
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-component nanodiscs via the thermal folding of amphiphilic graft copolymers with the adjusted flexibility of the main chain†
Tomoki
Nishimura
 *a,
Yusuke
Hatatani
b,
Mitsuru
Ando
c,
Yoshihiro
Sasaki
b and
Kazunari
Akiyoshi
*b
*a,
Yusuke
Hatatani
b,
Mitsuru
Ando
c,
Yoshihiro
Sasaki
b and
Kazunari
Akiyoshi
*b
aDepartment of Chemistry and Materials, Faculty of Textile Science and Technology, Shinshu University, 3-15-1, Tokida, Ueda, Nagano 386-8567, Japan. E-mail: nishimura_tomoki@shinshu-u.ac.jp
bDepartment of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
cDepartment of Regeneration Science and Engineering, Institute for Frontier Life and Medical Sciences, Kyoto University, Shogoin Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan
First published on 19th April 2022
Abstract
Nanodiscs have attracted considerable attention as structural scaffolds for membrane-protein research and as biomaterials in e.g. drug-delivery systems. However, conventional disc-fabrication methods are usually laborious, and disc fabrication via the self-assembly of amphiphiles is difficult. Herein, we report the formation of polymer nanodiscs based on the self-assembly of amphiphilic graft copolymers by adjusting the persistence length of the main chain. Amphiphilic graft copolymers with a series of different main-chain persistence lengths were prepared and these formed, depending on the persistence length, either rods, discs, or vesicles. Notably, polymer nanodiscs were formed upon heating a chilled polymer solution without the need for any additives, and the thus obtained nanodiscs were used to solubilize a membrane protein during cell-free protein synthesis. Given the simplicity of this disc-fabrication method and the ability of these discs to solubilize membrane proteins, this study considerably expands the fundamental and practical scope of graft-copolymer nanodiscs and demonstrates their utility as tools for studying the structure and function of membrane proteins.
Introduction
The self-assembly of amphiphiles represents a promising strategy to fabricate molecular assemblies with various morphologies, such as spherical and worm-like micelles, rods, lamellae, and vesicles.1–15 Among such assemblies, disc structures have a unique morphology that is characterized by a flat bilayer structure with a finite size. This structure allows discs to incorporate membrane proteins into their bilayer, and they are widely used as biomimetic membrane models to study the structure and function of membrane proteins.16–21 Recent advances in the application of discs have demonstrated the potential of nanodiscs as templates for the fabrication of inorganic nanoparticles,22–25 antigen-delivery carriers, i.e., vaccines,26–31 and bio-imaging materials.32–34Despite their important applications, the fabrication of nanodiscs via the self-assembly of amphiphiles remains extremely difficult. The major obstacle in nanodisc fabrication is that by definition, flat bilayer regions and highly curved edges must coexist in a single nanostructure,35 which makes nanodiscs a rare class of nanostructures. To achieve nanodisc structures, mixed systems of two or more phospholipids or surfactants have long been used.36–40 Lipid nanodiscs formed by mixtures of membrane-scaffold proteins and phospholipids41–44 or by mixtures of amphiphilic copolymers and phospholipids45–49 have been developed. Although these methods represent promising approaches toward the fabrication of nanodiscs, and even though the resulting nanodiscs have been used to study membrane proteins, these approaches suffer from difficulties associated with size control, multi-step fabrication procedures, or poor nanodisc stability. Block-copolymer self-assembly is an alternative approach to afford nanodiscs.50–58 Although this method is relatively simple compared to the aforementioned strategies, its drawbacks include the need for additives, difficulties associated with incorporating membrane proteins in the crystalline domains, and strong absorbance in the UV region of the hydrophobic segments. In summary, simple methods to produce practical nanodiscs to study membrane proteins remain virtually unknown.
Research into the fabrication of nanodiscs has so far only focused on the self-assembly of phospholipids, surfactants, and amphiphilic block copolymers, whereas the formation of nanodiscs based on the self-assembly of amphiphilic graft copolymers remains entirely unexplored. Amphiphilic graft copolymers, which consist of a continuous chain of amphiphilic units, form unique and well-defined molecular assemblies.59–66 Given that graft-copolymer self-assembly is achieved by folding of their main chains, we refer to amphiphilic graft polymers that fold into well-defined structures as macromolecular foldamers.65 A unique feature of the self-assembly of macromolecular foldamers is that the persistence length of the polymer main chain is one factor that controls the morphology of the resulting graft-copolymer assemblies.67 Therefore, we envisaged that if we could control the persistence length of the main chain of the amphiphilic graft polymers appropriately and then use the flexibility of the main chains to alter their packing in molecular assemblies, graft copolymer assemblies with flat bilayer areas and highly curved edges could be obtained without any additives.
Herein, we report that amphiphilic graft copolymers with a suitably adjusted persistence length of the main chain self-assemble into single-component nanodiscs in aqueous solution. The graft copolymers contain succinic-acid-functionalized poly(vinyl alcohol) (PVA) as the hydrophilic main chain and poly(propylene oxide)(PPO) as the hydrophobic and thermoresponsive graft chain (Fig. 1a). Amphiphilic graft copolymers with a series of different main-chain persistence lengths were prepared by adjusting the degree of succinic-acid substitution, and subsequently self-assembled into either vesicles, discs, or rods depending on the persistence length, which demonstrates that this method is a promising design strategy for various graft-copolymer assemblies (Fig. 1b). Notably, the nanodiscs can be prepared by simply heating the polymer solutions without any additives (Fig. 1c). We also demonstrate that the graft-copolymer nanodiscs act as solubilizing scaffolds for membrane proteins during a cell-free membrane-protein synthesis (Fig. 1d).
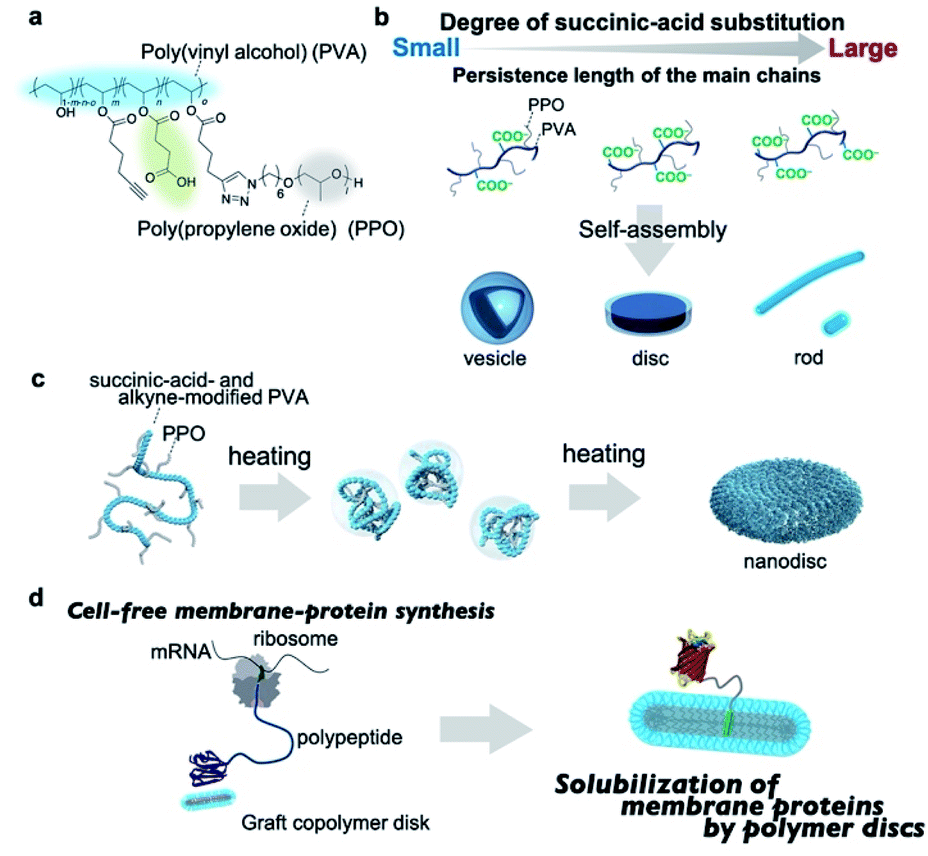 | ||
| Fig. 1 (a) Chemical structure of succinic-acid-functionalized poly(vinyl alcohol)(PVA)-g-poly(propylene oxide)(PPO). (b) Schematic illustration of the self-assembly of the graft copolymers into different nanostructures (vesicles, discs, rods, or short rods) depending on the persistence length of the main chain. (c) Schematic illustration of the thermal folding of the graft copolymer into nanodiscs. (d) Schematic illustration of the cell-free membrane-protein synthesis in the presence of the graft-copolymer nanodiscs. | ||
Results and discussion
Synthesis and characterization of succinic-acid-functionalized poly(vinyl alcohol) and the graft copolymers
We chose poly(vinyl alcohol) (PVA) as the hydrophilic main chain because it is a highly flexible polymer with hydroxyl groups that can be chemically modified with ease.68 The average molecular weight and the racemo content of the used PVA were ∼9 × 104 g mol−1 and 56%, respectively (Fig. S1†). To obtain hydrophilic PVA derivatives with a series of persistence lengths, we attempted to control the persistence length by adjusting the degree of succinic-acid substitution; the synthetic route is outlined in Scheme S1.† 5-Hexynoic acid-modified PVA was synthesized by the coupling of 5-hexynoic acid and PVA in the presence of dicyclohexyl carbodiimide. Due to the low coupling efficiency, we used an excess amount of 5-hexynoic acid against monomer unit of PVA to achieve the desired degree of the substitution of 5-hexynoic acid. The alkyne-functionalized PVA (Fig. S2†) was treated with succinic-acid anhydride in the presence of pyridine at 45 °C for 24 h, followed by dialysis against aqueous solutions and lyophilization to afford succinic-acid- and alkyne-modified PVAs (Table S1, Fig. S3–S7†). The degree of substitution (DS) of the succinic acid group was controlled by the molar feed ratio of succinic anhydride and hexynoic acid-modified PVA. To investigate the effect of the degree of succinic-acid substitution on the persistence length of the resulting polymers, we evaluated the persistence length using size-exclusion chromatography with multi-angle light scattering (SEC-MALS). The plots of the molecular weights versus the radius of gyration were fitted using the Benoit–Doty equation to determine the persistence length (p) of the polymers (Fig. S8†). As shown in Table 1, the persistence length of the graft copolymers increased from 1.3 nm to 2.7 nm with increasing degree of succinic-acid substitution, which indicates that the stiffness of the main chains increases with increasing degree of substitution. The persistence length of semi-flexible polyelectrolytes is given by the sum of the intrinsic persistence length (q0) and the electrostatic persistence length (qe).69 The latter is directly related to the ionic strength according to the Odijk–Skolnick–Fixman theory.70 To clarify the factors responsible for the increase in persistence length, we calculated the qe values of succinic-acid-modified PVAs under the following conditions: the ionic strength was set to 0.15 M and the succinic acid was fully deprotonated. The approximate qe values were less than 0.1 nm for all polymers (Table S2;† for details of the calculations, see ESI†), which can be rationalized in terms of the electrostatic shield effect; in the presence of a sufficient number of ions, the electrostatic repulsion of the carboxylate anions in the polymers will be suppressed. Accordingly, the contribution of qe is practically negligible. The main reason for the increase in the persistence length is, therefore, the increased steric demand of the main chain with increasing chemical modification. Overall, these data demonstrate that the persistence length of the main chain can be controlled by adjusting its chemical modification.| Polymer | M w (g mol−1) | M w/Mn | p (nm) | M L (nm−1) |
|---|---|---|---|---|
| PVA-COOH4 | 9.2 × 104 | 1.6 | 1.3 | 210 |
| PVA-COOH7 | 9.6 × 104 | 1.7 | 1.5 | 220 |
| PVA-COOH10 | 1.1 × 105 | 1.7 | 1.6 | 233 |
| PVA-COOH13 | 1.1 × 105 | 1.9 | 2.0 | 254 |
| PVA-COOH23 | 1.5 × 105 | 2.0 | 2.7 | 303 |
Self-assembly of amphiphilic graft copolymers is affected by the degree of substitution of the graft chain. We previously developed amphiphilic graft copolymers (e.g., pullulan-g-PPO, dextran-g-PPO) with the DS of the PPO group of 10 per 100 repeating units of polysaccharides.65,67 In contrast, PVA is used as the hydrophilic main chain instead of polysaccharides in this study. The length of the repeating unit of PVA (ca. 0.25 nm) is about half the length of glucose (ca. 0.5 nm), which is the repeating unit of pullulan and dextran. Therefore, the DS of the PPO group of 5 PPO per 100 repeating units of PVA was targeted to achieve comparable hydrophilic segment lengths and was fixed throughout the experiments so that the self-assembly behavior of the graft polymers would not be affected by differences in the degree of PPO substitution. In order for the DS of the PPO group to be 5 PPO per 100 repeat units of PVA, the DS of hexynoic acid should be higher than DS = 5. For this reason, the DS of hexynoic acid was targeted to DS = 6. Succinic-acid-functionalized PVA-g-PPO was synthesized via a “graft onto” method using a copper-catalyzed Huisgen reaction between azide-functionalized PPO [Mn(NMR) = 2.0 × 103 g mol−1; ĐM = 1.05]71 and alkyne- and succinic-acid-modified PVA. The degree of the substitution of PPO group was controlled by the molar feed ratio of the alkyne and carboxylic groups functionalized PVA and N3-PPO due to the high coupling efficiency of the click reaction. The graft copolymers are referred to as PVA-COOHi-g-PPO. Here, i indicates the degree of succinic-acid-group substitution per 100 repeating units of PVA. Detailed synthetic procedures are provided in the ESI,† together with representative 1H NMR spectra and size-exclusion chromatograms (Figs. S9–S15†).
Thermo- and pH-responsive behavior of the graft copolymers in aqueous solution
The graft copolymers have two stimulus-responsive groups, i.e., the carboxylic group and the PPO group. The former exhibits pH-responsive behavior, while the latter shows lower critical solution temperature (LCST)-like behavior in aqueous solution. Thus, we examined their stimulus-responsive behavior in aqueous solutions to determine the solution conditions in order to investigate their self-assembly behavior. We first conducted potentiometric titrations of the graft-copolymer solutions at 25 °C to determine their apparent pKa values. The initial pH value of the polymer solution was adjusted to 3 using an HCl solution (0.1 M), and the solution was titrated with a NaOH solution (0.01 M). The resulting titration curves showed broad pH responses in the range of pH = 3–9 (Fig. S16†). The apparent pKa values (5.5–6.1) were estimated based on the half-equivalence points. We then investigated the thermoresponsive behavior of the graft copolymers in a phosphate-buffered saline (PBS) solution (pH = 7.4; 0.15 M NaCl) using differential scanning calorimetry (DSC). DSC measurements of the polymer solutions (10 mg mL−1) showed an endothermic peak at around 15 °C during the heating process and an exothermic peak at around 11 °C during the cooling process (Fig. S17†). These peaks are derived from the dehydration and hydration of the PPO segments in the graft copolymers. We further investigated the effect of the polymer concentration on the phase-transition temperatures. As shown in Fig. S18,† the transition temperatures of the graft copolymers at the polymer concentration of 1.0 mg mL−1 are around 13–19 °C, suggesting that the graft copolymers self-assemble into nanoparticles above 25 °C.Self-assembly behavior of the graft copolymers in aqueous solution
Having determined the apparent pKa values and phase-transition temperatures of the polymers, we next examined their self-assembly behavior above the phase-transition temperature in the PBS buffer. The polymers were added to the PBS buffer and cooled below their phase-transition temperature with an ice bath. The resulting polymer solutions were incubated for at least 30 min at 25 °C. Since the pKa values of the polymers were in the range of 5.5–6.1, the carboxylic acid groups in the graft copolymers were almost completely deprotonated in the PBS buffer (pH = 7.4). The size-distribution curves obtained from dynamic light scattering (DLS) measurements of the representative graft-copolymer solutions (PVA–COOH4–g-PPO, PVA–COOH7–g-PPO, and PVA–COOH13–g-PPO) showed unimodal peaks, and the average hydrodynamic diameters of the graft copolymer assemblies were in the range of 40–70 nm (Fig. 2a–c). We next evaluated the weight-average molar masses and the radii of gyration (Rg) of the assemblies using field-flow fluctuation with multiangle light scattering (FFF-MALS; Fig. S19, and Table S3†).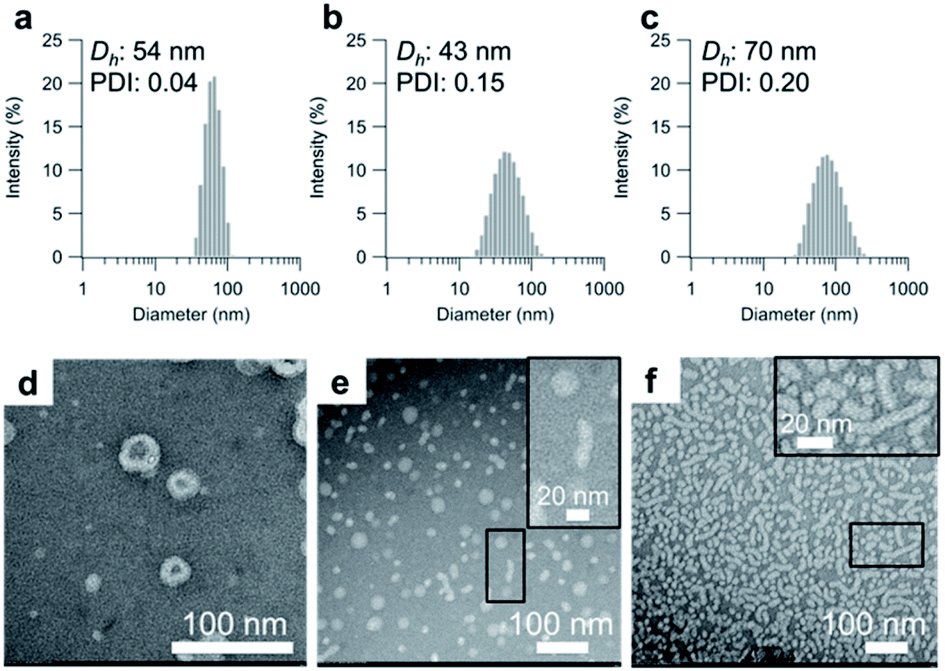 | ||
| Fig. 2 Size distribution of the self-assembled particles of (a) PVA–COOH4–g-PPO, (b) PVA–COOH7–g-PPO, and (c) PVA–COOH13–g-PPO in the PBS buffer. Concentration: [polymers] = 1.0 mg mL−1. TEM images of the self-assembled particles in (d) a PVA–COOH4–g-PPO solution, (e) a PVA–COOH7–g-PPO solution, and (f) a PVA–COOH13–g-PPO solution using negative staining with phosphotungstic acid. Inset: enlarged TEM images of the regions in the black circles; concentration: [polymers] = 1.0 mg mL−1. | ||
The molar masses of the PVA–COOH4–g-PPO, PVA–COOH7–g-PPO, and PVA–COOH13–g-PPO assemblies were calculated to be 1.8 × 107, 8.6 × 106, and 2.9 × 107 g mol−1, respectively. Based on the molar mass of the graft copolymers (1.6 × 105 g mol−1), the average aggregation numbers of the PVA–COOH4–g-PPO, PVA–COOH7–g-PPO, and PVA–COOH13–g-PPO assemblies were calculated to be 113, 54, and 181, respectively. The differences in size and aggregation numbers of the graft copolymer assemblies imply that the graft copolymers self-assemble into different nanostructures. The Rg values found for the PVA–COOH4–g-PPO (30 nm) and PVA–COOH7–g-PPO (22 nm) assemblies were almost identical to their hydrodynamic radii (PVA–COOH4–g-PPO: 27 nm; PVA–COOH7–g-PPO: 22 nm). On the other hand, the Rg of the PVA–COOH13–g-PPO assemblies (77 nm) was 2.2 times greater than its Rh (35 nm), suggesting that PVA–COOH13–g-PPO self-assembled into cylindrical structures.72,73 Transmission electron microscopy (TEM) images of a PVA–COOH4–g-PPO solution showed spherical structures with a circular border (Fig. 2d), which could originate from bilayer membrane structures. Conversely, the TEM image of a PVA–COOH7–g-PPO solution showed spherical and elongated objects with an average size of 40 nm (Fig. 2e), and that of a PVA–COOH13–g-PPO solution revealed cylindrical structures with lengths of 20–100 nm (Fig. 2f), which is in good agreement with the prediction from the MALS measurements. Since it is difficult to conclude the nanostructures of the self-assemblies from the TEM images, we next conducted small-angle X-ray scattering (SAXS) measurements. SAXS is widely recognized as a characterization technique for self-assembled nanoobjects in solutions. The scattering images are derived from hundreds of thousands of self-assemblies, and thus, SAXS data generally gives structural information with statistically high averages compared with that from TEM analysis. The SAXS profile of a PVA–COOH4–g-PPO solution was quite similar to those of previously reported pullulan–g-PPO and dextran–g-PPO polymer vesicle solutions (Fig. 3a).65,67 The scattering intensities in the low-q region decayed as a function of q−2 for PVA–COOH7–g-PPO (Fig. 3b) and of q−1 for PVA–COOH13–g-PPO (Fig. 3c). These data suggest the presence of thin plate assemblies such as discs and cylindrical assemblies, respectively. Using structural information based on the TEM observations and SAXS analyses, we fitted the SAXS profiles of the PVA–COOH4–g-PPO, PVA–COOH7–g-PPO, and PVA–COOH13–g-PPO assemblies using a spherical bilayer vesicle, core–shell disc, and core–shell cylinder model, respectively. The models fit the SAXS profiles of the graft-copolymer assemblies over the entire q range. Overall, these data confirmed that PVA–COOH4–g-PPO, PVA–COOH7–g-PPO, and PVA–COOH13–g-PPO self-assembled into spherical vesicles, discs, and cylindrical micelles, respectively. The obtained fitting parameters and the structural information of the self-assemblies are summarized in Fig. 3d–f and Table S4.† The average diameter of the vesicles and discs was 43 nm and 49 nm, respectively, while the average length of the cylindrical micelles was 44 nm. These values are in good agreement with the results obtained from the DLS measurements (Fig. 2a–c). To further validate the formation of nanodiscs, we performed AFM and cryo-TEM observations. As shown in Fig. S20,† nanoparticles with a uniform height in the range of 8 to 10 nm can be observed. The height is consistent with the thickness of nanodiscs revealed by SAXS analysis. Cryo-TEM image of the self-assembly of PVA–COOH7–g-PPO also showed spherical and elongated assemblies (Fig. S21†). The length of the elongated assemblies was ca. 9 nm in the short axis direction and 35 nm in the long axis direction, which was in good agreement with the diameters and the thickness of nanodiscs obtained from the SAXS analysis. Accordingly, the elongated assemblies and the circular assemblies seen in the TEM images are nanodiscs oriented perpendicular or parallel to the electron beam, respectively (Fig. 2e and S21†). Together, we concluded that PVA–COOH7–g-PPO self-assembled into nanodiscs. The stability of the nanodiscs in a PBS buffer solution (pH = 7.4, [NaCl] = 0.15 M) was also evaluated (Fig. S22†). The sizes of the nanodiscs remained essentially constant after one week, suggesting that the nanodiscs are stable in the PBS buffer solution.
 | ||
| Fig. 3 SAXS profiles (open circles) of self-assembled particles in (a) PVA–COOH4–g-PPO, (b) PVA–COOH7–g-PPO, and (c) PVA–COOH13–g-PPO solutions. The colored solid lines show the theoretical curves obtained using (a) a bilayer-vesicle model, (b) a core–shell-disc model (green), and (c) a core–shell-cylinder model (blue). Structural parameters of (d) the spherical vesicles, (e) the core–shell discs, and (f) the core–shell cylinders; concentration: [polymers] = 1.0 mg mL−1. | ||
To obtain further insight into the relationship between the persistence length of the main chain and the morphologies of the graft-copolymer assemblies, we also prepared PVA–COOH10–g-PPO and PVA–COOH23–g-PPO graft copolymers (persistence length for PVA-COOH10: 1.6 nm; PVA-COOH23: 2.7 nm; Table 1) and examined their self-assembly behavior in the PBS buffer. We found that PVA–COOH10–g-PPO self-assembled into nanodiscs with an average diameter of 35 nm (Fig. S23 and Table S5†). This data suggests that graft copolymers with a main-chain persistence length of around 1.5–1.6 nm form nanodiscs in aqueous solution. Interestingly, PVA–COOH23–g-PPO formed short rods with an average length and diameter of 24 nm and 12 nm, respectively (Fig. S24, and Table S6†). In contrast, cylindrical micelles with an average length of 44 nm were formed by the self-assembly of PHPMA-g-PPO and PVA–COOH13–g-PPO, which have main chains with a persistence length of 2.0 nm. With increasing persistence length of the main chain, the average length of the cylindrical micelles decreases. These results suggest that the persistence length also influences the length of the cylindrical micelles and that the length of cylindrical micelles could potentially be controlled by adjusting the main-chain persistence length. However, this is beyond the scope of this study, and further studies are needed to reach meaningful conclusions regarding this aspect.
To better understand the graft copolymer self-assembly, the persistence length of the main chains and the morphologies of the graft copolymer assemblies are summarized in Fig. 4.65,67,74 Graft copolymers with main-chain persistence lengths of less than 1.6 nm form plate-like structures, i.e., vesicles or discs. Conversely, in the case of main chains with a persistence length of around 2–2.7 nm, the graft copolymers self-assemble into cylindrical structures. With further increasing persistence length of the main chain (>2.9 nm), the graft copolymers formed short rods and spherical micelles. These results can be explained as follows. The main-chain extension of the molecular assemblies depends on the persistence length of the main chain, and the surface area of the hydrophilic region varies with the persistence length of the hydrophilic main chains.
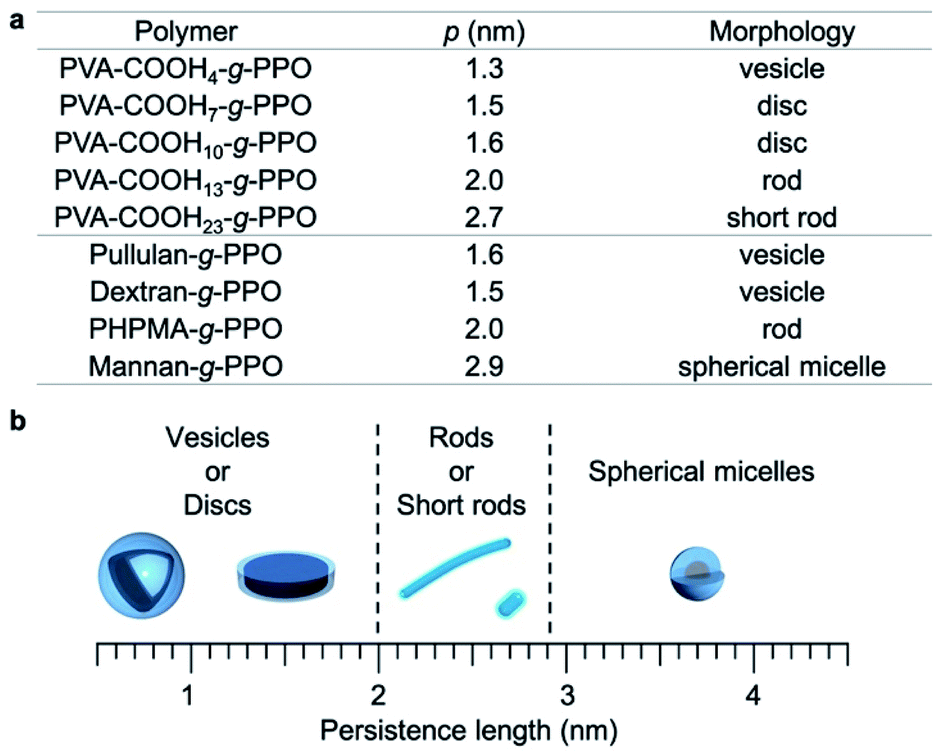 | ||
| Fig. 4 Relationship between the persistence length (p) of the hydrophilic main chains and the morphologies of the graft-copolymer assemblies. | ||
This results in a change of the packing parameter of the graft copolymers, which facilitate the morphological change of the molecular assemblies. Overall, it can be concluded that the morphologies of the molecular assemblies can be manipulated by adjusting the persistence length of the main chain under the same conditions via changing its degree of substitution.
Molecular mechanism for the formation of nanodiscs
The above studies confirmed that the morphologies of the amphiphilic graft copolymer assemblies can be tailored by adjusting the persistence length of the main chain. Disc-like nanostructures are not thermodynamically stable morphologies due to their large curvature at the peripheral regions. To reduce the unfavorable edge energy, mixed systems, e.g., mixtures of an amphiphile with long hydrophobic segments and an amphiphile with short hydrophobic segments, or membrane-scaffold proteins that cover the hydrophobic peripheral regions are often used. In contrast to these systems, the PVA–COOH7–g-PPO graft copolymers self-assembled into nanodiscs without any additives. We attribute this difference to the flexibility of the graft copolymers in the discs. In the self-assembly of block copolymers and low-molecular-weight amphiphiles, the thermodynamic packing in the assemblies, i.e., the extension and conformation of the hydrophilic and hydrophobic segments, is unequivocally determined by their degree of polymerization and/or chemical structures, and they usually form assemblies that contain a single curvature; conversely, it is difficult to form morphologies with large differences in curvature such as discs. Graft copolymers, on the other hand, consist of a continuous chain of amphiphilic units, and the flexibility of the main chain allows them to exhibit multiple packing modes in molecular assemblies. This allows the hydrophilic chains to fold compactly in the bilayer membrane of the discs, while simultaneously spreading widely to form a high curvature at the edges (Fig. S25†). Therefore, the graft copolymer can form discs without additives.In our previous study, we reported that the morphology of graft-copolymer assemblies can be manipulated by adjusting the persistence length of the main chains.67 Amphiphilic graft copolymers composed of pullulan or dextran (persistence length: 1.5–1.6 nm) as the main chain with a comparable degree of PPO substitution (Mn(NMR) = 2.0 × 103 g mol−1) self-assembled into spherical vesicles. When poly(2-hydroxypropyl methacrylamide) (PHPMA), whose persistence length (2.0 nm) is longer than those of pullulan and dextran, was used as the main chain, the graft copolymer formed cylindrical micelles. In the case of mannan-g-PPO, in which the more rigid mannan (persistence length: 2.9 nm) was used as the main chain, spherical micelles were formed. This variable self-assembly behavior is driven by the differences in the extension of the polymer main chain, which is controlled by the persistence length. The surface areas of the hydrophilic region of the amphiphilic units of the graft polymers vary, and thus, the graft polymers self-assemble into different morphologies. However, although the persistence length of PVA–COOH7 is very similar to those of pullulan and dextran, the morphologies of the resulting self-assemblies were different (PVA–COOH7–g-PPO: discs; pullulan-g-PPO and dextran-g-PPO: vesicles). In terms of chemical structure, a distinct difference is whether the polymers are negatively charged or neutral, which may lead to the differences in the self-assembled structures. We previously reported that pullulan-g-PPO forms disc-like intermediates, which fuse to form polymer vesicles.65 Based on the formation of disc-like structures by the self-assembly of pullulan-g-PPO, we predicted that PVA–COOH7–g-PPO would also form disc-like structures as intermediate assemblies. However, we expected that the negatively charged PVA-based discs would not fuse due to electrostatic repulsion and thus remain in nanodisc form. Assuming that this hypothesis is correct, we anticipated that we could prepare a PVA–COOH7–g-PPO polymer vesicle solution by reducing the electrostatic repulsion via increasing the ionic strength. To validate this hypothesis, we prepared a solution of PVA–COOH7–g-PPO that contained 300 mM NaCl. The resulting TEM image showed spherical objects with a thick circular border, and the SAXS profile of the polymer solution revealed two distinct Bragg peaks at q = 0.46 and 0.92 nm−1 (Fig. S26†). The q-values of these peaks exhibited a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, suggesting that they correspond to lamellar structures. These results clearly indicate that under higher-ionic-strength conditions, PVA–COOH7–g-PPO self-assembled into multilamellar vesicles. Accordingly, it seems feasible to conclude that the electrostatic repulsion between negatively charged disc-like intermediates inhibits their fusion and thus forms stable nanodiscs. Overall, the nanodiscs can be prepared by simply heating the graft copolymer solutions, and three factors are critical to fabricate nanodiscs based on graft copolymer self-assembly: (1) a grafted polymer structure; (2) appropriate flexibility of the main chain; (3) a negative charge.
:2 ratio, suggesting that they correspond to lamellar structures. These results clearly indicate that under higher-ionic-strength conditions, PVA–COOH7–g-PPO self-assembled into multilamellar vesicles. Accordingly, it seems feasible to conclude that the electrostatic repulsion between negatively charged disc-like intermediates inhibits their fusion and thus forms stable nanodiscs. Overall, the nanodiscs can be prepared by simply heating the graft copolymer solutions, and three factors are critical to fabricate nanodiscs based on graft copolymer self-assembly: (1) a grafted polymer structure; (2) appropriate flexibility of the main chain; (3) a negative charge.
In vitro membrane-protein synthesis in the presence of graft-copolymer nanodiscs
After confirming the formation of the graft-copolymer nanodiscs via adjusting the persistence length of the main chain, we then turned our attention to the possibility of using the graft-copolymer nanodiscs as a solubilizer for membrane proteins. Based on the non-crystallinity and flexibility of PPO, we anticipated that membrane proteins could be incorporated into the nanodiscs by adjusting the thickness of the hydrophobic layer to the size of the membrane proteins.75 To verify this possibility, we attempted to synthesize a membrane protein, i.e., tag red fluorescence protein (tagRFP)-fused transmembrane domain of platelet-derived growth factor receptor (PDGFR-TMD),76 in the presence of nanodiscs using a cell-free membrane-protein synthesis system (Fig. 5a).77–79 We chose a tagRFP-PDGFR-TMD fusion membrane protein because direct quantification of the properly folded membrane protein can be achieved by measuring the fluorescence intensity (Fig. 5b).80 The cell-free synthesis of tagRFP-PDGFR-TMD was performed for 4 h at 37 °C in the presence of PVA–COOH7–g-PPO or PVA–COOH10–g-PPO graft copolymer nanodiscs (0.22 mg mL−1). As a control, the membrane–protein synthesis was carried out in the absence of the nanodiscs. Western blot analysis was conducted to confirm that the target tagRFP-PDGFR-TMD (theoretical MW: 33.4 kDa) was produced. As shown in Fig. S17,† the bands were observed at approximately the same position as the 36 kDa protein-size marker, suggesting that the target membrane protein was successfully synthesized. We then evaluated whether the membrane proteins were solubilized in the nanodiscs and folded correctly. As shown in Fig. 5c, the fluorescence intensities of tagRFP in the reaction solution in the presence of the nanodiscs were four to five times higher than that of the solution without the nanodiscs. In the absence of nanodiscs, the lack of a scaffold to solubilize the produced TMDs may lead to aggregation, which results in low fluorescence intensity. On the other hand, in the presence of the nanodiscs, the produced TMD can be solubilized by incorporation into the nanodiscs, and thus, the fluorescence intensities increased. To evaluate the advantages of this nanodisc system over another scaffold system, we also conducted the cell-free membrane–protein synthesis in the presence of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) liposomes. We observed that the fluorescence intensity of tagRFP remained constant at DOPC concentrations of up to 1 mM (Fig. S28†). From the average diameter (140 nm) of the liposomes and thickness (3.7 nm) of the bilayer membranes, the surface area and aggregation number of the liposomes were calculated to be 6.1 × 104 nm3 and 1.6 × 105, respectively. The sum of the surface area of the liposomes (2.3 × 1020 nm3) at a concentration of 1 mM was calculated and compared to the sum of the surface area of PVA–COOH10–g-PPO graft copolymer nanodiscs (5.1 × 1019 nm3) at a concentration of 0.22 mg mL−1. Although the total surface area of the nanodiscs was less than one-fifth of that of the liposomes, the solubilization ability of the membrane protein of the nanodiscs was comparable to that of the liposomes, demonstrating their superior ability for solubilizing membrane proteins. A method that shares the same purpose as our approach is the SMA-lipid particles (SMALP) technology.81–83 However, our system has several advantages that SMALP technology. First, because of the use of cell-free protein synthesis, our system enables to incorporate only target membrane proteins into the nanodiscs. In contrast, cells overexpressing membrane proteins are used in the SMALP method, which means that other membrane proteins are included, resulting in complicating the purification process. Second, the in situ synthesis and incorporation of membrane proteins into the nanodiscs allows for rapid functional evaluation of the membrane proteins. However, at present, it is not clear how the membrane proteins are incorporated into the nanodiscs. The elucidation of the three-dimensional structure of the membrane proteins in the nanodiscs will therefore constitute an important future direction of research. Moreover, the solubilization capacity of the nanodiscs for other membrane proteins must be determined to understand their applicability. Overall, we have demonstrated that the graft copolymer nanodiscs can act as a solubilizer for membrane proteins.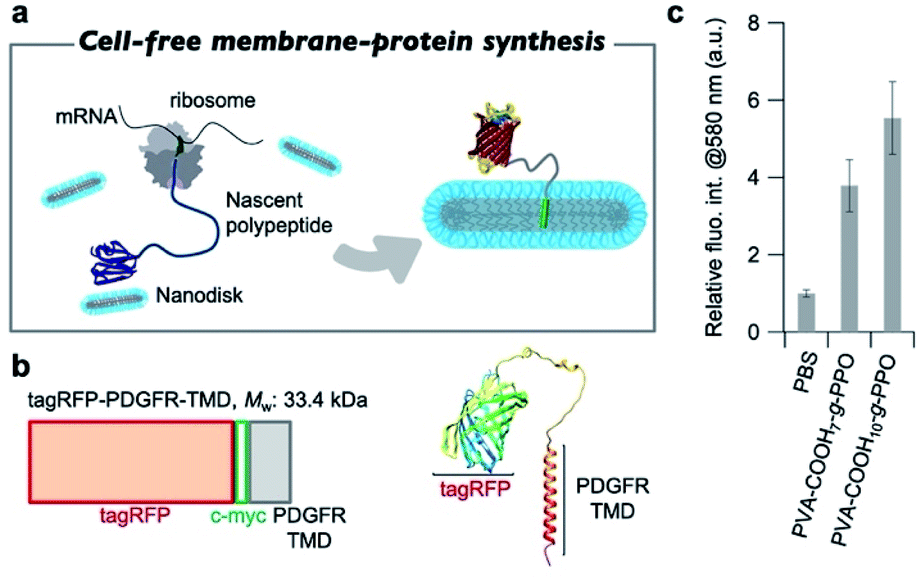 | ||
| Fig. 5 (a) Schematic illustration of cell-free membrane–protein synthesis in the presence of the graft-copolymer nanodiscs. (b) Illustration of the tagRFP-PDGFR-TMD gene and the predicted structure of the tagRFP-PDGFR-TMD. (c) Fluorescence intensity of tagRFP-PDGFR-TMD at 580 nm in PBS buffer at 37 °C in the reaction solution without the nanodiscs, in the presence of PVA–COOH7–g-PPO, and in the presence of PVA–COOH10–g-PPO; concentration: [polymers] = 0.22 mg mL−1; results represent mean values ± SD (n = 3). | ||
Conclusions
In summary, we prepared amphiphilic graft copolymers that consist of succinic-acid-modified-poly(vinyl alcohol)-graft-poly(propylene oxide) with a series of different main-chain persistence lengths by adjusting the degree of succinic-acid substitution. In aqueous solution, the graft copolymers self-assembled into spherical vesicles, discs, and cylindrical micelles with increasing persistence length of the main chain. Given that similar graft copolymers with a less flexible polymer (e.g., mannan) form spherical micelles, adjusting the persistence length of the amphiphilic graft copolymers enables the fabrication of polymer assemblies with desired morphologies. It should be noted here that the self-assembly of graft copolymers by simply warming a solution of the polymer with a main-chain persistence length of 1.5–1.6 nm can afford a single-component nanodisc without the addition of any additives. Moreover, membrane proteins can be incorporated spontaneously into the graft-copolymer discs during cell-free membrane–protein synthesis, and thus, the nanodiscs act as a solubilizer for membrane proteins. This system has several advantages compared to previously described methods such as mixed systems. First, the preparation of nanodiscs is simple; special equipment, expensive chemicals, or detergents are not needed. Secondly, the graft copolymers do not absorb ultraviolet or visible light, and spectroscopic absorbance interference does not occur between the discs and the membrane proteins. Thus, such graft-copolymer nanodiscs may have potential as a novel tool for studying the function of membrane proteins, and we envision that our study will inspire further research into graft-copolymer self-assembly and macromolecular foldamers.Data availability
Experimental data is available in the ESI† online.Author contributions
Conceptualization and supervision: TN, YS, KA; synthetic work, physical measurements and cell-free protein synthesis experiments: YH, MA, TN; writing: TN, YH: All authors commented on, and approved the final version of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS in the form of Grants-in-Aid for Scientific Research (S: 16H06313; B: 18H01845, 22H02140), the Asahi Glass Foundation, the MEXT Leading Initiative for Excellent Young Researchers, JST FOREST Program (JPMJFR201P), and MEXT Promotion of Distinctive Joint Research Center Program (JPMXP0621467946). SAXS experiments were conducted at the BL40B2 beamline of SPring-8 under proposal numbers 2020A0524, 2020A1070, and 2021A1065.Notes and references
- R. Nagarajan and E. Ruckenstein, Langmuir, 1991, 7, 2934–2969 CrossRef CAS
.
- Y. L. Chen, S. Chen, C. Frank and J. Israelachvili, J. Colloid Interface Sci., 1992, 153, 244–265 CrossRef CAS
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS PubMed
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS PubMed
- J. R. Howse, R. A. L. Jones, G. Battaglia, R. E. Ducker, G. J. Leggett and A. J. Ryan, Nat. Mater., 2009, 8, 507–511 CrossRef CAS PubMed
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed
- I. W. Hamley, Soft Matter, 2011, 7, 4122–4138 RSC
- X. Zhang and C. Wang, Chem. Soc. Rev., 2011, 40, 94–101 RSC
- C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618 CrossRef CAS PubMed
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC
- M. Ramanathan, L. K. Shrestha, T. Mori, Q. Ji, J. P. Hill and K. Ariga, Phys. Chem. Chem. Phys., 2013, 15, 10580–10611 RSC
- R. J. Williams, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2015, 6, 2998–3008 RSC
- H. M. G. Barriga, M. N. Holme and M. M. Stevens, Angew. Chem., Int. Ed., 2019, 58, 2958–2978 CrossRef CAS
- L. MacFarlane, C. Zhao, J. Cai, H. Qiu and I. Manners, Chem. Sci., 2021, 12, 4661–4682 RSC
- U. H. N. Dürr, M. Gildenberg and A. Ramamoorthy, Chem. Rev., 2012, 112, 6054–6074 CrossRef PubMed
- I. G. Denisov and S. G. Sligar, Nat. Struct. Mol. Biol., 2016, 23, 481–486 CrossRef CAS PubMed
- J. E. Rouck, J. E. Krapf, J. Roy, H. C. Huff and A. Das, FEBS Lett., 2017, 591, 2057–2088 CrossRef CAS PubMed
- I. G. Denisov and S. G. Sligar, Chem. Rev., 2017, 117, 4669–4713 CrossRef CAS PubMed
- M. L. Nasr, D. Baptista, M. Strauss, Z.-Y. J. Sun, S. Grigoriu, S. Huser, A. Plückthun, F. Hagn, T. Walz, J. M. Hogle and G. Wagner, Nat. Methods, 2017, 14, 49–52 CrossRef CAS PubMed
- S. G. Sligar and I. G. Denisov, Protein Sci., 2021, 30, 297–315 CrossRef CAS
- Y. Song, R. M. Dorin, R. M. Garcia, Y.-B. Jiang, H. Wang, P. Li, Y. Qiu, F. v. Swol, J. E. Miller and J. A. Shelnutt, J. Am. Chem. Soc., 2008, 130, 12602–12603 CrossRef CAS PubMed
- P. Mohanty, J. Lee, K. J. Glover and K. Landskron, Nanoscale Res. Lett., 2010, 6, 61 CrossRef PubMed
- R. M. Garcia, Y. Song, R. M. Dorin, H. Wang, A. M. Moreno, Y.-B. Jiang, Y. Tian, Y. Qiu, C. J. Medforth, E. N. Coker, F. van Swol, J. E. Miller and J. A. Shelnutt, Phys. Chem. Chem. Phys., 2011, 13, 4846–4852 RSC
- H. Sharma and E. E. Dormidontova, ACS Nano, 2017, 11, 3651–3661 CrossRef CAS PubMed
- R. Kuai, L. J. Ochyl, K. S. Bahjat, A. Schwendeman and J. J. Moon, Nat. Mater., 2017, 16, 489–496 CrossRef CAS PubMed
- R. Kuai, X. Sun, W. Yuan, Y. Xu, A. Schwendeman and J. J. Moon, Bioconjugate Chem., 2018, 29, 771–775 CrossRef CAS PubMed
- H.-J. Park, R. Kuai, E. J. Jeon, Y. Seo, Y. Jung, J. J. Moon, A. Schwendeman and S.-W. Cho, Biomaterials, 2018, 161, 69–80 CrossRef CAS PubMed
- R. Kuai, W. Yuan, S. Son, J. Nam, Y. Xu, Y. Fan, A. Schwendeman and J. J. Moon, Sci. Adv., 2018, 4, eaao1736 CrossRef PubMed
- P. Kadiyala, D. Li, F. M. Nuñez, D. Altshuler, R. Doherty, R. Kuai, M. Yu, N. Kamran, M. Edwards, J. J. Moon, P. R. Lowenstein, M. G. Castro and A. Schwendeman, ACS Nano, 2019, 13, 1365–1384 CAS
- L. Scheetz, P. Kadiyala, X. Sun, S. Son, A. Hassani Najafabadi, M. Aikins, P. R. Lowenstein, A. Schwendeman, M. G. Castro and J. J. Moon, Clin. Cancer Res., 2020, 26, 4369–4380 CrossRef CAS
- C. E. Carney, I. L. Lenov, C. J. Baker, K. W. MacRenaris, A. L. Eckermann, S. G. Sligar and T. J. Meade, Bioconjugate Chem., 2015, 26, 899–905 CrossRef CAS PubMed
- X. Mu, Y. Lu, F. Wu, Y. Wei, H. Ma, Y. Zhao, J. Sun, S. Liu, X. Zhou and Z. Li, Adv. Mater., 2020, 32, 1906711 CrossRef CAS PubMed
- P. Wong, L. Li, J. Chea, W. Hu, E. Poku, T. Ebner, N. Bowles, J. Y. C. Wong, P. J. Yazaki, S. Sligar and J. E. Shively, Bioconjugate Chem., 2020, 31, 743–753 CrossRef CAS PubMed
- S. J. Holder and N. A. J. M. Sommerdijk, Polym. Chem., 2011, 2, 1018–1028 RSC
- T. Zemb, M. Dubois, B. Demé and T. Gulik-Krzywicki, Science, 1999, 283, 816–819 CrossRef CAS PubMed
- H.-T. Jung, S. Y. Lee, E. W. Kaler, B. Coldren and J. A. Zasadzinski, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15318–15322 CrossRef CAS PubMed
- R. S. Prosser, F. Evanics, J. L. Kitevski and M. S. Al-Abdul-Wahid, Biochemistry, 2006, 45, 8453–8465 CrossRef CAS PubMed
- R. Matsui, M. Ohtani, K. Yamada, T. Hikima, M. Takata, T. Nakamura, H. Koshino, Y. Ishida and T. Aida, Angew. Chem., Int. Ed., 2015, 54, 13284–13288 CrossRef CAS PubMed
- N. Hassan, J. M. Ruso and Á. Piñeiro, Langmuir, 2011, 27, 9719–9728 CrossRef CAS PubMed
- I. G. Denisov, Y. V. Grinkova, A. A. Lazarides and S. G. Sligar, J. Am. Chem. Soc., 2004, 126, 3477–3487 CrossRef CAS PubMed
- V. L. Marin, T. H. Bayburt, S. G. Sligar and M. Mrksich, Angew. Chem., Int. Ed., 2007, 46, 8796–8798 CrossRef CAS PubMed
- N. T. Johansen, F. G. Tidemand, T. T. T. N. Nguyen, K. D. Rand, M. C. Pedersen and L. Arleth, FEBS J., 2019, 286, 1734–1751 CrossRef CAS PubMed
- P. Stepien, B. Augustyn, C. Poojari, W. Galan, A. Polit, I. Vattulainen, A. Wisnieska-Becker and T. Rog, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183420 CrossRef CAS PubMed
- T. J. Knowles, R. Finka, C. Smith, Y.-P. Lin, T. Dafforn and M. Overduin, J. Am. Chem. Soc., 2009, 131, 7484–7485 CrossRef CAS PubMed
- A. O. Oluwole, B. Danielczak, A. Meister, J. O. Babalola, C. Vargas and S. Keller, Angew. Chem., Int. Ed., 2017, 56, 1919–1924 CrossRef CAS PubMed
- K. Yasuhara, J. Arakida, T. Ravula, S. K. Ramadugu, B. Sahoo, J.-i. Kikuchi and A. Ramamoorthy, J. Am. Chem. Soc., 2017, 139, 18657–18663 CrossRef CAS PubMed
- Z. Stroud, S. C. L. Hall and T. R. Dafforn, Methods, 2018, 147, 106–117 CrossRef CAS PubMed
- L. E. Ball, L. J. Riley, W. Hadasha, R. Pfukwa, C. J. I. Smith, T. R. Dafforn and B. Klumperman, Biomacromolecules, 2021, 22, 763–772 CrossRef CAS PubMed
- A. Ramzi, M. Prager, D. Richter, V. Efstratiadis, N. Hadjichristidis, R. N. Young and J. B. Allgaier, Macromolecules, 1997, 30, 7171–7182 CrossRef CAS
- A. Constancis, R. Meyrueix, N. Bryson, S. Huille, J.-M. Grosselin, T. Gulik-Krzywicki and G. Soula, J. Colloid Interface Sci., 1999, 217, 357–368 CrossRef CAS PubMed
- T. P. Lodge, M. A. Hillmyer, Z. Zhou and Y. Talmon, Macromolecules, 2004, 37, 6680–6682 CrossRef CAS
- Z. Li, Z. Chen, H. Cui, K. Hales, K. Qi, K. L. Wooley and D. J. Pochan, Langmuir, 2005, 21, 7533–7539 CrossRef CAS PubMed
- W. F. Edmonds, Z. Li, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2006, 39, 4526–4530 CrossRef CAS
- H. Cui, Z. Chen, K. L. Wooley and D. J. Pochan, Macromolecules, 2006, 39, 6599–6607 CrossRef CAS
- L. Yin and M. A. Hillmyer, Macromolecules, 2011, 44, 3021–3028 CrossRef CAS
- S. Venkataraman, A. L. Lee, H. T. Maune, J. L. Hedrick, V. M. Prabhu and Y. Y. Yang, Macromolecules, 2013, 46, 4839–4846 CrossRef CAS
- X. Lin, X. He, C. Hu, Y. Chen, Y. Mai and S. Lin, Polym. Chem., 2016, 7, 2815–2820 RSC
- K. Akiyoshi, S. Deguchi, N. Moriguchi, S. Yamaguchi and J. Sunamoto, Macromolecules, 1993, 26, 3062–3068 CrossRef CAS
- Y. Morishima, S. Nomura, T. Ikeda, M. Seki and M. Kamachi, Macromolecules, 1995, 28, 2874–2881 CrossRef CAS
- T. Mes, R. van der Weegen, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 5085–5089 CrossRef CAS PubMed
- Y. Hirai, T. Terashima, M. Takenaka and M. Sawamoto, Macromolecules, 2016, 49, 5084–5091 CrossRef CAS
- H. Frisch, J. P. Menzel, F. R. Bloesser, D. E. Marschner, K. Mundsinger and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140, 9551–9557 CrossRef CAS PubMed
- S. Imai, M. Takenaka, M. Sawamoto and T. Terashima, J. Am. Chem. Soc., 2019, 141, 511–519 CrossRef CAS
- T. Nishimura, S. Shishi, Y. Sasaki and K. Akiyoshi, J. Am. Chem. Soc., 2020, 142, 11784–11790 CrossRef CAS PubMed
- Y. Kimura, S. Imai, M. Takenaka and T. Terashima, Macromolecules, 2021, 54, 3987–3998 CrossRef CAS
- T. Nishimura, S. Fujii, K. Sakurai, Y. Sasaki and K. Akiyoshi, Macromolecules, 2021, 54, 7003–7009 CrossRef CAS
- H. Li, W. Zhang, W. Xu and X. Zhang, Macromolecules, 2000, 33, 465–469 CrossRef CAS
- J. Skolnick and M. Fixman, Macromolecules, 1977, 10, 944–948 CrossRef CAS
- M. Tricot, Macromolecules, 1984, 17, 1698–1704 CrossRef CAS
- T. Nishimura, S. Hirose, Y. Sasaki and K. Akiyoshi, J. Am. Chem. Soc., 2020, 142, 154–161 CrossRef CAS PubMed
- J. R. Mishic and M. R. Fisch, J. Chem. Phys., 1990, 92, 3222–3229 CrossRef CAS
- S. M. Stephany, T. M. Kole and M. R. Fisch, J. Phys. Chem., 1994, 98, 11126–11128 CrossRef CAS
- J. Yang and T. Sato, Polymers, 2020, 12, 1266 CrossRef CAS PubMed
- J.-F. Le Meins, O. Sandre and S. Lecommandoux, Eur. Phys. J. E, 2011, 34, 14 CrossRef PubMed
- P.-H. Chen, X. Chen and X. He, Biochim. Biophys. Acta, Proteins Proteomics, 2013, 1834, 2176–2186 CrossRef CAS
- Y. Shimizu, A. Inoue, Y. Tomari, T. Suzuki, T. Yokogawa, K. Nishikawa and T. Ueda, Nat. Biotechnol., 2001, 19, 751–755 CrossRef CAS PubMed
- M. Ando, M. Akiyama, D. Okuno, M. Hirano, T. Ide, S. Sawada, Y. Sasaki and K. Akiyoshi, Biomater. Sci., 2016, 4, 258–264 RSC
- M. Ando, S. Schikula, Y. Sasaki and K. Akiyoshi, Adv. Sci., 2018, 5, 1800524 CrossRef PubMed
- E. M. Merzlyak, J. Goedhart, D. Shcherbo, M. E. Bulina, A. S. Shcheglov, A. F. Fradkov, A. Gaintzeva, K. A. Lukyanov, S. Lukyanov, T. W. J. Gadella and D. M. Chudakov, Nat. Methods, 2007, 4, 555–557 CrossRef CAS PubMed
- M. Parmar, S. Rawson, C. A. Scarff, A. Goldman, T. R. Dafforn, S. P. Muench and V. L. G. Postis, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 378–383 CrossRef CAS PubMed
- M. Orwick-Rydmark, J. E. Lovett, A. Graziadei, L. Lindholm, M. R. Hicks and A. Watts, Nano Lett., 2012, 12, 4687–4692 CrossRef CAS
- J. M. Dörr, M. C. Koorengevel, M. Schäfer, A. V. Prokofyev, S. Scheidelaar, E. A. W. van der Cruijsen, T. R. Dafforn, M. Baldus and J. A. Killian, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18607 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01674e |
| This journal is © The Royal Society of Chemistry 2022 |