
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature-responsive Pickering high internal phase emulsions for recyclable efficient interfacial biocatalysis†
Chao
Wang
 ab,
Hui
Chi
a,
Fan
Zhang
ab,
Xinyue
Wang
ab,
Jiarui
Wang
ab,
Hao
Zhang
a,
Ying
Liu
a,
Xiaona
Huang
a,
Yungang
Bai
*a,
Kun
Xu
*a and
Pixin
Wang
ab
ab,
Hui
Chi
a,
Fan
Zhang
ab,
Xinyue
Wang
ab,
Jiarui
Wang
ab,
Hao
Zhang
a,
Ying
Liu
a,
Xiaona
Huang
a,
Yungang
Bai
*a,
Kun
Xu
*a and
Pixin
Wang
ab
aKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, PR China. E-mail: byungang@ciac.ac.cn; xukun@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei 230026, PR China
First published on 10th June 2022
Abstract
The field of biocatalysis is expanding owing to the increasing demand for efficient low-cost green chemical processes. However, a feasible strategy for achieving product separation, enzyme recovery, and high catalytic efficiency in biocatalysis remains elusive. Herein, we present thermoresponsive Pickering high internal phase emulsions (HIPEs) as controllable scaffolds for efficient biocatalysis; these HIPEs demonstrate a transition between emulsification and demulsification depending on temperature. Ultra-high-surface-area Pickering HIPEs were stabilized by Candida antarctica lipase B immobilized on starch particles modified with butyl glycidyl ether and glycidyl trimethyl ammonium chloride, thus simplifying the separation and reuse processes and significantly improving the catalytic efficiency. In addition, the switching temperature can be precisely tuned by adjusting the degree of substitution of the modified starches to meet the temperature demands of various enzymes. We believe that this system provides a green platform for various interfacial biocatalytic processes of industrial interest.
Introduction
Enzymes, which are natural biocatalysts with high specificity and high efficiency, are widely used in biotechnology and industry and have achieved great success in catalysing the production of essential compounds under mild and sustainable conditions.1–5 In general, enzymatic catalysis mainly occurs at the oil/water interface because enzymes need an aqueous environment for their activities, whereas most substrates are oil-soluble.6,7 Therefore, enzymes are often used in two-phase organic solvent/water systems. However, the reaction rate and efficiency obtained by such systems are extremely limited by the interface area and mass transfer resistance.8,9 Furthermore, high-speed shear stirring and long-term exposure to the oil/water interface lead to irreversible denaturation and inactivation of enzymes.10To address these limitations, Pickering emulsions, which are stabilised by colloidal particles, are emerging as an innovative platform for enhancing the biocatalytic process in biphasic systems because of their advantageous properties, such as large interfaces, short molecule diffusion distances, and low toxicity of particles.11–16 Yang et al. used a Pickering emulsion stabilized by lipase-immobilized alginate gel microparticles coated with silanized titania nanoparticles for biphasic catalysis.14 Not only did this catalytic system exhibit good circulation ability, but the emulsion also exhibited good storage stability. By adding a small amount of a solid-particle emulsifier, Wei et al. converted a traditional biphasic enzymatic reaction into an oil-in-water Pickering emulsion system that did not require stirring and having a large reaction interface area and short molecular distance.17 The Candida antarctica lipase B (CALB)-catalyzed kinetic resolution hydrolysis of racemic esters showed better reaction efficiency and enantioselectivity in this system than in the traditional two-phase system. Clearly, Pickering emulsions play a positive role in enzyme catalysis. However, once the reaction is complete, the processes of separating and recirculating the enzyme, usually centrifugation and filtration, respectively, are required. These processes are time-consuming and can lead to enzyme depletion and inactivation because Pickering emulsions containing a dense layer of particles at the interface are extremely stable.18–20 In addition, product separation and emulsion recovery remain problematic.13,21 Thus, for sustainable and green biocatalysis in Pickering emulsions, efficient separation of products, recycling of catalysts, and recovery of emulsions are the major objectives.15
Fortunately, recently developed ‘smart emulsifiers’ that respond to external stimuli, such as pH,22–24 temperature,25,26 light27 and chemicals,28–30 enable on-demand switching control of emulsification and demulsification, thus facilitating the separation and recovery of catalysts and making the process more sustainable and environment-friendly. For example, Li et al. designed a light-responsive Pickering emulsion stabilised by silica microspheres functionalized with surface-loaded Pd and an azobenzene-based ionic liquid surfactant. This system can switch between emulsification and demulsification in response to UV and visible light, successfully simplifying the process of product separation and catalyst recovery.31 Compared to traditional catalysts, biocatalysts have certain advantages; for example, they can catalyse reactions at room temperature and normal pressure, can achieve high reaction rates, and are available at lower prices. The main disadvantage of biocatalysts is that they are vulnerable to external conditions, such as the temperature and pH range of the reaction.32,33 For successful biocatalysis, in addition to controllable switching properties, it is necessary to ensure the activity and stability of enzymes when stabilization/destabilization occurs. Therefore, relatively mild external stimuli must be considered while designing recyclable biocatalytic systems to ensure high efficiency when applied in reaction cycles.
High internal phase emulsions (HIPEs), usually defined as highly concentrated emulsions in which the volume fraction of the internal phase exceeds 0.74,34 have shown exceptional performance in applications like porous materials,35,36 templates for gels,37 and nutraceutical containers26,38 owing to their unique structure and high internal phase ratio. In theory, biocatalysis in an HIPE system should be more efficient than that in a conventional emulsion system because of the considerably large specific surface area of HIPEs.39 However, there have only been a few reports on enzymatic catalysis based on HIPEs in recent years. This is probably due to the high viscoelasticity of HIPEs and the extreme difficulty in separating enzymes and products from the reaction mixture using a straightforward and basic method like centrifugation and filtration.40–42 Moreover, centrifugation is a popular method for preparing HIPEs in many cases, rather than an ideal demulsification method.43,44 The bridging effect of emulsifier particles and the cross-linking networks of HIPEs also increase the barriers to demulsification, thus making relatively mild methods ineffective without any strong external interference, such as the addition of an acid or a base.45,46 However, drastic changes in the external environment will make it difficult to ensure enzyme activity. Therefore, although HIPEs show potential for application in biocatalysis, triggering HIPEs for on-demand demulsification remains challenging.
It would be of great significance for the field of biocatalysis if stimulus-responsive Pickering HIPEs could be constructed to simplify the separation and reuse processes, while simultaneously improving the catalytic efficiency. To the best of our knowledge, no related report has been published to date. Herein, we present a switchable Pickering HIPE as a controllable scaffold for efficient biocatalysis, which shows a transition between emulsification and demulsification depending on temperature (Scheme 1). Such thermoresponsive Pickering HIPEs are stabilized by Candida antarctica lipase B (CALB) immobilized on starch particles modified with butyl glycidyl ether (BGE) and glycidyl trimethyl ammonium chloride (GTAC). The main purpose of using BGE modification is to endow starch particles with thermoresponsive properties based on the reversible change in the hydrogen bond and solvation shell of the 2-hydroxy-3-butoxypropyl groups at different temperatures.47 The introduction of GTAC makes the starch particles positively charged and allows them to bind tightly with CALB via electrostatic forces. We used electrostatic interactions rather than covalent bonding to immobilize the enzymes to protect the enzyme structure from damage. Immobilizing lipase on starch particles has two main purposes: one is to make lipase adsorbed with starch particles at the interface to shorten the mass transfer distance and improve biocatalytic efficiency; the other is to simplify the reuse of the enzyme by fixing lipase on starch particles. In addition, starch is a green natural biopolymer that is an ideal candidate for biocatalysis because of its biodegradability, drivability, high availability, and low cost.48
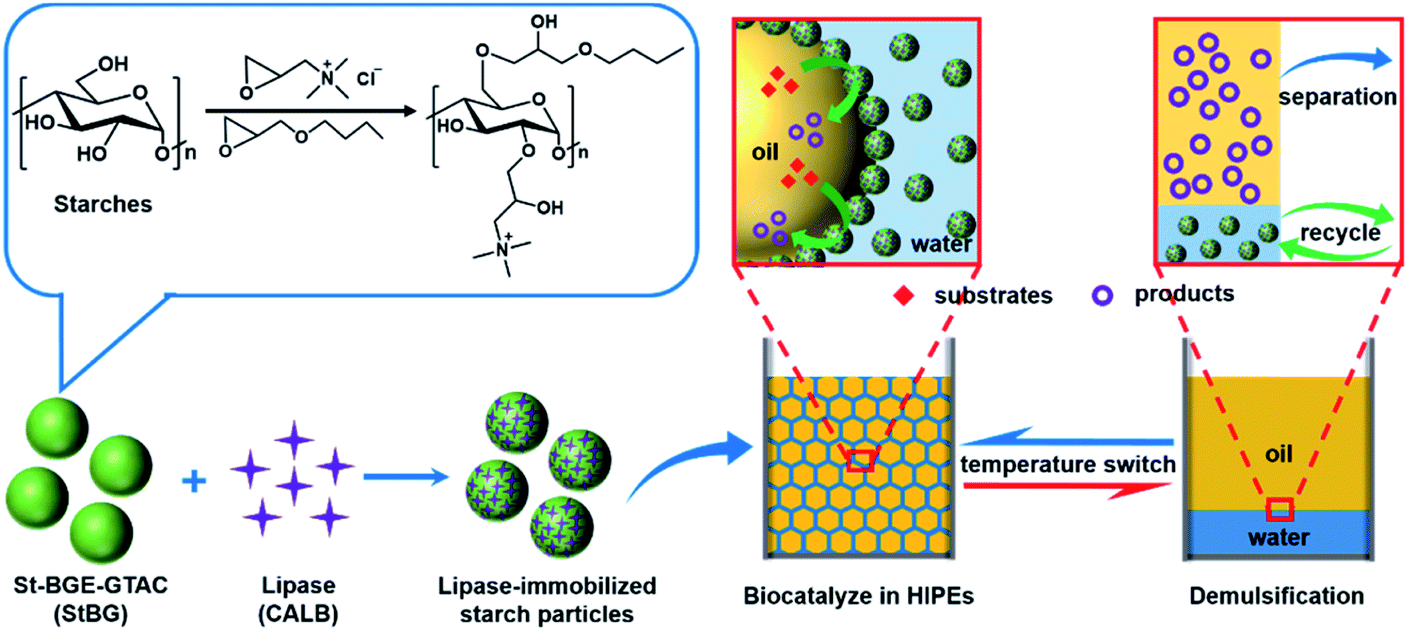 | ||
| Scheme 1 Schematic of thermoresponsive Pickering high-internal-phase emulsions (HIPEs) stabilised by lipase-immobilised starch particles applied to recycle interfacial biocatalysis. Included in the diagram are the modification method and chemical structure of starch (St) with butyl glycidyl ether (BGE) and glycidyl trimethyl ammonium chloride (GTAC), represented by StBG, lipase immobilisation (Candida antarctica lipase B [CALB]), temperature-switchable HIPEs (emulsification/demulsification), and the biocatalysis, product separation and lipase recovery processes. | ||
Experimental
Experimental procedures and characterization data are provided in the ESI.†Results and discussion
The synthesis and 1H NMR and FT-IR characterisations of the starch-based particles are described in the ESI (Fig. S1 and S2†). Both FT-IR and 1H NMR spectra confirmed that BGE and GTAC were successfully bonded to the starch molecules by etherification. The introduction of BGE and GTAC not only functionalized the starch particles but also improved their hydrophobicity, which allowed them to adsorb at the interface to stabilize the emulsion; natural starches are strongly hydrophilic owing to the presence of a large number of hydroxyl groups (Fig. S3†). We synthesised modified starches with different degrees of substitution (DS) and named them StBG-n. The details are presented in Table S1.† We consider StBG-3 as an example in the following discussion.The modified starch (StBG-3) was dispersed in water at a concentration of 1 wt% and mixed with the same volume of 50 mM Tris–HCl at room temperature (25 °C). A colloidal starch particle dispersion solution was obtained by the electrostatic interaction between StBG and trometamol. The morphology of the obtained colloidal particles is shown in Fig. 1a, and the average diameter was 275 nm, as determined by dynamic light scattering (Fig. 1b). A gel-like emulsion was then obtained by homogenizing the dispersion with eight times the volume of hexane. The results of conductivity measurements (σ = 6.47 S m−1) and drop test experiments showed that StBG particles successfully stabilized the o/w Pickering HIPEs with an oil![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water ratio of 8:2. The emulsion droplets presented a special irregular polyhedral shape (Fig. 1c) owing to the extrusion of droplets, with the volume of the inner phase exceeding the maximum packing density. This is an obvious characteristic of HIPEs.39 Rheological characterization of HIPEs showed that the storage modulus was higher than the corresponding loss modulus in the test frequency range of 0.1–100 rad s−1, indicating that the prepared emulsions exhibited elastic or solid-like behaviour and confirmed the formation of the gel network (Fig. 1d).40
:water ratio of 8:2. The emulsion droplets presented a special irregular polyhedral shape (Fig. 1c) owing to the extrusion of droplets, with the volume of the inner phase exceeding the maximum packing density. This is an obvious characteristic of HIPEs.39 Rheological characterization of HIPEs showed that the storage modulus was higher than the corresponding loss modulus in the test frequency range of 0.1–100 rad s−1, indicating that the prepared emulsions exhibited elastic or solid-like behaviour and confirmed the formation of the gel network (Fig. 1d).40
 | ||
| Fig. 1 (a) Microscopic appearance of StBG-3 at 25 °C (T < lower critical solution temperature [LCST]) and 50 °C (T > LCST) observed by field emission scanning electron microscopy. (b) Size distribution of StBG-3 at different temperatures determined by dynamic light scattering. The sample concentration was 0.5 wt% and the dispersion medium was 50 mM Tris–HCl buffer. (c) Photograph and laser confocal images of HIPEs stabilised by 0.5 wt% StBG-3 with a water/oil (water/hexane) ratio of 2:8. Details of the homogenisation are given in the ESI.† Scale bar: 50 μm. (d) The rheological characterization of the HIPEs which was performed at room temperature with a rheometer by a plate–plate mode at varied frequencies (0.1 to 10 Hz). | ||
As expected, the prepared Pickering HIPEs exhibited excellent thermoresponses. Specifically, the emulsion state was maintained at low temperatures and was immediately demulsified after heating to a certain temperature; this process was completely reversible (Fig. 2a). After ten cycles, stable gel-like HIPEs could still be obtained, and the size and microstructure of the emulsion did not change significantly (Fig. S4†). As stabilisers, StBG particles play an important role in the switching process of the Pickering HIPEs. The hydrogen bonds between the modified starch macromolecules and the water molecules were strengthened or weakened in response to the changing temperature (Fig. S5†), which corresponded to the transition of the interfacial activity of the StBG particles. This behaviour was verified by the change in the water/hexane interfacial tension at different temperatures (Fig. 2b and c). Compared to the pure water/hexane interface, the addition of StBG particles significantly reduced the interfacial tension at lower temperatures. However, the effect of StBG particles on reducing this interfacial tension became weaker with increasing temperature, until the interfacial activity was almost completely lost (from 4.40 mN m−1 at 5 °C to 36 mN m−1 at 60 °C). As the temperature increased, the hydrogen bonds between the modified starch and the water molecules were weakened, and the solvated layer was destroyed, resulting in aggregation of the StBG particles (Fig. 1a, b and S6†) and the inability to be adsorbed at the interface. Therefore, the switching ‘on’ and ‘off’ of emulsion was closely related to the phase transition behaviour of StBG particles (Fig. 2d and e). Specifically, emulsions stabilized by StBG particles can be emulsified at temperatures below the lower critical solution temperature (LCST), partially demulsified at a ‘window temperature’, and demulsified at temperatures above the LCST. The LCST was defined as the temperature at which the UV transmittance of an aqueous dispersion of 0.5 wt% StBG at 500 nm decreased by 50% during the heating process. This phenomenon occurred for all StBGs with different DS values (Fig. S7 and S8†). This also enabled the switching of the emulsion to be quantified and provided an accurate reference for the prediction of the emulsification and demulsification temperatures. Therefore, in this study, we refer to LCST as the switching temperature. The GTAC group has stronger hydrophilicity than BGE; therefore, by adjusting the DS of GTAC, the switching temperature can be precisely tailored to meet the temperature requirements of various enzymes. The detailed results are summarised in Table S1.† Pickering HIPE as a biocatalytic support is also crucial for ensuring that the enzyme is not damaged during the entire process and that it maintains a high-activity state. In this study, we used StBG-3 as a stabiliser and CALB as a biocatalyst to investigate the specific performance of thermoresponsive Pickering HIPEs in biocatalysis. In this study, starch particles with and without enzymes were named StBG and lipase@StBG, respectively.
 | ||
| Fig. 2 (a) Photographs of the reversible emulsification/demulsification process of HIPEs stabilised by StBG-3. (b) Time dependence of interfacial tension at the water/hexane interface at different temperatures, in which the aqueous phase contained 0.5 wt% StBG-3 particles. (c) Equilibrium interfacial tension at different temperatures. (d) Transmittance changes at 500 nm for 0.5 wt% aqueous dispersions of StBG-3 at a heating rate of 1 °C min−1 from 0 to 80 °C. (e) Photographs of emulsions stabilized by StBG-3 at different temperatures. Unless otherwise mentioned, all emulsions in this experiment were obtained at a water/hexane ratio of 2:8, emulsifier concentration of 0.5 wt%, and homogenization at 1000 r min−1 for 120 s. | ||
The quaternary amine groups derived from GTAC on the StBG colloidal particles provided a strong positive electrostatic charge, and the zeta potential of the particles was maintained above +30 mV between pH 3 and 11. On the other hand, CALB was electronegative at pH 7.4 (−4.5 mV). Therefore, CALB readily combined with the quaternary amine groups owing to electrostatic interactions (Fig. 3a). Therefore, lipase@StBG was obtained by dissolving a certain amount of CALB (0.5 mg mL−1) in a dispersion of StBG particles in 50 mM Tris–HCl buffer (pH 7.4). Note that HIPE is a tightly packed structure (Fig. 1c) and it is difficult to observe the distribution of lipase in the emulsion (adsorbed at the interface with starch particles or dispersed in the aqueous phase). Therefore, in order to more clearly observe and confirm the distribution of lipase in the emulsion, we designed a group of control experiments with the water/oil ratio of 5:5. Confocal laser scanning microscopy (CLSM) of the emulsions stabilized by fluorescent-labelled lipase@StBG showed that most of the enzymes were adsorbed at the o/w interface (Fig. 3b). As a control, emulsions stabilized by starches modified only with BGE (StB) which were electronegative at all pH levels (Fig. S9†), and CLSM showed that the enzymes were completely dispersed in the aqueous phase (Fig. S10†). This phenomenon indicated that lipase CALB was indeed immobilized on the StBG particles and that the enzyme was also fixed to the interface when the particles stabilized the emulsions. Compared with StBG, the temperature responsiveness of lipase@StBG was not affected, except for its larger size (Fig. S11 and S12†). This may be due to electrostatic interactions that facilitate aggregation of StBG, but do not affect the emulsification and demulsification of the Pickering HIPEs. This was because the amphiphilicity of the particles was not significantly changed by lipase.
 | ||
| Fig. 3 (a) Zeta potentials of 0.5 wt% StBG-3 aqueous dispersions at pH 3–12 and 0.5 mg mL−1 CALB at pH 7.4 measured by electrophoretic light scattering at 25 °C. (b) Confocal laser scanning microscopy images of emulsions stabilised by fluorescent-labelled lipase@StBG-3; the w/o ratio was 5:5. (c) Hydrolysis conversation rate of hexyl hexanoate for different biocatalytic systems; the ratios within parentheses represent the water/oil ratios. (d) Specific activities of lipase under the five conditions shown in (c), which were measured within 30 min. (e) Visual depiction of the recyclable route for biocatalysis; the aqueous phase was a lipase@StBG dispersion containing 0.5 mg mL−1 of CALB and 0.5 wt% of StBG-3; the organic phase was composed of solvent (hexane) and substrate (100 mM hexyl hexanoate). (f) Chart of the conversion due to hydrolysis (green) and specific activity of lipase (red) for every cycle of biocatalysis illustrated in (e). | ||
Subsequently, the biocatalytic performance of lipase@StBG in o/w Pickering HIPEs with a water/hexane ratio of 2:8 was investigated using the hydrolysis of hexyl hexanoate [lipase@StBG (2:8)]. Four other systems were examined as references. First, free lipase was dissolved in water and mixed with eight times the volume of the hexane-containing substrate by continuous stirring for biphase interfacial catalysis [free lipase (2:8)]. Second, a free lipase solution with an equal volume of hexane was added to the conventional emulsions stabilized with the anionic surfactant sodium dodecyl sulfate (SDS) [lipase + SDS (5:5)]. Third, lipase@StBG particles were used to stabilize ordinary Pickering emulsions at a water/hexane ratio of 5:5 [lipase@StBG (5:5)]. Fourth, free lipase was dissolved in the aqueous phase of Pickering HIPEs stabilized by St-BGE (no immobilized enzyme) [lipase + StB (2:8)]. Except for the free lipase system, which required stirring, all other systems were left standing at room temperature (25 °C), and the lipase and substrate concentrations were the same. Fig. 3c shows the conversion rates of reactants to products in the five cases. All other systems had significantly higher conversions than the free lipase (2:8) system, even though they were unstirred and had lower o/w ratios [lipase + SDS (5:5) and lipase@StBG (5:5)]. In emulsions stabilized by surface-active molecules (e.g. surfactants or polymers), one of the phases is divided into micrometre-sized droplets dispersed within the other incompatible liquid, which dramatically increases the interfacial area available for chemical reactions compared to typical two-phase systems. Therefore, agitation is unnecessary because each droplet acts as a microcompartment that enhances the interaction between the catalyst and reactant. Among the aforementioned emulsions, the systems with the enzyme immobilized on the stabilizer performed better in interfacial biocatalysis, with higher reaction rates and conversions. This indicated that lipase was adsorbed at the interface with the stabilizer, which simultaneously reduced the mass transfer distance and enhanced the reaction rate. Moreover, owing to the high specific surface area supported by HIPE, the conversion in the lipase@StBG (2:8) system reached 80% within 2.5 h, which was significantly greater than those in other systems. The results showed that immobilizing the enzyme on the stabilizer particles and increasing the volume fraction of the internal phase are effective methods for enhancing catalytic activity in emulsion biocatalysis systems. Further evidence for the same was provided by the specific catalytic activity of the lipases, as shown in Fig. 3d. Specifically, the specific activity of the Pickering HIPEs system in which the enzyme was fixed on starch particles was 10.8867 U mg−1, which was 16, 14, 5.7, and 4.7 times greater, respectively, than those in the common emulsion system (lipase + SDS), typical two-phase systems (free lipase), the Pickering non-HIPE system with enzymes immobilized on starch particles (lipase@StBG), and the free enzyme Pickering HIPEs system (lipase + StB).
To reveal the facile separation and recyclability of thermoresponsive Pickering HIPEs stabilized by lipase@StBG particles, hydrolysis of hexyl hexanoate in water/hexane Pickering HIPEs was performed for 10 cycles, as shown in Fig. 3e. The entire catalytic reaction was performed at 25 °C for 6 h, and the emulsion was transferred to a water bath at 50 °C when the reaction reached equilibrium. After demulsification, the organic phase containing the product was removed directly and a new substrate was added before emulsification at 25 °C for another cycle. During the entire cyclic catalytic process, traditional methods that consume time and energy, such as centrifugal filtration, are completely avoided. The separation of products and recovery of lipase-immobilized StBG particles can only be achieved by changing the ambient temperature. Notably, after demulsification, CLSM revealed that the enzymes originally on the interface were preferentially returned to the aqueous phase (light green) rather than to the organic phase (dark), which ensured that there was no loss of enzymes during the cycle (Fig. S13†). As shown in Fig. 3f, there was very little deactivation of lipase immobilized on the StBG particles during the hydrolysis process. The conversion of hexyl hexanoate was maintained above 80% in all cycles, and the specific activity of lipase remained as high as 10.1022 U mg−1 after 10 cycles. The precise tailoring of the switching temperature not only provides an ideal catalytic environment for enzymes but also ensures that enzymes will not be damaged by external stimuli during the cycle. Moreover, the separation and recovery processes greatly avoided enzyme loss during cycling.
Conclusions
Herein, we described the construction of enzyme-immobilized starch particles that stabilize Pickering HIPEs for application in biphasic biocatalysis. Highly hydrophilic starches were modified using BGE and GTAC to achieve suitable wettability and stabilize the target o/w Pickering HIPEs. The prepared Pickering HIPEs were temperature-responsive and could be destabilized, enabling the complete separation of the aqueous and organic phases upon heating. Moreover, these emulsions could be restored at lower temperatures after homogenization. The switching temperature can also be precisely tuned by adjusting the degree of GTAC substitution to meet different enzyme temperature requirements. Based on these Pickering HIPEs with on-demand demulsification, lipase-catalysed hydrolysis of hexyl hexanoate was investigated as a model. The Pickering HIPEs stabilized by lipase-immobilized starch particles demonstrated better performance in biocatalysis than the other systems because they provided a shorter mass transfer distance and a larger specific surface area. Moreover, the process of separating products and recovering enzymes was facile, bypassing traditional energy- and time-consuming methods, such as centrifugation and filtration. This is because the thermoresponsive Pickering HIPEs can be easily triggered to induce demulsification by changing the temperature. This high-performance Pickering emulsion system provides a green platform for various interfacial biocatalytic processes of industrial interest.Data availability
All experimental data and detailed procedures are available in the ESI.†Author contributions
Chao Wang: data curation, formal analysis, investigation and writing of the original draft. Hui Chi: formal analysis and project administration. Fan Zhang: formal analysis and investigation. Xinyue Wang: validation. Jiarui Wang: investigation. Hao Zhang: investigation. Ying Liu: validation. Xiaona Huang: formal analysis. Yungang Bai: funding acquisition, project administration, and validation. Kun Xu: funding acquisition, formal analysis, project administration, and validation. Pixin Wang: funding acquisition, formal analysis, and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Jilin Province Science and Technology Development Project Foundation (grant no. 20200708061YY) and Capital Care Foundation in Jilin Province Budget (grant no. 20210201124GX).Notes and references
- F. Hollmann, I. W. C. E. Arends, K. Buehler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226–265 RSC.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236–6249 RSC.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- X. M. Huang, Z. J. Luo, J. Guo, Q. J. Ruan, J. M. Wang and X. Q. Yang, J. Agric. Food Chem., 2020, 68, 8890–8899 CrossRef CAS PubMed.
- H. Jiang, L. Liu, Y. Li, S. Yin and T. Ngai, ACS Appl. Mater. Interfaces, 2020, 12, 4989–4997 CrossRef CAS PubMed.
- S. Wiese, A. C. Spiess and W. Richtering, Angew. Chem., Int. Ed., 2013, 52, 576–579 CrossRef CAS PubMed.
- P. Tundo and A. Perosa, Chem. Soc. Rev., 2007, 36, 532–550 RSC.
- K. Piradashvili, E. M. Alexandrino, F. R. Wurm and K. Landfester, Chem. Rev., 2016, 116, 2141–2169 CrossRef CAS PubMed.
- S. Crossley, J. Faria, M. Shen and D. E. Resasco, Science, 2010, 327, 68–72 CrossRef CAS PubMed.
- J. Shi, X. Wang, W. Zhang, Z. Jiang, Y. Liang, Y. Zhu and C. Zhang, Adv. Funct. Mater., 2013, 23, 1450–1458 CrossRef CAS.
- M. Pera-Titus, L. Leclercq, J. M. Clacens, F. De Campo and V. Nardello-Rataj, Angew. Chem., Int. Ed., 2015, 54, 2006–2021 CrossRef CAS PubMed.
- X. Yang, Y. Wang, R. Bai, H. Ma, W. Wang, H. Sun, Y. Dong, F. Qu, Q. Tang, T. Guo, B. P. Binks and T. Meng, Green Chem., 2019, 21, 2229–2233 RSC.
- A. M. B. Rodriguez and B. P. Binks, Soft Matter, 2020, 16, 10221–10243 RSC.
- F. Chang, C. M. Vis, W. Ciptonugroho and P. C. A. Bruijnincx, Green Chem., 2021, 23, 2575–2594 RSC.
- L. Wei, M. Zhang, X. Zhang, H. Xin and H. Yang, ACS Sustainable Chem. Eng., 2016, 4, 6838–6843 CrossRef CAS.
- Z. Ao, Z. Yang, J. Wang, G. Zhang and T. Ngai, Langmuir, 2009, 25, 2572–2574 CrossRef CAS PubMed.
- J. Li and H. D. Stover, Langmuir, 2010, 26, 15554–15560 CrossRef CAS PubMed.
- Z. Niu, J. He, T. P. Russell and Q. Wang, Angew. Chem., Int. Ed., 2010, 49, 10052–10066 CrossRef CAS PubMed.
- Z. Chen, C. Zhao, E. Ju, H. Ji, J. Ren, B. P. Binks and X. Qu, Adv. Mater., 2016, 28, 1682–1688 CrossRef CAS PubMed.
- Q. Chen, X. Cao, H. Liu, W. Zhou, L. Qin and Z. An, Polym. Chem., 2013, 4, 4092–4102 RSC.
- W. Li, B. Ju and S. Zhang, Carbohydr. Polym., 2020, 229, 115401 CrossRef CAS PubMed.
- W. Wijaya, P. Van der Meeren, C. H. Wijaya and A. R. Patel, Food Funct., 2017, 8, 584–594 RSC.
- S. Tsuji and H. Kawaguchi, Langmuir, 2008, 24, 3300–3305 CrossRef CAS PubMed.
- C. Wang, X. Pei, J. Tan, T. Zhang, K. Zhai, F. Zhang, Y. Bai, Y. Deng, B. Zhang, Y. Wang, Y. Tan, K. Xu and P. Wang, Int. J. Biol. Macromol., 2020, 146, 171–178 CrossRef CAS PubMed.
- Z. Chen, L. Zhou, W. Bing, Z. Zhang, Z. Li, J. Ren and X. Qu, J. Am. Chem. Soc., 2014, 136, 7498–7504 CrossRef CAS PubMed.
- M. Quesada, C. Muniesa and P. Botella, Chem. Mat., 2013, 25, 2597–2602 CrossRef CAS.
- S. Yu, D. Zhang, J. Jiang, Z. Cui, W. Xia, B. P. Binks and H. Yang, Green Chem., 2019, 21, 4062–4068 RSC.
- J. Jiang, S. Yu, W. Zhang, H. Zhang, Z. Cui, W. Xia and B. P. Binks, Angew. Chem., Int. Ed., 2021, 60, 11793–11798 CrossRef CAS PubMed.
- Z. Li, Y. Shi, A. Zhu, Y. Zhao, H. Wang, B. P. Binks and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 3928–3933 CrossRef CAS PubMed.
- F. Hasan, A. A. Shah and A. Hameed, Enzyme Microb. Technol., 2006, 39, 235–251 CrossRef CAS.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- J. M. Williams, Langmuir, 1991, 7, 1370–1377 CrossRef CAS.
- W. Zhu, Y. Zhu, C. Zhou and S. Zhang, RSC Adv., 2019, 9, 18909–18916 RSC.
- M. S. Silverstein, Prog. Polym. Sci., 2014, 39, 199–234 CrossRef CAS.
- S. Kovačič and M. S. Silverstein, Polym. Chem., 2017, 8, 6319–6328 RSC.
- H. Tan, G. Sun, W. Lin, C. Mu and T. Ngai, ACS Appl. Mater. Interfaces, 2014, 6, 13977–13984 CrossRef CAS PubMed.
- K. J. Lissant, J. Colloid Interface Sci., 1966, 22, 462–468 CrossRef CAS.
- R. Pal, Food Hydrocolloids, 2006, 20, 997–1005 CrossRef CAS.
- X. Li, X. Xu, L. Song, A. Bi, C. Wu, Y. Ma, M. Du and B. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 45493–45503 CrossRef CAS PubMed.
- Y. Zhu, S. Huan, L. Bai, A. Ketola, X. Shi, X. Zhang, J. A. Ketoja and O. J. Rojas, ACS Appl. Mater. Interfaces, 2020, 12, 11240–11251 CrossRef CAS PubMed.
- R. Foudazi, S. Qavi, I. Masalova and A. Y. Malkin, Adv. Colloid Interface Sci., 2015, 220, 78–91 CrossRef CAS PubMed.
- T. Zeng, Z. L. Wu, J. Y. Zhu, S. W. Yin, C. H. Tang, L. Y. Wu and X. Q. Yang, Food Chem., 2017, 231, 122–130 CrossRef CAS PubMed.
- M. N. Lee, H. K. Chan and A. Mohraz, Langmuir, 2012, 28, 3085–3091 CrossRef CAS PubMed.
- Y. Chevalier and M.-A. Bolzinger, Colloids Surf., A, 2013, 439, 23–34 CrossRef CAS.
- B. Ju, D. Yan and S. Zhang, Carbohydr. Polym., 2012, 87, 1404–1409 CrossRef CAS.
- Y. Tan, K. Xu, L. Li, C. Liu, C. Song and P. Wang, ACS Appl. Mater. Interfaces, 2009, 1, 956–959 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01746f |
| This journal is © The Royal Society of Chemistry 2022 |