
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirected, nickel-catalyzed 1,2-alkylsulfenylation of alkenyl carbonyl compounds†
Zi-Qi
Li
,
Wen-Ji
He
,
Hui-Qi
Ni
and
Keary M.
Engle
 *
*
Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. E-mail: keary@scripps.edu
First published on 2nd May 2022
Abstract
We report a regioselective, nickel-catalyzed syn-1,2-carbosulfenylation of non-conjugated alkenyl carbonyl compounds with alkyl/arylzinc nucleophiles and tailored N–S electrophiles. This method allows the simultaneous installation of a variety of C(sp3) and S(Ar) (or Se(Ar)) groups onto unactivated alkenes, which complements previously developed 1,2-carbosulfenylation methodology in which only C(sp2) nucleophiles are compatible. A bidentate directing auxiliary controls regioselectivity, promotes high syn-stereoselectivity with a variety of E- and Z-internal alkenes, and enables the use of an array of electrophilic sulfenyl (and seleno) electrophiles. Among compatible electrophiles, those with N-alkyl-benzamide leaving groups were found to be especially effective, as determined through comprehensive structure–reactivity mapping.
Introduction
Organosulfur compounds possess unique bioactivity and electronic properties and thus find applications in drug discovery1 and as functional materials.2 While catalytic two-component C–S bond formation3 through cross-coupling4 and C–H functionalization5 reactions has been extensively studied during the past few decades, multicomponent C–S bond-forming protocols remain underdeveloped.6 In particular, reactions that merge an alkene, a carbon nucleophile, and a sulfur electrophile together in a programmable fashion are appealing. Pioneering reports on alkene carbosulfenylation by Trost,7 Denmark,8 and others9 harness the oxidative properties of sulfenium ion reagents for the generation of a thiiranium ion intermediate, which subsequently undergoes nucleophilic ring opening to account for the anti-selectivity of the reaction (Scheme 1A, left panel).10 Regioselectivity is controlled by alkene substitution patterns. While these methods are useful to access some sulfur-containing products, the intrinsic reactivity of the thiiranium ion intermediate introduces inherent limitations with respect to functional group compatibility and has hampered the development of a general three-component version of this methodology. Namely, only a small collection of sulfenyl groups (–SMe, –SPh) and carbogenic nucleophiles (cyanide, acetylide, and organozinc reagents)7,9d have been reported to participate in three-component couplings. To complement these methodologies with a syn-selective counterpart that has broad electrophile and nucleophile scope would be highly enabling.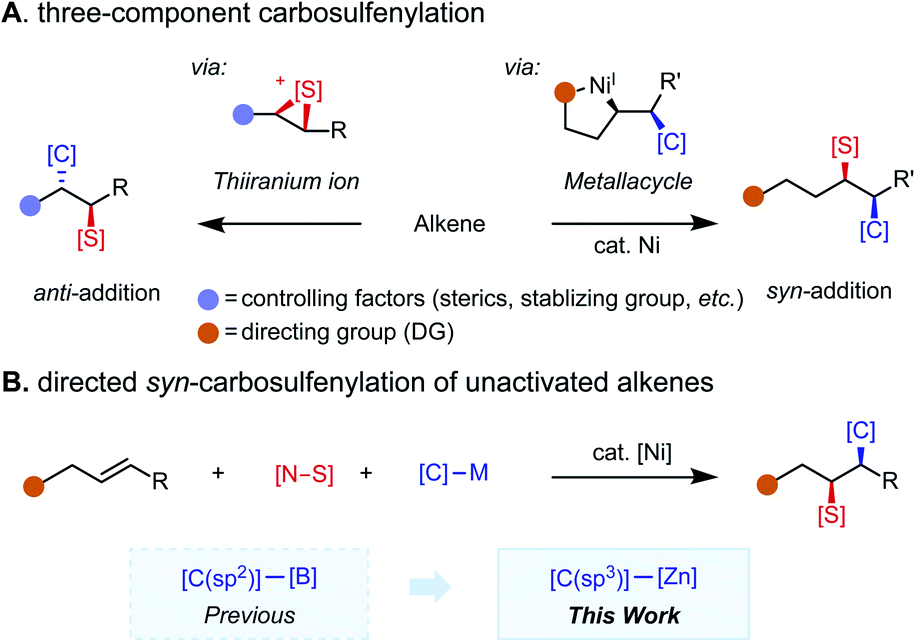 | ||
| Scheme 1 Background and synopsis of current work. | ||
Nickel-catalyzed redox-neutral 1,2-difunctionalization11,12 of unactivated alkenes has emerged as an attractive means of quickly assembling structurally complex products from feedstock alkenes, a nucleophile, and an electrophile in a regio- and stereoselective fashion. The presence of a directing group leads to formation of a metallacycle intermediate, which controls regioselectivity and suppresses potential side reactions; diastereoselectivity is dictated by the inner-sphere migratory insertion mechanism (Scheme 1A, right panel). While 1,2-dicarbofunctionalization13–15 has been extensively studied, 1,2-carboheterofunctionalization remains less explored and is largely limited to transformations introducing metalloid (semimetal) elements, namely borylative and silylative functionalizations, where the new C–B(Si) bond is formed via migratory insertion.16 For non-metal elements (N, S, Se, etc.), C–heteroatom bond formation is challenging, often requiring reductive elimination to take place from a high-valent Ni(III) intermediate.17 Hence, a strategy for intercepting the alkylnickel(I) intermediate formed upon migratory insertion with a heteroatom electrophile of interest is key to this type of transformation. Specifically, in the case of carbosulfenylation reactions, potential pitfalls include competitive β-hydride elimination caused by less reactive sulfur electrophiles and oxidative dinucleophile coupling caused by over-reactive sulfur electrophiles. With weakly coordinating directing groups, specially tailored sulfur electrophiles have been found to enable selective coupling at a specific point along the reaction coordinate,17e namely in oxidative addition with an alkylnickel(I) intermediate. As a complementary approach, we envisioned that strongly coordinating directing groups/auxiliaries may accommodate more diverse sulfur electrophiles, as examined in this study.
Recently, we reported a syn-(hetero)arylsulfenylation of unactivated alkenes with (hetero)arylboronic acid neopentyl ester nucleophiles that enables installation of a C(sp2) carbogenic group.17e Complementing this previous study by expanding the nucleophile scope to include C(sp3) partners is appealing given the potential for simultaneous C(sp3)–C(sp3) and C(sp3)–S formation (Scheme 1B). In this transformation, syn-selectivity is proposed to arise from the inner-sphere migratory insertion mechanism. Critical to the success of the present study is the use of a bidentate directing auxiliary (8-aminoquinoline, AQ) and the identification of a family of N-alkyl-N-(arylsulfenyl)benzamide sulfur electrophiles.
Results and discussion
Initial attempts to directly extend our previous method for (hetero)arylsulfenylation of alkenes17e containing native directing groups14 from C(sp2) to C(sp3) nucleophiles were unsuccessful (see ESI† for details), with catalyst deactivation being the main cause. Based on our previous work, we envisioned that a strongly coordinating bidentate directing auxiliary would better accommodate alkyl nucleophiles, particularly alkylzinc reagents.13,17a We thus launched our investigation by seeking to identify an optimal N–S electrophile using β,γ-unsaturated amide 1 as the pilot alkene substrate and commercially available diethylzinc as the standard nucleophile (Scheme 2). After extensive screening, tractable conditions were established with 10 mol% Ni(COD)(DMFU) as catalyst18 and THF (0.1 M) as solvent. A batchwise addition protocol was employed to avoid reagent decomposition and ensure high substrate conversion (see ESI† for details). Under these conditions, we surveyed a wide range of sulfenylating agents. First, reagents with N-alkyl sulfonamide leaving groups were tested, given their unique effectiveness in our earlier work.17e The electronic properties of the arenesulfonyl group (as in S1–S3) did not affect the yield. A slight increase in steric bulk from methyl to ethyl on the N-alkyl vector proved deleterious to the reaction (S4), while increasing steric encumbrance on the arenesulfonyl group exhibited no effect on the reaction (S5). An N-alkyl-alkylsulfonamide leaving group gave 76% yield (S6). We then turned our attention to N–S electrophiles with amide leaving groups. Caprolactam and N-acetyl aniline leaving groups gave the desired product in moderate yields (S7–S8). Next, a series of N-alkyl-N-(arylsulfenyl)benzamide family of sulfur electrophiles were evaluated. Electronic effects within this class of reagents were tested by varying the substituents on the para-position (S9–S13). The commercially available N-methyl benzamide leaving group (S10) gave the highest yield of 90%. When using an in situ ligation protocol with Ni(COD)2 as pre-catalyst and DMFU as ligand, the desired product was obtained in slightly diminished yield (83%). The X-ray crystal structure of S10 revealed a slightly non-pyramidalized nitrogen center and an N–S bond of 1.692 Å.17e Increasing the steric encumbrance on either the benzoyl (S14) or the N-alkyl (S15–S17) fragment gave diminished yields. Interestingly, S18–S21 with BDEs ranging from 39.6 kcal mol−1 to 79.6 kcal mol−1 all furnished the desired product in moderate to good yields (58–86%). Notably, commercially available disulfides also gave reasonably good yields (typically within 25% of the optimal N–S reagent family) and thus represent a cost-effective alternative (see ESI† for details). The effectiveness of a structurally diverse collection of sulfenylating agents in this reaction stands in contrast to our previous findings on arylsulfenylation of alkenes with native directing groups, where efficient three-component coupling is only achieved with N–S electrophiles with a narrow range of properties. Our current hypothesis is that the origin of this difference is the stabilizing nature of the bidentate directing group, which rigidifies the key 5,5-nickelabicycle intermediate, thereby suppressing side reactions and making the intermediate sufficiently long-lived to engage coupling partners with diverse reactivity profiles. | ||
| Scheme 2 Optimization of sulfur electrophilea. aReaction conditions: Ni(COD)(DMFU)/1/Et2Zn/[N–S] = 0.01/0.1/0.15/0.18 (mmol). Batchwise addition: (1) [N–S]/Et2Zn = 0.12/0.1 (mmol), 30 min; (2) then [N–S]/Et2Zn = 0.12/0.1 (mmol), 20 h. Reaction was run in THF (0.1 M) at 60 °C. All percentages represent 1H NMR yields with CH2Br2 as internal standard. DMFU = dimethyl fumarate. bResult in parentheses obtained with Ni(COD)2 (10 mol%) as pre-catalyst and DMFU (20 mol%) as ligand. | ||
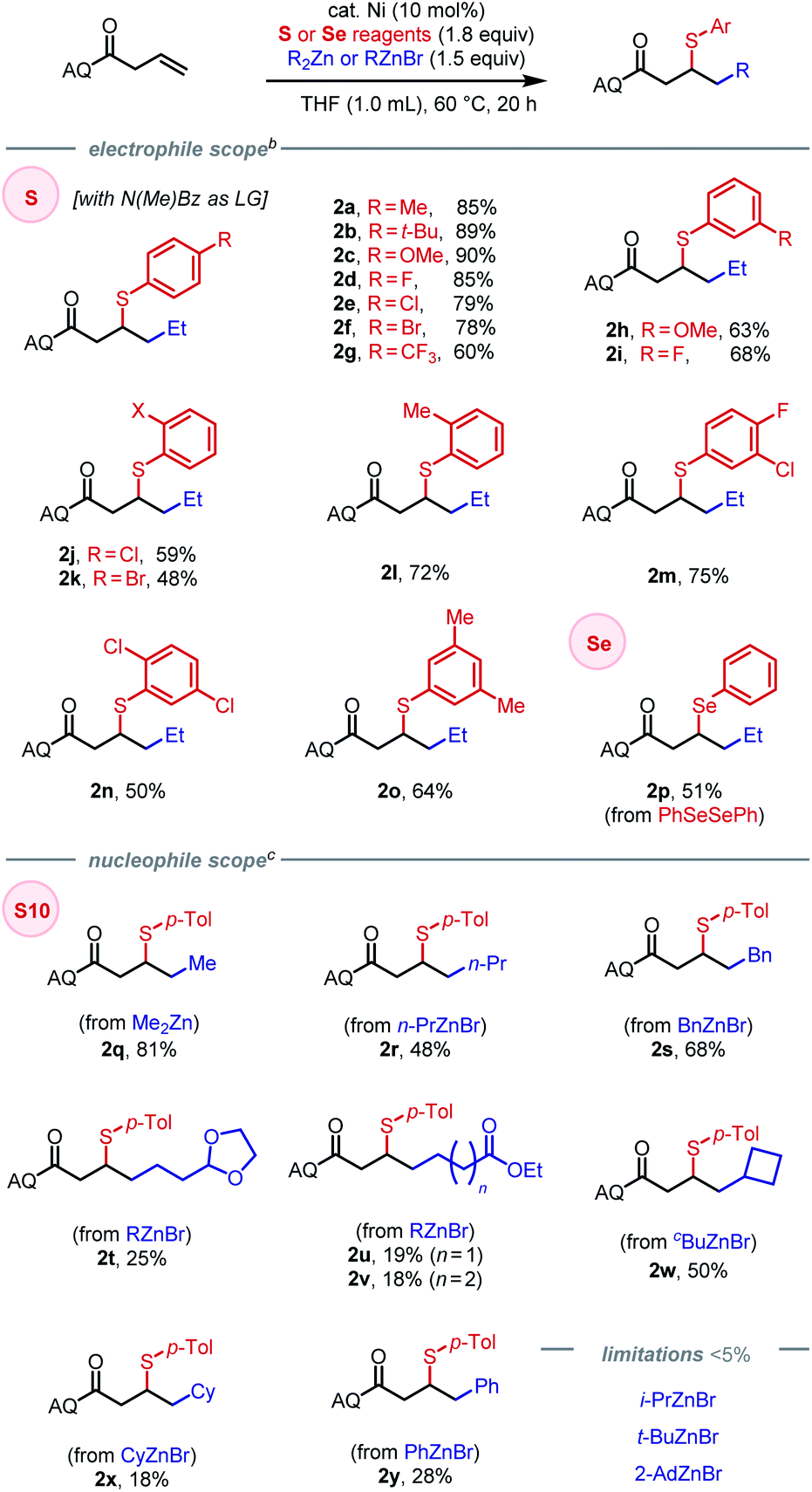
Having identified an effective leaving group, our focus then shifted to exploring the scope of the method with respect to each of the three components, namely electrophiles, nucleophiles (Table 1), and alkene substrates (Table 2). Using N-methyl-benzamide as leaving group, N–S electrophiles analogous to S10 were used to examine the electrophile scope. We first evaluated para-substituted arylsulfenyl coupling partners with different electronic properties and found that higher yield was obtained with N–S electrophiles that bear an electron-donating substituent (2a–2g). Moderate yields were obtained with reagents bearing meta-substituents (2h–2i). Ortho-substitution gave diminished yields due to steric hindrance (2j–2l). Arylsulfenyl units with diverse substitution patterns were tolerated, giving moderate to good yields (2m–2o). Preliminary data showed limited success (<5% of the desired product) with alkylsulfenyl electrophiles. To our delight, 1,2-carboselenation was also achieved using diphenyl diselenide as an electrophile (2p).
| a Reactions performed on 0.1 mmol scale. Percentages represent isolated yields. Batchwise addition: (1) [E]/[Nuc] = 0.12/0.1 (mmol), 30 min; then (2) [E]/[Nuc] = 0.12/0.1 (mmol), 20 h. b Reaction conditions: Ni(COD)(DMFU)/1/Et2Zn/[N–S] = 0.01/0.1/0.15/0.18 (mmol). c Reaction conditions: Ni(COD)2/1/RZnX/S10 = 0.01/0.1/0.15/0.18 (mmol). For 2q, Ni(COD)(DMFU)/1/Me2Zn/S10 = 0.01/0.1/0.15/0.18 (mmol). |
|---|
|
| a Reactions performed on 0.1 mmol scale. Percentages represent isolated yields. Batchwise addition: (1) [E]/[Nuc] = 0.12/0.1 (mmol), 30 min; then (2) [E]/[Nuc] = 0.12/0.1 (mmol), 20 h. b Reaction conditions: Ni(COD)(DMFU)/alkene/Et2Zn/S1 = 0.01/0.1/0.15/0.18 (mmol). c Reaction conditions: Ni(COD)(DMFU)/alkene/Et2Zn/S10 = 0.01/0.1/0.15/0.18 (mmol). |
|---|

|
Compatible nucleophiles include commercially available diorganozinc or organozinc bromide reagents, though some examples provided modest yields. When using Me2Zn as nucleophile under the established conditions, 2q was obtained in 81% yield. However, applying the same protocol to alkylzinc bromide reagents was lower-yielding, likely stemming from their attenuated reactivity. In these cases, yields could be slightly improved by using Ni(COD)2 as pre-catalyst in place of Ni(COD)(DMFU) (see SI for details). The origin of this difference in pre-catalyst performance is unclear at this time. Primary alkylzinc bromide reagents with n-propyl or benzyl groups provided the corresponding products in (2r–2s) in moderate yields. Synthetically useful functional groups such as dioxolane (2t) and ethyl esters (2u–2v) were tolerated as well, albeit in diminished yields. Cyclic secondary alkyl nucleophiles, such as cyclobutyl and cyclohexyl groups, could be introduced in moderate yields (2w–2x). Due to steric hindrance or susceptibility to β-hydride elimination, acyclic secondary and tertiary alkylzinc reagents were incompatible in the reaction, as was the extremely hindered cyclic secondary alkyl coupling partner, 2-AdZnBr. Using phenylzinc bromide as nucleophile, 1,2-arylsulfenylation could be achieved in 28% yield (2y).
A series of alkenyl amide substrates with different substitution patterns were evaluated. Alkenes with α-substituents benefited from using the more reactive sulfonamide-derived N–S electrophile S1, giving the corresponding products in moderate to good yields (3a–3f), with greater steric encumbrance leading to lower yields and slightly higher diastereoselectivity. We then tested internal alkenes with both Z and E configurations with the expectation that the stereochemistry of the product would provide insight into the mechanism of the reaction. First, using (E)-3-pentenoic acid derived substrate, 3g was obtained in 89% yield and >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoselectivity. The relative configuration was confirmed by X-ray crystallography, indicating a syn-addition process that arises from a migratory insertion mechanism. Interestingly, using the reaction to form 3g as a model system, we found that changing the electrophile to S18 or S19 (featuring vastly different BDE) gave the corresponding product with consistent diastereoselectivity (>20:1) albeit in lower yields, 84% and 70%, respectively (see SI for details). The opposite diastereomer was obtained with the combination of (E)-3-hexenoic-acid-derived substrate and dimethylzinc nucleophile (3h). Using (Z)-alkene as substrates, the desired products could be obtained in moderate to good yields and high diastereoselectivity (3i–3l). Alkenes bearing masked –OH and –NH2 moieties were tolerated as well (3m–3p), with –NHBoc, as in 3n, led to diminished yield due to the incompatibility with zinc reagent. The present method shows high diastereoselectivity with both E and Z alkenes compared to our previous study on 1,2-(hetero)arylsulfenylation using monodentate directing group where only the more reactive Z alkenes gave consistently high diastereoselectivity (10–20:1). Our current hypothesis is that the 5,5-nickelabicycle intermediate that arises through use of a bidentate auxiliary is key to suppressing the homolysis/recombination pathway that erodes d.r. with weakly coordinating monodentate directing groups.
:1 diastereoselectivity. The relative configuration was confirmed by X-ray crystallography, indicating a syn-addition process that arises from a migratory insertion mechanism. Interestingly, using the reaction to form 3g as a model system, we found that changing the electrophile to S18 or S19 (featuring vastly different BDE) gave the corresponding product with consistent diastereoselectivity (>20:1) albeit in lower yields, 84% and 70%, respectively (see SI for details). The opposite diastereomer was obtained with the combination of (E)-3-hexenoic-acid-derived substrate and dimethylzinc nucleophile (3h). Using (Z)-alkene as substrates, the desired products could be obtained in moderate to good yields and high diastereoselectivity (3i–3l). Alkenes bearing masked –OH and –NH2 moieties were tolerated as well (3m–3p), with –NHBoc, as in 3n, led to diminished yield due to the incompatibility with zinc reagent. The present method shows high diastereoselectivity with both E and Z alkenes compared to our previous study on 1,2-(hetero)arylsulfenylation using monodentate directing group where only the more reactive Z alkenes gave consistently high diastereoselectivity (10–20:1). Our current hypothesis is that the 5,5-nickelabicycle intermediate that arises through use of a bidentate auxiliary is key to suppressing the homolysis/recombination pathway that erodes d.r. with weakly coordinating monodentate directing groups.
Subsequently, a large-scale experiment was performed to demonstrate the practicality of this methodology (Scheme 3A). Alkylsulfenylation of alkene substrate 1 offered 2a in 70% isolated yield using an in situ DMFU ligation procedure, compared with 85% on 0.1 mmol scale (Table 1). Deprotection of the aminoquinoline directing group in using 6 M HCl afforded 4 in 84% yield with arylsulfenyl group intact (Scheme 3B).
 | ||
| Scheme 3 Large-scale experiment and deprotection of directing auxiliarya. aLarge-scale reaction performed with: Ni(COD)2/DMFU/1/Et2Zn/[N–S] = 0.25/0.5/2.5/3.75/4.5 (mmol). Batchwise addition: (1) [N–S]/[Et2Zn] = 3.0/2.5 (mmol), 30 min; then (2) [N–S]/[Et2Zn] = 1.5/1.25 (mmol), 20 h. Deprotection of directing group experiment performed with 2a (0.1 mmol). Percentages represent isolated yields. | ||
Conclusions
In summary, a series of sulfenyl (and seleno) electrophiles were identified to enable the 1,2-alkylsulfenylation and -selenation of unactivated alkenes with alkylzinc reagents as C(sp3) nucleophiles. Among the effective sulfenylating agents, an N-alkyl-N-(arylsulfenyl)benzamide family of sulfur electrophiles was identified through systematic structure–reactivity analysis. The reaction was enabled by a removable bidentate directing auxiliary that controls the regio- and diastereochemical outcome of this reaction. High syn-selectivity derived from an inner-sphere migratory insertion mechanism was obtained with a variety of E- and Z-internal alkenes.Data availability
All experimental procedures and data related to this study can be found in the ESI.† Original NMR data for the products (in MNova format) are included in a master ZIP file.Author contributions
Z.-Q. L. and K. M. E. conceived the project. Z.-Q. L. optimized the reaction conditions. Z.-Q. L. and W.-J. H. evaluated the scope. Z.-Q. L., W.-J. H., and H.-Q. N. synthesized the starting materials. Z.-Q. L. and K. M. E. wrote the manuscript with input from the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Foundation (CHE-1800280). We thank Bristol Myers Squibb for a Graduate Fellowship (Z.-Q. L.) and Nankai University College of Chemistry for an International Research Scholarship (W.-J. H.). We thank Dr Nana Kim for providing the nickel pre-catalysts used in this study. Dr Milan Gembicky and Dr Jake B. Bailey are acknowledged for X-ray crystallographic analysis. Quynh Nguyen Wong and Brittany Sanchez (Scripps Research Automated Synthesis Facility) are acknowledged for HRMS analysis. We thank Camille Z. Rubel for carefully proofreading this manuscript.References
- (a) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed; (b) C. Zhao, K. P. Rakesh, L. Ravidar, W.-Y. Fang and H.-L. Qin, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS PubMed.
- K. Takimiya, I. Osaka, T. Mori and M. Nakano, Acc. Chem. Res., 2014, 47, 1493–1502 CrossRef CAS PubMed.
- For reviews, see: (a) T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS PubMed; (b) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed . For representative recent reports, see:; (c) Y. Fang, T. Rogge, L. Ackermann, S.-Y. Wang and S.-J. Ji, Nat. Commun., 2018, 9, 2240 CrossRef PubMed; (d) D. Liu, H.-X. Ma, P. Fang and T.-S. Mei, Angew. Chem., Int. Ed., 2019, 58, 5033–5037 CrossRef CAS PubMed; (e) N. W. J. Ang and L. Ackermann, Chem.–Eur. J., 2021, 27, 4883–4887 CrossRef CAS PubMed; (f) T. Delcaillau, P. Boehm and B. Morandi, J. Am. Chem. Soc., 2021, 143, 3723–3728 CrossRef CAS PubMed; (g) T. Delcaillau and B. Morandi, Chem.–Eur. J., 2021, 27, 11823–11826 CrossRef CAS PubMed.
- G. Mann, D. Baranano, J. F. Hartwig, A. L. Rheingold and I. A. Guzei, J. Am. Chem. Soc., 1998, 120, 9205–9219 CrossRef CAS.
- C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC.
- (a) R. Li, Y. Zhou, K.-Y. Yoon, Z. Dong and G. Dong, Nat. Commun., 2019, 10, 3555 CrossRef PubMed; (b) W. Cai and Z. Gu, Org. Lett., 2019, 21, 3204–3209 CrossRef CAS PubMed.
- (a) B. M. Trost, T. Shibata and S. J. Martin, J. Am. Chem. Soc., 1982, 104, 3228–3230 CrossRef CAS; (b) B. M. Trost and S. J. Martin, J. Am. Chem. Soc., 1984, 106, 4263–4265 CrossRef CAS.
- (a) S. E. Denmark and A. Jaunet, J. Am. Chem. Soc., 2013, 135, 6419–6422 CrossRef CAS PubMed; (b) S. E. Denmark and A. Jaunet, J. Org. Chem., 2014, 79, 140–171 CrossRef CAS PubMed; (c) S. E. Denmark, E. Hartmann, D. J. P. Kornfilt and H. Wang, Nat. Chem., 2014, 6, 1056–1064 CrossRef CAS PubMed; (d) S. E. Denmark and H. M. Chi, J. Am. Chem. Soc., 2014, 136, 3655–3663 CrossRef CAS PubMed; (e) Z. Tao, K. A. Robb, J. L. Panger and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 15621–15625 CrossRef CAS PubMed; (f) Z. Tao, K. A. Robb, K. Zhao and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 3569–3573 CrossRef CAS PubMed . For a recent review, see:; (g) A. Matviitsuk, J. L. Panger and S. E. Denmark, Angew. Chem., Int. Ed., 2020, 59, 19796–19819 CrossRef CAS PubMed.
- (a) J. Luo, Q. Cao, X. Cao and X. Zhao, Nat. Commun., 2018, 9, 527 CrossRef PubMed; (b) X.-F. Song, A.-H. Ye, Y.-Y. Xie, J.-W. Dong, C. Chen, Y. Zhang and Z.-M. Chen, Org. Lett., 2019, 21, 9550–9554 CrossRef CAS PubMed; (c) Y.-Y. Xie, Z.-M. Chen, H.-Y. Luo, H. Shao, Y.-Q. Tu, X. Bao, R.-F. Cao, S.-Y. Zhang and J.-M. Tian, Angew. Chem., Int. Ed., 2019, 58, 12491–12496 CrossRef CAS PubMed; (d) M. Tang, S. Han, S. Huang, S. Huang and L.-G. Xie, Org. Lett., 2020, 22, 9729–9734 CrossRef CAS PubMed.
- For selected examples using heteroatom nucleophiles, see: (a) S. E. Denmark and H. M. Chi, J. Am. Chem. Soc., 2014, 136, 8915–8918 CrossRef CAS PubMed; (b) S. E. Denmark, S. Rossi, M. P. Webster and H. Wang, J. Am. Chem. Soc., 2014, 136, 13016–13028 CrossRef CAS PubMed; (c) A. Matviitsuk and S. E. Denmark, Angew. Chem., Int. Ed., 2019, 58, 12486–12490 CrossRef CAS PubMed; (d) A. Roth and S. E. Denmark, J. Am. Chem. Soc., 2019, 141, 13767–13771 CrossRef CAS PubMed.
- (a) J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287–4296 RSC; (b) J. Diccianni, Q. Lin and T. Diao, Acc. Chem. Res., 2020, 53, 906–919 CrossRef CAS PubMed; (c) X. Qi and T. Diao, ACS Catal., 2020, 10, 8542–8556 CrossRef CAS PubMed.
- (a) R. Giri and S. KC, J. Org. Chem., 2018, 83, 3013–3022 CrossRef CAS PubMed; (b) S. Zhu, X. Zhao, H. Li and L. Chu, Chem. Soc. Rev., 2021, 50, 10836–10856 RSC; (c) L. M. Wickham and R. Giri, Acc. Chem. Res., 2021, 54, 3415–3437 CrossRef CAS PubMed.
- For selected examples using directing auxiliaries: (a) J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657–10660 CrossRef CAS PubMed; (b) J. Derosa, V. A. van der Puyl, V. T. Tran, M. Liu and K. M. Engle, Chem. Sci., 2018, 9, 5278–5283 RSC; (c) P. Basnet, R. K. Dhungana, S. Thapa, B. Shrestha, S. KC, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2018, 140, 7782–7786 CrossRef CAS PubMed; (d) W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600–607 RSC.
- For selected examples using native directing functional groups: (a) J. Derosa, R. Kleinmans, V. T. Tran, M. K. Karunananda, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 17878–17883 CrossRef CAS PubMed; (b) J. Derosa, T. Kang, V. T. Tran, S. R. Wisniewski, M. K. Karunananda, T. C. Jankins, K. L. Xu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 1201–1205 CrossRef CAS PubMed; (c) V. T. Tran, Z.-Q. Li, T. J. Gallagher, J. Derosa, P. Liu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 7029–7034 CrossRef CAS PubMed; (d) O. Apolinar, V. T. Tran, N. Kim, M. A. Schmidt, J. Derosa and K. M. Engle, ACS Catal., 2020, 10, 14234–14239 CrossRef CAS; (e) R. K. Dhungana, V. Aryal, D. Niroula, R. R. Sapkota, M. G. Lakomy and R. Giri, Angew. Chem., Int. Ed., 2021, 60, 19092–19096 CrossRef CAS PubMed; (f) R. Kleinmans, O. Apolinar, J. Derosa, M. K. Karunananda, Z.-Q. Li, V. T. Tran, S. R. Wisniewski and K. M. Engle, Org. Lett., 2021, 23, 5311–5316 CrossRef CAS PubMed.
- H. Wang, C.-F. Liu, R. T. Martin, O. Gutierrez and M. J. Koh, Nat. Chem., 2022, 14, 188–195 CrossRef CAS PubMed.
- (a) K. M. Logan, S. R. Sardini, S. D. White and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 159–162 CrossRef CAS PubMed; (b) S. R. Sardini, A. L. Lambright, G. L. Trammel, H. H. Omer, P. Liu and M. K. Brown, J. Am. Chem. Soc., 2019, 141, 9391–9400 CrossRef PubMed; (c) L.-A. Chen, A. R. Lear, P. Gao and M. K. Brown, Angew. Chem., Int. Ed., 2019, 58, 10956–10960 CrossRef CAS PubMed; (d) S. Joung, A. M. Bergmann and M. K. Brown, Chem. Sci., 2019, 10, 10944–10947 RSC; (e) Z. Zhang and X. Hu, ACS Catal., 2020, 10, 777–782 CrossRef CAS; (f) A. L. Lambright, Y. Liu, I. A. Joyner, K. M. Logan and M. K. Brown, Org. Lett., 2021, 23, 612–616 CrossRef CAS PubMed; (g) D. Ni and M. K. Brown, ACS Catal., 2021, 11, 1858–1862 CrossRef CAS PubMed; (h) A. K. Simlandy, S. R. Sardini and M. K. Brown, Chem. Sci., 2021, 12, 5517–5521 RSC; (i) G. L. Trammel, R. Kuniyil, P. F. Crook, P. Liu and M. K. Brown, J. Am. Chem. Soc., 2021, 143, 16502–16511 CrossRef CAS PubMed; (j) J. Wang, Z. Duan, X. Liu, S. Dong, K. Chen and J. Li, Angew. Chem., Int. Ed., 2022, 61, e202202379 CAS.
- (a) V. A. van der Puyl, J. Derosa and K. M. Engle, ACS Catal., 2019, 9, 224–229 CrossRef CAS; (b) T. Kang, N. Kim, P. T. Cheng, H. Zhang, K. Foo and K. M. Engle, J. Am. Chem. Soc., 2021, 143, 13962–13970 CrossRef CAS PubMed; (c) T. Kang, J. M. González, Z.-Q. Li, K. Foo, P. T. W. Cheng and K. M. Engle, ACS Catal., 2022, 12, 3890–3896 CrossRef CAS; (d) L. Xie, S. Wang, L. Zhang, L. Zhao, C. Luo, L. Mu, X. Wang and C. Wang, Nat. Commun., 2021, 12, 6280 CrossRef CAS PubMed; (e) Z.-Q. Li, Y. Cao, T. Kang and K. M. Engle, J. Am. Chem. Soc., 2022, 144, 7189–7197 CrossRef CAS PubMed , A related example was published while this manuscript was under review:; (f) L. Zhu, X. Meng, L. Xie, W. Li, Q. Shen, L. Zhang and C. Wang, Org. Chem. Front., 2022 10.1039/D1032QO00396A.
- (a) N. Kim, V. T. Tran, O. Apolinar, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, Synlett, 2021, 32, 1570–1754 CrossRef CAS; (b) V. T. Tran, N. Kim, C. Z. Rubel, X. Wu, T. Kang, T. C. Jankins, Z.-Q. Li, M. V. Joannou, S. Ayers, M. Gembicky, J. Bailey, E. J. Sturgell, B. B. Sanchez, J. S. Chen, S. Lin, M. D. Eastgate, S. R. Wisniewski and K. M. Engle, ChemRxiv, 2022 DOI:10.26434/chemrxiv-2022-7zjvh.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2154909 and 2149662. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01563c |
| This journal is © The Royal Society of Chemistry 2022 |