologies have been invented to take on the challenge, which can be mainly divided into four categories (Scheme 1b). The addition of nucleophiles into structurally privileged α-ketiminoesters can possibly produce enantioenriched α-QAAs (Scheme 1b, path a);3 the umpolung variant of this strategy would enable the coupling of a Schiff-base-derived nucleophile and an activated carbon electrophile towards the target molecules (Scheme 1b, path b).4 These protocols typically require privileged amino acid-based starting materials. Alternative methods include the asymmetric nucleophilic attack of a suitable electrophilic amino reagent (Scheme 1b, path c),5 a Strecker-type reaction (Scheme 1b, path d)6 and some other inventive approaches.7 These elegant approaches have greatly advanced the synthesis of enantioenriched α-QAAs (Scheme 1b).3–6
Amines are versatile and commercially available. The direct use of amines as an amino source would offer a concise and modular approach for the synthesis of invaluable α-QAAs. In this respect, cyclic α-QAAs could be accessed from amines in spite of the distinct limitations on substituent patterns.8 The Gaunt group recently developed a modular approach for the synthesis of racemic α-amino acids via photochemistry with amines.9 With this background in mind, we speculate that the direct asymmetric coupling of amines and ester-bonded tertiary carbon electrophiles would be the most straightforward and versatile approach for the synthesis of enantioenriched α-QAAs; however, this streamlined reaction remains a formidable unsolved challenge (Scheme 1c). Such a challenge mainly stems from the fact that an ester-bonded tertiary carbon typically works as a nucleophile and thus is difficult to couple with an amine nucleophile via C–N bond formation, in addition to the introduction of high enantioselectivity in building a hindered carbon center.
The Cu-catalyzed asymmetric propargylic substitution reaction (Cu-APR) has showcased great potential in the synthesis of α-tertiary propargylic compounds (Scheme 1d, R′ = H).10 With the introduction of hydrogen-bonding assistance, the asymmetric construction of an α-alkyl quaternary stereocenter could be accomplished (Scheme 1d, R′ = alkyl) despite the requirement of cyclic precursors.11 However, in the presence of an extra-strong electron-withdrawing group in the α-position, the Cu-APR is elusive and quite problematic. The underlying challenges become quickly evident from the fact that exceedingly rare examples have been reported on this topic, although the construction of quaternary stereocenters continues to be a challenging and attractive area. In 2016, the Nishibayashi group realized the asymmetric substitution of CF3-functionalized propargylic perfluorobenzoates with indole as a nucleophile, and this work represents the only related example in Cu-catalyzed chemistry; the privileged perfluorobenzoate leaving group proved to be crucial, suggesting the distinctiveness of such reactions.12 Our group recently reported the asymmetric synthesis of α-amino ketones from the amination of a carefully designed cyclic carbonate; the reaction was proposed to proceed by the intermediacy of a Cu-bounded zwitterionic enolate species (Scheme 1e).11d The planar sp2-C hybridization in the enolate fragment was proposed to be beneficial for the enantio-discrimination process. Enlightened by this work, we envisioned that the utilization of substrates equipped with ester groups in the Cu-APR may work similarly to offer a cheaper and operationally friendly solution towards the modular and versatile synthesis of chiral α-QAAs and simultaneously solve the challenge for the first asymmetric synthesis of α-QAAs via an amine nucleophilic approach. The dangling alkynyl group is versatile13 and is ready for bioorthogonal usage,14 adding a further bonus to the resultant products. Moreover, the ester group is also versatile, and can be transformed into aldehyde, alcohol, and carboxylic acid products through simple experimental operations. Although synthetically promising, preliminary tests indicate the fast decomposition of the ester-bonded propargylic starting material at rt under normal Cu-APR conditions without any of the target amino esters noted (Scheme 1f, R = Ph). It has been widely accepted10 that the Cu-APR is mediated by T1 derived from the deprotonation of S, followed by the nucleophilic attack of T2 or T3 (Scheme 1f). We theorize that the input of the ester group makes the intermediates T1–T3 super reactive and unstable, consequently leading to the fast decomposition of the propargylic species due to their intermolecular coupling reactions before the external amine nucleophilic attack (Scheme 1f). This is probably why this area has remained unnaturally silent so far.
Results and discussion
We commenced the study by reacting compound S1 and aniline in THF at room temperature in the presence of L1 and copper catalyst (Fig. 1a). 19F NMR indicated the complete decomposition of S1 with the target product P1 undetectable, and a similar outcome was observed even at −40 °C. The complete decomposition of S1 also took place with the use of other solvents, including toluene, DCM, ACN and DMF (Fig. 1a), despite these solvents being frequently applied in Cu-catalyzed propargylation reactions.15 No reaction was observed in the absence of any base. Fortunately, the target product P1 was isolated in 78% yield when we changed the solvent to methanol despite of the low enantioselectivity (Fig. 1a). We propose that the alcohol solvent is the key to stabilizing the reactive ester-bonded electrophilic copper intermediates T1–T3 through hydrogen bonding interactions (Scheme 1d).16,17 We herein communicate the successful realization of such a Cu-APR enabling the streamlined synthesis of enantioenriched protected α-QAAs with high efficiency, and this case also represents the first amine nucleophilic approach resulting in such invaluable products (Fig. 1b).18 The reaction between S3 (Fig. 2, R1 = Ph) and aniline was used as a model for condition screening. Extensive experiments19 have indicated that the utilization of a newly developed chiral MeO-functionalized box-ligand L in a mixed solvent TFE/ACN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) at lower temperatures (−30 to −40 °C) is vital for producing product 1 with both high chemo- and enantioselectivity (85% yield, 94% ee; Fig. 2). The presence of an electron-donating group on the backbone of the ligand was believed to better stabilize the Cu-allenylidene intermediate,17 which is beneficial for the subsequent enantioselective amine attack; such an effect was probably related to the big electronic difference between the aryl and ester substituents, which were attached on the planar α-C with an sp2-hybridization in the key intermediate.17 A positive nonlinear relationship between the enantiopurity of the product and that of the corresponding ligand L was observed, suggesting that this reaction probably proceeded through the intermediacy of a dinuclear Cu-complex;13a,15,17 this is different from the monomeric-Cu-intermediated process that was proposed in our previous zwitterionic enolate study (Scheme 1e).11d,17 With satisfactory reaction conditions established, we then turned to investigate the generality of the reactions (Fig. 2). To our great joy, the present catalytic system tolerated quite well a broad range of either amines (1–20) or propargylic esters (21–37) towards the formation of the target enantioenriched products in high efficiency (most >70% yield and >91% ee). A number of substrates featuring either the electron-withdrawing (5–9, 14, 23–26, 28–29) or -donating (2–4, 11, 15, 17–19, 21–22, 27, 30–31) functionalities on the para-, ortho- or meta-position of the aryl substituents are applicable for the reactions. The styrene-functionalized product (10) could be easily obtained in this system, which is generally challenging in methods based on photocatalysis, and the presence of the styrene group may facilitate the usage of the corresponding product in polymeric chemistry. The absolute (S)-configuration of these enantioenriched α-QAAs can be deduced from the X-ray analysis of compound 1 (Fig. 2, inset). Notably, the present catalytic system was feasible for the preparation of α-QAAs with a tertiary amino group, as exemplified by the successful isolation of product 20, which was impossible via the approaches summarized in Scheme 1b.3–6 The synthetically challenging α,α-dialkylated α-QAAs9 could be synthesized smoothly from the products (34–36), albeit with relatively lower enantioselectivity, which may be due to steric reasons. The steric effect was further evidenced by the fact that the utilization of the substrate equipped with the bulkier isopropyl group resulted in higher enantioselectivity (35vs.36). Additional screening19 with a methyl-substituted substrate suggested that the reaction performed in methanol at 0 °C under otherwise identical conditions gave rise to compound 34 with improved enantioselectivity (39% ee vs. 62% ee), while similar improvements were not observed for the reaction at 0 °C in methanol using the propyl-substituted substrates;20 these results indicate that the solvent and electronic factors also affect the enantio-discrimination process. The introduction of the alkynyl functionality in the target product (37) also proved feasible; this is especially interesting because the enantio-discrimination between two structurally similar linear groups through an otherwise activation mode is quite challenging. The creation of boronated (12) or brominated (6, 24 and 29) enantioenriched α-QAAs signifies the potential of these products to be used in Suzuki coupling reactions, correspondingly expanding the diversity of the amino acids. Those amines containing pharmaceutically relevant groups such as benzodioxole (17) and indole fragment (19) were appropriate reaction partners, as well as the naphthyl-decorated substrates (16 and 32). The use of alkyl amines resulted in the decomposition of the propargylic precursor, suggesting some limitations of the current approach as well as the idiosyncratic nature of the Cu-APR with the use of the propargyl precursor bearing an extra strong electron-withdrawing group.21 Further attempts with the use of HOOC-bonded propargylic substrates or Ph-substituted internal alkynyl substrates failed to afford the desired products.
:3) at lower temperatures (−30 to −40 °C) is vital for producing product 1 with both high chemo- and enantioselectivity (85% yield, 94% ee; Fig. 2). The presence of an electron-donating group on the backbone of the ligand was believed to better stabilize the Cu-allenylidene intermediate,17 which is beneficial for the subsequent enantioselective amine attack; such an effect was probably related to the big electronic difference between the aryl and ester substituents, which were attached on the planar α-C with an sp2-hybridization in the key intermediate.17 A positive nonlinear relationship between the enantiopurity of the product and that of the corresponding ligand L was observed, suggesting that this reaction probably proceeded through the intermediacy of a dinuclear Cu-complex;13a,15,17 this is different from the monomeric-Cu-intermediated process that was proposed in our previous zwitterionic enolate study (Scheme 1e).11d,17 With satisfactory reaction conditions established, we then turned to investigate the generality of the reactions (Fig. 2). To our great joy, the present catalytic system tolerated quite well a broad range of either amines (1–20) or propargylic esters (21–37) towards the formation of the target enantioenriched products in high efficiency (most >70% yield and >91% ee). A number of substrates featuring either the electron-withdrawing (5–9, 14, 23–26, 28–29) or -donating (2–4, 11, 15, 17–19, 21–22, 27, 30–31) functionalities on the para-, ortho- or meta-position of the aryl substituents are applicable for the reactions. The styrene-functionalized product (10) could be easily obtained in this system, which is generally challenging in methods based on photocatalysis, and the presence of the styrene group may facilitate the usage of the corresponding product in polymeric chemistry. The absolute (S)-configuration of these enantioenriched α-QAAs can be deduced from the X-ray analysis of compound 1 (Fig. 2, inset). Notably, the present catalytic system was feasible for the preparation of α-QAAs with a tertiary amino group, as exemplified by the successful isolation of product 20, which was impossible via the approaches summarized in Scheme 1b.3–6 The synthetically challenging α,α-dialkylated α-QAAs9 could be synthesized smoothly from the products (34–36), albeit with relatively lower enantioselectivity, which may be due to steric reasons. The steric effect was further evidenced by the fact that the utilization of the substrate equipped with the bulkier isopropyl group resulted in higher enantioselectivity (35vs.36). Additional screening19 with a methyl-substituted substrate suggested that the reaction performed in methanol at 0 °C under otherwise identical conditions gave rise to compound 34 with improved enantioselectivity (39% ee vs. 62% ee), while similar improvements were not observed for the reaction at 0 °C in methanol using the propyl-substituted substrates;20 these results indicate that the solvent and electronic factors also affect the enantio-discrimination process. The introduction of the alkynyl functionality in the target product (37) also proved feasible; this is especially interesting because the enantio-discrimination between two structurally similar linear groups through an otherwise activation mode is quite challenging. The creation of boronated (12) or brominated (6, 24 and 29) enantioenriched α-QAAs signifies the potential of these products to be used in Suzuki coupling reactions, correspondingly expanding the diversity of the amino acids. Those amines containing pharmaceutically relevant groups such as benzodioxole (17) and indole fragment (19) were appropriate reaction partners, as well as the naphthyl-decorated substrates (16 and 32). The use of alkyl amines resulted in the decomposition of the propargylic precursor, suggesting some limitations of the current approach as well as the idiosyncratic nature of the Cu-APR with the use of the propargyl precursor bearing an extra strong electron-withdrawing group.21 Further attempts with the use of HOOC-bonded propargylic substrates or Ph-substituted internal alkynyl substrates failed to afford the desired products.
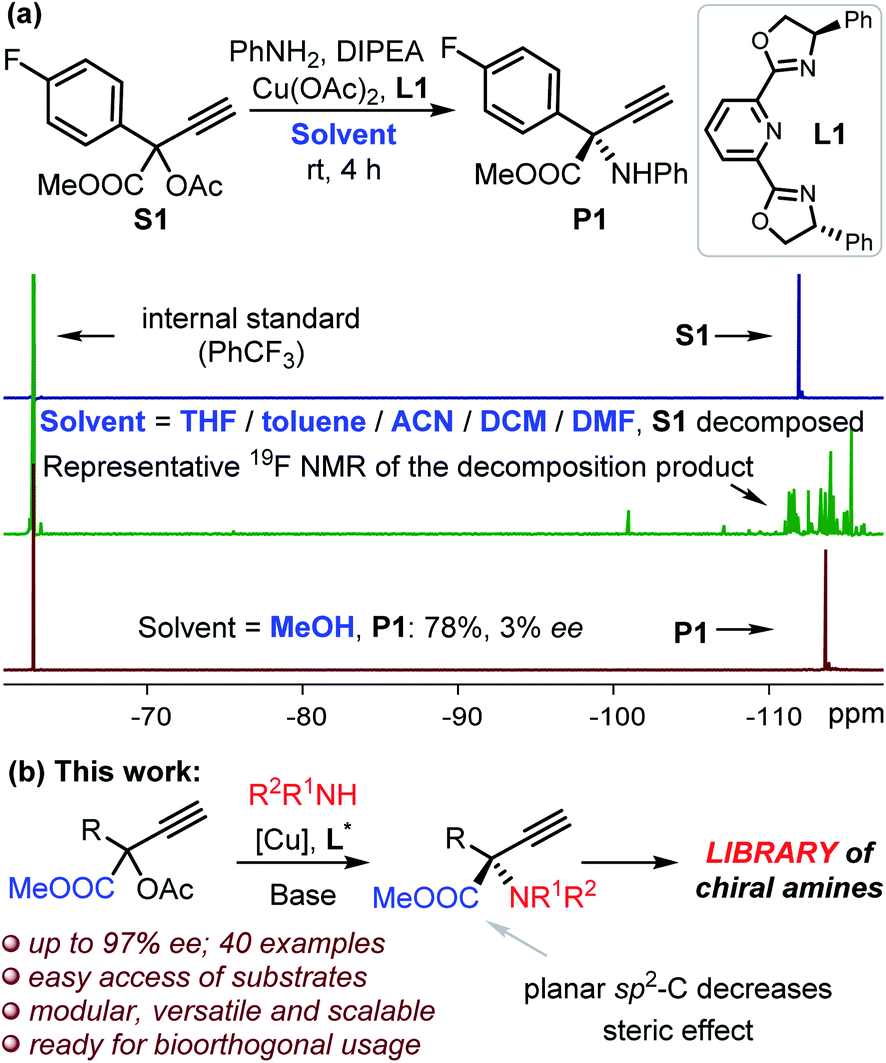 | ||
| Fig. 1 Cu-catalyzed elusive propargylation of aryl amines: (a) the reaction outcomes of the propargylation of aniline with S1 in different solvents on the basis of 19F NMR; (b) the asymmetric propargylation of amines towards the formation of protected α-quaternary amino acids. | ||
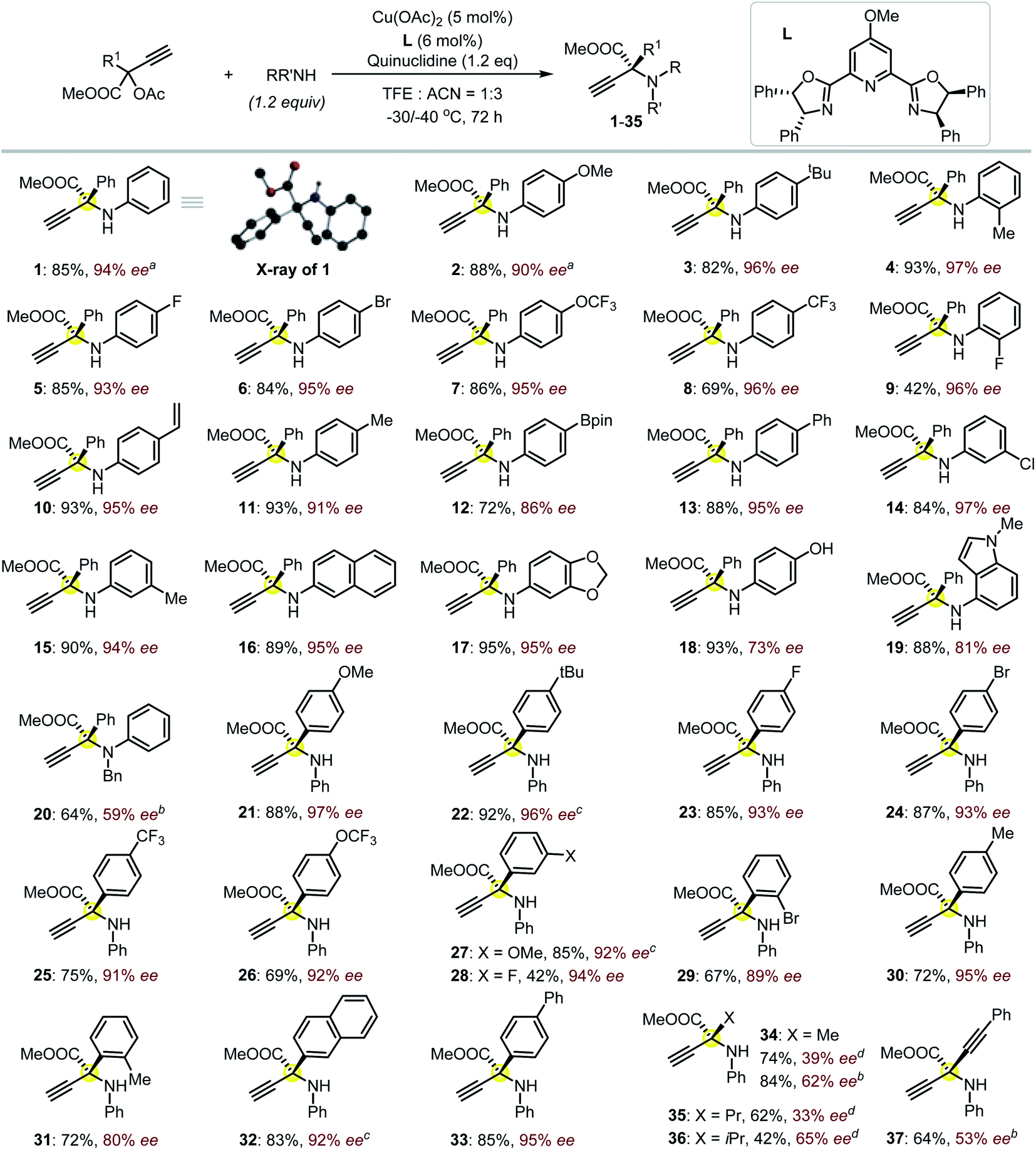 | ||
| Fig. 2 The investigation of the reaction scope. Isolated yields were reported, and the ee value was evaluated by HPLC equipped with a chiral column. The reactions were performed on a 0.1 mmol scale at −30 °C for amino esters 3–19 and −40 °C for amino esters 21, 23–26, 28–31 and 33; other conditions are noted. Inset is the solid state of product 1 (CDCC-2092791) with only the hydrogen attached to the amino group shown for clarity. a−40 °C. bUsing methanol as a solvent and reaction at 0 °C for 24 h. c−30 °C. d0 °C for 24 h. | ||
These reactions could be performed on a larger scale, as suggested from the gram-scale synthesis of product 1 (Fig. 3a). Remarkably, in addition to the well-known excellent application potential of amino acids in biologics,1,14 these α-QAAs (Fig. 2) could also be transformed into precious amines via easy experiments with the retention of high enantioselectivities typically >92% ee (Fig. 3b). For instance, the hydrolysis of product 1 under basic conditions generated the free amino acid 38 in high yield. The selective reduction of the ester group in product 1 could be controlled, resulting in either β-amino alcohol 40 or the otherwise quite synthetically challenging α-amino aldehyde 39. The use of Sonogashira coupling enabled quick access to amino acid-functionalized internal alkynes, as demonstrated by the preparation of product 41. The terminal alkynyl group facilitated the construction of an enantioenriched product 42 containing an indole skeleton. The use of different Pd catalysts enabled the controllable reduction of the dangling alkynyl group towards the formation of protected ethyl or vinyl amino acids 43 and 44. A further note is that the compound similar to 44 is remarkably challenging to access with other methodologies. The access to the primary amine 45 was feasible through an oxidative C–N cleavage from amino ester 2.22 These examples (Fig. 3b) showcase the great potential of the access to a library of enantioenriched amines with high enantioselectivities from the as-prepared protected α-QAAs 1–37.
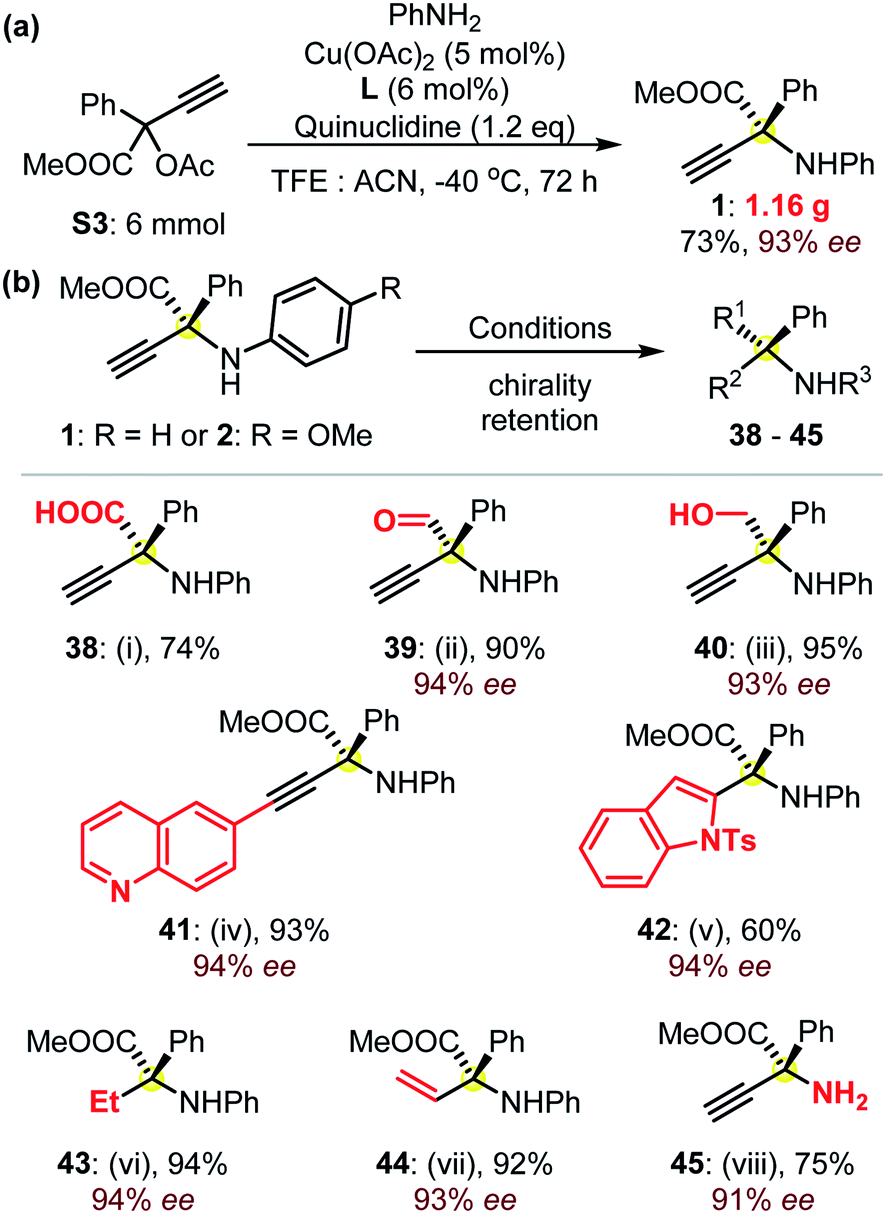 | ||
| Fig. 3 (a) The synthesis of the protected amino acid 1 at the gram-scale, and (b) synthetic applications of product 1: (i) LiOH (3.0 eq.), MeOH/H2O, rt, 24 h; (ii) DIBAL-H (2.5 eq.), toluene, −78 °C, 2 h; (iii) LiAlH4 (2.0 eq.), THF, 0 °C, 4 h; (iv) 6-iodoquinoline (1.1 eq.), Pd(PPh3)2Cl2 (5 mol%), CuI (5 mol%), TEA (3.0 eq.) THF, rt, 24 h; (v) N-tosyl-2-iodoaniline (1.0 eq.), Pd(PPh3)2Cl2 (5 mol%), CuI (5 mol%), TMG (3.0 eq.), DMF, 40 °C, 24 h; (vi) Pd/C (10 mol%), H2, EtOH, rt, 4 h; (vii) Lindlar catalyst (2 mol%), H2, EtOH, rt, 30 min; (viii) from protected amino acid 2, CAN (3.0 eq.), MeCN/H2O, 0 °C, 2 h. Please see the ESI† for details. | ||
The protected α-QAAs could undergo Click cyclization as indicated by the derivatization of the bioactive pharmaceutical zidovudine (Fig. 4a). The functionalization of other commercially available drugs such as procaine and aminoglutethimide with this protocol could be easily realized (Fig. 4b). The use of amino acids in peptide synthesis also proved feasible (Fig. 4c). The synthetic application of this Cu-APR methodology was further evidenced by the formal synthesis of protected α-ethyl norvaline 52 (Fig. 4d). Under the standard conditions, the symmetric amination of the propargylic precursor S16 afforded compound 51, followed by an oxidative C–N cleavage and reduction sequence to give rise to the target product 52. α-Ethylnorvaline was proven to be a bioactive compound and required an 11-step sequence to synthesize in a previous report.2b,c This practical improvement further verified the usefulness of our methodology in synthetic chemistry.
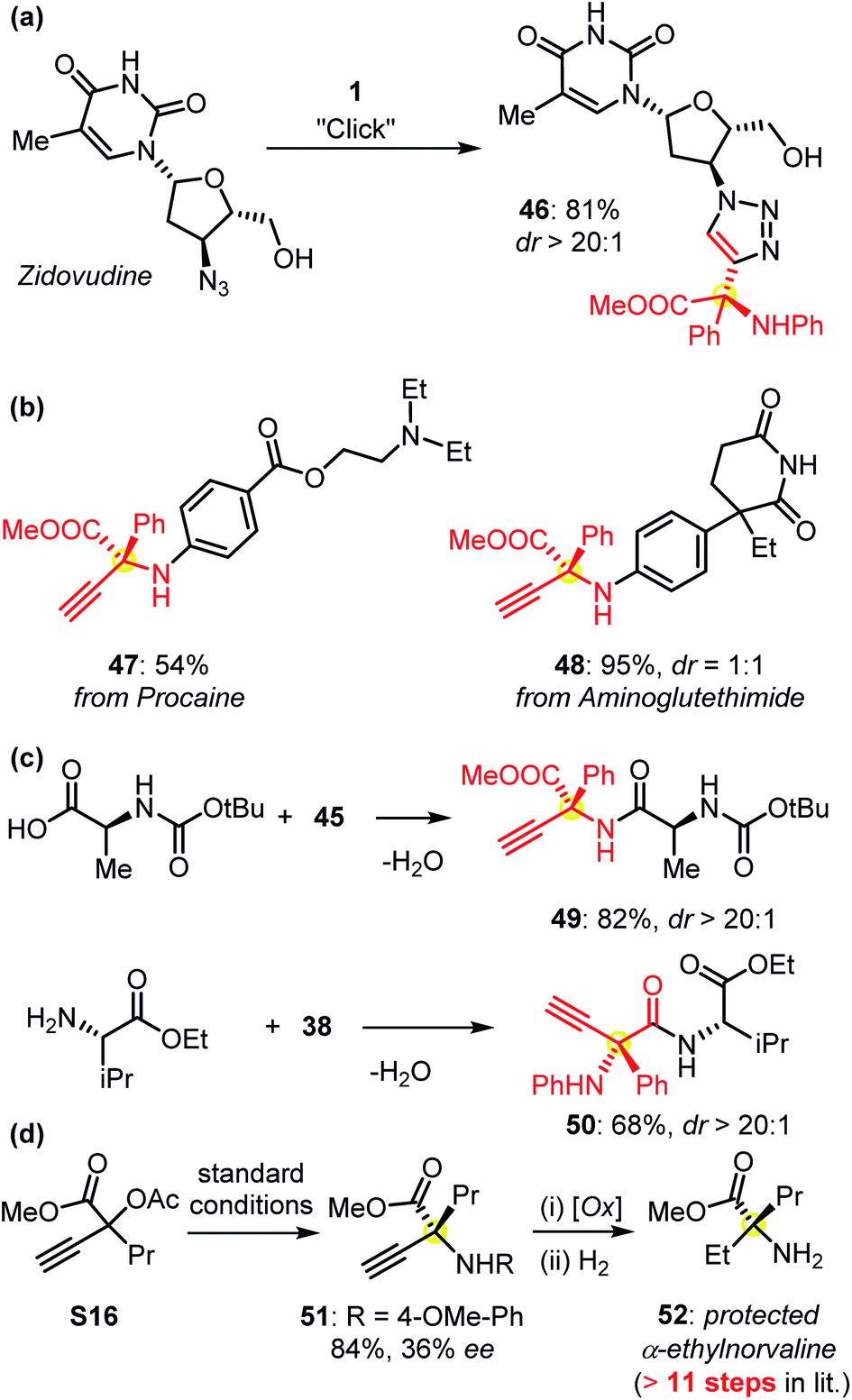 | ||
| Fig. 4 (a and b) The derivatization of commercially available drug molecules; (c) the syntheses of enantioenriched peptides 49 and 50 from products 45 and 38, respectively; (d) a concise route for the synthesis of protected bioactive α-ethyl norvaline 52versus a multiple-step pathway reported previously. Please see the ESI† for details. | ||
Conclusions
In conclusion, we herein report the first amine nucleophilic approach for the modular and straightforward synthesis of invaluable enantioenriched protected α-quaternary amino acids with high efficiency. This practical protocol features wide functional group tolerance and high enantioselectivity introduction with typically >90% ee, and is amendable to the modification of commercially available bioactive molecules. The resultant protected amino acids could be readily converted into a number of precious enantioenriched amines, which are otherwise synthetically quite challenging, including those of α-amino aldehyde, peptides or protected α-vinyl amino acids with >92% ee in excellent yields. This protocol could be utilized for the synthesis of protected bioactive α-ethylnorvaline in 3 steps, a significant advancement in comparison to an 11-step sequence reported previously.Data availability
The ESI† includes experimental details, HPLC data, NMR data and HRMS data.Author contributions
TL performed all the experiments. WG supervised the project and wrote the first draft. All authors participated in the interpretation and analysis of the reaction results, and finalized the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the starting funding scheme of Xi'an Jiaotong University. We thank the Instrument Analysis Center of Xi'an Jiaotong University for their assistance with the HRMS analysis.References
- Selective review on amino acids: (a) J. B. Hedges and K. S. Ryan, Chem. Rev., 2020, 120, 3161–3209 CrossRef CAS PubMed; (b) J. Moschner, V. Stulberg, R. Fernandes, S. Huhmann, J. Leppkes and B. Koksch, Chem. Rev., 2019, 119, 10718–10801 CrossRef CAS PubMed; (c) B. Eftekhari-Sis and M. Zirak, Chem. Rev., 2017, 117, 8326–8419 CrossRef CAS PubMed; (d) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775–8806 CrossRef CAS PubMed; (e) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626–2704 CrossRef CAS PubMed; (f) C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584–4671 CrossRef PubMed; (g) L. Aurelio, R. T. C. Brownlee and A. B. Hughes, Chem. Rev., 2004, 104, 5823–5846 CrossRef CAS PubMed; (h) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013–3028 CrossRef CAS PubMed; (i) V. A. Soloshonok and A. E. Sorochinsky, Synthesis, 2010, 2319–2344 CrossRef CAS; (j) M. S. Segundo and A. Correa, Synthesis, 2018, 50, 2853–2866 CrossRef; (k) A. E. Metz and M. C. Kozlowski, J. Org. Chem., 2015, 80, 1–7 CrossRef CAS PubMed.
- (a) C. Cativiela, M. Ordóñez and J. L. Viveros-Ceballos, Tetrahedron, 2020, 76, 130875 CrossRef CAS; (b) C. Cativiela, M. D. Diaz-de-Villegas and J. A. Gálvez, J. Org. Chem., 1994, 59, 2497–2505 CrossRef CAS; (c) M. Rainaldi, A. Moretto, C. Peggion, F. Formaggio, S. Mammi, E. Peggion, J. A. Galvez, M. D. Díaz-de-Villegas, C. Cativiela and C. Toniolo, Chem.–Eur. J., 2003, 9, 3567–3576 CrossRef CAS PubMed.
- Selective research articles, please see: (a) K. Morisaki, M. Sawa, R. Yonesaki, H. Morimoto, K. Mashima and T. Ohshima, J. Am. Chem. Soc., 2016, 138, 6194–6203 CrossRef CAS PubMed; (b) H. Wang, T. Jiang and M.-H. Xu, J. Am. Chem. Soc., 2013, 135, 971–974 CrossRef CAS PubMed; (c) P. Fu, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 5530–5541 CrossRef CAS PubMed; (d) U. Bhakta, P. V. Kattamuri, J. H. Siitonen, L. B. Alemany and L. Kürti, Org. Lett., 2019, 21, 9208–9211 CrossRef CAS PubMed; (e) P. V. Kattamuri, U. Bhakta, S. Siriwongsup, D.-H. Kwon, L. B. Alemany, M. Yousufuddin, D. H. Ess and L. Kürti, J. Org. Chem., 2019, 84, 7066–7099 CrossRef CAS PubMed; (f) The Kozlowski group reported a one-pot umpolung strategy for the synthesis of protected α-allyl-α-aryl α-amino acids in moderate enantioselectivities: J. M. Curto, J. S. Dickstein, S. Berritt and M. C. Kozlowski, Org. Lett., 2014, 16, 1948–1951 CrossRef CAS PubMed.
- Selective research articles, please refer to: (a) P. Chen and J. Zhang, Org. Lett., 2017, 19, 6550–6553 CrossRef CAS PubMed; (b) R. He, X. Huo, L. Zhao, F. Wang, L. Jiang, J. Liao and W. Zhang, J. Am. Chem. Soc., 2020, 142, 8097–8103 CrossRef CAS PubMed; (c) Q. Zhang, H. Yu, L. Shen, T. Tang, D. Dong, W. Chai and W. Zi, J. Am. Chem. Soc., 2019, 141, 14554–14559 CrossRef CAS PubMed; (d) C.-J. Wang, G. Liang, Z.-Y. Xue and F. Gao, J. Am. Chem. Soc., 2008, 130, 17250–17251 CrossRef CAS PubMed; (e) L. Chen, M.-J. Luo, F. Zhu, W. Wen and Q.-X. Guo, J. Am. Chem. Soc., 2019, 141, 5159–5163 CrossRef CAS PubMed; (f) X. Huo, J. Zhang, J. Fu, R. He and W. Zhang, J. Am. Chem. Soc., 2018, 140, 2080–2084 CrossRef CAS PubMed; (g) L. Wei, Q. Zhu, S.-M. Xu, X. Chang and C.-J. Wang, J. Am. Chem. Soc., 2018, 140, 1508–1513 CrossRef CAS PubMed; (h) X. Huo, R. He, J. Fu, J. Zhang, G. Yang and W. Zhang, J. Am. Chem. Soc., 2017, 139, 9819–9822 CrossRef CAS PubMed; (i) T. Wang, Z. Yu, D. L. Hoon, C. Y. Phee, Y. Lan and Y. Lu, J. Am. Chem. Soc., 2016, 138, 265–271 CrossRef CAS PubMed; (j) B. M. Trost and K. Dogra, J. Am. Chem. Soc., 2002, 124, 7256–7257 CrossRef CAS PubMed; (k) T. Ooi, M. Takeuchi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 2000, 122, 5228–5229 CrossRef CAS; (l) M. Wang, M. Zhou, L. Zhang, Z. Zhang and W. Zhang, Chem. Sci., 2020, 11, 4801–4807 RSC; (m) D. J. Sprague, A. Singh and J. N. Johnston, Chem. Sci., 2018, 9, 2336–2339 RSC; (n) A racemic version, for example: Y. Matsumoto, J. Sawamura, Y. Murata, T. Nishikata, R. Yazaki and T. Ohshima, J. Am. Chem. Soc., 2020, 142, 8498–8505 CrossRef CAS PubMed.
- (a) R. He, X. Wang, T. Hashimoto and K. Maruoka, Angew. Chem., Int. Ed., 2008, 47, 9466–9468 CrossRef CAS PubMed; (b) K. Ohmatsu, Y. Ando, T. Nakashima and T. Ooi, Chem, 2016, 1, 802–810 CrossRef CAS; (c) W. Chen, Y. Wang, X. Mi and S. Luo, Org. Lett., 2019, 21, 8178–8182 CrossRef CAS PubMed; (d) J. López-Cantarero, M. B. Cid, T. B. Poulsen, M. Bella, J. L. G. Ruano and K. A. Jørgensen, J. Org. Chem., 2007, 72, 7062–7065 CrossRef PubMed; (e) A racemic version, for example: D. J. Fisher, G. L. Burnett, R. Velasco and J. Read de Alaniz, J. Am. Chem. Soc., 2015, 137, 11614–11617 CrossRef CAS PubMed.
- (a) P. Vachal and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 10012–10014 CrossRef CAS PubMed; (b) S. Masumoto, H. Usuda, M. Suzuki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2003, 125, 5634–5635 CrossRef CAS PubMed; (c) J. Wang, X. Hu, J. Jiang, S. Gou, X. Huang, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2007, 46, 8468–8470 CrossRef CAS PubMed.
- (a) D. J. Leonard, J. W. Ward and J. Clayden, Nature, 2018, 562, 105–109 CrossRef CAS PubMed; (b) H. Abas, J. Mas-Roselló, M. M. Amer, D. J. Durand, R. R. Groleau, N. Fey and J. Clayden, Angew. Chem., Int. Ed., 2019, 58, 2418–2422 CrossRef CAS PubMed; (c) D. Zhang, J. Zhou, F. Xia, Z. Kang and W. Hu, Nat. Commun., 2015, 6, 5801–5808 CrossRef CAS PubMed; (d) K. Fesko, M. Uhl, J. Steinreiber, K. Gruber and H. Griengl, Angew. Chem., Int. Ed., 2010, 49, 121–124 CrossRef CAS PubMed; (e) Z.-L. Wu and Z.-Y. Li, J. Org. Chem., 2003, 68, 2479–2482 CrossRef CAS PubMed; (f) K. A. Teegardin, L. Gotcher and J. D. Weaver, Org. Lett., 2018, 20, 7239–7244 CrossRef CAS PubMed; (g) M. Baumann, I. R. Baxendale, S. V. Ley, N. Nikbin, C. D. Smith and J. P. Tierney, Org. Biomol. Chem., 2008, 6, 1577–1586 RSC.
- (a) C. Luo and Y. Huang, J. Am. Chem. Soc., 2013, 135, 8193–8196 CrossRef CAS PubMed; (b) A racemic version, please refer to: C. Jing, D. Xing, Y. Qian, T. Shi, Y. Zhao and W. Hu, Angew. Chem., Int. Ed., 2013, 52, 9289–9292 CrossRef CAS PubMed.
- The synthesis of α,α-dialkylated α-QAAs is quite challenging but useful, please see: J. H. Blackwell, R. Kumar and M. J. Gaunt, J. Am. Chem. Soc., 2021, 143, 1598–1609 CrossRef CAS PubMed.
- Selected reviews, please refer to: (a) Y. Nishibayashi, Synthesis, 2012, 489–503 CrossRef CAS; (b) R. J. Detz, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2009, 2009, 6263–6276 CrossRef; (c) R. Roy and S. Saha, RSC Adv., 2018, 8, 31129–31193 RSC; (d) C.-H. Ding and X.-L. Hou, Chem. Rev., 2011, 111, 1914–1937 CrossRef CAS PubMed; (e) One example of research article, please refer to: G. Hattori, H. Matsuzawa, Y. Miyake and Y. Nishibayashi, Angew. Chem., Int. Ed., 2008, 47, 3781–3783 CrossRef CAS PubMed.
- (a) J. E. Gómez, W. Guo, S. Gaspa and A. W. Kleij, Angew. Chem., Int. Ed., 2017, 56, 15035–15038 CrossRef PubMed; (b) L. Tian, L. Gong and X. Zhang, Adv. Synth. Catal., 2018, 360, 2055–2059 CrossRef CAS; (c) J. E. Gómez, À. Cristòfol and A. W. Kleij, Angew. Chem., Int. Ed., 2019, 58, 3903–3907 CrossRef PubMed; (d) W. Guo, L. Zuo, M. Cui, B. Yan and S. Ni, J. Am. Chem. Soc., 2021, 143, 7629–7634 CrossRef CAS PubMed , this method could not be used for the preparation of α-quaternary amino acids..
- K. Tsuchida, Y. Senda, K. Nakajima and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 9728–9732 CrossRef CAS PubMed . Please note that Cu-APRs typically use propargylic precursors with OBoc or OAc as leaving group (see ref. 10) which is not applicable in this CF3-substituted project (ref. 12)..
- (a) Alkynyl group is versatile toward the access to a range of functionalities, see representative example: R.-Z. Li, D.-Q. Liu and D. Niu, Nat. Catal., 2020, 3, 672–680 CrossRef CAS; (b) α-Amino esters are also versatile toward the formation of a range of use molecules, please refer to: N. Mateu, S. L. Kidd, L. Kalash, H. F. Sore, A. Madin, A. Bender and D. R. Spring, Chem.–Eur. J., 2018, 24, 13681–13687 CrossRef CAS PubMed.
- Propargylic compounds are readily for biorthogonal applications, please see representative articles: (a) C. G. Parker and M. R. Pratt, Cell, 2020, 180, 605–632 CrossRef CAS PubMed; (b) K. F. Suazo, K.-Y. Park and M. D. Distefano, Chem. Rev., 2021, 121, 7178–7248 CrossRef CAS PubMed.
- (a) Using DCM as solvent, please see: J. Song, Z.-J. Zhang and L.-Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 5212–5216 CrossRef CAS PubMed; (b) Using THF as solvent, please see: R.-Z. Li, H. Tang, L. Wan, X. Zhang, Z. Fu, J. Liu, S. Yang, D. Jia and D. Niu, Chem, 2017, 3, 834–845 CrossRef CAS; (c) Using toluene as solvent, please see: F.-L. Zhu, Y. Zou, D.-Y. Zhang, Y.-H. Wang, X.-H. Hu, S. Chen, J. Xu and X.-P. Hu, Angew. Chem., Int. Ed., 2014, 53, 1410–1414 CrossRef CAS PubMed; (d) Using DMA as solvent, please see ref. 13a..
- Hydrogen bonding assisted process was also proposed by Nishibayashi and coworkers, please see: G. Hattori, K. Sakata, H. Matsuzawa, Y. Tanabe, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2010, 132, 10592–10608 CrossRef CAS PubMed.
- Please see ESI† for our mechanism proposal.
- We applied a patent with ref. CN202111006086.5 for the preparation of α-amino acids with the present reported methodology. This patent was still under consideration and was not issued yet.
- Please refer to Tables S1–S3 in ESI† for the selected screening data.
- See ESI† for details.
- Alkyl amines were frequently used as nucleophiles in Cu-APR, please see: (a) J. Huang, H.-H. Kong, S.-J. Li, R.-J. Zhang, H.-D. Qian, D.-R. Li, J.-Y. He, Y.-N. Zheng and H. Xu, Chem. Commun., 2021, 57, 4674–4677 RSC; (b) Y. Imada, M. Yuasa, I. Nakamura and S.-I. Murahashi, J. Org. Chem., 1994, 59, 2282–2284 CrossRef CAS . Please also refer to ref. 16..
- The synthesis of unprotected α-QAAs is the continuous endeavour of synthetic chemists, representative references please refer to: (a) M. Sawa, K. Morisaki, Y. Kondo, H. Morimoto and T. Ohshima, Chem.–Eur. J., 2017, 23, 17022–17028 CrossRef CAS PubMed; (b) R. Yonesaki, Y. Kondo, W. Akkad, M. Sawa, K. Morisaki, H. Morimoto and T. Ohshima, Chem.–Eur. J., 2018, 24, 15211–15214 CrossRef CAS PubMed; (c) L. Peng, H. Wang and C. Guo, J. Am. Chem. Soc., 2021, 143, 6376–6381 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2092791. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02318k |
| This journal is © The Royal Society of Chemistry 2022 |