
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic oxidative dehydrogenation of N-heterocycles with nitrogen/phosphorus co-doped porous carbon materials†
Kangkang
Sun
ab,
Hongbin
Shan
a,
Rui
Ma
b,
Peng
Wang
b,
Helfried
Neumann
 *b,
Guo-Ping
Lu
*a and
Matthias
Beller
*b
*b,
Guo-Ping
Lu
*a and
Matthias
Beller
*b
aSchool of Chemistry and Chemical Engineering, Nanjing University of Science & Technology, Xiaolingwei 200, Nanjing 210094, P. R. China
bLeibniz-Institute for Catalysis, Albert-Einstein-Straße 29a, Rostock 18059, Germany. E-mail: Matthias.Beller@catalysis.de
First published on 17th May 2022
Abstract
A metal-free oxidative dehydrogenation of N-heterocycles utilizing a nitrogen/phosphorus co-doped porous carbon (NPCH) catalyst is reported. The optimal material is robust against traditional poisoning agents and shows high antioxidant resistance. It exhibits good catalytic performance for the synthesis of various quinoline, indole, isoquinoline, and quinoxaline ‘on-water’ under air atmosphere. The active sites in the NPCH catalyst are proposed to be phosphorus and nitrogen centers within the porous carbon network.
Introduction
In general, oxidative dehydrogenations of saturated and partially saturated heterocycles offer an efficient and straightforward route for the synthesis of functionalized aromatic N-heterocycles.1–7 As an example (iso)quinolines are of interest for many applications including dyes, paints, solvents, and corrosion inhibitors (Scheme 1A).8,9 In addition, many of these and related N-heterocycles are important building blocks for bio-active molecules and natural products, including pharmaceuticals and agrochemicals, such as anti-schizophrenia drug10 and coccidiostats.11 Thus, in the past a plethora of synthetic protocols involving dehydrogenations have been developed in this field. More specifically, homogeneous catalytic systems have been used for the oxidative dehydrogenation of saturated N-heterocycles (Scheme 1B). Unfortunately, many of these methods require complicated and expensive ligands and/or stoichiometric amounts of toxic oxidants such as TEMPO,12 DDQ,13 and tert-butyl hydroperoxide.14 In addition, product/catalyst separation and catalyst reusability remain difficult. To overcome these limitations, heterogeneous catalysts, such as Pd/C,15 Pd3Pb/MgO,16 Ni2Mn-LDH,17 Cu(0)/Al2O3,18 Co NCs/N–C,19 FeOx@NGr-C,20 and 2[PW]-OMS-2 complexes,21 were introduced (Scheme 1B). Obviously, an “ideal” heterogeneous catalyst for such transformations would be “metal-free” due to cost efficiency and possible advantages regarding toxicity as well as selectivity, e.g. for substrates containing halogen or other functional groups.22 In this respect, Shi and co-workers elegantly showed that specific polymaleimide materials (PMI) can acts as molecularly defined single-active site heterogeneous catalysts for oxidative dehydrogenations.23 Unfortunately, the recyclability and long-term stability of such materials is not known. In this respect, the preparation of more robust and thermally stable carbon-based materials is interesting, which are simply prepared by pyrolysis. In fact, such materials have shown promising applications as catalysts in different organic reactions,24–26 degradation of environmental pollutants, and electrochemical catalysis in the past decade.27–29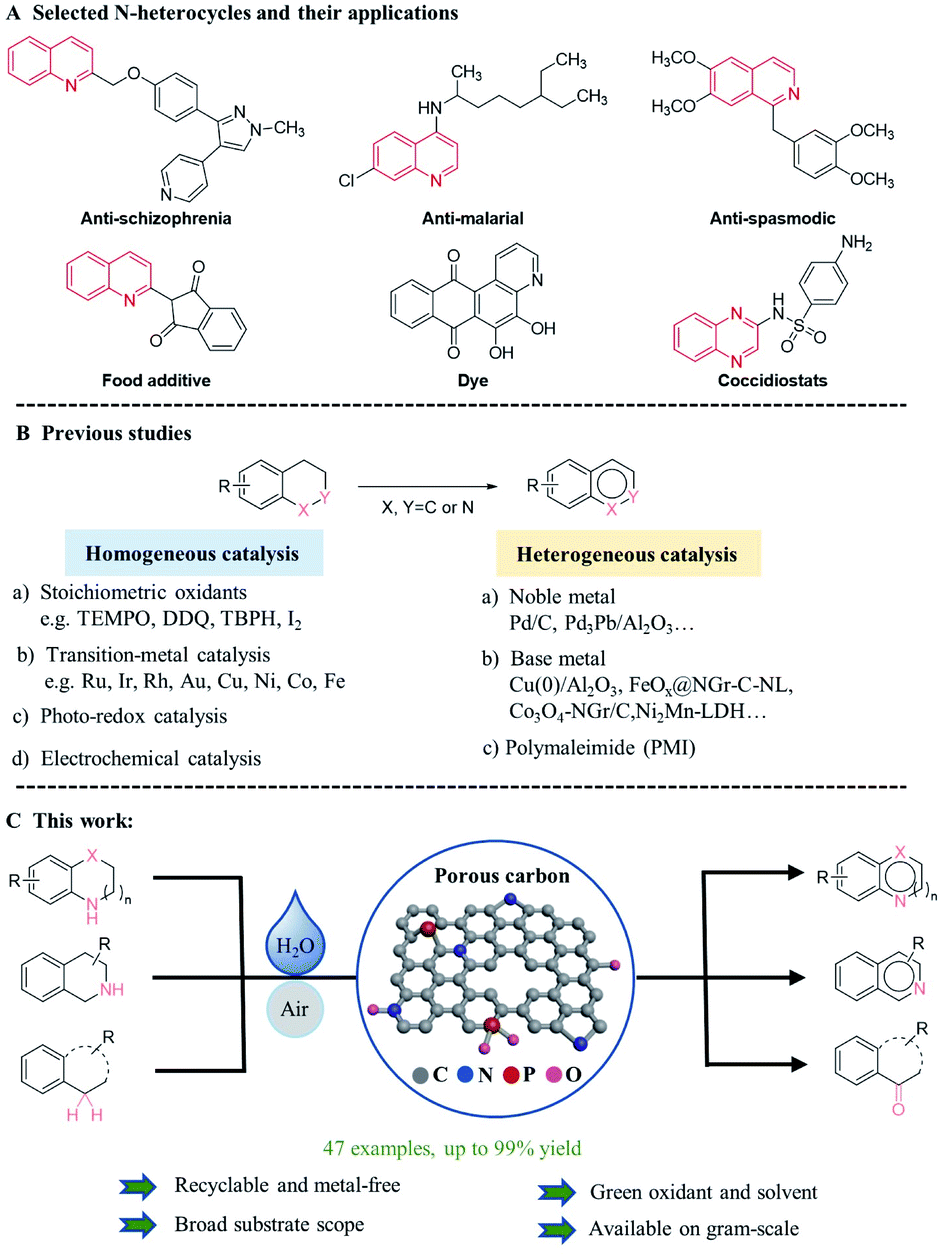 | ||
| Scheme 1 (A) Selected examples of bioactive N-heterocycles-containing molecules. (B) Overview of oxidative dehydrogenations of N-heterocycles. (C) This work. | ||
To improve the catalytic activity of the pristine carbon materials, heteroatom doping (B, N, P, S, etc.) is a promising approach, which can effectively modulate the electronic structures, induce defects in lattices and produce active catalytic centers.30 Specifically, N- as well as P-doped porous carbon materials have been applied for instance in photocatalytic oxidation of benzyl alcohol,31 reduction of nitroarenes,32 and hydrogen transfer reactions33 owing to their metal-like d-band electronic structure nearby to the doped heteroatom. In addition to these mono-doped materials, there is an increasing interest in the co-doping of different heteroatoms into the carbon framework, which can cause special electric redistribution by the synergistic effects of different dopants.34–36 Indeed, such materials showed promising performance in selected organic transformations.37,38
Based on our general interest in the preparation of novel materials and synthesis of heterocycles,39–43 we were curious to know whether such metal-free heterogeneous catalysts might be applicable for oxidative dehydrogenation of N-heterocycles. Notably, we expected an increased activity of the carbon materials via N,P-co-doping, as well as a stabilization against oxidative degradation compared to the parent materials. Here, we disclose a N,P co-doped porous carbon catalyst via pyrolysis of benzylamine modified zeolitic imidazolate frameworks (ZIF-8) and PPh3. Notably, control of the morphology and the crystal size of the ZIF-8 precursor is achieved by employing benzylamine as a crystallization modulator. The obtained co-doped porous carbon catalyst (NPCH) is robust against poisoning, highly stable against self-oxidation, and exhibits good performance in oxidative dehydrogenations of various N-heterocycles using ‘on-water’ conditions44–46 in air atmosphere (Scheme 1C).
Results and discussion
Catalyst synthesis
For the preparation of the potential catalyst materials, 2-methylimidazole and benzylamine were dissolved in water, and then the mixture was added to zinc nitrate hexahydrate solution and stirred at room temperature for 4 hours to synthesize ZIF-8-Bn. The obtained ZIF-8-Bn was homogeneously mixed with different amounts of triphenylphosphine followed by pyrolysis at 900 °C in a N2 atmosphere to produce NPC-x (x = 150, 300, 450), where x denoted the mass of PPh3 added to one thousand milligrams of ZIF-8-Bn. During the pyrolysis process, Zn species evaporate above 800 °C and leave defects or nanopores in their places.47 After etching with 2 M hydrochloride acid (HCl) solution for 10 h to remove residual Zn components and washing, the resultant nitrogen/phosphorus co-doped porous carbon composite was collected as a black powder. For comparison, the catalyst NC-Bn with the sole N-dopant and NC synthesized by direct pyrolysis of ZIF-8 were also prepared with a similar procedure.Model reaction and catalyst optimization
All the prepared materials as well as some commercially available ones were tested in the oxidative dehydrogenation of 1,2,3,4-tetrahydroquinoline as a benchmark reaction. Typically, the catalytic reactions were carried out using only water as solvent at ambient atmospheric pressure of air as “green” oxidant. At this point, it is worth mentioning that such ‘on-water’ reactions are also attractive due to the unique nature of this green solvent.48–50 The yield of quinoline 2a in the presence of various catalysts is shown in Table 1. Obviously, a blank experiment (without any catalyst) revealed no quinoline formation (Table 1, entry 1). Comparing the performance of nitrogen-doped carbon NC derived from ZIF-8 without benzylamine modification with the benzylamine-derived NC-Bn, showed the importance of this modification (Table 1, entries 2 and 3). Interestingly, further introduction of different amounts of P atoms into the benzylamine-derived material improved the yield of quinoline 2a to 87% (Table 1, entries 4–6). Testing the reaction in different organic solvents, such as CH3CN, DMF, toluene, MeOH, 1,4-dioxane, and DMSO, showed lower product yields compared to water (Table 1, entries 7–12).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | T (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 15 mg catalyst, 0.1 MPa air, 1.5 mL H2O, 24 h, the most active catalyst is shown in bold. b The yields of products were determined by GC analysis using hexadecane as an internal standard. c The catalyst was prepared at 800 °C. d 5 mg catalyst, 12 h. | ||||
| 1 | — | H2O | 80 | nr |
| 2 | NC | H2O | 80 | 3 |
| 3 | NC-Bn | H2O | 80 | 55 |
| 4 | NPC-150 | H2O | 80 | 79 |
| 5 | NPC-300 | H2O | 80 | 87 |
| 6 | NPC-450 | H2O | 80 | 82 |
| 7 | NPC-300 | CH3CN | 80 | 52 |
| 8 | NPC-300 | DMF | 80 | 29 |
| 9 | NPC-300 | Toluene | 80 | 41 |
| 10 | NPC-300 | MeOH | 80 | 59 |
| 11 | NPC-300 | 1,4-Dioxane | 80 | 34 |
| 12 | NPC-300 | DMSO | 80 | 53 |
| 13c | NPC-300 | H2O | 80 | 16 |
| 14 | NPCH | H2O | 80 | >99 |
| 15 | NPCH | H 2 O | 70 | 97 |
| 16 | NPCH | H2O | 60 | 83 |
| 17d | NPCH | H2O | 110 | 88 |
| 18 | NPCH | H 2 O | 120 | >99 |
| 19 | Zn(NO3)2·6H2O | H2O | 80 | nr |
| 20 | ZnO | H2O | 80 | nr |
| 21 | C-L(lignin) | H2O | 80 | 12 |
| 22 | Activated carbon | H2O | 80 | 8 |
Notably, zinc is a sacrificial template for the formation of porous structure, which is of great significance for the absorption and transport of reactants, thereby boosting the catalytic process. In the optimal catalyst material, the contents of Zn and P were determined by ICP-OES to be 1.1 wt% and 0.86 wt%, respectively (Table S1†).
To understand the effect of the remaining Zn species, an N,P-co-doped carbon material with larger amount on zinc present was prepared by pyrolysis at 800 °C. It is well-known that this temperature is not high enough to remove residual Zn components via sublimation. Using the latter material in the benchmark reaction the yield of 2a is low, which indicates that the residual Zn atoms have a detrimental influence on quinoline formation (Table 1, entry 13). To remove the small amounts of residual Zn components and improve the dispersibility of the catalyst in water, the material pyrolyzed at 900 °C was washed with 2 M HCl solution. The obtained NPCH (Zn: 0.56 wt%, Table S1†) showed excellent performance for this reaction, even at 60 °C (Table 1, entries 14–16). More importantly, quinoline can also be obtained in high yields by reducing the amount of catalyst and increasing the reaction temperature (Table 1, entries 17, 18). Control experiments with commercially available ZnO and Zn(NO3)2·6H2O displayed no activity and formation of quinoline (Table 1, entries 19 and 20).
Mechanistic investigations
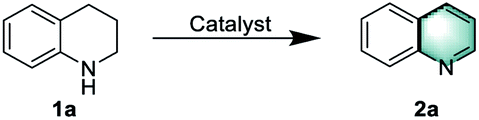
To gain information on the active sites of the catalyst material as well as to evaluate its robustness against deactivation, several experiments were performed with thiophene, thiourea, and KSCN as potentially binding molecules, which are well-known poisoning reagents for catalysts with metal-centered active sites.51 Thus, 2 eq of thiophene, thiourea, KSCN, or high concentration of the KSCN (5 eq) were added into the reaction mixture; however the yield of quinoline decreased only slightly (Fig. 1). This again indicates that the remaining zinc traces in the catalyst are not the active sites. Finally, for comparison, we evaluated the performance of commercially available activated carbon and other porous carbon-based heterogeneous catalysts (Table 1, entries 21 and 22), but none of them showed significant activity under our reaction conditions.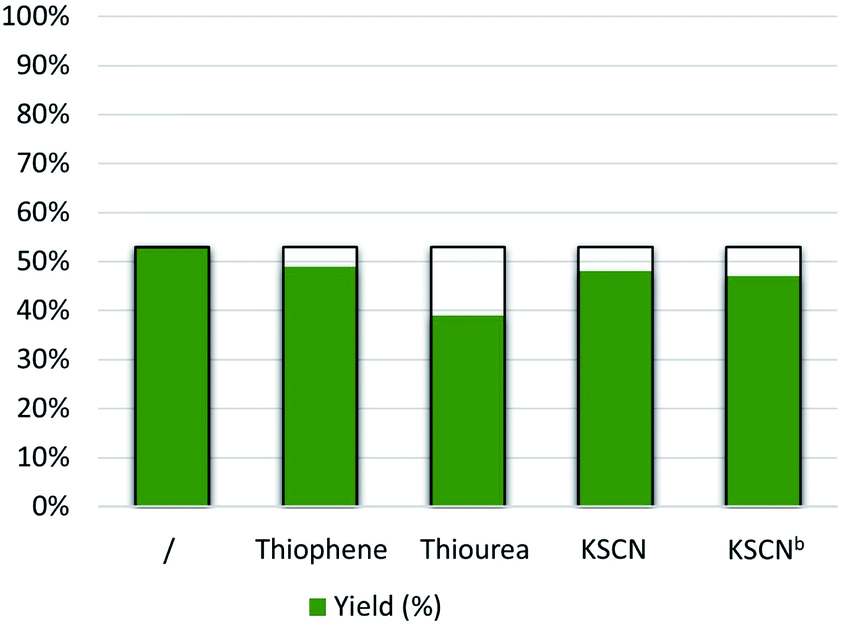 | ||
| Fig. 1 NPCH-catalyzed oxidative dehydrogenation of 1,2,3,4-tetrahydroquinoline in the presence of S-containing reagents. aReaction conditions: 1a (0.2 mmol), 5 mg catalyst, 110 °C, 0.1 MPa air, 1.5 mL H2O, 6 h, 2 eq of poisoning reagents to the total amount of zinc; b5 eq of poisoning reagents to the total amount of zinc. | ||
Next, the kinetic progress of the model reaction was examined under the optimal conditions. As shown in Fig. 2, 1a is rapidly converted to 2a within the first 4 hours.
 | ||
| Fig. 2 Kinetic profile of the model reaction: The X-axis represents reaction time, and the Y-axis represents product distribution. Reaction conditions: 1a (0.2 mmol), 5 mg catalyst, 120 °C, 0.1 MPa air, 1.5 mL H2O. | ||
In the following eight hours, the yield of desired product 2a increased steadily, and reached 99% yield after 12 h. In this kinetic profile no side products and intermediates could be observed. To get more detailed insight into the mechanism of the reaction, several control experiments were carried out (Scheme S1†). At first, the model reaction was performed in the presence of BHT (2,6-di-tert-butyl-4-methylphenol), a well-known radical scavenger, and no significant reduction in 2a yield occurred. In agreement with this observation no radical signal is detected by EPR (electron paramagnetic resonance) measurements. These results indicate that radical processes are likely not involved in this transformation. Using N-methyl-1,2,3,4-tetrahydroquinoline or 1,2,2,4,7-pentamethyl-1,2,3,4-tetrahydroquinoline as substrates for the reaction gave no desired products (Scheme S1B†). Apparently, the presence of both N–H bond and the α-H at C-2 position plays a decisive role in the oxidative dehydrogenation of N-heterocycles. Based on the above results and previous reports,52–54 we propose the following mechanism for the present process (Scheme S2†): Firstly, the substrate 1a and oxygen molecules are absorbed and thereby activated on the surface of the N,P-co-doped carbon material (NPCH). The surface-bound N-heterocycles will undergo a dehydrogenation reaction to give the intermediate 1a′. This intermediate can undergo isomerization followed by subsequent dehydrogenation to give the complete dehydrogenated product 2a. Following previous suggestions,28,54 the mechanism of this catalytic oxidative dehydrogenation may be similar to that of laccase oxidation of phenol, in which O2 can be directly reduced to water by a four-electron transfer process.
Catalyst characterization
To understand the relationship between the structural properties and catalytic activity, a series of characterization measurements were performed. Compared with pure ZIF-8, the X-ray powder diffraction (XRD) patterns of the ZIF-8-Bn matched the simulated ones very well (Fig. S1†).This confirms the ability of benzylamine additive to effectively improve the crystallinity of the material and thereby helps to control the particle size and morphology. More specifically, ZIF-8-Bn exhibits a uniform rhombic dodecahedral morphology with small particle size of around 190 nm (Fig. 3B), while ZIF-8 is an irregular sheet-like structure with larger diameter (Fig. 3A). After pyrolysis at 900 °C under N2 atmosphere and washing with HCl, the morphology of the ZIF-8-BnP particles remained basically unchanged (Fig. 3C and D). The average particle sizes of NPC-300 and NPCH are 182 nm and 180 nm, respectively (Fig. S7†). Meanwhile, elemental mapping of NPCH revealed the homogeneous dispersion of C, N, O and P species over the entire nanoarchitecture (Fig. 3E). X-ray diffraction (XRD) and Raman spectra were conducted to examine the graphitic features and structural defects of the catalysts. As shown in Fig. S1,† the two diffraction peaks around 25° and 43° correspond to the (002) and (101) planes of graphite (JCPDS no. 41-1487), respectively.55 Raman spectra of the samples present the G band at 1587 cm−1, which is a characteristic feature of graphitic carbon, while the D band at 1339 cm−1 corresponds to defective carbon structures.56 Notably, the Raman spectra are sensitive to the subtle structural changes of porous carbon materials. A higher peak intensity ratio of the D-band to G-band (ID/IG) in NPC-300 indicates the generation of large amounts of defects, which suggests that the doping with P atoms increases the structural disorder of the carbon material (Fig. S2†), similar to previous findings.57 The ID/IG ratio decreases after HCl etching, but it is still higher than that of the catalyst without phosphorus doping. We assume these defects facilitate the chemisorption mode of substrates and O2 and allows for easier O–O bond cleavage, thereby improving the catalytic performance.58
 | ||
| Fig. 3 Representative electron microscopy images. (A) SEM image of ZIF-8; (B) SEM image of ZIF-8-Bn; (C) TEM image of NPC-300; (D) TEM image of NPCH; (E) EDS mapping images (C, N, O and P) of NPCH. | ||
Nitrogen adsorption–desorption measurements were applied to investigate the NPCH catalyst in terms of the BET specific surface areas and hierarchically porous structure. As shown in Fig. 4, the BET surface area of NC-Bn is 1047 m2 g−1, and the total pore volume is 0.72 cm3 g−1, which is much larger than the material obtained by direct pyrolysis of ZIF-8 without adding benzylamine (BET surface area: 572 m2 g; pore volume: 0.26 cm3 g−1).
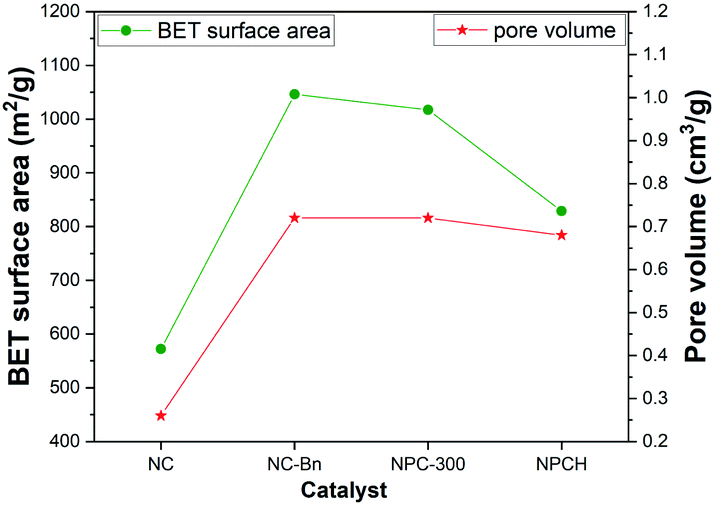 | ||
| Fig. 4 BET surface area and pore volume of the different materials. | ||
The physisorption isotherms of the NPCH catalyst shows type IV isotherms with high-pressure region hysteresis loops, demonstrating the coexistence of micro- and mesopores in the catalyst. The surface area and pore size distribution of NPCH and NC-Bn are similar, indicating that the introduction of triphenylphosphine and the washing step with hydrochloric acid will not destroy the pore structure of the catalyst (Fig. S3 and Table S2†). We assume that the highly porous structure can effectively absorb air and reactants, while the large surface area provides additional active sites for the desired oxidation reaction.
The chemical valence and composition of the NPCH composites were characterized by XPS measurements. As shown in Fig. S4,† the survey spectra is mainly composed of C, N, O, whereas the signals of P are not traceable. The undetected XPS signals of P can be attributed to the low concentration, as determined by ICP-OES (P in NPCH, 0.68 wt%) and EDS. In addition, The O/C ratios in the NPCH and NPC-300 are 0.06 and 0.05 (Fig. S4 and Table S3†), respectively. The higher oxygen content may provide the NPCH with higher wettability (Fig. S5†). Good wettability is beneficial for the dispersion of the catalyst in water, thereby promoting the progress of the reaction.59
The detailed high-resolution XPS analysis of C, N, O and P in NPCH were also recorded as shown in Fig. 5. For C 1s spectra, the peaks located at 284.6 eV, 285.4 eV, 286.7 eV, and 290.0 eV can be attributed to C–C, C–O/C–P, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and O–C–O, respectively (Fig. 5A).60 Four kinds of N species are observed in the NPCH that can be assigned to oxidized N (403.7 eV), graphitic N (400.9), pyrrolic N (399.0 eV), and pyridinic N (398.1 eV) in Fig. 5B. These N atoms may give the catalyst rich Lewis base position, which is believed to facilitate the oxidative dehydrogenation process.61 The XPS P 2p spectrum in NPCH shows deconvoluted peaks at 132.8 eV and 134.0 eV, which correspond to P–C and P–O types of species (Fig. 5D). In addition, the XPS pattern of O 1s in Fig. 5C can be fitted into three main peaks located at 531.3 eV, 532.1 eV, and 533.2 eV, which can be ascribed to CO/PO, C–O/P–O–C, and C–OH/P–OH bonds at the surface.62,63 In general, the XPS results confirmed that N and P heteroatoms were successful doped into the carbon environment, which play a significant role during the catalytic process.
O, and O–C–O, respectively (Fig. 5A).60 Four kinds of N species are observed in the NPCH that can be assigned to oxidized N (403.7 eV), graphitic N (400.9), pyrrolic N (399.0 eV), and pyridinic N (398.1 eV) in Fig. 5B. These N atoms may give the catalyst rich Lewis base position, which is believed to facilitate the oxidative dehydrogenation process.61 The XPS P 2p spectrum in NPCH shows deconvoluted peaks at 132.8 eV and 134.0 eV, which correspond to P–C and P–O types of species (Fig. 5D). In addition, the XPS pattern of O 1s in Fig. 5C can be fitted into three main peaks located at 531.3 eV, 532.1 eV, and 533.2 eV, which can be ascribed to CO/PO, C–O/P–O–C, and C–OH/P–OH bonds at the surface.62,63 In general, the XPS results confirmed that N and P heteroatoms were successful doped into the carbon environment, which play a significant role during the catalytic process.
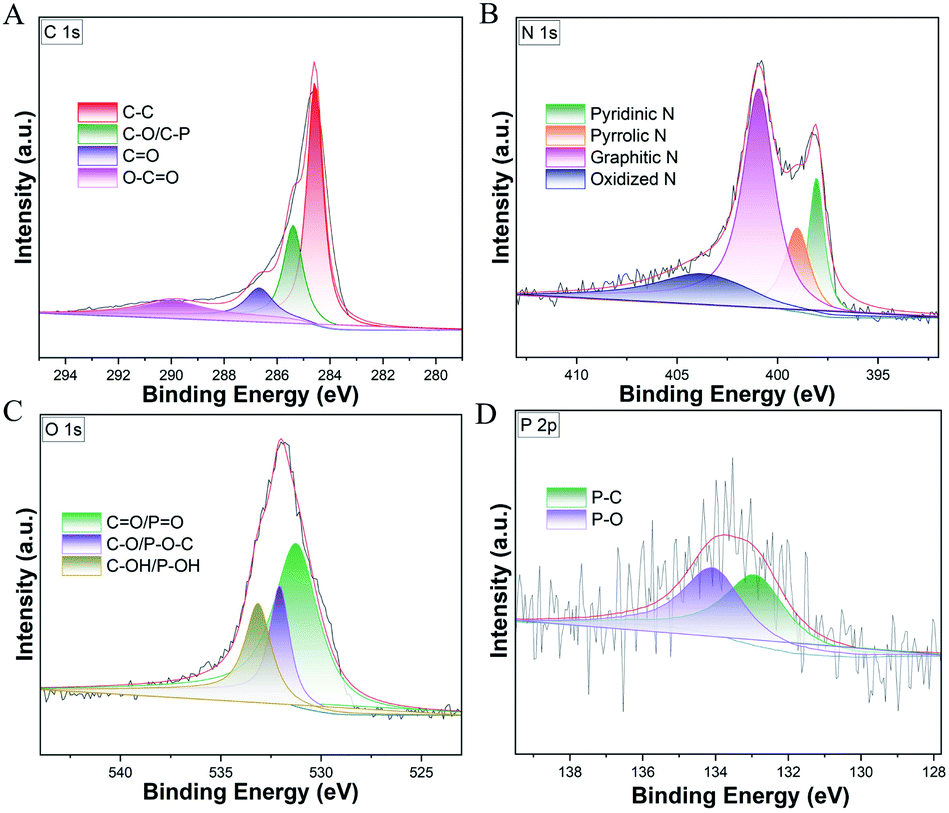 | ||
| Fig. 5 XPS spectra of NPCH. (A) C 1s; (B) N 1s; (C) O 1s; (D) P 2p. | ||
Substrate scope
With the optimized conditions in hand, the reactivity of various substituted quinolines was explored (Scheme 2). Initially, tetrahydroquinolines bearing methyl or methoxy substituents on different positions afforded the corresponding heteroarenes in good yields (2a–2h). When the N-ring was substituted by 2-phenyl group, the dehydrogenation also proceeded well to afford 2m in 73% yield. Similarly, the oxidative dehydrogenation of N-heterocycles with electron-withdrawing groups such as –F, –Cl, Br, –NO2 or –CF3 provided the desired products efficiently (2i–2l and 2p).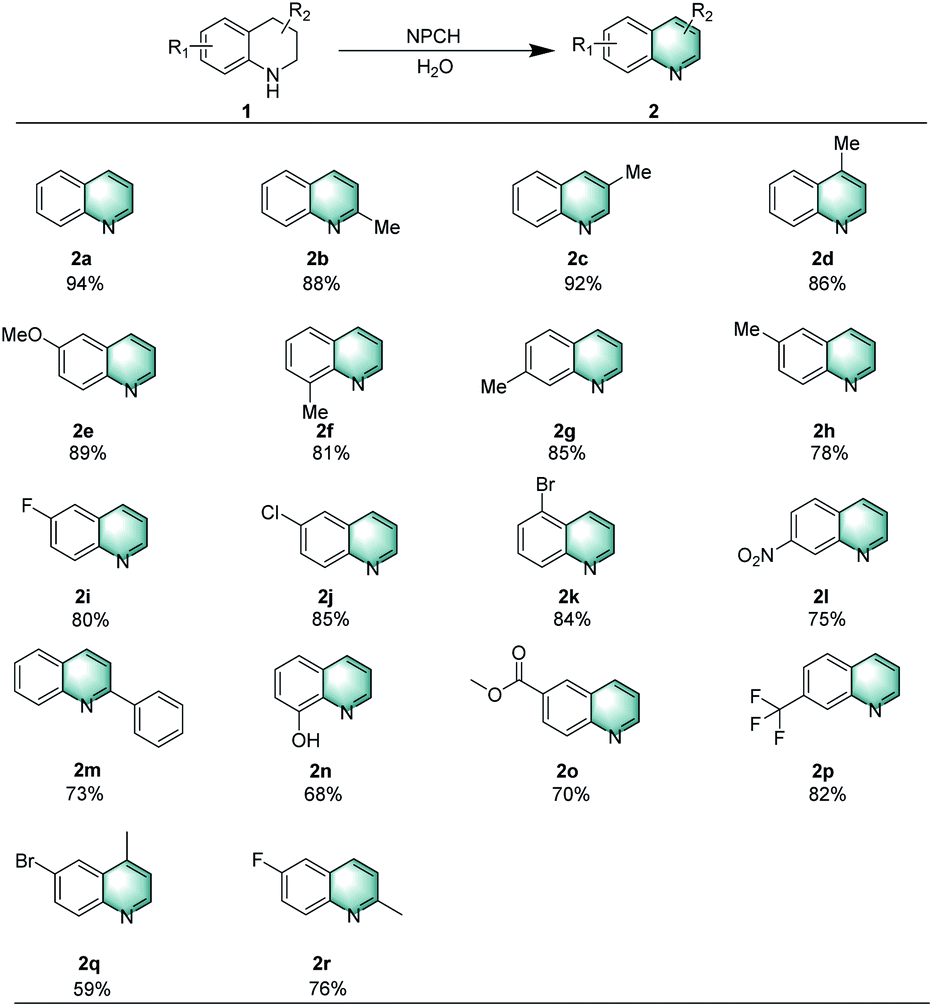 | ||
| Scheme 2 Oxidation of tetrahydroquinolines catalyzed by NPCH. Reaction conditions: 1 (0.2 mmol), 5 mg catalyst (NPCH), 120 °C, 0.1 MPa air, 1.5 mL H2O, 12 h, isolated yields. | ||
In addition, hydroxyl- and ester-substituents are compatible with these reaction conditions (2n, 2o). Finally, di-substituted tetrahydroquinolines were treated under these conditions, leading to the formation of dehydrogenative products 2q and 2r in 59% and 76% isolated yields, respectively.
Apart from tetrahydroquinolines, indolines can be smoothly dehydrogenated under the standard conditions using air as oxidant in water. The results shown in Scheme 3 confirmed that a range of indolines with different substituents (–CH3, –OCH3, –Cl, –Br, –F, NO2, etc.) gave the desired products in moderate to good yields (4a–4h). Furthermore, substrates with cyano and ester groups, afforded indoles 4i, 4j in 88% and 90% yields, respectively. It is noteworthy that more demanding substrates, such as 7-bromo-5-nitroindoline, 5-bromo-2-methyl-indoline, 2,3-dimethyl-indoline, and 2,3-dihydro-7-azaindole, are successfully converted to the corresponding indoles (4k–4n).
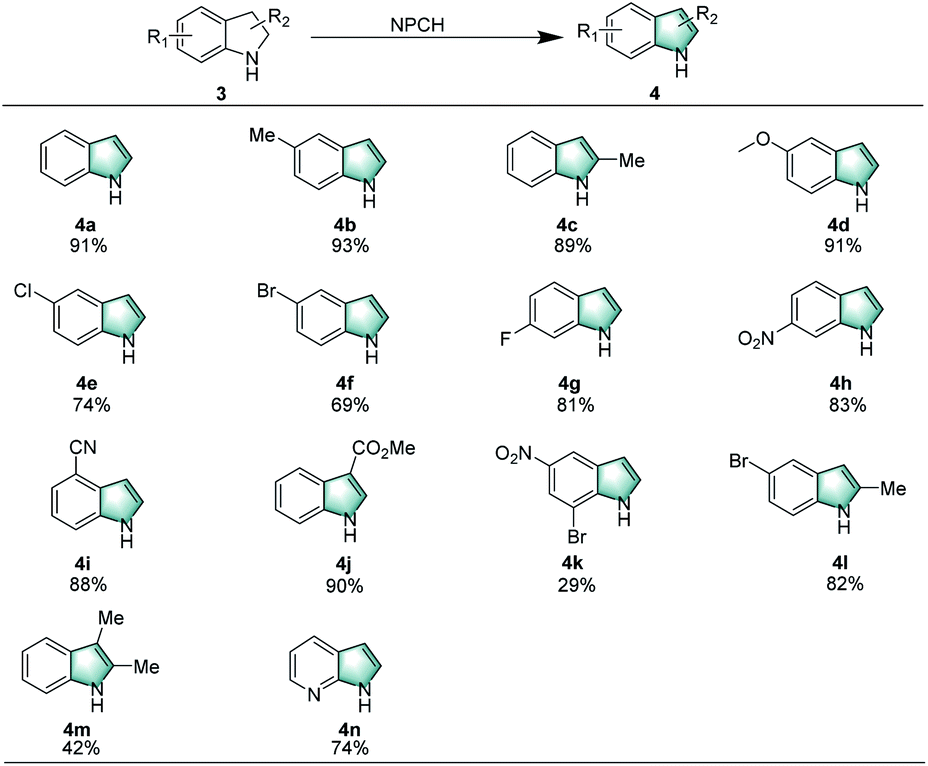 | ||
| Scheme 3 Oxidation of indolines catalyzed by NPCH. Reaction conditions: 3 (0.2 mmol), 5 mg catalyst (NPCH), 120 °C, 0.1 MPa air, 1.5 mL H2O, 12 h, isolated yields. | ||
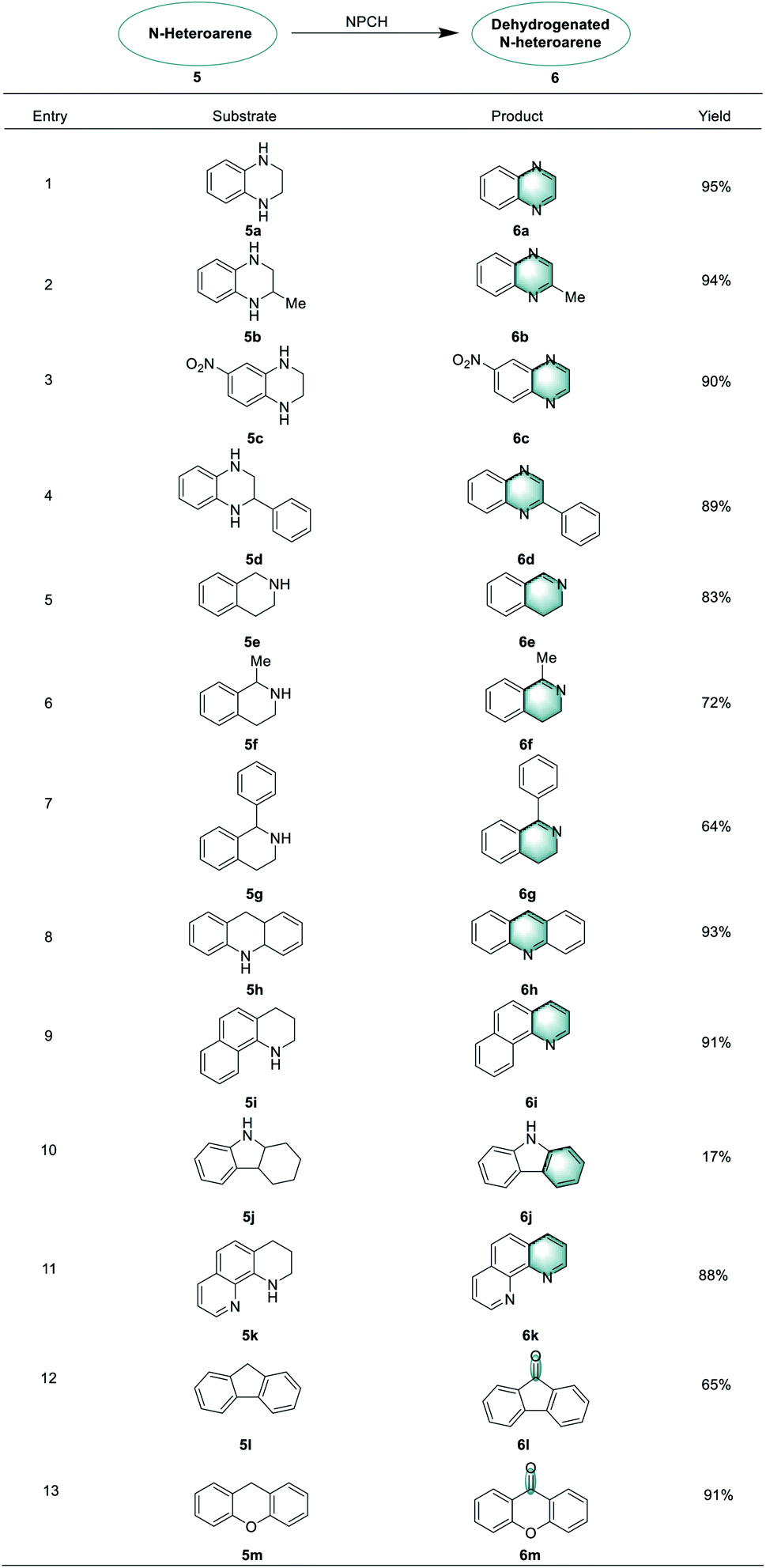
Next, this novel protocol was applied for the synthesis of other N-heteroarene derivatives, and the results are listed in Table 2. Oxidative dehydrogenation reactions of 1,2,3,4-tetrahydroquinoxalines proceeded smoothly to give the corresponding quinoxaline products in 89–95% yields (6a–6d).
| a Reaction conditions: 5 (0.2 mmol), 5 mg catalyst (NPCH), 120 °C, 0.1 MPa air, 1.5 mL H2O, 12 h, isolated yields. |
|---|
|
Interestingly, tetrahydroisoquinolines behaved differently in this reaction compared to tetrahydroquinolines and partial dehydrogenation products were obtained along with complete dehydrogenation products (6e–6g).14,64,65 In addition, acridine, 1,2,3,4-tetrahydrobenzo[h]quinoline, and 1,2,3,4-tetrahydro-1,10-phenanthroline could be oxidized to form the desired products in good yields under standard conditions (6h–6k). Furthermore, the NPCH catalyst can be successfully applied for benzylic C–H oxidations. Here, 9-fluorenone and xanth-9-one products (6l, 6m) could be obtained from 9H-fluorene and xanthene.66–68
Synthetic applications
Finally, we showcased the applicability of the catalytic system for the preparation of selected pharmaceutically relevant molecules. When probing the reactivity of 5,6,6a,11b-tetrahydro-7H-indeno[2,1-c]quinoline 5n, dehydrogenation and benzyl oxidation occurred to provide product 7H-indeno[2,1-c]quinolin-7-one 6n in 64% yield, which can be used to synthesize topoisomerase inhibitor TAS-102. Notably, dehydrogenation of 5o selectively yielded 6o, a precursor for nM5-lipoxygenase inhibitor (Scheme 4).69,70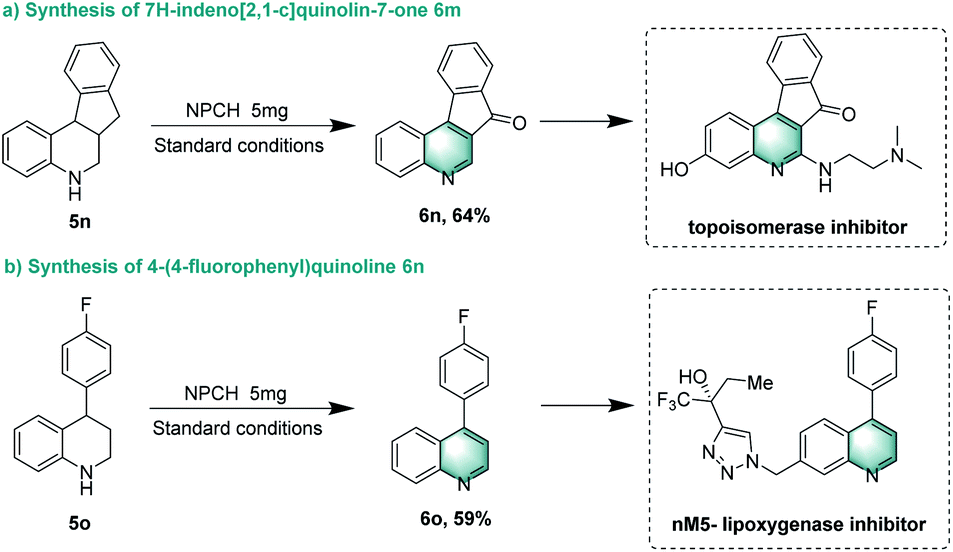 | ||
| Scheme 4 Synthesis of two pharmaceutically relevant molecules. Reaction conditions: 0.2 mmol of substrate, 1.5 mL H2O, 5 mg NPCH, 0.1 MPa air, 120 °C, 12 h. | ||
To demonstrate the synthetic utility of this catalytic system further on, NPCH was used for the gram-scale synthesis of various N-heterocycles. Despite the limitation of oxygen transfer from gas phase to liquid phase, for all the tested substrates high yields were obtained on multi g-scale also with lower catalyst usage (Scheme 5).
 | ||
| Scheme 5 Gram-scale synthesis. Reaction conditions: 20 mmol of substrate, 100 mL H2O, 200 mg NPCH, 20 bar of air, 100 °C, 12 h. | ||
Catalyst recycling
Finally, the recycling and reusability of our optimal material was tested at full and half conversions. As shown in Fig. 6, the catalytic activity of NPCH remained high after five runs and only a slight decrease in quinoline yield is observed, also demonstrating the durability of this catalyst system. In addition, the XRD pattern, TEM image and Raman spectra of the recycled NPCH showed no significant difference compared to the fresh samples. However, the BET surface area and pore volume of the recycled catalyst decreased slightly after six cycles (BET surface area: 662 m2 g; pore volume: 0.50 cm3 g−1), which may account for the reduced quinoline yield (Fig. S1, S6, and Table S2†).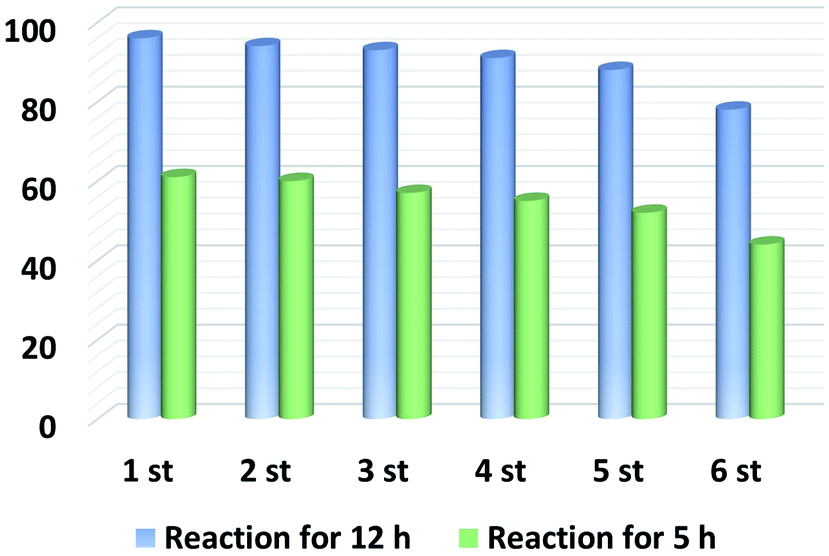 | ||
| Fig. 6 Recyclability of NPCH for the synthesis of quinoline. Reaction conditions: 1a (20 mmol), 200 mg catalyst, 100 °C, 20 bar air, 100 mL H2O, the yield was determined by GC analysis using hexadecane as an internal standard. | ||
Conclusions
In conclusion, an N,P-co-doped carbon material NPCH has been developed as a metal-free heterogeneous catalyst for the oxidative dehydrogenation of N-heterocycles. The optimal catalyst displays high activity for the dehydrogenation of various 1,2,3,4-tetrahydroquinoline, tetrahydroisoquinolines, indoline, and tetrahydroquinoxaline derivatives to give the desired heteroarenes with excellent selectivity. The synthetic utility of our protocol is demonstrated for the synthesis of two pharmaceutically relevant intermediates. Notably, the material shows high tolerance towards typical poisoning reagents and can be easily recycled/reused with only slight loss in activity. We believe this metal-free oxidation with air and ‘on-water’ complements the existing synthetic tools for a green and sustainable synthesis of functionalized aromatic N-heterocycles.Data availability
All experimental data associated with this work is available in the ESI.†Author contributions
M. B., G. L., and K. S. conceived and designed the experiments. K. S. and H. S. performed the experiments and analyzed the data. H. N., R. M., and P. W. participated in the discussions and supported the project. M. B., G. L., and K. S. prepared the manuscript with feedback from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Fundamental Research Funds for the Central Universities (30920021120), the China Scholarship Council (scholarship to K.S.) for financial support. We also thank the analytical staff at the Leibniz-Institut für Katalyse e.V. (LIKAT), for characterizations and acknowledge funding from the state of Mecklenburg-Western Pomerania and the BMBF.References
- J. Wu, D. Talwar, S. Johnston, M. Yan and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 6983–6987 CrossRef CAS PubMed.
- M. K. Sahoo and E. Balaraman, Green Chem., 2019, 21, 2119–2128 RSC.
- Y. Wu, Z. Chen, W. C. Cheong, C. Zhang, L. Zheng, W. Yan, R. Yu, C. Chen and Y. Li, Chem. Sci., 2019, 10, 5345–5352 RSC.
- D. Zhang, T. Iwai and M. Sawamura, Org. Lett., 2020, 22, 5240–5245 CrossRef CAS PubMed.
- S. Srinath, R. Abinaya, A. Prasanth, M. Mariappan, R. Sridhar and B. Baskar, Green Chem., 2020, 22, 2575–2587 RSC.
- R. Yang, S. Yue, W. Tan, Y. Xie and H. Cai, J. Org. Chem., 2020, 85, 7501–7509 CrossRef CAS PubMed.
- W. Chen, H. Tang, W. Wang, Q. Fu and J. Luo, Adv. Synth. Catal., 2020, 362, 3905–3911 CrossRef CAS.
- P. Tarnow, C. Zordick, A. Bottke, B. Fischer, F. Kuhne, T. Tralau and A. Luch, Chem. Res. Toxicol., 2020, 33, 742–750 Search PubMed.
- T. Tsuchiya, Y. Takagi and H. Yamada, Bioorg. Med. Chem. Lett., 2000, 10, 203–207 CrossRef CAS PubMed.
- P. R. Verhoest, D. S. Chapin, M. Corman, K. Fonseca, J. F. Harms, X. Hou, E. S. Marr, F. S. Menniti, F. Nelson, R. O'Connor, J. Pandit, C. Proulx-Lafrance, A. W. Schmidt, C. J. Schmidt, J. A. Suiciak and S. Liras, J. Med. Chem., 2009, 52, 5188–5196 CrossRef CAS PubMed.
- T. Hille, T. Irrgang and R. Kempe, Chem.–Eur. J., 2014, 20, 5569–5572 CrossRef CAS PubMed.
- X. W. Zhang, G. Q. Jiang, S. H. Lei, X. H. Shan, J. P. Qu and Y. B. Kang, Org. Lett., 2021, 23, 1611–1615 CrossRef PubMed.
- A. E. Wendlandt and S. S. Stahl, Angew. Chem., Int. Ed., 2015, 54, 14638–14658 CrossRef CAS PubMed.
- H. Choi and M. P. Doyle, Chem. Commun., 2007, 745–747 RSC.
- T. Tanaka, K.-i. Okunaga and M. Hayashi, Tetrahedron Lett., 2010, 51, 4633–4635 CrossRef CAS.
- S. Furukawa, A. Suga and T. Komatsu, Chem. Commun., 2014, 50, 3277–3280 RSC.
- W. Zhou, Q. Tao, F. a. Sun, X. Cao, J. Qian, J. Xu, M. He, Q. Chen and J. Xiao, J. Catal., 2018, 361, 1–11 CrossRef CAS.
- D. Damodara, R. Arundhathi and P. R. Likhar, Adv. Synth. Catal., 2014, 356, 189–198 CrossRef CAS.
- Y. Han, Z. Wang, R. Xu, W. Zhang, W. Chen, L. Zheng, J. Zhang, J. Luo, K. Wu, Y. Zhu, C. Chen, Q. Peng, Q. Liu, P. Hu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 11262–11266 CrossRef CAS PubMed.
- X. Cui, Y. Li, S. Bachmann, M. Scalone, A. E. Surkus, K. Junge, C. Topf and M. Beller, J. Am. Chem. Soc., 2015, 137, 10652–10658 CrossRef CAS PubMed.
- X. Bi, T. Tang, X. Meng, M. Gou, X. Liu and P. Zhao, Catal. Sci. Technol., 2020, 10, 360–371 RSC.
- S. Mukhopadhyay, G. Rothenberg, D. Gitis and Y. Sasson, Org. Lett., 2000, 2, 211–214 CrossRef CAS PubMed.
- Y. Zhang, S. Pang, Z. Wei, H. Jiao, X. Dai, H. Wang and F. Shi, Nat. Commun., 2018, 9, 1465 CrossRef PubMed.
- Y. Wang, H. Arandiyan, J. Scott, A. Bagheri, H. Dai and R. Amal, J. Mater. Chem. A, 2017, 5, 8825–8846 RSC.
- T. Rui, G. Lu, X. Zhao, X. Cao and Z. Chen, Chin. Chem. Lett., 2021, 32, 685–690 CrossRef CAS.
- G. Lu, H. Shan, Y. Lin, K. Zhang, B. Zhou, Q. Zhong and P. Wang, J. Mater. Chem. A, 2021, 9, 25128–25135 RSC.
- F. Qin, X. Tian, Z. Guo and W. Shen, ACS Sustainable Chem. Eng., 2018, 6, 15708–15719 CrossRef CAS.
- Y. Lin, F. Wang, J. Yu, X. Zhang and G. Lu, J. Hazard. Mater., 2022, 425, 127763 CrossRef CAS PubMed.
- A. B. Jorge, R. Jervis, A. P. Periasamy, M. Qiao, J. Y. Feng, L. N. Tran and M. M. Titirici, Adv. Energy Mater., 2020, 10, 1902494 CrossRef CAS.
- H. Wang, Y. Shao, S. Mei, Y. Lu, M. Zhang, J. K. Sun, K. Matyjaszewski, M. Antonietti and J. Yuan, Chem. Rev., 2020, 120, 9363–9419 CrossRef CAS PubMed.
- Y. Chen, J. Zhang, M. Zhang and X. Wang, Chem. Sci., 2013, 4, 3244–3248 RSC.
- X. Chen, Q. Shen, Z. Li, W. Wan, J. Chen and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 654–666 CrossRef CAS PubMed.
- H. Yang, X. Cui, X. Dai, Y. Deng and F. Shi, Nat. Commun., 2015, 6, 6478 CrossRef PubMed.
- W. Ai, Z. Luo, J. Jiang, J. Zhu, Z. Du, Z. Fan, L. Xie, H. Zhang, W. Huang and T. Yu, Adv. Mater., 2014, 26, 6186–6192 CrossRef CAS PubMed.
- G. Lu, K. Sun, Y. Lin, Q. Du, J. Zhang, K. Wang and P. Wang, Nano Res., 2022, 15, 603–611 CrossRef CAS.
- K. Sun, D. Li, G. Lu and C. Cai, ChemCatChem, 2020, 13, 373–381 CrossRef.
- X. Xie, J. Shi, Y. Pu, Z. Wang, L. L. Zhang, J. X. Wang and D. Wang, J. Colloid Interface Sci., 2020, 571, 100–108 CrossRef CAS PubMed.
- H. Zhang, C. Zhang, Y. Zhang, P. Cui, Y. Zhang, L. Wang, H. Wang and Y. Gao, Appl. Surf. Sci., 2019, 487, 616–624 CrossRef CAS.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M. M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- K. Sun, H. Shan, G. P. Lu, C. Cai and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 25188–25202 CrossRef CAS PubMed.
- K. Sun, S. Chen, Z. Li, G. Lu and C. Cai, Green Chem., 2019, 21, 1602–1608 RSC.
- K. Sun, J. Sun, G. Lu and C. Cai, Green Chem., 2019, 21, 4334–4340 RSC.
- D. B. Eremin and V. V. Fokin, J. Am. Chem. Soc., 2021, 143, 18374–18379 CrossRef CAS PubMed.
- L. Shao, Q. Xie, H. Sun, L. Xiao and W. Li, Synthesis, 2017, 49, 4845–4852 CrossRef.
- S. B. Gohain, M. Basumatary, P. K. Boruah, M. R. Das and A. J. Thakur, Green Chem., 2020, 22, 170–179 RSC.
- C. Tang and T. Asefa, J. Mater. Chem. A, 2020, 8, 12285–12290 RSC.
- T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed.
- L. Lin, Y. Ge, H. Zhang, M. Wang, D. Xiao and D. Ma, JACS Au, 2021, 1, 1834–1848 CrossRef CAS PubMed.
- C. J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68–82 RSC.
- C. Xie, L. Lin, L. Huang, Z. Wang, Z. Jiang, Z. Zhang and B. Han, Nat. Commun., 2021, 12, 4823 CrossRef CAS PubMed.
- C. Liu, C. Zhu, Y. Cai and H. Jiang, Angew. Chem., Int. Ed., 2021, 60, 12038–12045 CrossRef CAS PubMed.
- G. Jaiswal, M. Subaramanian, M. K. Sahoo and E. Balaraman, ChemCatChem, 2019, 11, 2449–2457 CrossRef CAS.
- V. P. Hitaishi, R. Clement, L. Quattrocchi, P. Parent, D. Duche, L. Zuily, M. Ilbert, E. Lojou and I. Mazurenko, J. Am. Chem. Soc., 2020, 142, 1394–1405 CrossRef CAS PubMed.
- S. Liu, S. Chen, A. Yu, Y. Hu, B. Yu, H. Wang, P. Peng and F.-F. Li, Green Chem., 2020, 22, 7839–7847 RSC.
- F. Tang, L. Wang, G. Zhang, M. Zhang and Y.-N. Liu, Ind. Eng. Chem. Res., 2019, 58, 5543–5551 CrossRef CAS.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6, 7084–7091 CrossRef CAS PubMed.
- C. Hu and L. Dai, Adv. Mater., 2019, 31, e1804672 CrossRef PubMed.
- X.-N. Tang, C.-Z. Liu, X.-R. Chen, Y.-Q. Deng, X.-H. Chen, J.-J. Shao and Q.-H. Yang, Carbon, 2019, 146, 147–154 CrossRef CAS.
- S. Wang, H. Jang, J. Wang, Z. Wu, X. Liu and J. Cho, ChemSusChem, 2019, 12, 830–838 CrossRef CAS PubMed.
- L. Yang, X. Zeng, W. Wang and D. Cao, Adv. Funct. Mater., 2018, 28, 1704537 CrossRef.
- M. A. Patel, F. Luo, M. R. Khoshi, E. Rabie, Q. Zhang, C. R. Flach, R. Mendelsohn, E. Garfunkel, M. Szostak and H. He, ACS Nano, 2016, 10, 2305–2315 CrossRef CAS PubMed.
- X. Hu, M. Fan, Y. Zhu, Q. Zhu, Q. Song and Z. Dong, Green Chem., 2019, 21, 5274–5283 RSC.
- Y. Wu, H. Yi and A. Lei, ACS Catal., 2018, 8, 1192–1196 CrossRef CAS.
- D. V. Jawale, E. Gravel, N. Shah, V. Dauvois, H. Li, I. N. Namboothiri and E. Doris, Chem.–Eur. J., 2015, 21, 7039–7042 CrossRef CAS PubMed.
- X. H. Li, J. S. Chen, X. Wang, J. Sun and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 8074–8077 CrossRef CAS PubMed.
- X. Feng, Y. Pi, Y. Song, Z. Xu, Z. Li and W. Lin, ACS Catal., 2021, 11, 1024–1032 CrossRef CAS.
- Y. Wang, P. Li, J. Wang, Z. Liu, Y. Wang, Y. Lu, Y. Liu, L. Duan, W. Li, S. Sarina, H. Zhu and J. Liu, Catal. Sci. Technol., 2021, 11, 4429–4438 RSC.
- D. Delorme, D. Dube, Y. Ducharme, E. L. Grimm, R. Friesen and C. Lepine, (Merck Frosst, Canada), US Pat. 5552437, Sept 3, 1996.
- T. Utsugi, K. Aoyagi, T. Asao, S. Okazaki, Y. Aoyagi, M. Sano, K. Wierzba and Y. Yamada, Jpn. J. Cancer Res., 1997, 88, 992 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01838a |
| This journal is © The Royal Society of Chemistry 2022 |