
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcylhydrazone-based supramolecular assemblies undergoing a converse sol-to-gel transition on trans → cis photoisomerization†
Zhao
Gao‡
a,
Fei
Yan‡
a,
Lulu
Shi
a,
Yifei
Han
b,
Shuai
Qiu
a,
Juan
Zhang
a,
Feng
Wang
 b,
Si
Wu
b and
Wei
Tian
*a
b,
Si
Wu
b and
Wei
Tian
*a
aShaanxi Key Laboratory of Macromolecular Science and Technology, MOE Key Laboratory of Material Physics and Chemistry under Extraordinary Conditions, School of Chemistry and Chemical Engineering, Northwestern Polytechnical University, Xi'an 710072, P. R. China. E-mail: happytw_3000@nwpu.edu.cn
bDepartment of Polymer Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
First published on 16th June 2022
Abstract
Photoisomeric supramolecular assemblies have drawn enormous attention in recent years. Although it is a general rule that photoisomerization from a less to a more distorted isomer causes the destruction of assemblies, this photoisomerization process inducing a converse transition from irregular aggregates to regular assemblies is still a great challenge. Here, we report a converse sol-to-gel transition derived from the planar to nonplanar photoisomer conversion, which is in sharp contrast to the conventional light-induced gel collapse. A well-designed acylhydrazone-linked monomer is exploited as a photoisomer to realize the above-mentioned phase transition. In the monomer, imine is responsible for trans–cis interconversion and amide generates intermolecular hydrogen bonds enabling the photoisomerization-driven self-assembly. The counterintuitive feature of the sol-to-gel transition is ascribed to the partial trans → cis photoisomerization of acylhydrazone causing changes in stacking mode of monomers. Furthermore, the reversible phase transition is applied in the valves formed in situ in microfluidic devices, providing fascinating potential for miniature materials.
Introduction
Photoisomerization is a process that reversibly converts one isomer into another with a clean and remote light source, which has been applied in a broad range of systems, including photoactuators, sustained drug release systems, information storage, photochromic materials, and photogated devices.1–9 Photoisomeric supramolecular assemblies, as an elegant combination of reversible photoisomers and dynamic non-covalent interactions, possess various intriguing properties, such as degradability, shape memory, and phase transition.10–14 Generally, photoisomerization from a relatively planar isomer to a distorted one gives rise to the disassembly of supramolecular systems, which is ascribed to the steric hindrance of the nonplanar isomers failing to orderly stack with each other (Fig. 1A). For instance, based on trans → cis photoisomerization, azobenzene-containing molecules induced a solid to isotropic liquid phase transition,14–18 and cyanostilbene derivatives caused not only a liquid crystal to an amorphous phase transition,19,20 but also a gel collapse (gel-to-sol transition).21 Nevertheless, although a few examples of transition from a less to a more ordered state upon photoirradiation have been reported in liquid crystal systems,22,23 the trans → cis photoisomerization-induced converse transition from irregular aggregates to regular assemblies is far less reported so far and still a great challenge (Fig. 1B). This is mainly because the bent-shaped (cis)-isomers loosely stack and consequently lead to weaker intermolecular interactions. However, this converse transition could enrich the categories of supramolecular photochemical phase transitions. Moreover, the formation of light-induced regular assemblies (such as gelation) perfectly meets the requirements for in situ gelation in a sealed environment, such as in the microfluidics field. Thus, the development of ingenious methods to enable the formation of light-induced regular assemblies from trans → cis conversion is of pivotal importance for both a fundamental viewpoint and potential applications.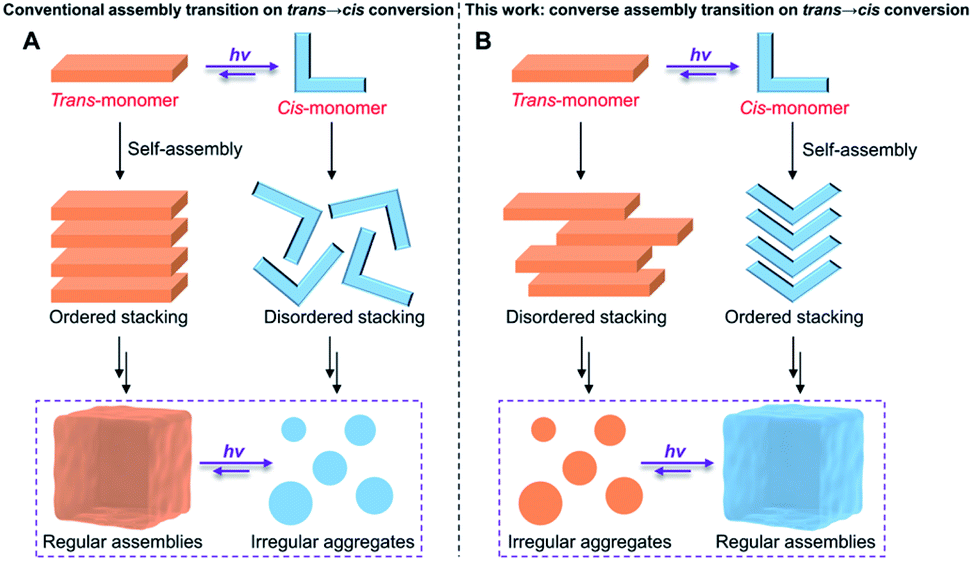 | ||
| Fig. 1 (A) The conventional self-assembly behaviors of trans and cis monomers, and (B) the converse self-assembly and the transition on trans → cis photoisomerization. | ||
An effective strategy to realize the above photoisomerization-induced transition from irregular aggregates to regular assemblies is to seek photoisomers with unique structures. Acylhydrazones,24–27 a novel class of photoisomers based on the imine structural unit (E–Z interconversion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond),28–30 have recently been regarded as promising candidates for photoswitches because of their facile synthesis, high stability, and fatigue resistance. More importantly, differing from other photoisomers such as azobenzene31–33 and cyanostilbene,34–37 the amide in acylhydrazone is an excellent fragment to generate a hydrogen bond, which is known as a strong driving force for constructing regular assemblies.38–40 On the other hand, the aggregated nature of supramolecular assemblies usually causes incomplete photochemical conversion,41–43 further leading to the coexistence of E- and Z-form isomers in the final state of supramolecular systems, which makes it possible to manipulate the stacking mode of the two isomers. Benefitting from all these essential advantages, we envision that, acylhydrazone-functionalized monomers could be exploited to realize the geometrical isomerism induced converse assembly transition on trans → cis conversion (Fig. 1B), in a supramolecular co-assembly manner by photo-regulating the balance between isomerization and aggregation.
N bond),28–30 have recently been regarded as promising candidates for photoswitches because of their facile synthesis, high stability, and fatigue resistance. More importantly, differing from other photoisomers such as azobenzene31–33 and cyanostilbene,34–37 the amide in acylhydrazone is an excellent fragment to generate a hydrogen bond, which is known as a strong driving force for constructing regular assemblies.38–40 On the other hand, the aggregated nature of supramolecular assemblies usually causes incomplete photochemical conversion,41–43 further leading to the coexistence of E- and Z-form isomers in the final state of supramolecular systems, which makes it possible to manipulate the stacking mode of the two isomers. Benefitting from all these essential advantages, we envision that, acylhydrazone-functionalized monomers could be exploited to realize the geometrical isomerism induced converse assembly transition on trans → cis conversion (Fig. 1B), in a supramolecular co-assembly manner by photo-regulating the balance between isomerization and aggregation.
Based on these considerations, we herein realize a converse sol-to-gel transition derived from the trans → cis photoisomerization of a low-molecular-weight acylhydrazone-linked monomer (Fig. 2), which is in sharp contrast to the previously reported light-induced gel collapse.44,45 Specifically, a tadpole-like monomer 1 consisting of a pyrene head and hydrophobic tail linked by an acylhydrazone unit, is first designed and synthesized. It can undergo a reversible E–Z photoisomerization process in both the monomeric and assembled states. Upon irradiating the concentrated decane solution of 1-E, a sol-to-gel transition is realized. This phase transition is ascribed to the co-assembly of 1-E and partial photoisomerization product 1-Z, mainly driven by the emergence of intermolecular hydrogen bonds between the two adjacent monomers. On this basis, the light-induced sol-to-gel transition is successfully employed to establish the valves of microfluidic channels based on a reversible and straightforward gelation process.
 | ||
| Fig. 2 Schematic illustration of the photoisomerization of monomer 1, and a cartoon representation for the proposed self-assembly behaviors of 1-E and 1-Z, and its trans → cis photoisomerization induced supramolecular sol-to-gel transition. | ||
Results and discussion
Photoisomerization of the acylhydrazone-linked monomer
We first investigated the photoisomerization behavior of 1 in the monomeric state. In dilute dichloroethane (DCE, c = 5.0 × 10−5 M), the UV-vis spectra of the pure (E)-isomer of 1 exhibited vibronically split absorption bands in the region of 325–425 nm (Fig. 3A and ESI Fig. S1†). Upon irradiation with 365 nm light for 50 s, the maximum absorption wavelength (λmax) of 1-E blue-shifted from around 374 nm to 346 nm with concomitant two isosbestic points at 315 and 351 nm, indicative of the photoisomerization from 1-E to the photostationary state (Z)-isomer of 1 (1-ZPSS). Irradiation of 1-ZPSS with a shorter wavelength light of 254 nm regenerated the initial (E)-isomer, which was confirmed by the complete restoration of the absorption bands (ESI Fig. S1†). With successive 365 nm and 254 nm light irradiation, the photoisomerization of 1 could be reversibly switched multiple times without noticeable degradation in performance (Fig. 3A and ESI Fig. S2†). Furthermore, based on the quantitative analysis of typical 1H NMR resonances of 1-E in C2D2Cl4, 1-E to 1-ZPSS photoisomerization was about 57% complete, while the back-isomerization was almost entirely complete within 30 min (>98% yield, Fig. 3B). The time-dependent absorbance curve conformed to the pseudo-first-order kinetics, providing an E–Z photoisomerization rate (kobs) of 0.058 s−1 (ESI Fig. S3†). kobs was related to the concentration of 1-E, which increased as the concentration increased. In addition to 254 nm light irradiation, heating could also trigger the back-isomerization (ESI Fig. S4†). In comparison, the reference compound 2 (varying acylhydrazone to ester bond) was unaffected by light irradiation, as evidenced by the complete lack of changes in the UV-vis and fluorescence signals (Fig. 3A, inset, and ESI Fig. S5†).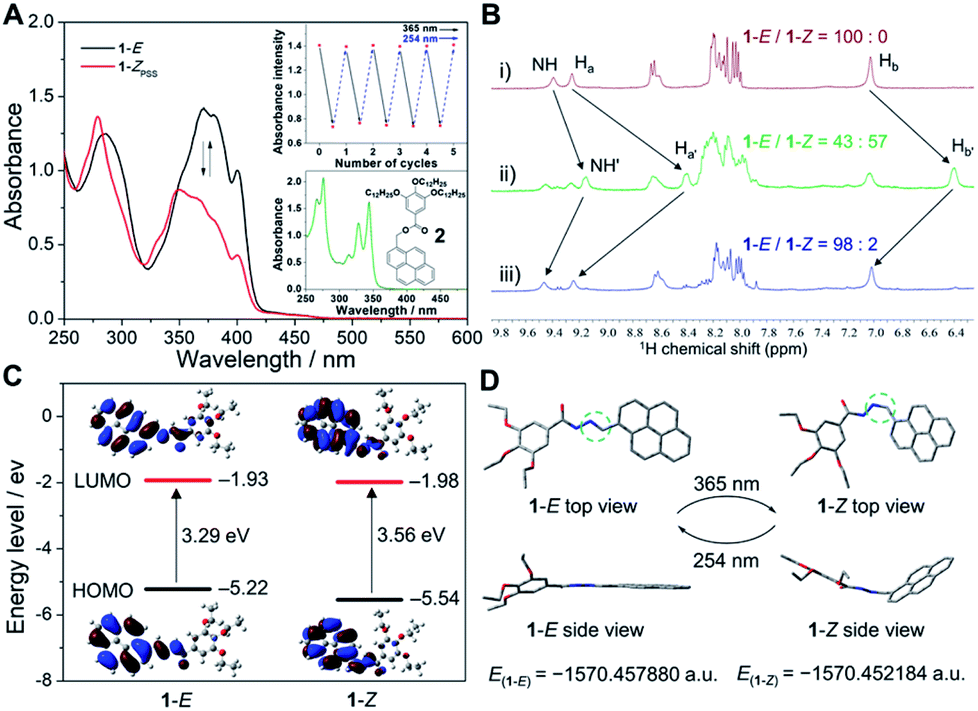 | ||
| Fig. 3 Photoisomerization studies of monomer 1. (A) UV-vis spectral change of 1-E (5.0 × 10−5 M in DCE) upon successive 365 nm (irradiation intensities: 0.98 mW cm−2) and 254 nm (irradiation intensities: 0.34 mW cm−2) light irradiation. Inset: multicycle experiments by monitoring the absorbance intensity changes at 373 nm. UV-vis spectral change of the control compound 2 (5.0 × 10−5 M in DCE) upon 365 nm light irradiation. (B) Partial 1H NMR spectra (C2D2Cl4, 298 K, 2.00 mM) of 1-E: (i) before irradiation, (ii) conversion to 1-ZPSS after 365 nm light irradiation for 60 min, and (iii) recovery to 1-E upon 254 nm light irradiation for 30 min. (C) The calculated energy levels and corresponding frontier molecular orbital diagrams of 1-E and 1-Z via DFT computation. (D) The side view and top view of the optimized structures and related Gibbs-free energy of 1-E and 1-Z based on DFT calculations at the B3LYP/6-311G(d) level. The long alkyl chains in 1-E and 1-Z are abbreviated as ethoxy groups to reduce computational costs. | ||
To get further insights into the relationship between acylhydrazone photoisomerization and molecular structures, quantum chemical calculations on both isomers of 1 were employed. Based on the comparative analysis of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of 1-E and 1-Z in the ground state (Fig. 3C),46 the calculated energy gap of 1-E (3.29 eV, λ = 376 nm) was relatively lower than that of 1-Z (3.56 eV, λ = 348 nm), which were consistent with the λmax values measured by the UV-vis spectra. The HOMO and LUMO electron densities were localized at the pyrene moieties for 1-E, reflective of the transition from π to π* orbitals. The similar phenomenon was observed for 1-Z. For the optimized structures of 1-E and 1-Z based on density functional theory (DFT) calculations at the B3LYP/6-311G(d) level, the total energy (E) values were calculated to be −1570.457880 and −1570.452184 a.u., respectively (Fig. 3D). It was found that 1-E was more stable with approximately 15.0 kJ mol−1 lower energy relative to 1-Z, because of the more twisted molecular structure of 1-Z.
Light-induced sol-to-gel transition
The assembly behaviors of monomer 1-E before photoirradiation were first studied. As is well known, hydrocarbon solvents such as decane are the common poor solvents for rod-coil-type monomers to form ordered supramolecular assemblies.47–49 When switching the solvent from DCE to decane, the UV-vis absorption spectra of 1-E showed an emergence of a shoulder band located between 400 and 475 nm (ESI Fig. S6†). Simultaneously, the intensity of the blue emission band at 412 nm corresponding to the monomeric pyrene units slightly decreased, along with the appearance of a new emission band at 520 nm (ESI Fig. S6†). The red-shifted structureless band was attributed to the excimer emission of the stacked pyrene units.50–52 Unfortunately, no noticeable aggregation of 1-E could be detected by the Tyndall effect experiment, and dynamic light scattering (DLS) and transmission electron microscopy (TEM) measurements (ESI Fig. S7†). Even on increasing the concentration up to 20 mM, no gels were visualized during the heating–cooling cycle, and remained unchanged in a dark place for several days (ESI Fig. S8†). Accordingly, no large-sized supramolecular assemblies were formed for 1-E in decane.Surprisingly, when the concentrated decane solution of 1-E was exposed to 365 nm light, the viscosity gradually increased and fluidity was lost. Subsequently, a yellow transparent gel was finally formed within 60 min (Fig. 4A and ESI Fig. S9†). Such results are in sharp contrast to the previously reported light-induced gel collapse.21,44,45,53 To acquire the detailed gelation process, viscosity experiments were then performed. Upon irradiating the decane solution of 1-E (10.0 mM), the specific viscosity was almost unchanged for the first 1.5 min and then increased sharply (Fig. 4B), indicating the favorable transition from monomeric species to higher-molecular-weight supramolecular assemblies. By varying the concentration, the viscosity transition time and intensity were finely adjusted. The critical gelation concentration was measured to be 6 mM. DLS analysis for 1-E under light irradiation showed the size enlargement phenomenon, revealing the light-induced supramolecular assembly behaviors (ESI Fig. S10†). In sharp contrast, no apparent viscosity and DLS hydrodynamic diameter (Dh) changes occurred for the reference compound 2 (Fig. 4B and ESI Fig. S10†). Furthermore, the light-induced sol-to-gel transition of 1-E was observed by microscopic morphologies. Depending on the TEM images, short nanofibers were observed for 1-E after irradiation for 10 min (Fig. 4C and ESI Fig. S11†). After 30 min, longer fibers with several microns in length were formed. It is thus envisioned that these one-dimensional fibers are mutually entangled to create three-dimensional fibrous networks that thereby entrap a large number of solvent molecules, which lays the basis for the formation of supramolecular gels.
 | ||
| Fig. 4 Light-induced sol-to-gel transition of 1-E. (A) Photographs of the gelation process of 1-E (10 mM) in decane upon 365 nm (irradiation intensity: 11.9 mW cm−2) photoirradiation for 0, 25, 45 and 60 min. (B) Specific viscosity of 1-E (1.0, 1.5, 3.0, 10.0 mM) and 2 (10.0 mM) versus the photoirradiation time (298 K). (C) TEM images of 1-E (2.0 × 10−4 M in decane) before and after photoirradiation for 10, 20 and 30 min. (D) and (E) G′ (black symbols) and G′′ (red symbols) for the light-induced gel derived from 1-E as a function of (D) oscillation frequency at 293 K with a strain of 0.2%; and (E) time at 293 K with a frequency of 1 Hz and a strain of 0.2%. (F) Small-angle XRD patterns of 1-E before and after photoirradiation. | ||
The properties of the light-induced gel were further proved by means of rheological experiments. The storage modulus (G′) value was remarkably higher than the loss modulus (G′′) value at all tested frequencies (Fig. 4D), which verified the formation of true supramolecular gels. Additionally, the resulting organogels were stable enough once they were formed. The conclusion was confirmed using the oscillation time curves, indicating that the values of G′ and G′′ remained constant over a long time (∼30 min, Fig. 4E). The stacking pattern of the light-induced gel was identified by small-angle X-ray diffraction (XRD) measurements. Specifically, no regular stacking formed for 1-E before irradiation, as evidenced by the lack of reflection signals in the XRD pattern (Fig. 4F). Upon irradiating 1-E with 365 nm light, the diffractogram showed three reflections with d-spacing values in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/√3:1/√4, assigned to the (100), (110), and (200) signals of the hexagonal columnar (Colh) stacking. The lattice parameter (α = 2d100/√3 = 3.78 nm) calculated from the diffraction maxima indicated that the diffraction units are comparable to the stacking model diameter of light-irradiated 1-E (3.89 nm, Fig. 4F, inset, and ESI Fig. S12†). Accordingly, the regular columnar stacks got together to form a long-range-ordered supramolecular gel. Interestingly, the gel switched back to the sol upon 254 nm light irradiation. When the sol was irradiated with 365 nm light again, the gel was regained (ESI Fig. S13†). Additionally, thermal stimulus (∼45 °C) could also trigger the liquefaction process, and the sol remained even on cooling to room temperature.
:1/√3:1/√4, assigned to the (100), (110), and (200) signals of the hexagonal columnar (Colh) stacking. The lattice parameter (α = 2d100/√3 = 3.78 nm) calculated from the diffraction maxima indicated that the diffraction units are comparable to the stacking model diameter of light-irradiated 1-E (3.89 nm, Fig. 4F, inset, and ESI Fig. S12†). Accordingly, the regular columnar stacks got together to form a long-range-ordered supramolecular gel. Interestingly, the gel switched back to the sol upon 254 nm light irradiation. When the sol was irradiated with 365 nm light again, the gel was regained (ESI Fig. S13†). Additionally, thermal stimulus (∼45 °C) could also trigger the liquefaction process, and the sol remained even on cooling to room temperature.
For the purpose of investigating the stability of light-induced organogels from the thermodynamic standpoint, we turned to elucidate the assembly mechanism of the supramolecular assemblies. The organogels were diluted to an appropriate concentration for spectral measurements. Temperature-dependent UV-vis spectra were then recorded. In detail, upon gradually decreasing the temperature at a rate of 60 K h−1, the absorption spectra of the diluted gel of light-irradiated 1-E showed two isosbestic points at 300 and 399 nm (Fig. 5A, inset), indicative of the transition between monomeric and assembled states. A non-sigmoidal cooling curve was obtained by monitoring the fraction of aggregated species (αagg) against temperature (Fig. 5A and ESI Fig. S14†), suggesting that the supramolecular assembly process followed a cooperative nucleation–elongation mechanism.54–57 The Meijer–Schenning–van-der-Schoot mathematical model54 was then employed for the non-linear fitting of the resulting curves. A Te (critical elongation temperature) value of 310.4 K and a Ka (dimensionless equilibrium constant of the activation step at Te) value of 8.6 × 10−5 were obtained. Moreover, on the basis of the modified Van't Hoff plot, ΔG0 (Gibbs free energy) was calculated to be −26.2 kJ mol−1 at 298 K (Fig. 5B). Such results manifest that organogels derived from light-induced supramolecular assemblies possess considerable stability.
 | ||
| Fig. 5 Supramolecular assembly mechanism studies. (A) αagg as a function of temperature for light irradiated 1-E in decane (5.0 × 10−5 M, λ = 367 nm). The solid lines denote the respective fitting curves. Inset: temperature-dependent UV-vis spectra. The arrows represent the signal variations upon decreasing the temperature. (B) Van't Hoff plot for light irradiated 1-E in decane. The red line denotes the linear fitting curve. | ||
Insights into the sol-to-gel transition mechanism
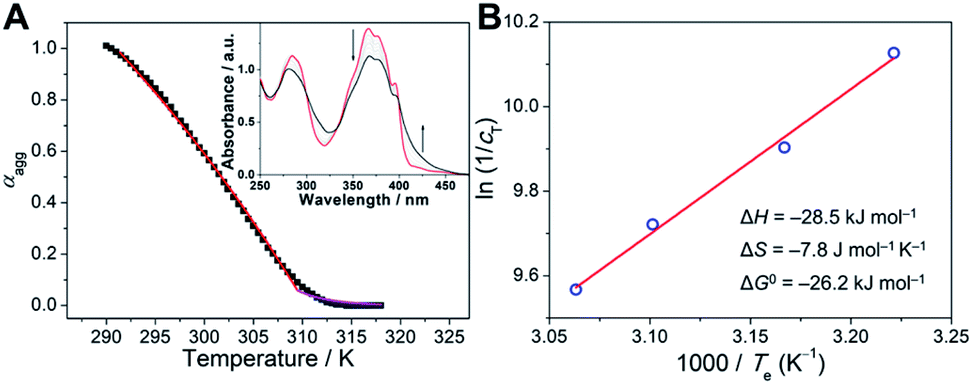
For a deeper understanding of the origin of the gelation behaviors triggered by photoirradiation, UV-vis spectra of 1-E (5.0 × 10−5 M) in decane upon exposure to 365 nm light were first recorded. In detail, the original absorbance signal decreased in intensity, while a blue-shifted band centered at 345 nm gradually turned into the main band, with two isosbestic points at 322 and 353 nm (ESI Fig. S16†). The results implied that the trans → cis photoisomerization occurred not only in the monomeric state (DCE solution), but also in the assembled state. Due to the steric hindrance effect of stacked pyrene units, kobs in decane was slower than that in DCE (0.034 s−1versus 0.058 s−1, ESI Fig. S17†). It is noteworthy that the trans → cis conversion of 1-E at a relatively higher concentration cannot reach the conversion level at a lower concentration, even on prolonging the irradiation time (ESI Fig. S16†). Approximately 40% relative conversion was achieved at 0.2 mM compared to the lower concentration condition (Fig. 6A). Thus, we rationalized that the supramolecular gel formation is ascribed to the partial photoisomerization of 1-E, that is, the participation of 1-Z. | ||
| Fig. 6 Insights into the light-induced sol-to-gel transition mechanism of 1-E. (A) The relative E → Z conversion of 1 (black dots: 5.0 × 10−5 M, red dots: 2.0 × 10−4 M) in decane upon 365 nm light irradiation, by monitoring the absorbance changes at 379 nm. (B) DLS hydrodynamic diameter variations upon gradual addition of 1-ZPSS (2.0 × 10−4 M) into the freshly prepared decane solution of 1-E (2.0 × 10−4 M) and pure decane solvent, respectively. (C) SEM image of 10 mol% 1-ZPSS in 1-E (2.0 × 10−4 M in decane). Insets display the enlarged P- and M-type helical fibers. (D) Optimized structures with color-mapped IGM isosurface graphs of octamers of 1-E (i) before, and (ii) after 365 nm light irradiation. The long side chains are simplified to ethoxy groups. The green color isosurfaces represent weak non-covalent interactions (such as van der Waals and π–π stacking interactions), while blue color isosurfaces in (ii) denote stronger attraction (such as hydrogen bonds). The color scale at the bottom represents the relative strength of non-covalent interactions. | ||
We executed the titration experiments to further investigate the effect of light-irradiated product 1-ZPSS on the formation of supramolecular co-assemblies. Upon progressively adding 1-ZPSS into the decane solution of 1-E (2.0 × 10−4 M), the Dh value of assemblies determined by the DLS measurements gradually increased (Fig. 6B and ESI Fig. S18†). Only 10 mol% of 1-ZPSS increased the Dh value from around 0.6 nm to 825 nm. As a comparison, no apparent enhancement of the Dh value was visualized when 1-ZPSS was introduced into the pure decane solvent (Fig. 6B and ESI Fig. S18†). It thus indicated that the large-sized aggregates were formed by the co-assembly of 1-E with its light-irradiated product 1-ZPSS. The conclusion was further verified by the morphology measurements. Intertangled long fibers were observed in the TEM images when adding 1-ZPSS into 1-E (ESI Fig. S19†), similar to that of 1-E exposed to 365 nm light for 30 min (Fig. 4C). Interestingly, the enlarged TEM and scanning electron microscopy (SEM) images of the co-assemblies 1-Z/1-E showed that the fibers appeared as distinct right-handed (P) and left-handed (M) helical structures (Fig. 6C and ESI Fig. S19–S21†). Therefore, it is envisioned that the structurally distorted 1-Z displays a considerable capacity to regulate the dynamic stacking structures of the co-assemblies 1-Z/1-E to form well-defined supramolecular helical aggregates.
The detailed stacking mode for the light-induced supramolecular assemblies was then sought to elucidate. Depending on the 1H measurements, the NH protons of acylhydrazone displayed upfield and downfield shifts upon 365 nm light irradiation (ESI Fig. S22†). The significant upfield shift from NH to NH′ was assigned to the conversion from 1-E to 1-ZPSS. The downfield shift of the unisomerized NH proton revealed the presence of hydrogen bond interactions after photoirradiation. The hydrogen bonds were then verified through Fourier transform infrared (FTIR) spectra experiments. The N–H stretching vibration signal shifted from 3391 and 3195 cm−1 to a lower frequency (3172 cm−1) upon irradiating 1-E with 365 nm light (ESI Fig. S23†), while the CO band shifted from 1643 cm−1 to 1641 cm−1, indicating the generation of intermolecular hydrogen bonds between neighboring acylhydrazone CO and N–H bonds.45,58,59 Moreover, proton competition experiments were performed to evaluate the significance of hydrogen bonds for the light-induced supramolecular gel formation. Upon adding trace amounts of DMF into the gel system, the gel-to-sol transition was observed (ESI Fig. S24†), manifesting that the intermolecular hydrogen bonds were disturbed by the protic solvent of DMF, and therefore no fibrous structures formed under this condition.
To visually observe the non-covalent interaction variations upon partial photoisomerization from 1-E to 1-Z, the independent gradient model (IGM) methods60 of theoretical calculations were applied. The optimized structures with color-mapped IGM isosurfaces of the octamers of 1-E before and after photoirradiation are shown in Fig. 6D. In the IGM analysis, large-scope green isosurface regions existed between the pyrene moieties of the interdigitated self-assembly of 1-E, which was attributed to the π–π interactions (Fig. 6D(i)). After photoirradiation, the stacking structure transformed from the interdigitated way into the head-to-head manner. The emerged hydrogen bonds (Fig. 6D(ii), blue isosurface in the red frame) as well as π–π stacking and van der Waals interactions between 1-E and 1-Z synergistically stabilized the light-induced supramolecular assemblies. The average binding energies between the adjacent monomers increased from −7.69 kcal mol−1 of 1-E to −20.79 kcal mol−1 of light-irradiated 1-E.
Overall, these experimental and theoretical results unambiguously evidence that supramolecular gel formation is ascribed to the partial photoisomerization of 1-E causing the changes in stacking mode of the co-assembly 1-Z/1-E, mainly driven by the intermolecular hydrogen bonds.
In situ sol-to-gel transition generated valves
Microfluidic technologies have been recently regarded as an advanced tool to investigate non-equilibrium assemblies and controlled reactive processes in self-assembly research.61–64 Introducing stimuli-responsive materials as a valve would realize the non-contact microfluidic flow control,65–68 probably producing simplified and versatile microfluidic devices. Nevertheless, most reported valves were based on temperature- and electro-responsive systems,69,70 which are not easy to achieve flow control in situ and are comparatively complicated. Considering that the organogels derived from the light-induced sol-to-gel transition of 1-E can be finely triggered by remote and contactless light, they may be potential candidates for developing in situ generated valves in the microfluidics field.On this basis, a three-layer microfluidic device was designed and fabricated, in which two circular chambers in the Y-shaped microchannel of the fluidic layer were used to fill the light-induced gel (Fig. 7A and B). Before employing 1 as the light-responsive valve, the decane fluid could pass through both directions of the microchannel (Fig. 7C(i)). The left side of the sealed microchannel was first filled with the pre-prepared high viscosity sol of 1 by injection, and the gel-based valve was then created by irradiating the sol with 365 nm light in situ. Thus, the left microchannel was blocked and the fluid could only pass through the right side (Fig. 7C(ii)). The fluid could be well maintained for several hours (even upon increasing the flow rate to around 20 μL min−1), suggesting that the valve possesses enough stability. When the gel-based valve was irradiated with 254 nm light, the gel-to-sol transition occurred and then the sealability weakened, resulting in the recovery of bidirectional flow (Fig. 7C(iii)). It requires only a small amount of the gel-to-sol transition to restore the flow.
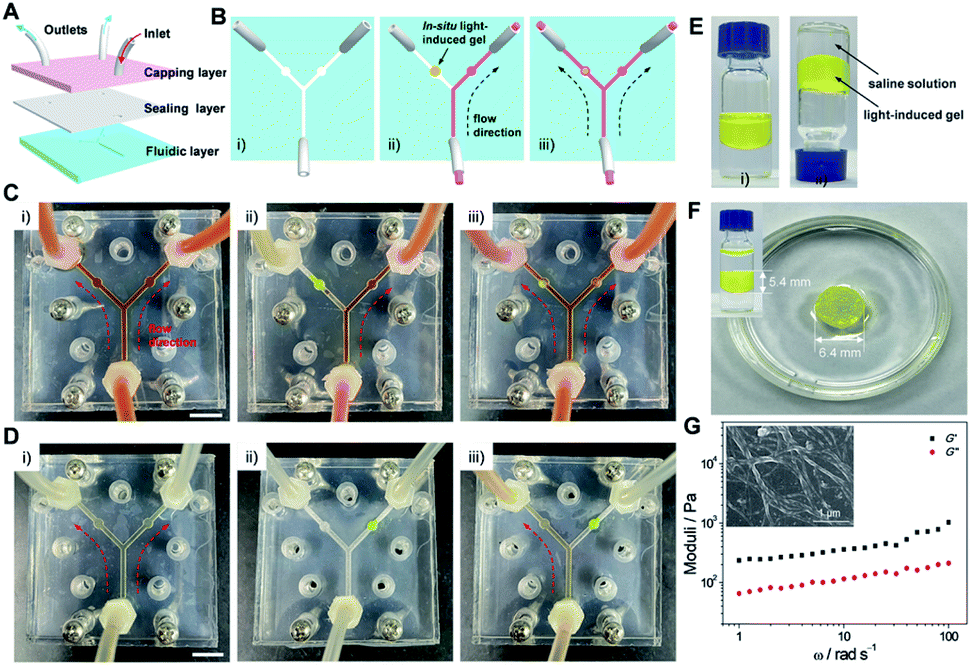 | ||
| Fig. 7 Performance of light-induced gel-based valves. (A) Schematic illustration of the microfluidic device containing three-layer chips used in the experiments. (B) Cartoon representation for the top view of the microfluidic device: (i) before usage, (ii) inletting flow when implemented 1-E (yellow dot) as the light-responsive valve, and (iii) the flow situation upon irradiation of the light-induced gel with 254 nm light. (C) Photographs of the microfluidic device: (i) inletting decane solvent without 1-E, (ii) no flow observed in the left side when the in situ formed gel is used as a valve within the sealed PMMA chips, and (iii) open valve of the left direction after irradiation of the gel. Scale bar: 10 mm. The fluid passing through the microfluidic device is colored red for easy visualization. The device was fastened with six sets of screws and nuts. (D) (i) Inletting the aqueous solution without 1-E, (ii) in situ formed gel in the right side (yellow dot), and (iii) no aqueous solution flow observed in the right side when the gel is used as a valve. Scale bar: 10 mm. (E) Light-induced sol-to-gel transition of 1-E in saline solution: (i) before and (ii) after 365 nm light irradiation. (F) The light-induced gel immersed in saline after two weeks. Inset of (F): the gel sandwiched between the saline solution. (G) Rheological measurements of the gel after immersion in saline for two weeks. Inset of (G): SEM image of the gel. | ||
Besides, it is worth noting that the kind of liquid can be varied from low polarity decane to high polarity water. As shown in Fig. 7D and ESI Fig. S25,† a similar flow control was achieved in the water-based environment. More importantly, the gel-based valves featured sufficient stability and sustainability in aqueous solution (even in saline). Specifically, 1-E could also undergo the light-induced sol-to-gel transition when mixed with saline solution (Fig. 7E). The gel did not swell or shrink after being immersed or sandwiched in saline for two weeks (Fig. 7F and ESI Fig. S26†). G′ and G′′ were determined to be 612.2 Pa and 140.5 Pa, respectively (Fig. 7G), which are comparable to the values of the freshly prepared gel (645.3 Pa and 151.3 Pa, Fig. 4D). The helical fibers were also maintained (Fig. 7G, inset). Thus, the light-induced gel-based valves provide efficient and flexible flow control in sealed microfluidic chips.
Conclusions
In summary, we have successfully established a converse light-induced sol-to-gel transition system derived from the trans → cis conversion of acylhydrazone-based supramolecular assemblies. The acylhydrazone-linked monomer 1 underwent a reversible photoisomerization process in both the monomeric and assembled states. A stable organogel was obtained upon irradiating the decane solution of 1-E with 365 nm light. This sol-to-gel transition is ascribed to the co-assembly of 1-E and light-irradiated product 1-ZPSS, mainly driven by the emergence of intermolecular hydrogen bonds between the two adjacent monomers in a head-to-head arrangement manner. We anticipate that, like acylhydrazone derivatives, other suitable stimuli-responsive molecules may manipulate the distinctive phase transition. Furthermore, the reversible sol-to-gel transition has been appropriately applied in gel-based valves formed in situ in microfluidic devices. The current work not only highlights the fundamental understanding of photoisomerization in supramolecular photochemistry, but opens up a new avenue towards remote-controlled miniature materials.Data availability
Detailed synthetic procedures, analytical data, and computational methods are provided in the ESI.†Author contributions
Z. G. and W. T. conceived the idea for this article. F. Y. and L. S. performed the experiments, analyzed the data, and produced the artwork under the direction of Z. G. Y. H. contributed to the theoretical calculations. S. Q. and J. Z. helped in the SEM measurements. F. W. and S. W. discussed the results and commented on the manuscript. The article was revised by Z. G. and W. T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22001213, 22022107, and 22071197), the Natural Science Basic Research Plan in Shaanxi Province of China (2020JQ-134 and 2020JC-20), and the Fundamental Research Funds for the Central Universities (G2020KY05304). We would like to thank the Analytical & Testing Center of Northwestern Polytechnical University for TEM and SEM tests.References
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- A. Bansal and Y. Zhang, Acc. Chem. Res., 2014, 47, 3052–3060 CrossRef CAS PubMed.
- C. Jia, A. Migliore, N. Xin, S. Huang, J. Wang, Q. Yang, S. Wang, H. Chen, D. Wang, B. Feng, Z. Liu, G. Zhang, D.-H. Qu, H. Tian, M. A. Ratner, H. Q. Xu, A. Nitzan and X. Guo, Science, 2016, 352, 1443–1445 CrossRef CAS PubMed.
- J. Lv, Y. Liu, J. Wei, E. Chen, L. Qin and Y. Yu, Nature, 2016, 537, 179–184 CrossRef CAS PubMed.
- D. D. Han, Y. L. Zhang, J. N. Ma, Y. Q. Liu, B. Han and H. B. Sun, Adv. Mater., 2016, 28, 8328–8343 CrossRef CAS PubMed.
- W. Ling and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- J. D. Harris, M. J. Moran and I. Aprahamian, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9414–9422 CrossRef CAS PubMed.
- K. Tashiro, K. Katayama, K. Tamaki, L. Pesce, N. Shimizu, H. Takagi, R. Haruki, M. J. Hollamby, G. M. Pavan and S. Yagai, Angew. Chem., Int. Ed., 2021, 60, 26986–26993 CrossRef CAS PubMed.
- Q. Yan, Z. Luo, K. Cai, Y. Ma and D. Zhao, Chem. Soc. Rev., 2014, 43, 4199–4221 RSC.
- M. Kathan and S. Hecht, Chem. Soc. Rev., 2017, 46, 5536–5550 RSC.
- A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600–2610 CrossRef CAS PubMed.
- T. Bian, Z. Chu and R. Klajn, Adv. Mater., 2020, 32, 1905866 CrossRef CAS.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- M. Hoshino, E. Uchida, Y. Norikane, R. Azumi, S. Nozawa, A. Tomita, T. Sato, S. Adachi and S. Koshihara, J. Am. Chem. Soc., 2014, 136, 9158–9164 CrossRef CAS PubMed.
- H. Zhou, C. Xue, P. Weis, Y. Suzuki, S. Huang, K. Koynov, G. K. Auernhammer, R. Berger, H. J. Butt and S. Wu, Nat. Chem., 2017, 9, 145–151 CrossRef CAS PubMed.
- W. C. Xu, S. Sun and S. Wu, Angew. Chem., Int. Ed., 2019, 58, 9712–9740 CrossRef CAS.
- M. Chen, B. Yao, M. Kappl, S. Liu, J. Yuan, R. Berger, F. Zhang, H. J. Butt, Y. Liu and S. Wu, Adv. Funct. Mater., 2020, 30, 1906752 CrossRef CAS.
- J. W. Chung, S. J. Yoon, B. K. An and S. Y. Park, J. Phys. Chem. C, 2013, 117, 11285–11291 CrossRef CAS.
- M. Martínez-Abadía, S. Varghese, R. Giménez and M. B. Ros, J. Mater. Chem. C, 2016, 4, 2886–2893 RSC.
- J. Seo, J. W. Chung, J. E. Kwon and S. Y. Park, Chem. Sci., 2014, 5, 4845–4850 RSC.
- T. Kosa, L. Sukhomlinova, L. Su, B. Taheri, T. J. White and T. J. Bunning, Nature, 2012, 485, 347–349 CrossRef CAS PubMed.
- M. J. Moran, M. Magrini, D. M. Walba and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 13623–13627 CrossRef CAS PubMed.
- D. J. van Dijken, P. Kovaricek, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS PubMed.
- I. Cvrtila, H. Fanlo-Virgós, G. Schaeffer, G. M. Santiago and S. Otto, J. Am. Chem. Soc., 2017, 139, 12459–12465 CrossRef CAS PubMed.
- Z. Kokan and M. J. Chmielewski, J. Am. Chem. Soc., 2018, 140, 16010–16014 CrossRef CAS PubMed.
- G. Men and J. M. Lehn, Chem. Sci., 2019, 10, 90–98 RSC.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- B. Shao and I. Aprahamian, Chem, 2020, 6, 2162–2173 CAS.
- S. Yang, J. D. Harris, A. Lambai, L. L. Jeliazkov, G. Mohanty, H. Zeng, A. Priimagi and I. Aprahamian, J. Am. Chem. Soc., 2021, 143, 16348–16353 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- S. Sun, S. Liang, W. C. Xu, G. Xu and S. Wu, Polym. Chem., 2019, 10, 4389–4401 RSC.
- J. V. Accardo, E. R. McClure, M. A. Mosquera and J. A. Kalow, J. Am. Chem. Soc., 2020, 142, 19969–19979 CrossRef CAS.
- B. K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544–554 CrossRef CAS PubMed.
- T. Dünnebacke, K. K. Kartha, J. M. Wahl, R. Q. Albuquerque and G. Fernández, Chem. Sci., 2020, 11, 10405–10413 RSC.
- Z. Gao, Z. Chen, Y. Han and F. Wang, Nanoscale Horiz., 2020, 5, 1081–1087 RSC.
- N. F. König, D. Mutruc and S. Hecht, J. Am. Chem. Soc., 2021, 143, 9162–9168 CrossRef PubMed.
- A. Mishra, S. Dhiman and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 2740–2756 CrossRef CAS PubMed.
- S. Yagai, Y. Kitamoto, S. Datta and B. Adhikari, Acc. Chem. Res., 2019, 52, 1325–1335 CrossRef CAS PubMed.
- I. Helmers, G. Ghosh, R. Q. Albuquerque and G. Fernández, Angew. Chem., Int. Ed., 2021, 60, 4368–4376 CrossRef CAS PubMed.
- X. D. Xu, J. Zhang, L. J. Chen, X. L. Zhao, D. X. Wang and H. B. Yang, Chem.–Eur. J., 2012, 18, 1659–1667 CrossRef CAS PubMed.
- H. Qian, Y. Y. Wang, D. S. Guo and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 1037–1040 CrossRef CAS PubMed.
- N. Bäumer, K. K. Kartha, N. K. Allampally, S. Yagai, R. Q. Albuquerque and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 15626–15630 CrossRef PubMed.
- S. Mondal, P. Chakraborty, P. Bairi, D. P. Chatterjee and A. K. Nandi, Chem. Commun., 2015, 51, 10680–10683 RSC.
- E. Weyandt, G. M. ter Huurne, G. Vantomme, A. J. Markvoort, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 6295–6303 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- Z. Chen, Y. Xue, M. Gui, C. Wang and F. Wang, Inorg. Chem., 2020, 59, 6481–6488 CrossRef CAS PubMed.
- J. Matern, N. Bäumer and G. Fernández, J. Am. Chem. Soc., 2021, 143, 7164–7175 CrossRef CAS PubMed.
- A. Sarkar, R. Sasmal, A. Das, A. Venugopal, S. S. Agasti and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 18209–18216 CrossRef CAS PubMed.
- M. Vybornyi, A. V. Rudnev, S. M. Langenegger, T. Wandlowski, G. Calzaferri and R. Häner, Angew. Chem., Int. Ed., 2013, 52, 11488–11493 CrossRef CAS PubMed.
- Q. Wang, Q. Zhang, Q. W. Zhang, X. Li, C. X. Zhao, T. Y. Xu, D. H. Qu and H. Tian, Nat. Commun., 2020, 11, 158 CrossRef PubMed.
- S. Hu, L. Hu, X. Zhu, Y. Wang and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 19451–19457 CrossRef CAS PubMed.
- S. Das, N. Okamura, S. Yagi and A. Ajayaghosh, J. Am. Chem. Soc., 2019, 141, 5635–5639 CrossRef CAS PubMed.
- M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS PubMed.
- Z. Gao, F. Yan, S. Qiu, Y. Han, F. Wang and W. Tian, Chem. Commun., 2020, 56, 9214–9217 RSC.
- Y. Han, Y. Yin, F. Wang and F. Wang, Angew. Chem., Int. Ed., 2021, 60, 14076–14082 CrossRef CAS PubMed.
- A. Sarkar, T. Behera, R. Sasmal, R. Capelli, C. Empereur-mot, J. Mahato, S. S. Agasti, G. M. Pavan, A. Chowdhury and S. J. George, J. Am. Chem. Soc., 2020, 142, 11528–11539 CrossRef CAS PubMed.
- H. Wang, Y. Zhang, Y. Chen, H. Pan, X. Ren and Z. Chen, Angew. Chem., Int. Ed., 2020, 59, 5185–5192 CrossRef CAS PubMed.
- T. Xiao, L. Zhou, X.-Q. Sun, F. Huang, C. Lin and L. Wang, Chin. Chem. Lett., 2020, 29, 1–9 CrossRef.
- C. Lefebvre, G. Rubez, H. Khartabil, J. C. Boisson, J. Contreras-García and E. Hénon, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 RSC.
- J. ter Schiphorst, S. Coleman, J. E. Stumpel, A. B. Azouz, D. Diamond and A. P. H. J. Schenning, Chem. Mater., 2015, 27, 5925–5931 CrossRef CAS.
- S. Coleman, J. T. Schiphorst, A. B. Azouz, S. Bakker, A. P. H. J. Schenning and D. Diamond, Sens. Actuators, B, 2017, 245, 81–86 CrossRef CAS.
- J. ter Schiphorst, J. Saez, D. Diamond, F. Benito-Lopez and A. P. H. J. Schenning, Lab Chip, 2018, 18, 699–709 RSC.
- S. Sevim, A. Sorrenti, C. Franco, S. Furukawa, S. Pané, A. J. demello and J. Puigmartí-Luis, Chem. Soc. Rev., 2018, 47, 3788–3803 RSC.
- S. Sugiura, A. Szilágyi, K. Sumaru, K. Hattori, T. Takagi, G. Filipcsei, M. Zrínyi and T. Kanamori, Lab Chip, 2009, 9, 196–198 RSC.
- S. Yamaguchi, S. Yamahira, K. Kikuchi, K. Sumaru, T. Kanamori and T. Nagamune, Angew. Chem., Int. Ed., 2012, 51, 128–131 CrossRef CAS PubMed.
- T. Satoh, S. Sugiura, K. Sumaru, S. Ozaki, S. Gomi, T. Kurakazu, Y. Oshima and T. Kanamori, Biomicrofluidics, 2014, 8, 024112 CrossRef PubMed.
- K. Sumaru, T. Takagi, K. Morishita and T. Kanamori, Biomacromolecules, 2018, 19, 2913–2922 CrossRef CAS PubMed.
- D. Kim and D. J. Beebe, Lab Chip, 2007, 7, 193–198 RSC.
- J. Puigmartí-Luis, M. Rubio-Martínez, I. Imaz, B. Z. Cvetković, L. Abad, A. P. del Pino, D. Maspoch and D. B. Amabilino, ACS Nano, 2014, 8, 818–826 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01657e |
| ‡ Z. Gao and F. Yan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |