DOI:
10.1039/D2SC02139K
(Edge Article)
Chem. Sci., 2022,
13, 7215-7223
Construction of a quaternary stereogenic center by asymmetric hydroformylation: a straightforward method to prepare chiral α-quaternary amino acids†
Received
14th April 2022
, Accepted 26th May 2022
First published on 6th June 2022
Abstract
The construction of chiral quaternary carbon stereocenters has been a long-standing challenge in organic chemistry. Particularly, α-quaternary amino acids that are of high importance in biochemistry still lack a straightforward synthetic method. We here reported a hydroformylation approach to access chiral quaternary stereogenic centers, which has been a long-standing challenge in transition metal catalysis. α,β-Unsaturated carboxylic acid derivatives undergo hydroformylation with a rhodium catalyst to generate an α-quaternary stereocenter under mild conditions. By using this method, a variety of chiral α-quaternary amino acids could be synthesized with satisfactory enantioselectivity. In-depth investigation revealed that the regioselectivity is dramatically influenced by the electronic properties of the substituents attached to the target C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. By applying NMR and DFT analyses, the chiral environment of a rhodium/Yanphos complex was depicted, based on which a substrate-catalyst interaction model was proposed.
C bond. By applying NMR and DFT analyses, the chiral environment of a rhodium/Yanphos complex was depicted, based on which a substrate-catalyst interaction model was proposed.
Instruction
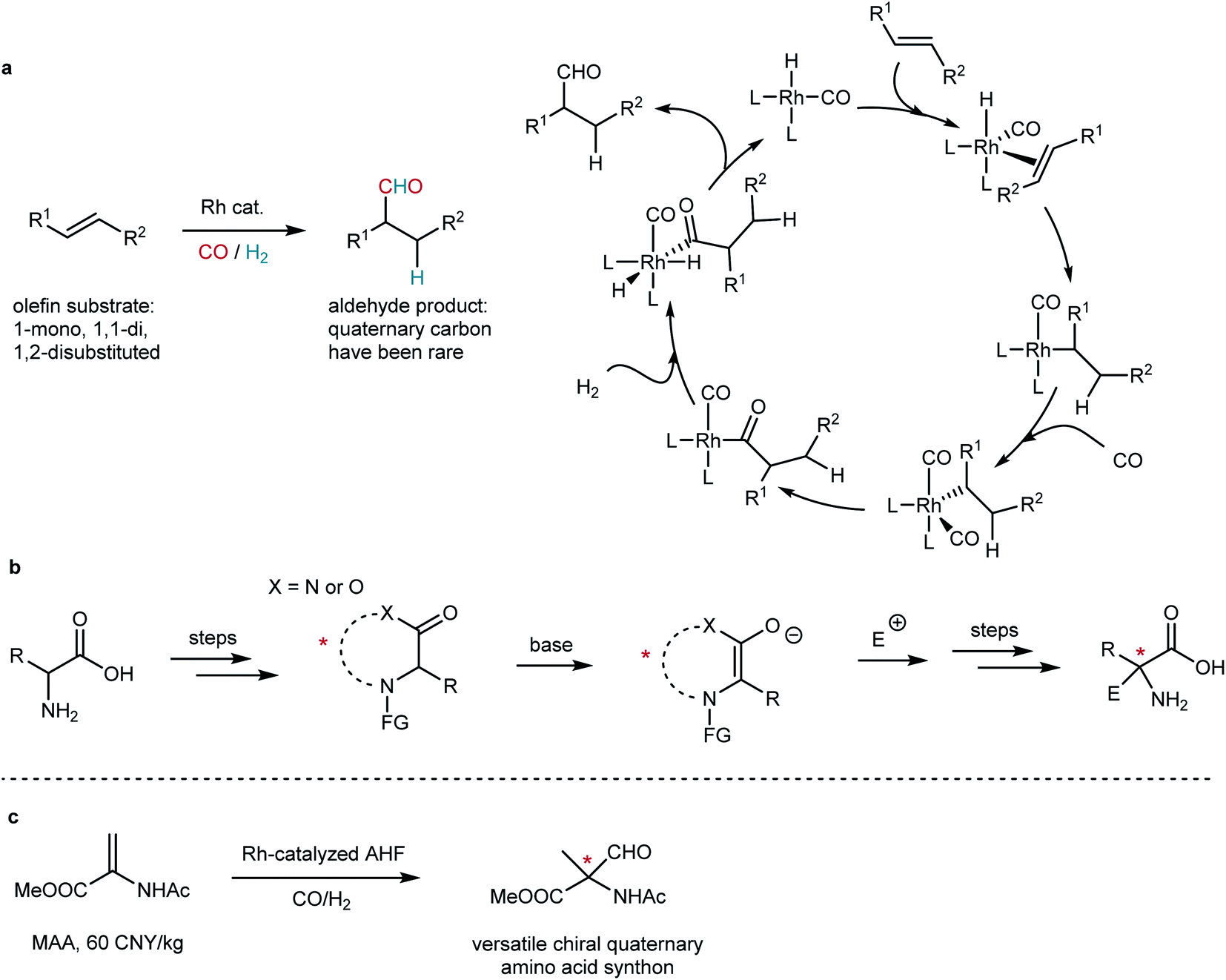
Chiral quaternary stereogenic centers are important structural motifs in both natural products and bioactive molecules. The construction of quaternary stereocenters has been attractive but challenging.1–4 Transition metal-catalyzed hydroformylation that adds a carbonyl group and a hydrogen atom to a CC bond has been a practical method to realize the hydrofunctionalization of olefins (Fig. 1a).5 Its asymmetric version, asymmetric hydroformylation (AHF), has been a powerful tool to build chiral molecules.6,7 We envisaged that rhodium-catalyzed AHF shall render a practical way to access quaternary stereogenic centers. However, AHF reactions were recognized as a formidable research area as many challenges remain in the way of this approach. Chemo-, regio- and enantioselectivity must be taken into consideration in a single catalytic reaction.8 According to Keulemans' empirical rule that claims “in hydroformylation, formyl groups are not produced at quaternary carbon centers”,9 the formation of quaternary carbon via hydroformylation has intrinsic difficulties. The selectivity and reactivity of hydroformylation reactions are very sensitive to steric and electronic properties of the substituents attached to CC bonds.8 To date, the construction of chiral quaternary aldehydes via AHF has been an unsolved problem.
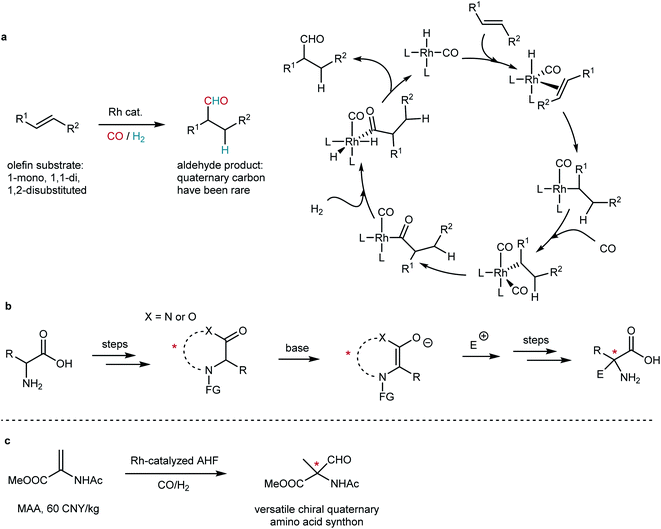 |
| | Fig. 1 Practical approaches to chiral quaternary α-amino acids. (a) Rh-catalyzed hydroformylation of olefin and its well-accepted mechanism. (b) Diastereoselective or enantioselective synthesis of quaternary amino acid derivatives via amino acid templates. (c) Our strategy of AHF of dehydroamino acid derivatives: a straightforward method. | |
Amino acids that bear a chiral quaternary stereogenic center at the α-position have great importance in biochemistry. As a family of non-natural amino acids, not only could they play an irreplaceable role in the modification of conformation of peptides and proteins,10 but they are also key intermediates for the synthesis of many bioactive compounds. The development of a methodology to approach chiral α-quaternary amino acids has been a long-standing challenge for organic synthesis11–16 and has still drawn great attention in the recent decade.17 The most widely applied approach has been the diastereoselective synthesis via amino acid templates (Fig. 1b).18 Such a strategy relies on the nucleophilicity of the enolate after deprotonation of the α-proton and the chiral environment created by either the concept of self-regeneration of stereocenters,19 memory of chirality20 or the introduction of an external chiral auxiliary.18 However, the formation of cyclic intermediates and removal of the protecting groups require extra steps, which lowers the atom efficiency and raises the cost. Therefore, a straightforward approach to chiral α-quaternary amino acids has always been in demand.
An early attempt was made by Gladiali and Pinna to carry out AHF of methyl N-acetamidoacrylate (MAA) with DIOP under rather harsh conditions, but the enantioselectivity was not satisfactory.21 The development toward the construction of chiral quaternary stereocenters via AHF has been slow,22 and only scattered reports by Alper,23 Buchwald,24 our group,25 and Landis and Schomaker26 were documented. Gratefully, previous investigation by the Landis group27–30 and our group31 of the regioselectivity of AHF provided guidance to tackle the challenges, and functionalized olefins that bear an electron-withdrawing group (EWG) show preference for the formation of branched aldehydes. The huge demand for direct construction of chiral α-quaternary amino acids and our expertise in AHF reactions spurred us to continuously explore this field to construct chiral quaternary stereocenters. Dehydroamino acid derivatives such as MAA are readily available fine chemicals. The straightforward transformation of such a type of functionalized olefins into versatile chiral α-quaternary amino acid synthons is of high synthetic value (Fig. 1c).
Results and discussion
The development of the method
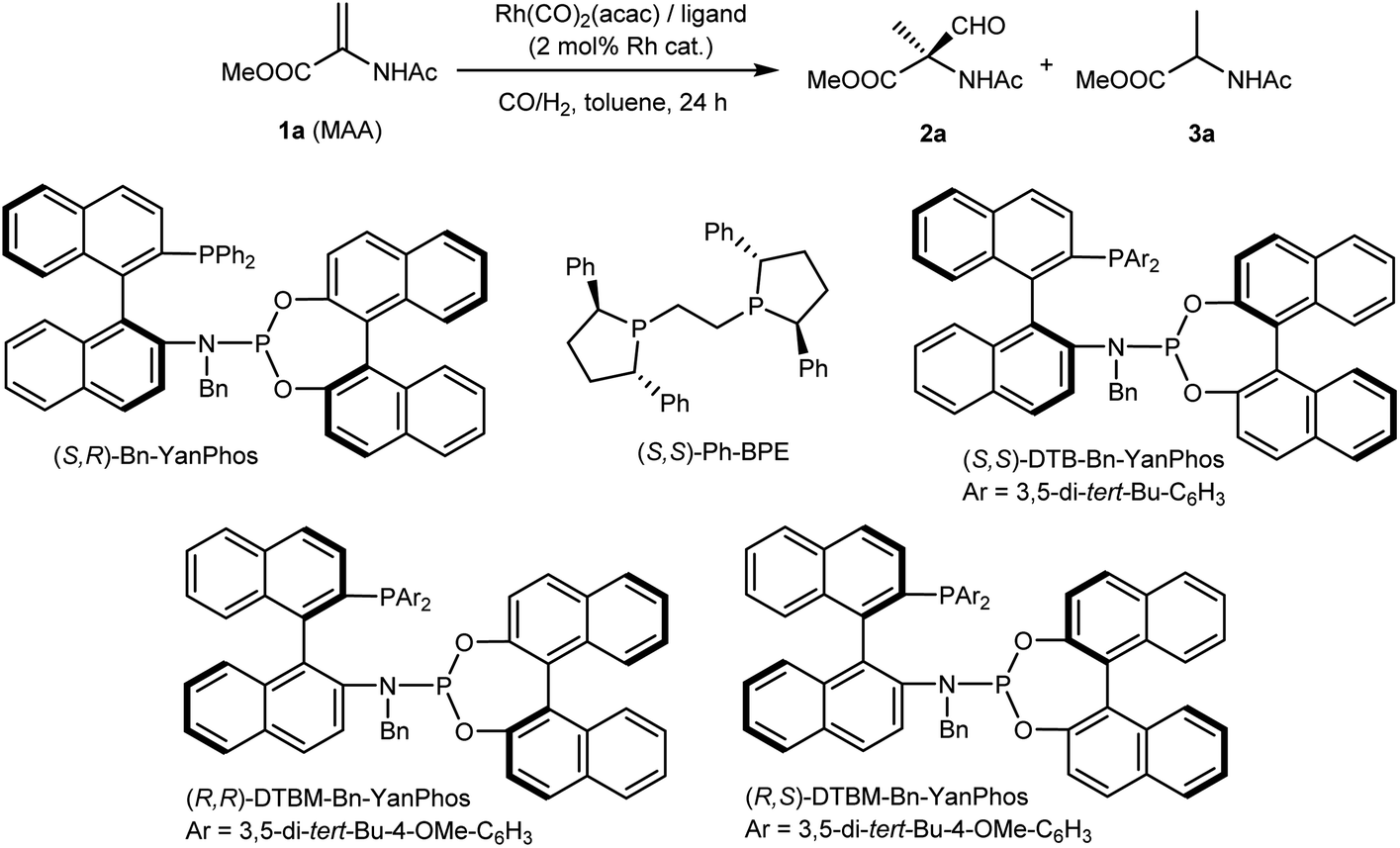
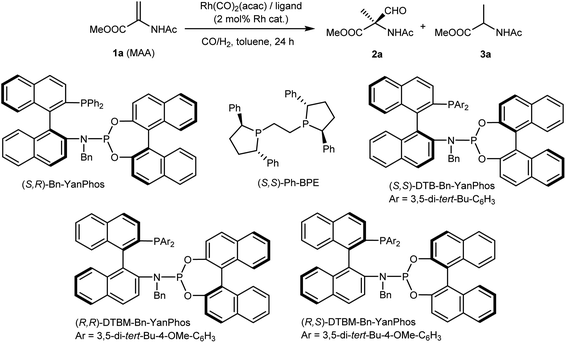
Our research on AHF to construct chiral amino acids commenced with the AHF of MAA, a cheap dehydroamino acid derivative. When we applied (S,R)-Yanphos,32 a family of hybrid33 phosphine–phosphoramidite ligands that were developed by our group and have successfully been applied in AHF reactions,34–40 MAA was consumed within 24 hours at 100 °C under a 10 bar syngas atmosphere. To our delight, this initial assessment yielded a quaternary aldehyde (branched selectivity), albeit a considerable ratio of the hydrogenation product (15%) and moderate enantioselectivity (79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :21 er) were observed. In comparison, Ph-BPE that has been applied in AHF reactions gave almost a racemic aldehyde. However, using the other diastereomer of Bn-Yanphos (R,R), though with increasing steric hindrance, did not improve the results (entry 3 and 4). Gratefully, the application of sterically demanding (R,S)-DTBM-Bn-Yanphos gave satisfactory chemoselectivity (96% aldehyde), regioselectivity (all quaternary carbon) and enantioselectivity (93:7 er). Based on this performance, further condition screening led to the following conclusion: lowering the ligand ratio to L/Rh = 1.5:1 and reducing the reaction temperature and pressure to 90 °C and 5 bar gave the best results. The increase in the yield of aldehyde in a lower syngas atmosphere (entry 9, 11 and 12) could rationalized by the assumption that lowering CO partial pressure could cause the higher concentration of the reactive (P–P)Rh(CO)H species.41 The formation of the hydrogenation product was suppressed at 3% in the reaction mixture, while the enantioselectivity reached 96:4 er.
:21 er) were observed. In comparison, Ph-BPE that has been applied in AHF reactions gave almost a racemic aldehyde. However, using the other diastereomer of Bn-Yanphos (R,R), though with increasing steric hindrance, did not improve the results (entry 3 and 4). Gratefully, the application of sterically demanding (R,S)-DTBM-Bn-Yanphos gave satisfactory chemoselectivity (96% aldehyde), regioselectivity (all quaternary carbon) and enantioselectivity (93:7 er). Based on this performance, further condition screening led to the following conclusion: lowering the ligand ratio to L/Rh = 1.5:1 and reducing the reaction temperature and pressure to 90 °C and 5 bar gave the best results. The increase in the yield of aldehyde in a lower syngas atmosphere (entry 9, 11 and 12) could rationalized by the assumption that lowering CO partial pressure could cause the higher concentration of the reactive (P–P)Rh(CO)H species.41 The formation of the hydrogenation product was suppressed at 3% in the reaction mixture, while the enantioselectivity reached 96:4 er.
Reaction scope and product elaboration
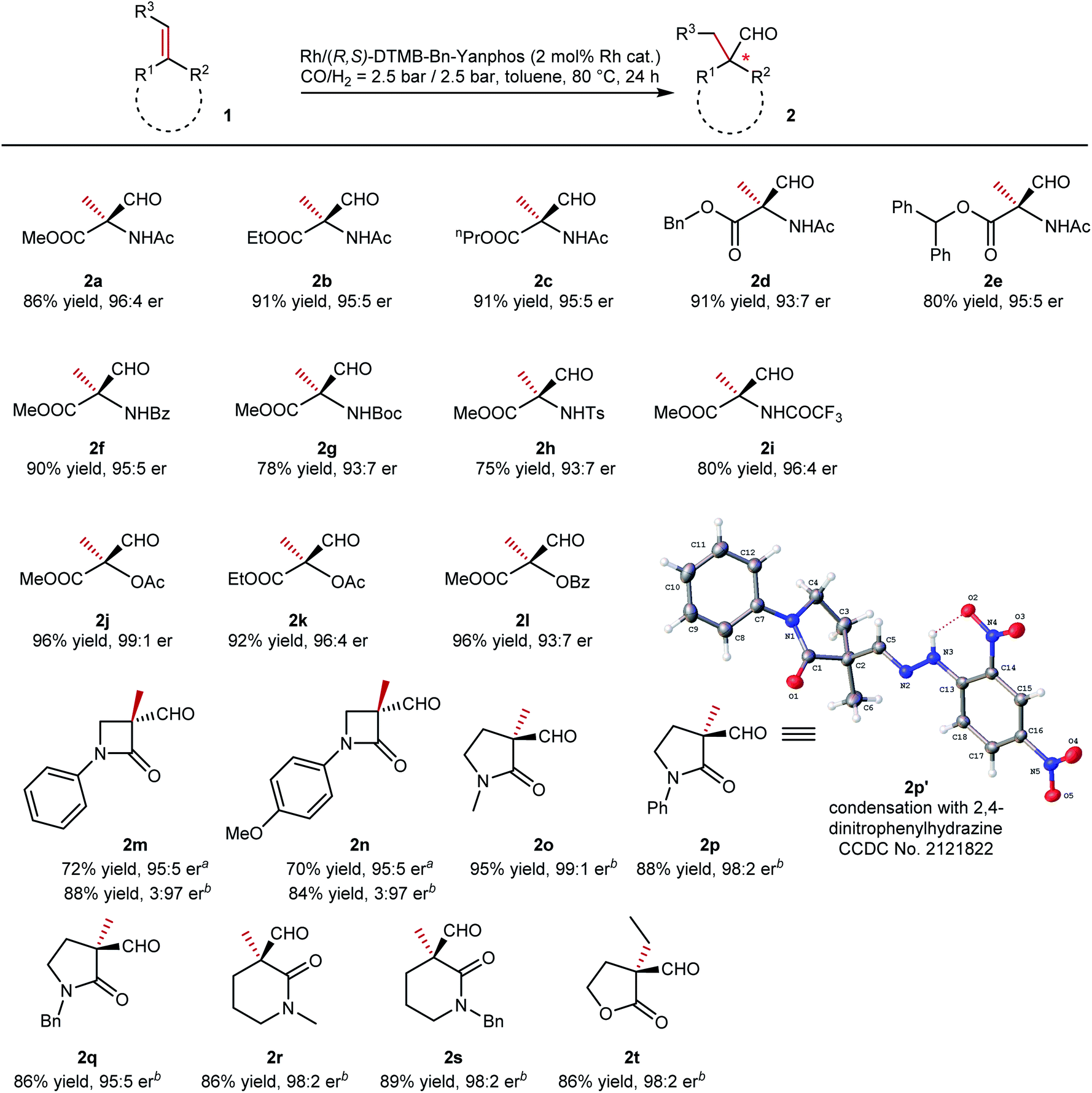
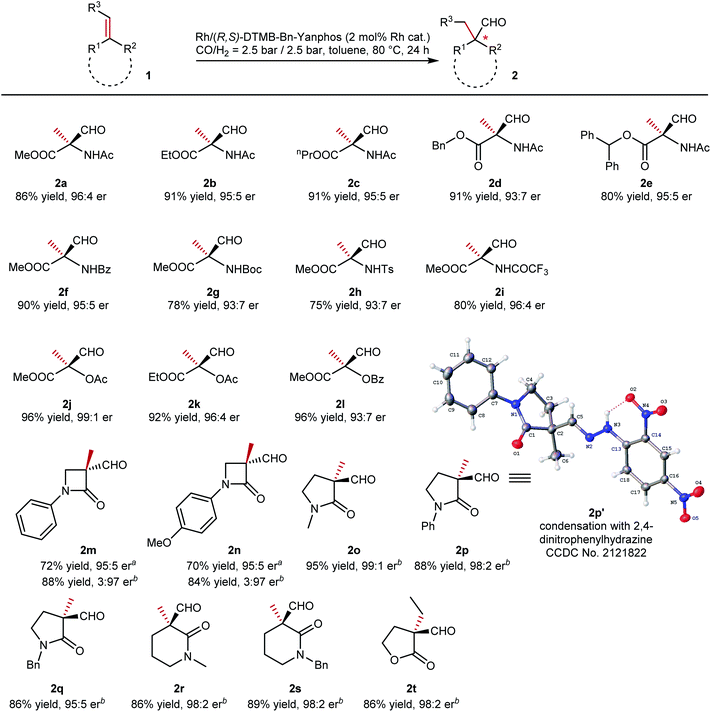
With the optimized conditions, we then evaluated the reaction scope of this methodology (summarized in Table 2). Various N-acetamidoacrylic acid esters were tested in this transformation, and quaternary aldehydes were obtained with efficient enantiodiscrimination (2a to 2e). Different substituents on the ester groups, from small methyl to bulky diphenylmethyl groups, did not bring significant influence on either yield or enantioselectivity. Altering the protecting group on the nitrogen atom of the dehydroamino acid ester is of synthetic merit. When benzoyl (2f, 90% yield, 95:5 er) and trifluoroacetyl (2i, 80% yield, 96:4 er) were introduced onto the substrate, the yield and er value were of the same level as that of the model substrate. The introduction of bulkier tert-butyl carbamate (Boc, 2g) and the tosyl (2h) group made the reaction slightly sluggish, and slightly eroded er values were observed in each case (93:7 er for both). Apart from enamines, enol esters were also tested (2j to 2l). The corresponding quaternary aldehydes were exclusively obtained. Lactams not only are an important core in many bioactive molecules42 but could also readily transform into amino acids and peptides.43,44 The exocyclic CC bond at the α-position of lactams was hydroformylated to quaternary aldehydes. β-Lactams that bear a four-membered ring were converted to aldehydes with high enantioselectivities (2m and 2n, 95:5 er), although the conversions still have room for improvement. To our surprise, better performances were observed for such cyclic substrates with a Rh/Ph-BPE complex (2m and 2n, 97:3 er, higher yields). Upon expansion of the ring size to five- or six-membered ring, both yields and enantioselectivities were increased (2o to 2s). Exocyclic conjugated lactams with both alkyl and aryl groups on the nitrogen atom gave chiral aldehydes with an α-quaternary stereocenter in high yields. Unfortunately, at this stage, only 1,1-disubstituted olefins could be efficiently hydroformylated under the optimized conditions, while 1,1,2-trisubstituted olefins gave poor reactivities. A further optimization of conditions seemed to be needed for such type of substrates. At the stage of initial screening of trisubstituted olefins, only exocyclic lactone 1t (E-configuration) yielded quaternary aldehyde 2t efficiently with a high er value.
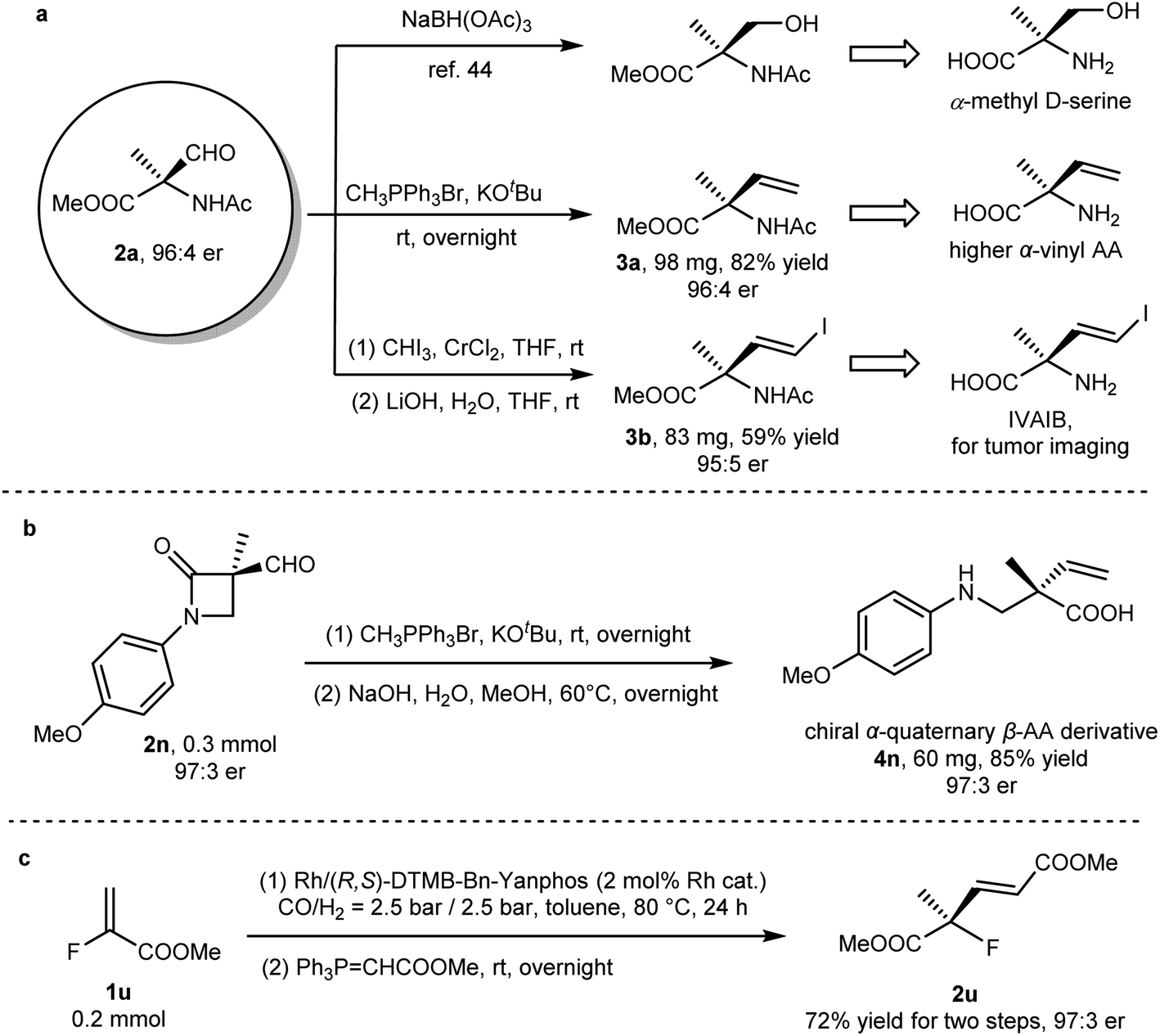
The α-formyl-α-amino acid derivative, as a versatile synthon, could be readily converted to other value-added chiral molecules bearing a quaternary carbon (Fig. 2a). The reduction of 2a yielded α-methyl serine, a valuable artificial amino acid.45 The reaction of this quaternary aldehyde with a phosphorus ylide yielded α-vinyl amino acid, a chiral compound that not only plays a role as a building block for more complicated artificial amino acids but also functions as a bioactive molecule.46 Tumor imaging reagents such as IVAIB47 and BVAIB48 could be rapidly prepared from 2a. To our delight, the optical purity was not eroded in the above-mentioned transformations. β-Lactam 2n easily undergoes olefination and hydrolysis to form β-amino acid with an α-quaternary stereocenter (Fig. 2b). In order to further demonstrate the synthetic potential of this method, such a synthetic method was coupled with other transformation such as Wittig olefination. A conjugated carboxylate (2u) with a quaternary stereocenter could be readily synthesized via AHF and the sequential Wittig reaction from methyl 2-fluoroacrylate (1u). Satisfactory yield (72%) and enantioselectivities (97:3 er) were recorded after two steps (Fig. 2c).
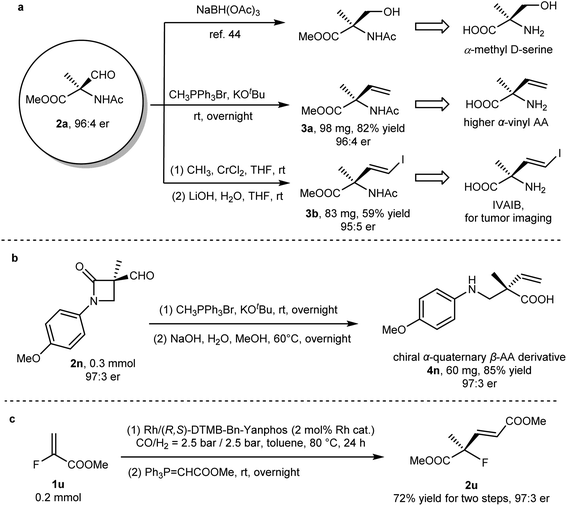 |
| | Fig. 2 Synthetic application of AHF in the construction of chiral quaternary stereocenters. (a) Elaboration on enantioenriched methyl α-formyl-N-acetyl-L-alaninate (2a) to value-added molecules. (b) Transformation of lactam 2n into α-quaternary β-amino acid. (c) Rapid sequential synthesis from methyl 2-fluoroacrylate (1u) to a chiral molecule with a quaternary stereocenter (2u). | |
Impact on regioselectivity from the substrate
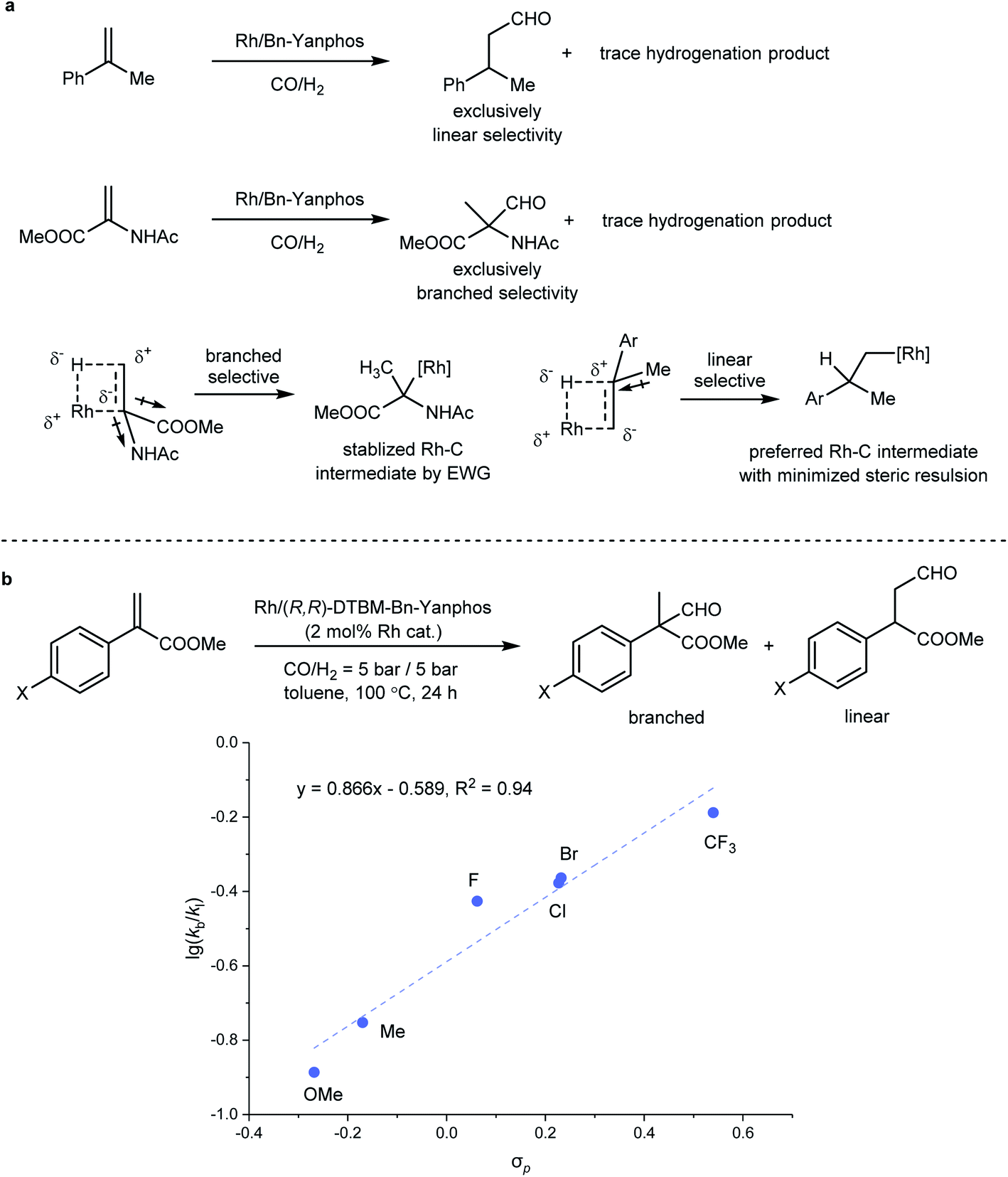
Regioselectivity has always been a focused topic in Rh-catalyzed hydroformylation.41 In previous studies by Landis29 and Clarke,49 the influence from the ligand and reaction temperature on the regioselectivity in Rh-catalyzed hydroformylation was investigated, but the regioselectivity of AHF also heavily depends on the steric and electronic features of the olefin substrate. In principle, due to steric repulsion of the substituent on the substrate and the rhodium catalyst, a linear product was favored in the hydroformylation of most terminal olefins. The migratory insertion of Rh–H to the CC bond generates an alkyl rhodium species, which is the regio- and enantiodetermining step. If an electron-withdrawing group (EWG) is incorporated, the alkyl rhodium intermediate could be stabilized via both inductive and resonance effects (Fig. 3a). In our previous study, AHF of 1,1-disubstituted olefin 1-methylstyrene yielded a linear aldehyde. In sharp comparison, AHF with MAA that is also categorized as 1,1-disubstituted olefin resulted in the formation of a branched aldehyde exclusively. Intrigued by such distinction, we chose methyl 1-arylacrylate as a target to study how the electronic properties influence the regioselectivity of hydroformylation. A series of substrates with a para-substituent underwent AHF under the same reaction conditions (Fig. 3b). The Hammett study revealed a positive linear correlation between the σp value and  that demonstrates the relative rates in the formation of both regiomers. The slope (0.866) of this regression line demonstrated high sensitivity of the regioselectivity towards the electronic properties of the substituents. In a study of the influence of the substrate steric effect on the regioselectivity, no conclusive impact from the steric hindrance was found on the selectivity (see the ESI† for details). Therefore, the electronic features of the substituents attached on the CC bond crucially contribute to determining the position where the formyl group attaches.
that demonstrates the relative rates in the formation of both regiomers. The slope (0.866) of this regression line demonstrated high sensitivity of the regioselectivity towards the electronic properties of the substituents. In a study of the influence of the substrate steric effect on the regioselectivity, no conclusive impact from the steric hindrance was found on the selectivity (see the ESI† for details). Therefore, the electronic features of the substituents attached on the CC bond crucially contribute to determining the position where the formyl group attaches.
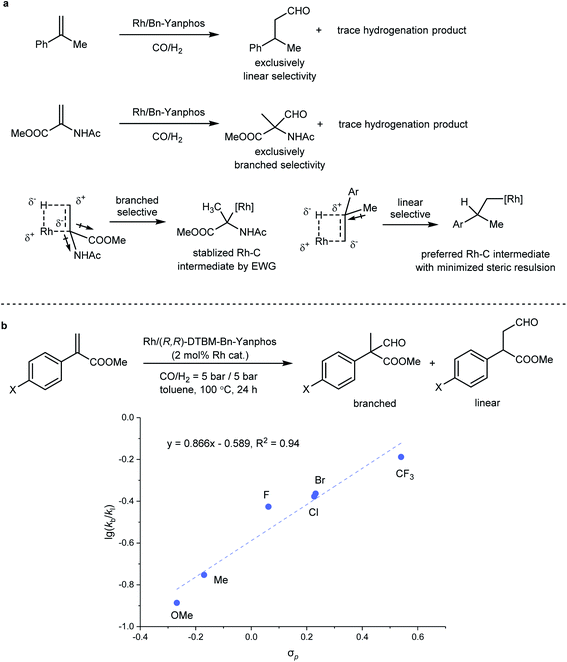 |
| | Fig. 3 Regioselectivity in Rh-catalyzed AHF. (a) Rationale for the AHF regioselectivity of 1-methylstyrene and MAA. (b) Hammett study on the influence from electronic properties on AHF of 1-arylstyrene. | |
Chiral environment of the catalyst and enantioinduction model
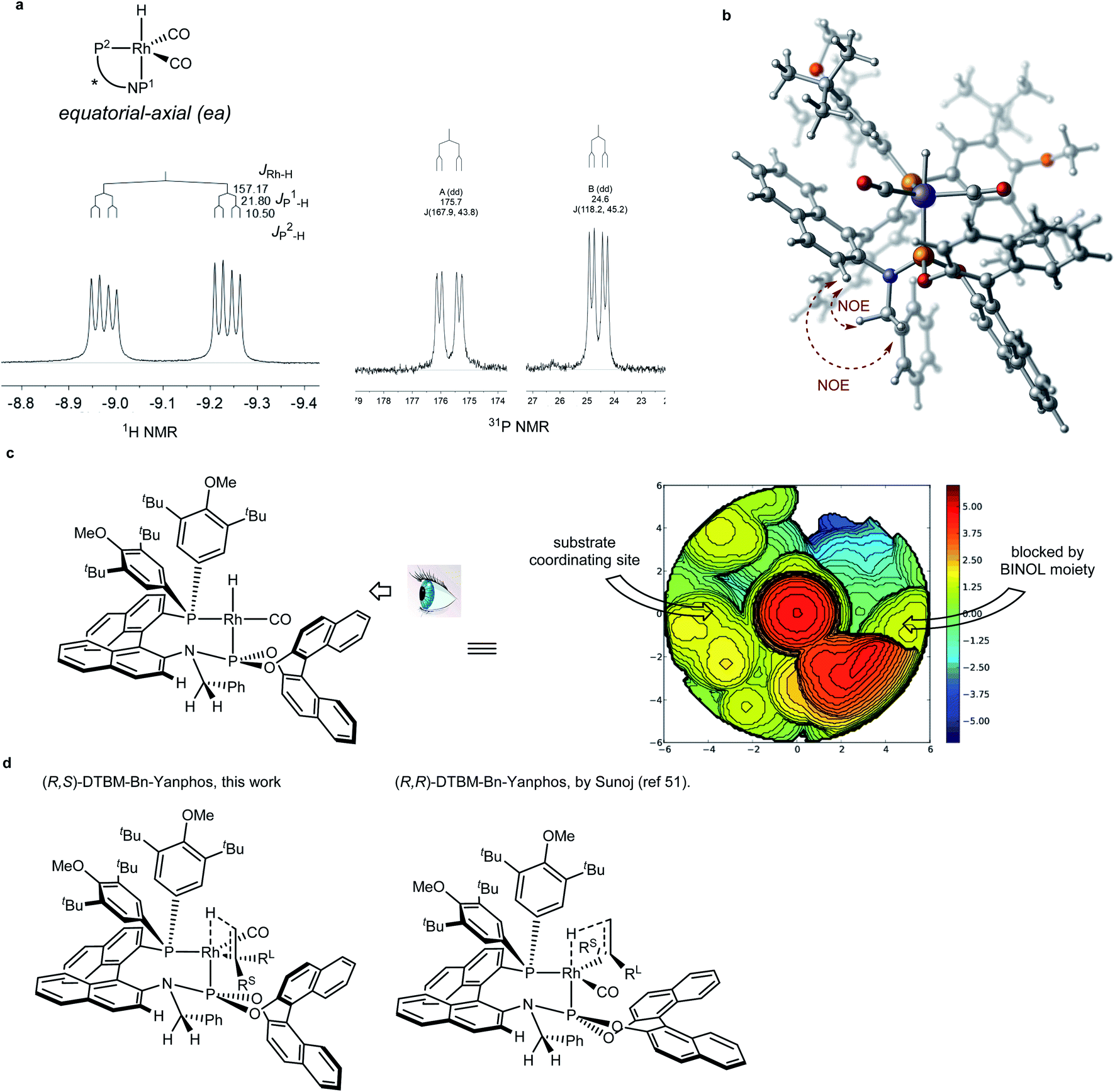
Compared to asymmetric hydrogenation reactions in which secondary interaction may assist the enantiomeric induction,50,51 the enantiodiscrimination in AHF has been believed to be as the result of steric repulsion.22 Our curiosity was drawn toward the chiral environment of the Rh/Yanphos catalyst and the enantioinduction model. Heating the Benzene-d6 solution of Rh(CO)2(acac) and (R,S)-DTBM-Bn-Yanphos in a syngas atmosphere at 60 °C gave full conversion to the dicarbonyl monohydride species Rh(Yanphos)(CO)2H. NMR analysis (Fig. 4a) showed an equatorial-axial configuration in which the P atom of the phosphoramidite moiety locates at the axial position. The NOE between both benzylic protons and the C–H proton adjacent to the amino linker confirmed the conformation of the benzyl group in the catalyst. Based on the hint from NMR spectroscopy, we optimized the structure of this complex by DFT calculations (Fig. 4b). The two diastereomers of the Yanphos ligand gave the opposite enantioselectivity (entry 4 and 5 in Table 1), which indicate the decisive role of the chiral BINOL frame of the phosphoramidite moiety. Based on our DFT calculations (see the ESI†) and Sunoj's computational research,52 we tentatively proposed the enantioinduction model as depicted in Fig. 4c. The rigid BINOL frame dictates where the substrate coordinates to the rhodium atom: changing the absolute configuration of BINOL leads to different coordinating sites for the substrate to approach (Fig. 4c, the complexation between the catalyst and substrate MAA was analyzed and is summarized in the ESI†). As the DTBM-phenyl group was believed to be highly sterically demanding, the small substituent on the substrate was forced to face the DTBM-phenyl group, and the large substituent was left to the other site (Fig. 4d).
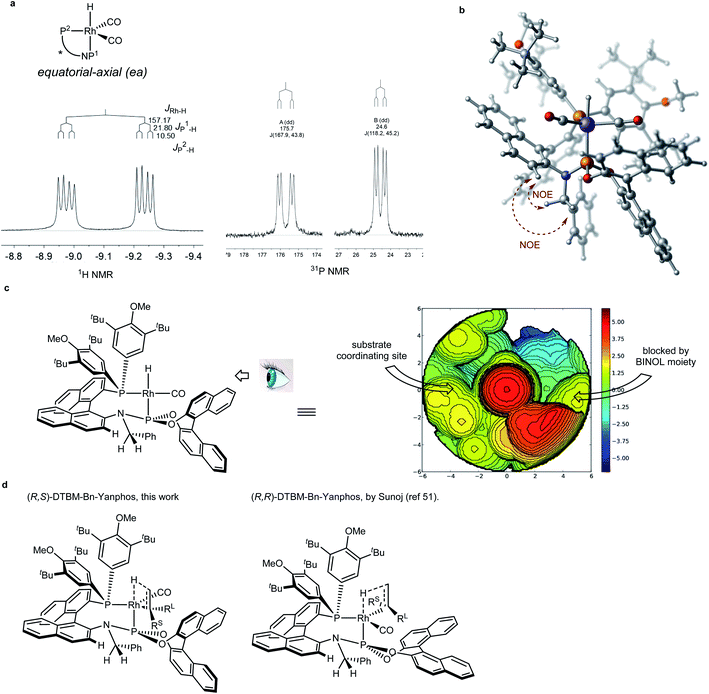 |
| | Fig. 4 Interpretation of the enantioinduction model. (a) NMR spectroscopy data supporting an equatorial-axial configuration of the Rh(Yanphos)(CO)2H complex. (b) NMR and DFT assisted optimization of the Rh(Yanphos)(CO)2H complex. (c) Topological map showing the steric environment of the reactive species Rh(Yanphos)(CO)H. (d) The model enantioinduction for both diastereomers of Yanphos in the enantiomeric determining step. | |
Table 1 Condition variation of Rh-catalyzed AHF of MAA
|
|
| Entry |
Ligand |
Rh/L |
T (°C) |
P
CO/PH2 (bar) |
Conversion |
er of 2a (R:S) |
|
2a
|
3a
|
| 1 |
(S,R)-Bn-Yanphos |
1:3 |
100 |
5/5 |
85% |
15% |
21:79 |
| 2 |
(S,S)-Ph-BPE |
1:3 |
100 |
5/5 |
76% |
24% |
52:48 |
| 3 |
(S,S)-DTB-Bn-Yanphos |
1:3 |
100 |
5/5 |
53% |
47% |
66:34 |
| 4 |
(R,R)-DTBM-Bn-Yanphos |
1:3 |
100 |
5/5 |
39% |
61% |
31:69 |
| 5 |
(R,S)-DTBM-Bn-Yanphos |
1:3 |
100 |
5/5 |
96% |
4% |
93.5:6.5 |
| 6 |
(R,S)-DTBM-Bn-Yanphos |
1:2 |
100 |
5/5 |
96% |
4% |
93:7 |
| 7 |
(R,S)-DTBM-Bn-Yanphos |
1:1.5 |
100 |
5/5 |
97% |
3% |
94:6 |
| 8 |
(R,S)-DTBM-Bn-Yanphos |
1:1.2 |
100 |
5/5 |
93% |
4% |
91:9 |
| 9 |
(R,S)-DTBM-Bn-Yanphos |
1:1.5 |
90 |
5/5 |
89% |
3% |
96:4 |
| 10 |
(R,S)-DTBM-Bn-Yanphos |
1:1.5 |
80 |
5/5 |
75% |
2% |
96:4 |
| 11 |
(R,S)-DTBM-Bn-Yanphos |
1:1.5 |
90 |
10/10 |
52% |
3% |
96:4 |
| 12 |
(R,S)-DTBM-Bn-Yanphos |
1:1.5 |
90 |
2.5/2.5 |
91% |
3% |
96:4 |
Table 2 Reaction scope of synthesis of chiral quaternary stereocenters via AHF
|
Reaction at 60 °C.
Using (R,R)-Ph-BPE as a ligand.
|
|
Conclusion
In summary, an α-quaternary amino acid was prepared via a Rh-catalyzed AHF reaction. Catalyzed by a rhodium complex, α-, β-, γ-, and δ-amino acids with an α-quaternary stereocenter could be prepared from simple starting materials. The regioselectivity and enantioselectivity in this transformation were satisfactory in each case. The Hammett study illustrated a high sensitivity of electronic properties of the olefin substituent towards the regioselectivity in AHF reactions. Based on the NMR spectroscopic data and DFT calculations, we depicted the chiral environment of the rhodium catalyst and built an enantioinduction model. This AHF approach renders a straightforward method to construct chiral quaternary aldehydes. We are looking forward to its wide application in synthetic chemistry.
Data availability
All data supporting the findings of this study are available within the article and its ESI.† The experimental procedures, characterization of the compounds and DFT calculations are available in the ESI.† The crystallographic data for compound 2p′ have been deposited at the Cambridge Crystallographic Data Centre, under deposition number CCDC 2121822.
Author contributions
D. Z. conducted all the experiments including the method development, exploration of the reaction scope and Hammett study. J. W. analyzed the structural features of the Rh/Yanphos complex and conducted the DFT calculations. J. W. conceived this project, designed the experiments and wrote the manuscript. X. Z. revised the manuscript and secured the funding. All the authors participated in the discussion and preparation of the manuscript.
Conflicts of interest
The authors declare no competing interests.
Abbreviations
| DIOP | 2,3-O-Isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane |
| NMR | Nuclear magnetic resonance |
| DFT | Density functional theory |
| er | Enantiomeric ratio |
| NOE | Nuclear overhauser effect |
Acknowledgements
We are grateful to Dr Fanping Huang (SUSTech) for the assistance in crystallography. This project was financially supported by Guangdong Provincial Key Laboratory of Catalysis (2020B121201002). The computational resources were supported on CHEM high-performance supercomputer clusters (CHEM-HPC) in SUSTech.
References
- J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593–4623 RSC.
- K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed.
- Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed.
- J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564–12580 CrossRef CAS PubMed.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- F. Agbossou, J.-F. Carpentier and A. Mortreux, Chem. Rev., 1995, 95, 2485–2506 CrossRef CAS.
- S. Chakrabortty, A. A. Almasalma and J. G. de Vries, Catal. Sci. Technol., 2021, 11, 5388–5411 RSC.
- B. Breit and W. Seiche, Synthesis, 2001, 2001, 0001–0036 CrossRef.
- A. I. M. Keulemans, A. Kwantes and T. van Bavel, Recl. Trav. Chim. Pays-Bas, 1948, 67, 298–308 CrossRef CAS.
- C. Toniolo, M. Crisma, F. Formaggio and C. Peggion, Pept. Sci., 2001, 60, 396–419 CrossRef CAS.
- C. Cativiela and M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry, 1998, 9, 3517–3599 CrossRef CAS.
- Y. Ohfune and T. Shinada, Eur. J. Org. Chem., 2005, 2005, 5127–5143 CrossRef.
- C. Cativiela and M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry, 2007, 18, 569–623 CrossRef CAS.
- A. E. Metz and M. C. Kozlowski, J. Org. Chem., 2015, 80, 1–7 CrossRef CAS PubMed.
- T. Boibessot, D. Bénimélis, P. Meffre and Z. Benfodda, Amino Acids, 2016, 48, 2081–2101 CrossRef CAS PubMed.
- R. A. Mosey, J. S. Fisk and J. J. Tepe, Tetrahedron: Asymmetry, 2008, 19, 2755–2762 CrossRef CAS.
- D. J. Leonard, J. W. Ward and J. Clayden, Nature, 2018, 562, 105–109 CrossRef CAS PubMed.
- C. Cativiela, M. Ordóñez and J. L. Viveros-Ceballos, Tetrahedron, 2020, 76, 130875 CrossRef CAS.
- D. Seebach, A. R. Sting and M. Hoffmann, Angew. Chem., Int. Ed., 1996, 35, 2708–2748 CrossRef CAS.
- M. Branca, S. Pena, R. Guillot, D. Gori, V. Alezra and C. Kouklovsky, J. Am. Chem. Soc., 2009, 131, 10711–10718 CrossRef CAS PubMed.
- S. Gladiali and L. Pinna, Tetrahedron: Asymmetry, 1990, 1, 693–696 CrossRef CAS.
- Y. Deng, H. Wang, Y. Sun and X. Wang, ACS Catal., 2015, 5, 6828–6837 CrossRef CAS.
- C. W. Lee and H. Alper, J. Org. Chem., 1995, 60, 499–503 CrossRef CAS.
- X. Wang and S. L. Buchwald, J. Org. Chem., 2013, 78, 3429–3433 CrossRef CAS PubMed.
- R. Tan, X. Zheng, B. Qu, C. A. Sader, K. R. Fandrick, C. H. Senanayake and X. Zhang, Org. Lett., 2016, 18, 3346–3349 CrossRef CAS PubMed.
- J. Eshon, F. Foarta, C. R. Landis and J. M. Schomaker, J. Org. Chem., 2018, 83, 10207–10220 CrossRef CAS PubMed.
- T. P. Clark, C. R. Landis, S. L. Freed, J. Klosin and K. A. Abboud, J. Am. Chem. Soc., 2005, 127, 5040–5042 CrossRef CAS PubMed.
- R. I. McDonald, G. W. Wong, R. P. Neupane, S. S. Stahl and C. R. Landis, J. Am. Chem. Soc., 2010, 132, 14027–14029 CrossRef CAS PubMed.
- E. R. Nelsen, A. C. Brezny and C. R. Landis, J. Am. Chem. Soc., 2015, 137, 14208–14219 CrossRef CAS PubMed.
- A. C. Brezny and C. R. Landis, J. Am. Chem. Soc., 2017, 139, 2778–2785 CrossRef CAS PubMed.
- X. Zhang, B. Cao, Y. Yan, S. Yu, B. Ji and X. Zhang, Chem.–Eur. J., 2010, 16, 871–877 CrossRef CAS PubMed.
- Y. Yan and X. Zhang, J. Am. Chem. Soc., 2006, 128, 7198–7202 CrossRef CAS PubMed.
- S. H. Chikkali, J. I. van der Vlugt and J. N. H. Reek, Coord. Chem. Rev., 2014, 262, 1–15 CrossRef CAS.
- X. Zhang, B. Cao, S. Yu and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 4047–4050 CrossRef CAS PubMed.
- C. Chen, S. Jin, Z. Zhang, B. Wei, H. Wang, K. Zhang, H. Lv, X.-Q. Dong and X. Zhang, J. Am. Chem. Soc., 2016, 138, 9017–9020 CrossRef CAS PubMed.
- C. You, B. Wei, X. Li, Y. Yang, Y. Liu, H. Lv and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 6511–6514 CrossRef CAS PubMed.
- C. You, S. Li, X. Li, J. Lan, Y. Yang, L. W. Chung, H. Lv and X. Zhang, J. Am. Chem. Soc., 2018, 140, 4977–4981 CrossRef CAS PubMed.
- C. You, S. Li, X. Li, H. Lv and X. Zhang, ACS Catal., 2019, 9, 8529–8533 CrossRef CAS.
- D. Zhang, C. You, X. Li, J. Wen and X. Zhang, Org. Lett., 2020, 22, 4523–4526 CrossRef CAS PubMed.
- S. Li, Z. Li, M. Li, L. He, X. Zhang and H. Lv, Nat. Comm., 2021, 12, 5279 CrossRef CAS PubMed.
-
P. W. N. M. van Leeuwen, C. P. Casey and G. T. Whiteker, Phosphines as ligands, in Rhodium Catalyzed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Springer Netherlands, Dordrecht, 2002, pp. 63–105 Search PubMed.
- G. R. Donowitz and G. L. Mandell, N. Engl. J. Med., 1988, 318, 490–500 CrossRef CAS PubMed.
- I. Ojima and F. Delaloge, Chem. Soc. Rev., 1997, 26, 377–386 RSC.
- L. M. Lima, B. N. M. d. Silva, G. Barbosa and E. J. Barreiro, Eur. J. Med. Chem., 2020, 208, 112829 CrossRef CAS PubMed.
- D. Obrecht, M. Altorfer, C. Lehmann, P. Schönholzer and K. Müller, J. Org. Chem., 1996, 61, 4080–4086 CrossRef CAS PubMed.
- D. B. Berkowitz, B. D. Charette, K. R. Karukurichi and J. M. McFadden, Tetrahedron: Asymmetry, 2006, 17, 869–882 CrossRef CAS PubMed.
- W. Yu, J. McConathy, J. Olson, V. M. Camp and M. M. Goodman, J. Med. Chem., 2007, 50, 6718–6721 CrossRef CAS PubMed.
- J. L. Bartels, C. Huang, A. Li, L. Yuan, K. Rich, J. McConathy and S. E. Lapi, J. Med. Chem., 2015, 58, 8542–8552 CrossRef CAS PubMed.
- M. L. Clarke and G. J. Roff, Chem.–Eur. J., 2006, 12, 7978–7986 CrossRef CAS PubMed.
- I. D. Gridnev and T. Imamoto, Chem. Commun., 2009, 7447–7464 RSC.
- P. A. Dub and J. C. Gordon, Nat. Rev. Chem., 2018, 2, 396–408 CrossRef CAS.
- Y. Dangat, S. Popli and R. B. Sunoj, J. Am. Chem. Soc., 2020, 142, 17079–17092 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab and
Xumu
Zhang
*ab and
Xumu
Zhang
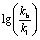 that demonstrates the relative rates in the formation of both regiomers. The slope (0.866) of this regression line demonstrated high sensitivity of the regioselectivity towards the electronic properties of the substituents. In a study of the influence of the substrate steric effect on the regioselectivity, no conclusive impact from the steric hindrance was found on the selectivity (see the ESI† for details). Therefore, the electronic features of the substituents attached on the C
that demonstrates the relative rates in the formation of both regiomers. The slope (0.866) of this regression line demonstrated high sensitivity of the regioselectivity towards the electronic properties of the substituents. In a study of the influence of the substrate steric effect on the regioselectivity, no conclusive impact from the steric hindrance was found on the selectivity (see the ESI† for details). Therefore, the electronic features of the substituents attached on the C