
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating the excited state chirality to fabricate high-performance-solid-state circularly polarized luminescence materials†
Jianlei
Han
 a,
Yonghong
Shi
ab,
Xue
Jin
a,
Xuefeng
Yang
ab and
Pengfei
Duan
*ab
a,
Yonghong
Shi
ab,
Xue
Jin
a,
Xuefeng
Yang
ab and
Pengfei
Duan
*ab
aCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, National Center for Nanoscience and Technology (NCNST), No. 11 ZhongGuanCun BeiYiTiao, Beijing 100190, P. R. China. E-mail: duanpf@nanoctr.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 27th April 2022
Abstract
Developing solid-state materials and greatly improving the luminescence dissymmetry factors (glum) are the key issues for the future oriented practical application in the field of circularly polarized luminescence (CPL). However, most of the solid-state CPL-active materials suffer from aggregation caused emission quenching and relatively small glum values, which intensively restrict the development and application. In this work, high-performance CPL-active solid-state materials were achieved by regulating the excited state chirality of a series of bi-pyrene based chiral emitters. Due to the reversible mechanochromic luminescence under external stimuli, their excited state chirality can also be switched. It was found that the pristine amorphous powder possessed weak but obvious chiroptical properties because of the inherently chiral structures. Mechanical grinding could switch the fluorescence color and eliminate the CPL activity. Subsequently, by carrying out solvent fumigation, instant crystallization with well-defined microcrystal formation occurred, which could activate the CPL emission. Due to the chiral supramolecular arrangement of chromophores in the crystalline state, the resulting excimer emission in microcrystals showed chirality amplification not only in the excited state but also in the ground state. These findings not only provide a new method to fabricate high-performance CPL-active solid-state materials, but also clarify the chirality origin of pyrene-excimer-based chiral luminophores in various states which showed the importance of CPL as a probe of excited state chirality.
Introduction
Chiral materials with circularly polarized luminescence (CPL) have been receiving increasing attention due to their unique optical properties.1 They can be used for many potential applications such as display devices,2–5 information encryption,6,7 chiral recognition,8–10 and asymmetric photocatalysis.11–13 Large luminescence dissymmetry factors (glum) are desired for their practical application. However, chiral lanthanide complexes with the largest glum values suffered from extremely low emission efficiency, which greatly restricted their practical applications.14 In recent years, more attention has been paid to CPL-active organic molecules due to their high luminescence efficiency, tailored luminescence wavelength, and excellent processability.15–18 A variety of CPL-active organic molecules have been developed to date. Unfortunately, it is still difficult to make a breakthrough in the core parameter of the glum value. It turns out to be an urgent task to boost the glum value in pure organic compounds. On the other hand, considering that most of the practical applications of CPL-active materials require solid-state materials, constructing highly efficient CPL-active solid-state materials is also important in this field. To overcome the two “pain points”, to date, the strategy of integrating the concept of aggregation-induced emission with chiral moieties could effectively solve the above problems.19–22 Still, solid-state CPL-active materials possessing a large glum value and high emission efficiency are very rare.23–27As CPL senses conformational and configurational information of chiral molecules and materials in the excited state, modulating the excited state chirality in turn would greatly affect CPL. In this work, by taking advantage of excimer formation, highly emissive characteristics in the solid state, and the reversible mechanochromic luminescence under external stimuli,28,29 we demonstrate the excellent CPL performance of chiral bi-pyrene compounds in the solid state. Here, a series of bi-pyrene chromophore based chiral emitters are designed and synthesized by introducing both axial and planar chiral moieties. Highly efficient fluorescence quantum yield (∼0.5) and CPL performance with glum values of up to 10−2 are realized in the solid state. It was found that short distance ordering of pyrene chromophores in the amorphous state could result in weak CPL activity, while in situ instant crystallization will significantly boost the CPL performance (Fig. 1).
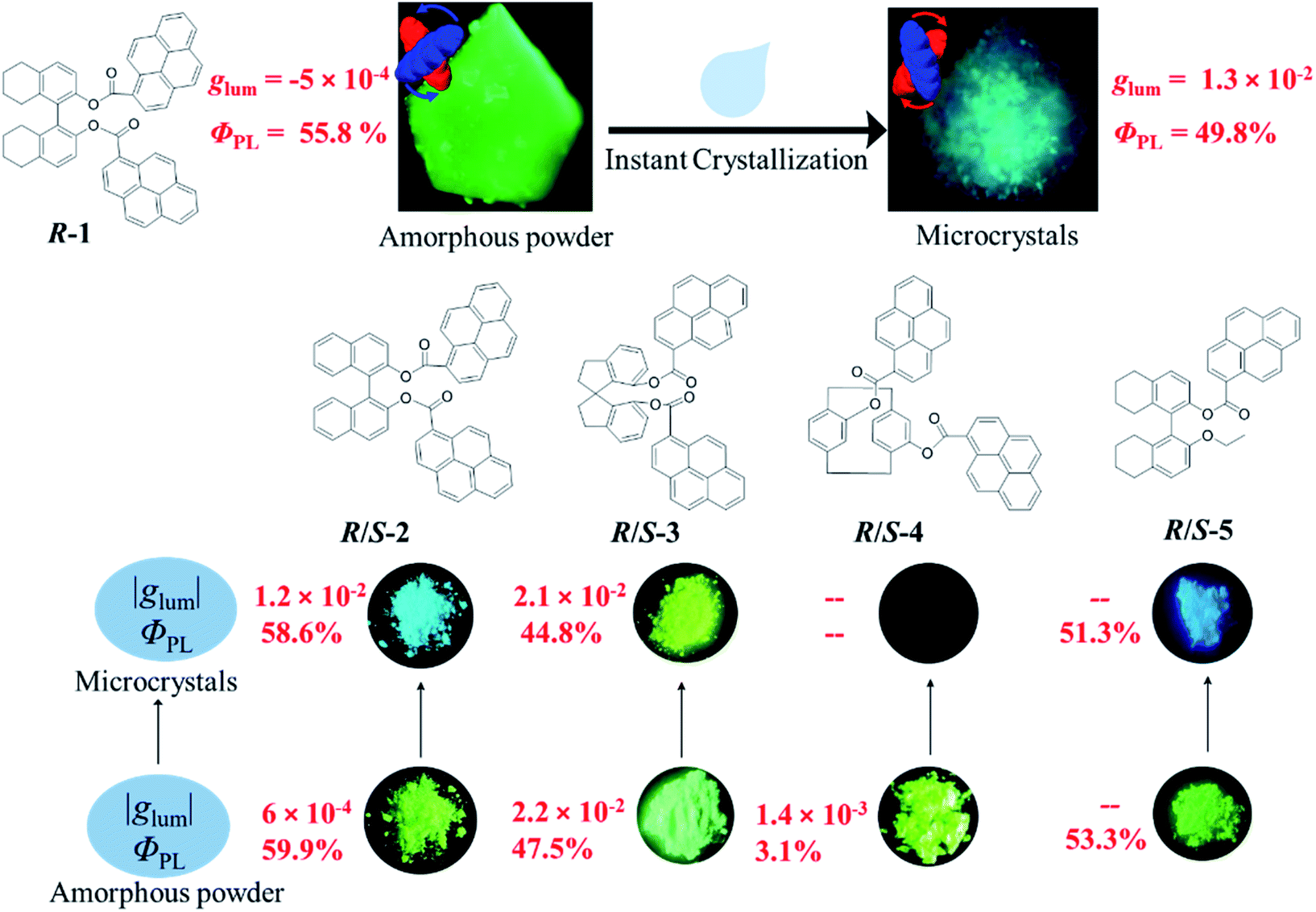 | ||
| Fig. 1 Chemical structures and emission photographs of pristine samples and the microcrystalline states of compounds 1–5. | ||
The excellent chiroptical performance originates from chiral supramolecular arrangement of chromophores in the crystalline state compared with the local ordered structure in the amorphous state. Additionally, grinding or scrubbing can completely destroy the ordered structure, shutting off the CPL emission. Interestingly, by carrying out solvent fuming (volatile solvents), in situ instant crystallization occurred, exhibiting strong CPL emission. Two static excimer states were found in the crystalline and amorphous states, which exhibit quite different excited state chiralities. Pre-associated intermolecular π dimers in the crystalline state are prone to form excimers with blue emission, while another static excimer exists in the amorphous state exhibiting greenish-yellow emission due to the close proximity of pyrene moieties. In addition, the excited state chirality of the dynamic excimer in concentrated solution was totally different from that of the static excimer in the solid state, though they shared the same photoluminescence (PL) behavior, which highlighted the importance of CPL as a probe of excited state chirality. Through the in situ instant crystallization process, the excited state chirality of the bi-pyrene emitters has been thoroughly modulated and investigated.
Results
The chiral bi-pyrene compounds were directly synthesized through the condensation reaction between 1-pyrenecarboxylic acid and the corresponding chiral dihydroxy compounds (Fig. 1 and the ESI†). Yellowish pristine samples were obtained for 1, 2, and 4 when concentrated in dichloromethane which was quite different from the white samples obtained after purification by using column chromatography on silica gel with an eluent of ethyl acetate and petroleum ether (Fig. 2a), while yellow sandy powder was observed for 3. For the following discussion, 1 was taken as an example. The white sample of 1 emitted sky-blue light, while greenish-yellow light was observed from the pristine powder under a UV-365 nm lamp (Fig. 2a). As color reflects the absorption spectral features in the visible region, the pristine sample showed a broad absorption peak beyond 500 nm, while a narrow peak was observed for the white sample (Fig. S1†), indicative of different molecular arrangements and/or intermolecular interactions. Compared with the dilute solution, which exhibited isolated monomer absorption with vibronic bands, both the solid samples exhibited broadened, structure-less red-shifted absorption spectra (Fig. S1†) resulting from strong coupling between chromophores. Then, the normalized PL spectra of these two samples were collected as displayed in Fig. 2b. The pristine sample showed emission at 507 nm, and the crystal sample exhibited a slightly broader peak ranging from 400 to 650 nm with a maximum wavelength of 472 nm. The fluorescence quantum yields of the pristine sample and microcrystals were determined to be 55.8% and 49.8% with the corresponding fluorescence lifetimes of 29.5 s and 33.9 s, respectively. The long fluorescence lifetimes and broad structure-less emission band indicated typical excimer emission of pyrene in both states.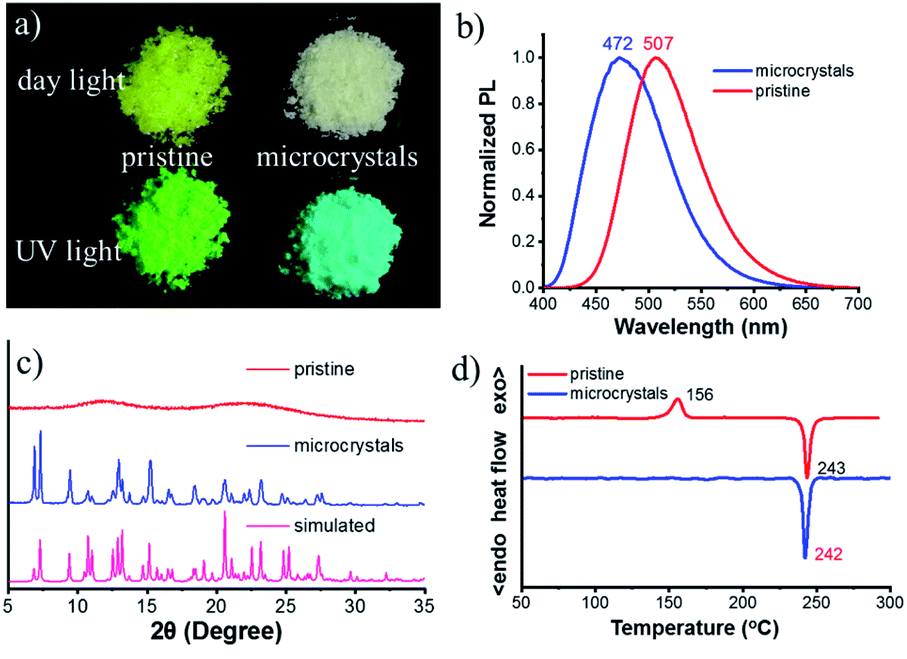 | ||
| Fig. 2 (a) Photographs of the pristine sample and microcrystals of S-1 under day light and UV irradiation (365 nm). (b) Normalized PL spectra of the pristine sample and microcrystals. (c) XRD patterns of the pristine sample, and microcrystals and the simulated pattern of single crystals. (d) DSC thermograms of the pristine sample and microcrystals. | ||
Interestingly, when ethyl acetate was added to S-1, part of the yellowish solid was dissolved, instantly accompanied by the appearance of a colorless little crystal-like solid (Fig. S2†). To slow down this transformation process, alcohol instead of ethyl acetate was used so that the process can be recorded by using a fluorescence microscope (Fig. 3, Movie S1† and the process induced by ethyl acetate/alcohol under an optical microscope is shown in Movie S2†). In the beginning, a small piece of the pentagonal block was used from the pristine sample (Fig. 3a and c). The sample was transparent, however, it turned out to be amorphous as shown by powder X-ray diffraction (PXRD) patterns (Fig. 2c) and polarized optical microscope (POM) images (Fig. S3†). After the addition of a drop of alcohol, thorn-like flakes with sky-blue light appeared on the edge of the green block in about 70 s (Fig. 3d). Gradually, more and more flakes sprang up and covered the entire surface (Fig. 3e). However, some green light can still be observed from the inner side with a pentagonal outline (Fig. 3f and g) due to the slow penetration of alcohol. In the end, the pentagonal block became thousands of fragments and the pentagonal outline persisted (Fig. 3b and h) (ethyl acetate induced transformation is shown in Fig. S4 and Movie S3†).
 | ||
| Fig. 3 Optical microscopy images of the pristine amorphous sample before solvent induced transformation (a) and microcrystals obtained after the transformation (b). (c–h) Fluorescence microscopy images of the crystallization process of the pristine sample with the aid of alcohol, which can slow down the crystallization speed. Fluorescence microscopy images of the initial state (c), at 70 s (d), 120 s (e), 120 s (f), 120 s (g), and final state (190 s). Scale bars, 20 μm (fluorescence microscopy images were from screenshots of Movie S1†). | ||
The white sample turned out to be crystalline (Fig. S3b, d and S5†) and its PXRD pattern accorded well with that of the simulated one based on crystal data (Fig. 2c). Next, thermal properties of these samples were determined by thermogravimetric analysis (TGA) (Fig. S6†) and differential scanning calorimetry (DSC) (Fig. 2d). The TGA results revealed that the onset temperature with 5% weight loss (Td) of these two samples was very similar (about 422 °C). DSC curves showed a thermo-induced phase transition at about 156 °C with an enthalpy-relaxation peak, which indicated that the amorphous phase crystallized upon heating. Fig. S7† showed that transformation from the amorphous state to the crystalline state could be realized by heating at 150 °C for 15 min, and the crystalline states from different transformation processes shared the same PL spectra and PXRD profiles (Fig. S8†).
Single crystals suitable for X-ray diffraction were obtained by slow solvent evaporation from the ethyl acetate/dichloromethane solution at room temperature. The crystal structure of S-1 is shown in Fig. S9.† The dihedral angle between the two naphthyl rings is 85.96° and the two pyrene moieties stretched away from each other with a dihedral angle of 78.26°, probably due to the steric repulsion and dipole–dipole interactions between the two ester groups. This also indicated that there are almost no intramolecular interactions between the two pyrene chromophores, which prevents the formation of the intramolecular excimer. However, intermolecular π–π interactions between pyrene rings on different molecules were observed along the b axis (3.404 Å and 3.427 Å, Fig. 4a), together with C–H⋯π interactions leading to molecular columns (Fig. S10†). These columns are brought together in the ac plane by weak C–H⋯π interactions (Fig. S11†) and form a herringbone array (Fig. 4b). This molecular packing pattern is prone to static excimer30 formation in the excited state, and even in the ground state, resulting in sky-blue emission at 470 nm (Fig. S12†), which was supported by fluorescence lifetime measurements (Fig. S13†).
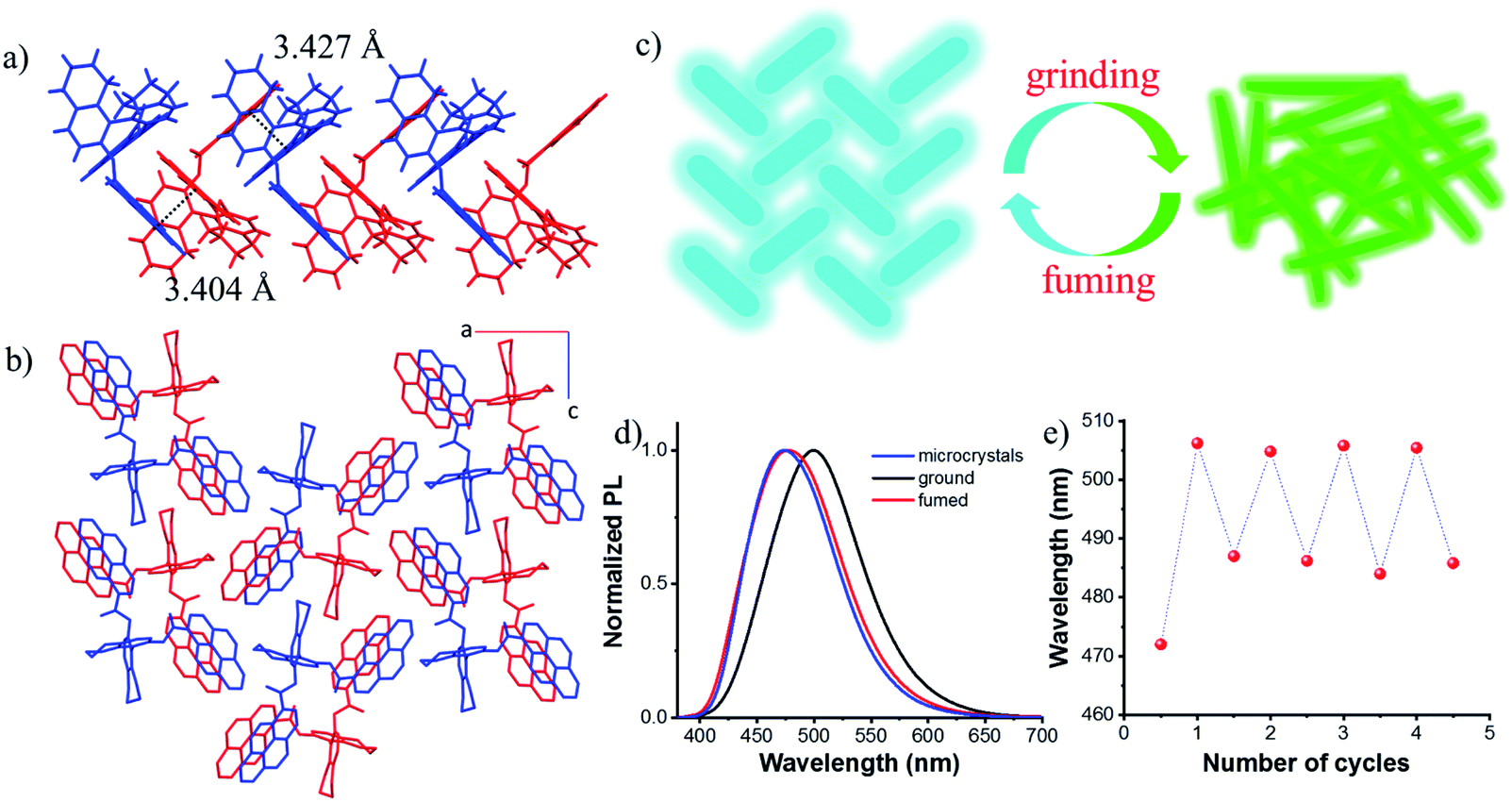 | ||
| Fig. 4 Intermolecular interactions (a) and the molecular packing structure (b) of S-1 (hydrogen atoms were omitted for clarity). (c) Proposed mechanochromic luminescence mechanism. (d) Normalized PL spectra of microcrystals, and ground and fumed samples. (e) Reversible switching of emission of S-1 by grinding/fuming cycles (λex = 360 nm). | ||
Due to the twisted conformation and weak π–π interactions, the molecular packing is relatively loose. Thus, crystals can be destroyed by external pressure. As shown in Fig. S14 and S15,† after grinding, the white crystals turned yellow (Fig. S14†), and the blue luminescence changed to intense greenish-yellow luminescence simultaneously. When exposed to ethyl acetate vapors, the ground powder recovered to the original blue luminescent solid (Fig. S15d†). Further grinding the blue luminescent solid afforded the greenish-yellow luminescence again (Fig. S15e†), and the blue luminescence would return with the help of drops of ethyl acetate (Fig. S15f†). PXRD patterns are shown in Fig. S16.† Sharp and intense diffraction peaks of the crystalline state disappeared after grinding, though some weak peaks remained due to the irregular and incomplete grinding. After fuming with ethyl acetate, a lot of sharp peaks reappeared which agreed well with those of microcrystals. It can be concluded that the amorphous sample is metastable and a stable crystalline state could be achieved easily by recrystallization either from solvent fuming or thermal annealing treatment as illustrated in Fig. 4c. After grinding, stronger intramolecular or intermolecular π–π interactions occurred between pyrene rings, enabling red-shifted emission from blue color to greenish-yellow color (Fig. 4d), thus, the excited state can be regulated. This reversible fluorescence change could be repeated many times due to the non-destructive nature of the grinding stimulus (Fig. 4e).
Almost the same molecular stacking arrangement was observed for S-2 which also belongs to an orthorhombic space group P212121 because of the same axial skeleton (Fig. S17†). Grinding and fuming induced PL and morphology switching properties are shown in Fig. S18.† The ground and fumed samples exhibited a high QY of 60% and 59%, respectively (Table S1†). Unlike the other bi-pyrene compounds, the yellow sandy powder of R-3 turned out to be in the crystalline state, which exhibited emission at 505 nm. After grinding, the amorphous state showed emission at 510 nm, while, a new state was observed when fumed with dichloromethane (Fig. S19†). Though there weren't large changes in the PL spectra for different states of R-3, clear distinctions in the emission color can be seen by the naked eye anyway (Fig. S19†). A butterfly-like structure was observed for R-3, with two wings (pyrene rings) spreading and stacking linearly with adjacent molecules (Fig. S20†). However, very weak emission at 465 nm was observed for the R-4 crystal, which cannot be seen by the naked eye (Fig. S21†). However, a quantum yield of 3% was observed for the ground sample. The stacking structure of the R-4 crystal was determined to be monoclinic, space group P21, and contained two discrete molecules (Fig. S22†). To explore the origin of poor emission of compound R-4, the π–π distance and the π–π overlapped area of pyrene in crystals were determined (Table S2†). It can be seen that a similar overlapped area (63%) was observed in the dimers of R-3 and R-4, while the π–π distance of R-3 is shorter than that of R-4. However, the QY of R-3 is very high compared with that of R-4. So, it's not the crystal stacking structure that accounts for the poor emission behavior. The para-cyclophane structure might be the reason, where para-cyclophane behaves as an electron acceptor, inducing transannular, intramolecular electron transfer, thus quenching the emission.31,32
Next, chiroptical properties were investigated. The ground state chirality cannot be detected for the pristine samples due to the strong scattering of powder. The R-1 pristine sample displayed right-handed circularly polarized luminescence and S-1 displayed the left-handed one with the calculated |glum| value of 5 × 10−4 (Fig. S23†). However, after grinding, no CPL signal can be detected (Fig. S24b†), which might be caused by the conformational change of the octahydro-binaphthyl skeleton33,34 and/or disturbance of ordering of chromophores. Grinding induced complete disorder of the molecular conformation, and circular dichroism (CD) signals were not detected either (Fig. S24a†), similar to the aggregation-annihilated circular dichroism effect.35,36 Interestingly, enhanced CD and CPL were observed for the crystalline state (Fig. 5a and b) after instant crystallization by solvent fuming37,38 (Fig. S25†), while the glum value was determined to be 0.013 at 470 nm. Regular arrangement or chiral supramolecular arrangement of chromophores in the crystalline phase may be the reason for the enhanced chiroptical properties.39,40 To eliminate artificial signals from the crystalline state, CPL tests were conducted by flipping and rotating the sample (Fig. S26†).41,42 The results showed the same CPL intensity, which ruled out the occurrence of artifacts. In addition, the signs of both CD and CPL for the crystalline state were opposite to those of pristine samples.
 | ||
| Fig. 5 g CD (a) and gCPL (b) spectra of R/S-1. (c) Molecular structures of compound 1, 2, and 3 and the emission pictures after grinding and fuming under a 365 nm lamp. Reversible CPL switching of R-1 (d), R-2 (e) and R-3 (f) by grinding/fuming cycles (ethyl acetate was used for the fuming process of compound 1 and 2, while dichloromethane was used for compound 3) (λex = 360 nm). | ||
As switchable emission can be achieved easily by grinding and fuming processes (Fig. 4e), the CPL emission could also be switched as shown in Fig. 5d. The R-1 pristine sample with a negative CPL signal (glum value is around −5 × 10−4) at 510 nm was used for the switching test. After grinding, the CPL signal disappeared. When the ground sample was treated with ethyl acetate vapor, instant crystallization occurred. The emission was switched to blue color with a positive CPL signal and the glum value was as large as ∼0.01. Moreover, CPL signals showed stable repeatability after several cycles between grinding and fuming treatment (Fig. 5d). Similar results were found for R/S-2 and R/S-3. The yellow amorphous pristine sample of R-2 emitted right-handed CPL with a glum value of −0.0016 (Fig. S27†). After grinding and fuming treatment, left-handed CPL was obtained for the crystalline state, which exhibited enhanced excited chirality (glum = 0.01 at 470 nm) (Fig. 5c, e and S28†). Interestingly, a very large glum value of about −0.0225 was observed at 505 nm for the R-3 pristine powder (Fig. S29†). After grinding and fuming treatment, a glum value of nearly the same magnitude but opposite sign was observed at 515 nm (Fig. 5c, f and S30†). Due to the very weak fluorescence quantum yield, CPL signals of R/S-4 in the crystalline state cannot be detected. However, enhanced CD signals in the crystalline state were observed (Fig. S31†).
To reveal the mechanism of the switchable CPL behavior between pristine and fumed samples, more detailed studies were carried out (Table S3†). First, the chiroptical properties of pure enantiomers were investigated by CD and CPL spectroscopy in THF solutions. CD spectra of R-1 and S-1 in THF revealed clear Cotton effects, and they are mirror images of each other (Fig. S32a†). The R-configuration exhibited a negative Cotton effect and a positive Cotton effect was observed for the S-configuration in the range from 300 to 400 nm, indicating successful chirality transfer from the axial skeleton to the pyrene chromophore. However, no CPL signals were observed in the monomer emission region, while strong positive CPL was observed for R-1 in the dynamic excimer emission region30 (Fig. S32b†). This meant that the excited state chirality differed from that of the ground state, indicating that sizable structural rearrangements occurred in the excited state due to the excimer formation.
Furthermore, CD and CPL from the cast film and aggregated states were investigated (Fig. S33†). When molecules aggregated either in the film or precipitate, the absorption spectra showed red-shift, resulting from the stronger intra and intermolecular interaction between pyrene rings (Fig. S34a†). Thus, the excimer emission at 508 nm should be attributed to the static excimer.30 As shown in Fig. S33,† CPL signs of R/S-1 in aggregated states followed the chirality of the ground state, and were the same as that of the pristine state (Fig. S23†), indicative of similar conformations in the ground and excited states (that's why CD spectra of aggregates were used for the ground state chirality shown in Fig. 5a). Compared with the aggregated state either in the film or precipitate, obvious amplification can be observed both from gCD and gCPL in the crystalline state (Fig. 5a and b), indicating the enhanced chiroptical properties in the crystalline state. In addition, even though both the aggregates and the concentrated solution of 1 shared the same excimer emission (Fig. S35a†), different excited state chiralities were observed (Fig. S32 and S33†). These results demonstrated that the different ground and excited state chiralities originated from the different molecular structures,43,44 which exhibited the advantage of CPL measurement over CD measurement.
Mono-pyrenyl derivatives (R/S-5) based on the octahydrobinaphthyl skeleton were synthesized and investigated as a comparative experiment. Switchable emission behavior was also observed between ground and fumed states (Fig. S39†). In contrast, the crystalline states showed no CPL signal compared with other bi-pyrene counterparts (Fig. S40†). Structural analysis showed that there were two discrete molecules with different conformations in the crystal based on the dihedral angle (θ) between the two octahydrobinaphthyl rings. One has M-helicity with a cisoid conformation (θ = 86.51°), and the other has P-helicity with a transoid conformation (θ = 96.05°)45,46 (Fig. S41†). Despite the same R-configuration, a racemic structure formed due to the opposite helical conformations which might result in the silence of CPL (Fig. S42†). It should be noted that the pyrene rings in the crystal had different orientations relative to the ethoxy groups, indicating greater motion ability in solution when crystallized. This might be due to the less rigid structure compared with other bi-pyrene compounds, demonstrating that double substituted skeletons were essential for achieving intense CPL in the crystalline state.
Conclusions
We demonstrated that high-performance CPL-active solid-state materials could be realized by instant crystallization from amorphous ground films, possessing both large glum values (up to 10−2) and high PL quantum yields (∼50%). We do so by thoroughly synthesizing and investigating 8 bi-pyrene based chiral emitters in the solid state. The instant crystallization process, resulting from the transformation of the amorphous state into the crystalline state could be visualized by using a microscope. Due to the chiral supramolecular arrangement of chromophores in the crystalline state, enhanced chiroptical properties were observed both in the ground and in the excited states. This work presented a simple yet effective approach for constructing high-performance CPL-active solid-state materials. The understanding of excimer-origin CPL would be beneficial for new molecular engineering strategies for designing highly efficient CPL-active materials too.Data availability
The data sets supporting this article have been uploaded as part of the ESI.†Author contributions
J. H. synthesized and characterised the compounds via crystallography, spectroscopy, microscopy and computations. Y. S., X. J. and X. Y. assisted with data analysis and discussion, reviewing and editing; P. D. directed the research and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21905065 and 52173159); the Beijing Municipal Science and Technology Commission (JQ21003); the National Key Basic R&D Research Program of Ministry of Science and Technology of the People's Republic of China (2021YFA1200303); Strategic Priority Research Program of the Chinese Academy of Sciences (XDB36000000).Notes and references
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1900110 CrossRef CAS PubMed.
- D.-W. Zhang, M. Li and C.-F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- L. Frédéric, A. Desmarchelier, R. Plais, L. Lavnevich, G. Muller, C. Villafuerte, G. Clavier, E. Quesnel, B. Racine, S. Meunier-Della-Gatta, J.-P. Dognon, P. Thuéry, J. Crassous, L. Favereau and G. Pieters, Adv. Funct. Mater., 2020, 30, 2004838 CrossRef PubMed.
- F. Song, Z. Xu, Q. Zhang, Z. Zhao, H. Zhang, W. Zhao, Z. Qiu, C. Qi, H. Zhang, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, Z. Zhao, A. Qin, D. Ma and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1800051 CrossRef.
- M. Li, Y.-F. Wang, D. Zhang, L. Duan and C.-F. Chen, Angew. Chem., Int. Ed., 2020, 59, 3500–3504 CrossRef CAS PubMed.
- C. Li, X. Yang, J. Han, W. Sun and P. Duan, Mater. Adv., 2021, 2, 3851–3855 RSC.
- M. Xu, C. Ma, J. Zhou, Y. Liu, X. Wu, S. Luo, W. Li, H. Yu, Y. Wang, Z. Chen, J. Li and S. Liu, J. Mater. Chem. C, 2019, 7, 13794–13802 RSC.
- Y. Imai, Y. Nakano, T. Kawai and J. Yuasa, Angew. Chem., Int. Ed., 2018, 57, 8973–8978 CrossRef CAS PubMed.
- R. Sethy, J. Kumar, R. Métivier, M. Louis, K. Nakatani, N. M. T. Mecheri, A. Subhakumari, K. G. Thomas, T. Kawai and T. Nakashima, Angew. Chem., Int. Ed., 2017, 56, 15053–15057 CrossRef CAS PubMed.
- Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS.
- D. Han, X. Yang, J. Han, J. Zhou, T. Jiao and P. Duan, Nat. Commun., 2020, 11, 5659 CrossRef CAS PubMed.
- T. Kawasaki, M. Sato, S. Ishiguro, T. Saito, Y. Morishita, I. Sato, H. Nishino, Y. Inoue and K. Soai, J. Am. Chem. Soc., 2005, 127, 3274–3275 CrossRef CAS PubMed.
- G. Yang, Y. Y. Xu, Z. D. Zhang, L. H. Wang, X. H. He, Q. J. Zhang, C. Y. Hong and G. Zou, Chem. Commun., 2017, 53, 1735–1738 RSC.
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- L. Arrico, L. Di Bari and F. Zinna, Chem.–Eur. J., 2021, 27, 2920–2934 CrossRef CAS PubMed.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.–Eur. J., 2015, 21, 13488–13500 CrossRef PubMed.
- J.-L. Ma, Q. Peng and C.-H. Zhao, Chem.–Eur. J., 2019, 25, 15441–15454 CrossRef CAS PubMed.
- Y. Deng, M. Wang, Y. Zhuang, S. Liu, W. Huang and Q. Zhao, Light: Sci. Appl., 2021, 10, 76 CrossRef CAS PubMed.
- H.-T. Feng, C. Liu, Q. Li, H. Zhang, J. W. Y. Lam and B. Z. Tang, ACS Mater. Lett., 2019, 1, 192–202 CrossRef CAS.
- A. Nitti and D. Pasini, Adv. Mater., 2020, 32, 1908021 CrossRef CAS PubMed.
- M.-M. Zhang, K. Li and S.-Q. Zang, Adv. Opt. Mater., 2020, 8, 1902152 CrossRef CAS.
- H. Zhang, J. Han, X. Jin and P. Duan, Angew. Chem., Int. Ed., 2021, 60, 4575–4580 CrossRef CAS PubMed.
- T. Zhao, J. Han, X. Jin, Y. Liu, M. Liu and P. Duan, Angew. Chem., Int. Ed., 2019, 58, 4978–4982 CrossRef CAS PubMed.
- S. Zheng, J. Han, X. Jin, Q. Ye, J. Zhou, P. Duan and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 22711–22716 CrossRef CAS PubMed.
- J. Liu, H. Su, L. Meng, Y. Zhao, C. Deng, J. C. Y. Ng, P. Lu, M. Faisal, J. W. Y. Lam, X. Huang, H. Wu, K. S. Wong and B. Z. Tang, Chem. Sci., 2012, 3, 2737–2747 RSC.
- Y. B. Tan, Y. Okayasu, S. Katao, Y. Nishikawa, F. Asanoma, M. Yamada, J. Yuasa and T. Kawai, J. Am. Chem. Soc., 2020, 142, 17653–17661 CrossRef CAS PubMed.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- Y. Li, Z. Ma, A. Li, W. Xu, Y. Wang, H. Jiang, K. Wang, Y. Zhao and X. Jia, ACS Appl. Mater. Interfaces, 2017, 9, 8910–8918 CrossRef CAS PubMed.
- S. Karuppannan and J.-C. Chambron, Chem.–Asian J., 2011, 6, 964–984 CrossRef CAS PubMed.
- W.-L. Wang, J. Xu, Z. Sun, X. Zhang, Y. Lu and Y.-H. Lai, Macromolecules, 2006, 39, 7277–7285 CrossRef CAS.
- W. Wang, J. Xu and Y.-H. Lai, Org. Lett., 2003, 5, 2765–2768 CrossRef CAS PubMed.
- Y. Wang, Y. Li, S. Liu, F. Li, C. Zhu, S. Li and Y. Cheng, Macromolecules, 2016, 49, 5444–5451 CrossRef CAS.
- L. Di Bari, G. Pescitelli and P. Salvadori, J. Am. Chem. Soc., 1999, 121, 7998–8004 CrossRef CAS.
- H. Zhang, H. Li, J. Wang, J. Sun, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2015, 3, 5162–5166 RSC.
- H. Zhang, X. Zheng, R. T. K. Kwok, J. Wang, N. L. C. Leung, L. Shi, J. Z. Sun, Z. Tang, J. W. Y. Lam, A. Qin and B. Z. Tang, Nat. Commun., 2018, 9, 4961 CrossRef PubMed.
- S. Ito, R. Sekine, M. Munakata, M. Asami, T. Tachikawa, D. Kaji, K. Mishima and Y. Imai, ChemPhotoChem, 2021, 5, 920–925 CrossRef CAS.
- M. Louis, R. Sethy, J. Kumar, S. Katao, R. Guillot, T. Nakashima, C. Allain, T. Kawai and R. Métivier, Chem. Sci., 2019, 10, 843–847 RSC.
- R. Hassey, E. J. Swain, N. I. Hammer, D. Venkataraman and M. D. Barnes, Science, 2006, 314, 1437–1439 CrossRef CAS PubMed.
- Z.-L. Tu, Z.-P. Yan, X. Liang, L. Chen, Z.-G. Wu, Y. Wang, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Adv. Sci., 2020, 7, 2000804 CrossRef CAS PubMed.
- W. Chen, Z. Tian, Y. Li, Y. Jiang, M. Liu and P. Duan, Chem.–Eur. J., 2018, 24, 17444–17448 CrossRef CAS PubMed.
- G. Albano, G. Pescitelli and L. Di Bari, Chem. Rev., 2020, 120, 10145–10243 CrossRef CAS PubMed.
- S. Ito, K. Ikeda, S. Nakanishi, Y. Imai and M. Asami, Chem. Commun., 2017, 53, 6323–6326 RSC.
- H. Okada, N. Hara, D. Kaji, M. Shizuma, M. Fujiki and Y. Imai, Phys. Chem. Chem. Phys., 2020, 22, 13862–13866 RSC.
- H. Hayasaka, T. Miyashita, M. Nakayama, K. Kuwada and K. Akagi, J. Am. Chem. Soc., 2012, 134, 3758–3765 CrossRef CAS PubMed.
- G. Proni, G. P. Spada, P. Lustenberger, R. Welti and F. Diederich, J. Org. Chem., 2000, 65, 5522–5527 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional photophysical data, crystal structures, X-ray crystallographic information and movies for the transformation process. CCDC 2104394–2104398. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01846b |
| This journal is © The Royal Society of Chemistry 2022 |