DOI:
10.1039/D2SC02077G
(Edge Article)
Chem. Sci., 2022,
13, 12440-12444
A light-gated regulation of the reaction site by a cucurbit[7]uril macrocycle†
Received
11th April 2022
, Accepted 10th October 2022
First published on 11th October 2022
Abstract
Competitive inhibition can be overcome by increasing the amount of catalyst in the reaction mixture. Here we present a pseudorotaxane system that circumvents this rule. A merocyanine inhibitor linked with the substrate obstructs the binding of the macrocyclic catalyst at the electrophilic reaction site preventing catalysis. Under UV light merocyanine is converted to the spiropyran form, losing its inhibition properties, thereby allowing the catalyst to bind the reaction center and promote the reaction. Moreover, when more than one nucleophile is present in the reaction mixture, the pseudorotaxane can scavenge a selected nucleophile and change the final product ratio. This work is a step forward in the development of new types of regulation in catalytic systems with remote control.
Introduction
Competitive inhibition is a widespread mechanism of regulation of catalytic activity, where an inhibitor molecule competes with a substrate for the active center.1 This type of inhibition can however be attenuated or completely stopped by increasing the amount of catalyst. This is due to the saturation of the competitive binder followed by the substrate–catalyst complex buildup. Here we present a way to switch off/on the inhibition regardless of the excess of the catalyst. For this, the substrate is merged with the inhibitor into one molecule. This design allows for the regulation not only the reaction rates but also product selectivity of external chemical reactions.
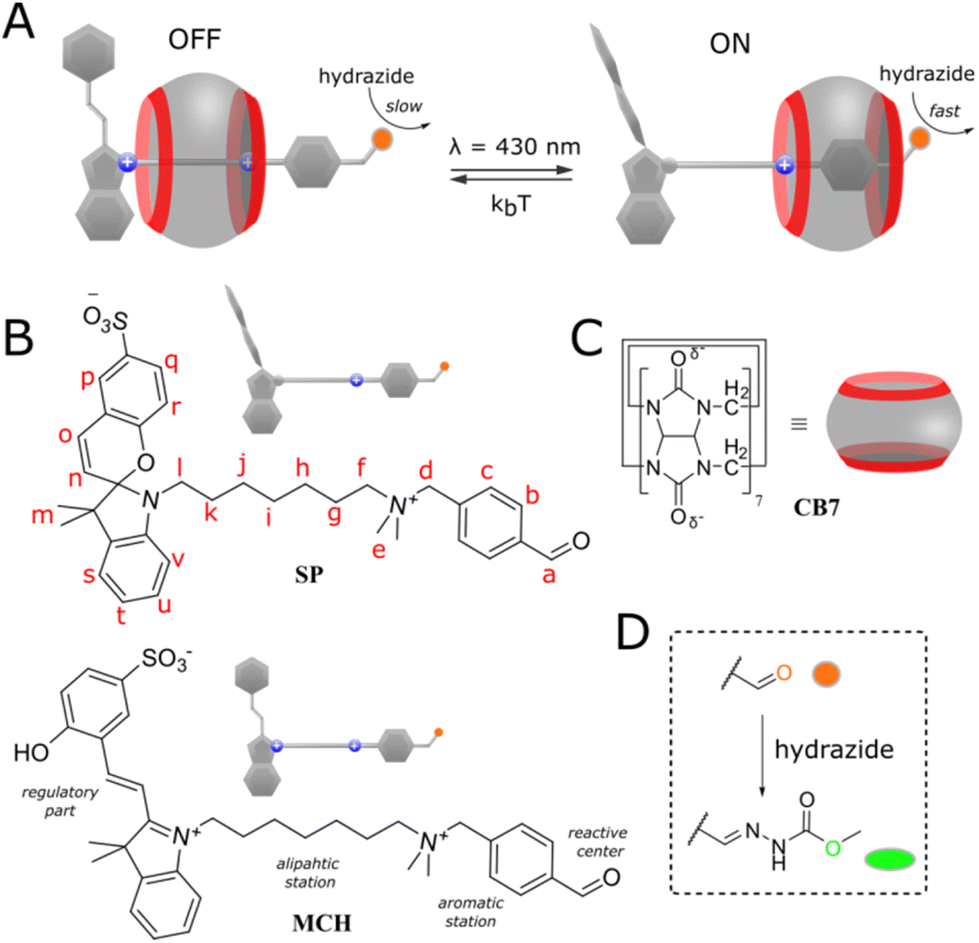
Our system is based on pseudorotaxane2 and depicted in Fig. 1A. It is composed of a cucurbit[7]uril macrocycle3 (Fig. 1C) having affinity to a molecular axis (Fig. 1B) containing two stations. The first station is benzaldehyde which also serves as a reaction site. The second heptyl station is terminated with spiropyran photoswitch4 as a regulator. Both stations are connected by a dimethylammonium group. The ammonium group confers solubility in water and keeps the macrocycle on the axle by the coulombic stabilization with partially negatively charged carbonyl rims. When the photoswitch is in the open merocyanine form (MCH, OFF state), the macrocycle binds preferentially the heptyl station due to the additional attraction with a positive charge on the indole ring, preventing the threading of the second macrocycle (for steric and electrostatic reasons), and therefore, has a limited influence on the condensation reaction of benzaldehyde with a nucleophile, i.e. hydrazide (Fig. 1D). Switching to the neutral spiropyran form (SP, ON state) cancels the electrostatic interaction of the macrocycle with the regulator and allows it to bind a more favorable benzaldehyde station, where it catalyzes the hydrazonation5 due to the stabilization of protonated reaction species.6 Details on the synthesis and isomerization behavior of the axle (UV-Vis spectra) can be found in the ESI (pp. S2–S22).†
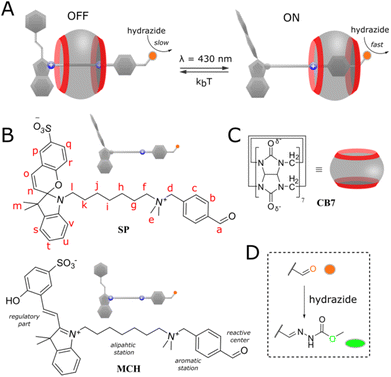 |
| | Fig. 1 (A) General presentation of the system; (B and C) chemical structures of the macrocycle and axle in different states; (D) condensation reaction occurring on the axle in the presence of methyl hydrazinocarboxylate. | |
Results and discussion
We began our study with the complexation study. The axle and CB7 in the dark (OFF state) were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at 5 mM concentration in acetonitrile-d3/D2O mixture with HCl added to keep the pD constant (≈3). NMR titrations showed the equimolar interaction and threading of the macrocycle on the axle (Fig. S32†). Since the direct determination of the association constant was impossible, displacement experiments using an ammonium benzaldehyde were carried out (pp. S23–S31, ESI†). This gave Ka = 8.8 × 104 ± 0.8 M−1 indicating high stability of the formed pseudorotaxane and that as little as 3% of it exists in the disassembled state (p. S26, ESI†). The detailed examination of the NMR spectra (Fig. 2A, top) showed that the macrocycle rests mostly on the aliphatic chain in close proximity to the stopper. This is evidenced by the upfield shifting of repeating methylene units, and downfield shifts of adjacent spiropyran protons. Upon illumination of the system with blue light (ON state), the macrocycle preferably docks to the benzaldehyde station with one rim located vis-à-vis the ammonium group, and the other out of the axle. This is manifested by upfield shifts of all protons up to the ammonium group, and downfield shift of a non-benzylic CH2 group next to it (Fig. 2A, bottom). The process is accompanied by a 4-fold decrease in the binding strength due to the cancellation of electrostatic attraction with indolium nitrogen atom. Note that not all shifts of the axle protons are consistent with the supposed position of the macrocycle. For instance, protons of the aromatic station remain shielded in either state. This indicates that the macrocycle performs the Brownian motion, and the apparent shift is the result of the average distribution of the macrocycle on both stations. It is estimated that the macrocycle in the ON state spends about 5 times more on the aromatic station than in the OFF state (p. S26, ESI†).
:1 ratio at 5 mM concentration in acetonitrile-d3/D2O mixture with HCl added to keep the pD constant (≈3). NMR titrations showed the equimolar interaction and threading of the macrocycle on the axle (Fig. S32†). Since the direct determination of the association constant was impossible, displacement experiments using an ammonium benzaldehyde were carried out (pp. S23–S31, ESI†). This gave Ka = 8.8 × 104 ± 0.8 M−1 indicating high stability of the formed pseudorotaxane and that as little as 3% of it exists in the disassembled state (p. S26, ESI†). The detailed examination of the NMR spectra (Fig. 2A, top) showed that the macrocycle rests mostly on the aliphatic chain in close proximity to the stopper. This is evidenced by the upfield shifting of repeating methylene units, and downfield shifts of adjacent spiropyran protons. Upon illumination of the system with blue light (ON state), the macrocycle preferably docks to the benzaldehyde station with one rim located vis-à-vis the ammonium group, and the other out of the axle. This is manifested by upfield shifts of all protons up to the ammonium group, and downfield shift of a non-benzylic CH2 group next to it (Fig. 2A, bottom). The process is accompanied by a 4-fold decrease in the binding strength due to the cancellation of electrostatic attraction with indolium nitrogen atom. Note that not all shifts of the axle protons are consistent with the supposed position of the macrocycle. For instance, protons of the aromatic station remain shielded in either state. This indicates that the macrocycle performs the Brownian motion, and the apparent shift is the result of the average distribution of the macrocycle on both stations. It is estimated that the macrocycle in the ON state spends about 5 times more on the aromatic station than in the OFF state (p. S26, ESI†).
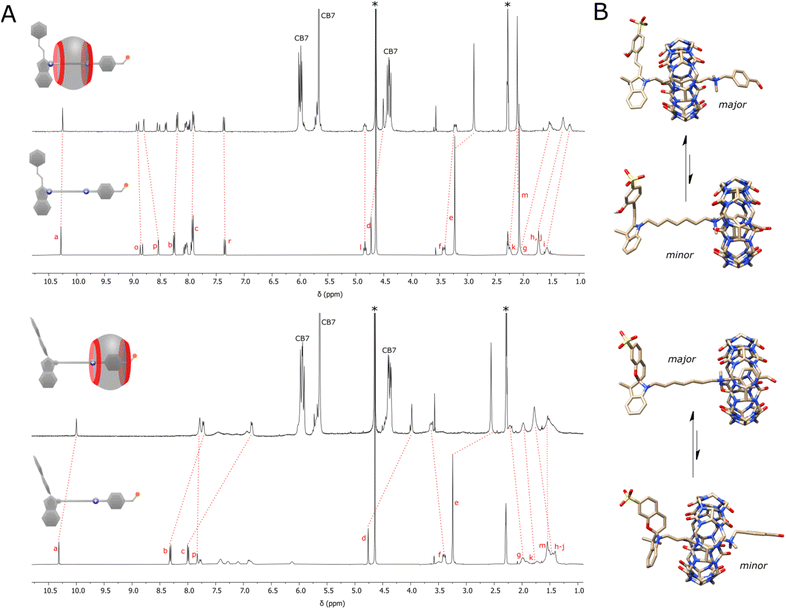 |
| | Fig. 2 (A) 1H NMR spectra showing the shifts of the proton signals of the axle (5 mM) alone and after the addition of the macrocycle (5 mM) in the dark (top) and under constant light illumination (bottom), acetonitrile-d3/D2O mixture (v/v ≈ 40:60), pD ≈ 3, 298 K. Residual solvents (water and acetonitrile) are denoted with asterisks. Lettering of signal corresponds to that shown in Fig. 1A. (B) Illustrations obtained after semi-empirical optimization at the PM6 level with the D3 dispersive term. | |
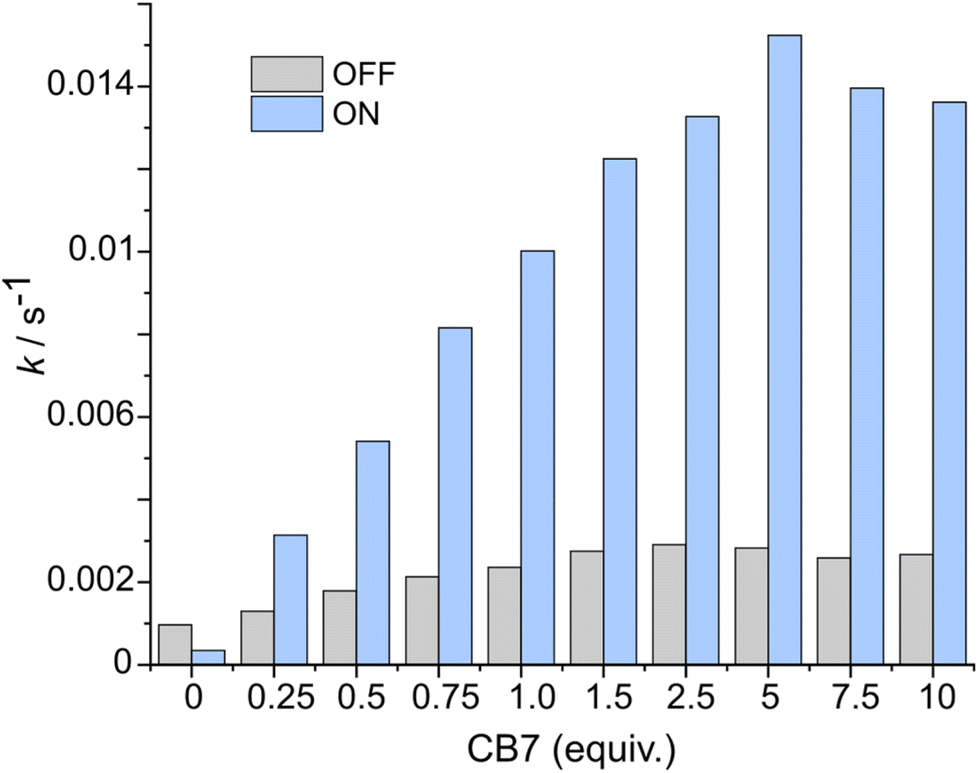
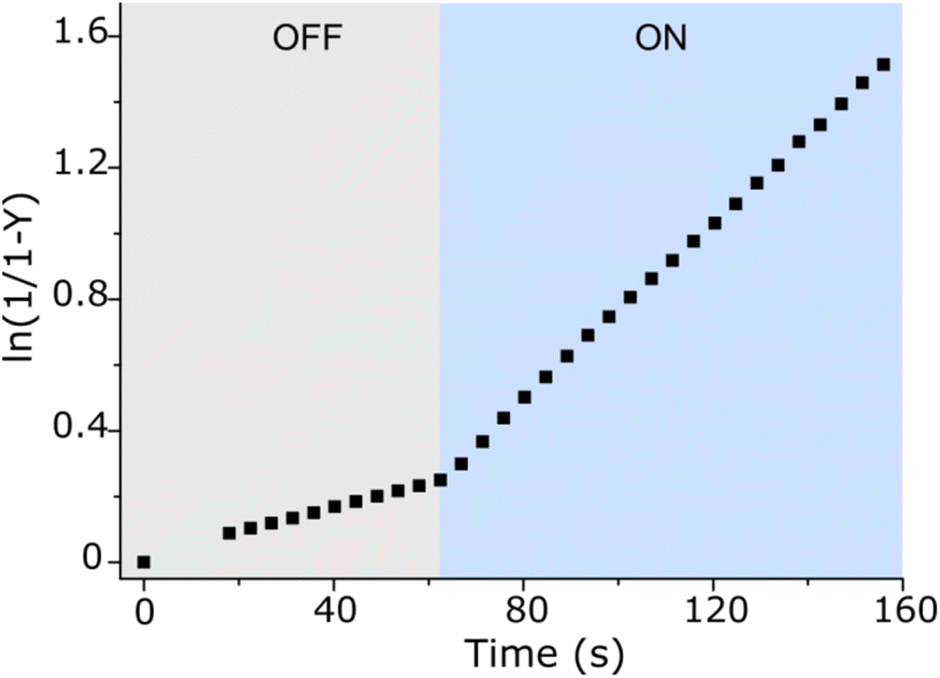
After the study of the interaction patterns of the axle with the macrocycle, we started catalytic experiments. Accordingly, we administered methyl hydrazinocarboxylate in 20 equiv. The employment of the excess of hydrazide (pKa ≈ 3.2, Fig. S39, ESI†) pursued three goals: (i) buffering of the system as photoswitching causes significant alteration of acidity;7 under these conditions (pH ≈ 3), hydrazonation, which is pH-sensitive,5 is neither too fast nor too slow, and the spiropyran ring does not undergo degradation; (ii) shifting the reaction equilibrium towards the hydrazonation product due to the reversibility of the process; (iii) downgrading the reaction order from 2° to pseudo 1° in respect to the axle, thereby facilitating the description of the reaction kinetics. Catalysis was studied by UV-Vis spectroscopy at different axle-macrocycle ratios under light and dark conditions (Fig. 3, pp. S32–S34, ESI†). As can be seen, in the OFF state, up to ca. 1 equiv. of CB7, the reaction rate slightly increased due to the partial formation of an active complex at the benzaldehyde station; however, when more CB7 was employed, the reaction rate remained practically unchanged due to the saturation of the aliphatic station with the macrocycle, and the inability of the second macrocycle to thread onto the axle. The distinct behavior of the system was observed under irradiation (ON state). The reaction rate increased steadily with increasing amount of CB7 until the stoppage at a 5:1 macrocycle-axle ratio, which corresponds to the presence 80% of the assembled pseudorotaxane (Table S2, SI†). At this ratio, the reaction sped up about 5.4 times compared to the dark (Table S3, ESI†). This is practically the same value we obtained for the relative population of the macrocycle on the aromatic station in each state (vide supra). That is, catalysis by the macrocycle occurs only when it resides on the aromatic station, as originally planned. Control experiments without macrocycle confirm this (Fig. 3, 0 equiv. of CB7), showing that the reaction in the dark is even faster, which is likely due to the intramolecular stabilization of protonated aldehyde by the sulfonic group.8 Importantly, the acceleration of the reaction can be done at any time (Fig. 4). Furthermore, after the reaction is complete, the system can be readily reinstated. It is enough to lower the pH (Fig. S42, ESI†), as the hydrazide detaches itself.
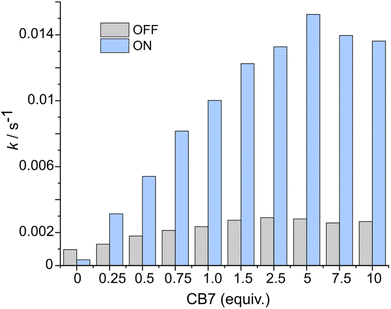 |
| | Fig. 3 Comparison of the rates of the hydrazonation of the axle (75 μM) at different amounts of CB7 in the dark (grey bars) and under constant light irradiation (blue bars), acetonitrile/H2O mixture (v/v ≈ 40:60), pH ≈ 3, 298 K. The determination of the rates is described in pp. S34–S35, ESI.† | |
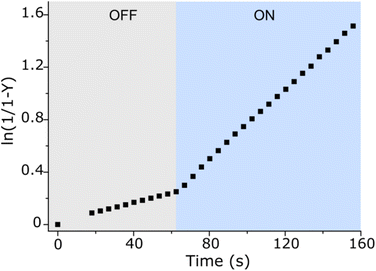 |
| | Fig. 4 Acceleration of the hydrazonation reaction under constant light irradiation (blue region) after the initial ≈60 s stay of the system (100 μM) in the dark (grey region), acetonitrile/H2O mixture (v/v ≈ 40:60), pH ≈ 3, 298 K. Note that Y stands for the product yield, and the first datapoint in the blue region corresponds to the steady state of the photoswitch. | |
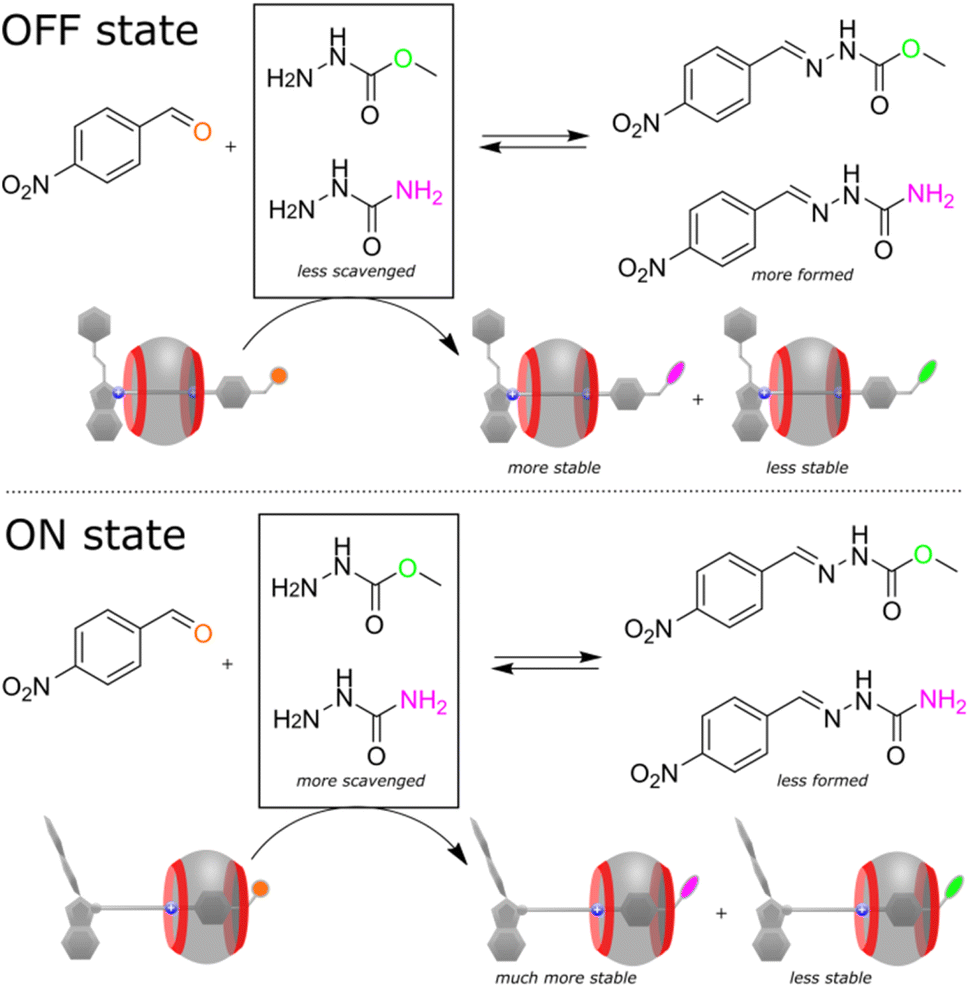
Considering the electrophilic character and tuneable reactivity of the pseudorotaxane, we tested it for scavenging nucleophiles from the reaction mixtures to affect the reaction outcomes. Recently, Hecht and co-workers demonstrated that a reacting diarylethene photoswitch can change the yield of a chemical reaction.9 We went further using our switching system to alter the reaction selectivity. As a proof-of-principle, we have chosen a condensation reaction between 4-nitrobenzaldehyde and the mixture of two hydrazides (the used early in this study methyl hydrazinocarboxylate and the additional semicarbazide, Fig. 5). In the presence of the disabled pseudorotaxane (in the dark), the reaction proceeds non-selectively affording the mixture of two hydrazones in a ratio of 2.6:1. However, after the activation with light, the axle preferentially reacts with semicarbazide, rendering the methyl hydrazine-carboxylate derivative as the predominant product (12:1). To unravel the mechanism of the selectivity change, we conducted a set of experiments. NMR showed that 4-nitrobenzaldehyde practically does not interact with CB7 (Fig. S45, ESI†), that is, the observed effect is solely the result of the pseudorotaxane operation. Further investigation revealed that the semicarbazide product of the axle binds CB7 slightly differently (Fig. S46, ESI†) and affords probably a more stable complex than one produced from methyl hydrazinocarboxylate. This ultimately leads to the depletion of the reaction mixture into semicarbazide, and the selective reaction of 4-nitrobenzaldehyde with the resulted excess of methyl hydrazinecarboxylate. In other words, the pseudorotaxane toggled by light is capable of shifting the thermodynamic equilibrium of two concurrent chemical reactions, which are inherently non-photoresponsive.
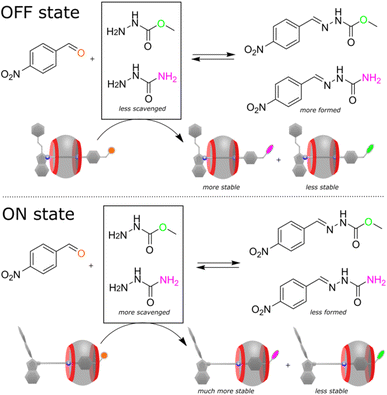 |
| | Fig. 5 Reaction of 4-nitrobenzaldehyde (1 equiv., 3.6 mM) with the mixture of two hydrazides (0.75 equiv. each) in the presence of the pseudorotaxane (1 equiv.), acetonitrile-d3/D2O mixture (v/v ≈ 40:60), pD ≈ 3, 298 K. In the OFF state, since CB7 ring spends more time on the distal heptyl station, the stabilities of the hydrazone products of the pseudorotaxane are comparable, resulting in lower consumption of semicarbazide by the pseudorotaxane and higher yield of the nitrobenzaldehyde product. Upon light irradiation (ON state), the interaction of CB7 ring with the semicarbazide residue increases the stability of the corresponding pseudorotaxane derivative, reducing the amount of semicarbazide and the nitrobenzaldehyde product. NMR spectra of the reaction mixtures and the substrate/product distributions are shown in the ESI (Fig. S43–S44, Table S4).† | |
Conclusions
In summary, we developed a new type of regulation of supramolecular catalysis. Photoswitchable inhibitor linked with substrate into one molecule impedes the increase in the reaction rate upon increasing the amount of catalyst. After deactivation of the inhibitor with light, the system starts to exhibit the typical catalysis enhancement until the saturation of the reaction site. Importantly, the prepared pseudorotaxane can regulate not only self-reaction but also the outcome of external reactions. When exposed to light it scavenges a selected nucleophile and improves the product selectivity. Ongoing research in our laboratory is aimed at improving and adapting the presented system for various purposes.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†
Author contributions
N. R. developed the model. V. S. conceptualized the project and wrote the manuscript. Both authors discussed the results and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financed from the National Science Centre of Poland (grant OPUS 18 no. 2019/35/B/ST4/01758). Computations were made possible thanks to PLGrid Infrastructure. The authors are also grateful to Grzegorz Sobczak for preparing the setup that allows kinetic measurements under irradiation.
References
-
J. E. House, Principles of Chemical Kinetics, Academic Press, Burlington, MA, USA, 2007 Search PubMed.
- For the use of (pseudo)rotaxanes in the regulation of catalysis see:
(a) V. Blanco, D. A. Leigh, U. Lewandowska, B. Lewandowski and V. Marcos, J. Am. Chem. Soc., 2014, 136, 15775–15780 CrossRef CAS PubMed;
(b) V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS PubMed;
(c) P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Nature, 2003, 424, 915–918 CrossRef CAS PubMed;
(d) K. Eichstaedt, J. Jaramillo-Garcia, D. A. Leigh, V. Marcos, S. Pisano and T. A. Singleton, J. Am. Chem. Soc., 2017, 139, 9376–9381 CrossRef CAS PubMed;
(e) J. Echavarren, M. A. Y. Gall, A. Haertsch, D. A. Leigh, J. T. J. Spence, D. J. Tetlow and C. Tian, J. Am. Chem. Soc., 2021, 143, 5158–5165 CrossRef CAS PubMed;
(f) M. Galli, J. E. M. Lewis and S. M. Goldup, Angew. Chem., Int. Ed., 2015, 54, 13545–13549 CrossRef CAS PubMed;
(g) S. Hoekman, M. O. Kitching, D. A. Leigh, M. Papmeyer and D. Roke, J. Am. Chem. Soc., 2015, 137, 7656–7659 CrossRef CAS PubMed;
(h) J. Beswick, V. Blanco, G. De Bo, D. A. Leigh, U. Lewandowska, B. Lewandowski and K. Mishiro, Chem. Sci., 2015, 6, 140–143 RSC;
(i) A. Martinez-Cuezva, C. Lopez-Leonardo, D. Bautista, M. Alajarin and J. Berna, J. Am. Chem. Soc., 2016, 138, 8726–8729 CrossRef CAS PubMed;
(j) Y. Cakmak, S. Erbas-Cakmak and D. A. Leigh, J. Am. Chem. Soc., 2016, 138, 1749–1751 CrossRef CAS PubMed;
(k) A. Martinez-Cuezva, A. Saura-Sanmartin, T. Nicolas-Garcia, C. Navarro, R.-A. Orenes, M. Alajarin and J. Berna, Chem. Sci., 2017, 8, 3775–3780 RSC;
(l) M. Dommaschk, J. Echavarren, D. A. Leigh, V. Marcos and T. A. Singleton, Angew. Chem., Int. Ed., 2019, 58, 14955–14958 CrossRef CAS PubMed;
(m) F. Modicom, E. M. G. Jamieson, E. Rochette and S. M. Goldup, Angew. Chem., Int. Ed., 2019, 58, 3875–3879 CrossRef CAS PubMed;
(n) M. Calles, J. Puigcerver, D. A. Alonso, M. Alajarin, A. Martinez-Cuezva and J. Berna, Chem. Sci., 2020, 11, 3629–3635 RSC;
(o) S. C. Rajappan, D. R. McCarthy, J. P. Campbell, J. B. Ferrell, M. Sharafi, O. Ambrozaite, J. Li and S. T. Schneebeli, Angew. Chem., Int. Ed., 2020, 59, 16668–16674 CrossRef CAS PubMed;
(p) C. Biagini, S. D. P. Fielden, D. A. Leigh, F. Schaufelberger, S. Di Stefano and D. Thomas, Angew. Chem., Int. Ed., 2019, 58, 9876–9880 CrossRef CAS PubMed;
(q) S. Borsley, D. A. Leigh and B. M. W. Roberts, J. Am. Chem. Soc., 2021, 143, 4414–4420 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- N. Rad, O. Danylyuk and V. Sashuk, Angew. Chem., Int. Ed., 2019, 58, 11340–11343 CrossRef CAS PubMed.
-
(a) V. Sashuk, H. Butkiewicz, M. Fiałkowski and O. Danylyuk, Chem. Commun., 2016, 52, 4191–4194 RSC;
(b) N. Rad and V. Sashuk, Chem. Commun., 2022, 58, 5249–5252 RSC.
-
(a) Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699–14703 CrossRef CAS PubMed;
(b) C. Berton, D. M. Busiello, S. Zamuner, R. Scopelliti, F. Fadaei-Tirani, K. Severin and C. Pezzato, Angew. Chem., Int. Ed., 2021, 58, 21737–21740 CrossRef PubMed;
(c) L. Wimberger, S. K. K. Prasad, M. D. Peeks, J. Andréasson, T. W. Schmidt and J. E. Beves, J. Am. Chem. Soc., 2021, 143, 20758–20768 CrossRef CAS PubMed.
- E. T. Kool, D.-H. Park and P. Crisalli, J. Am. Chem. Soc., 2013, 135, 17663–17666 CrossRef CAS PubMed.
- M. Kathan, F. Eisenreich, C. Jurissek, A. Dallmann, J. Gurke and S. Hecht, Nat. Chem., 2018, 10, 1031–1036 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization, isomerization, binding and kinetic studies. See DOI: https://doi.org/10.1039/d2sc02077g |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Volodymyr
Sashuk
and
Volodymyr
Sashuk