
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite specific NMR characterization of abeta-40 oligomers cross seeded by abeta-42 oligomers†
Han-Wen
Chang
 a,
Ho-I.
Ma
a,
Yi-Shan
Wu
a,
Ming-Che
Lee
a,
Eric
Chung-Yueh Yuan
a,
Shing-Jong
Huang
b,
Yu-Sheng
Cheng
a,
Meng-Hsin
Wu
c,
Ling-Hsien
Tu
c and
Jerry Chun Chung
Chan
*a
a,
Ho-I.
Ma
a,
Yi-Shan
Wu
a,
Ming-Che
Lee
a,
Eric
Chung-Yueh Yuan
a,
Shing-Jong
Huang
b,
Yu-Sheng
Cheng
a,
Meng-Hsin
Wu
c,
Ling-Hsien
Tu
c and
Jerry Chun Chung
Chan
*a
aDepartment of Chemistry, National Taiwan University, No. 1, Section 4, Roosevelt Road, Taipei, 10617, Taiwan. E-mail: chanjcc@ntu.edu.tw
bInstrumentation Center, National Taiwan University, No. 1, Section 4, Roosevelt Road, Taipei, 10617, Taiwan
cDepartment of Chemistry, National Taiwan Normal University, No. 88, Section 4, Ting-Chow Road, Taipei, 11677, Taiwan
First published on 22nd June 2022
Abstract
Extracellular accumulation of β amyloid peptides of 40 (Aβ40) and 42 residues (Aβ42) has been considered as one of the hallmarks in the pathology of Alzheimer's disease. In this work, we are able to prepare oligomeric aggregates of Aβ with uniform size and monomorphic structure. Our experimental design is to incubate Aβ peptides in reverse micelles (RMs) so that the peptides could aggregate only through a single nucleation process and the size of the oligomers is confined by the physical dimension of the reverse micelles. The hence obtained Aβ oligomers (AβOs) are 23 nm in diameter and they belong to the category of high molecular-weight (MW) oligomers. The solid-state NMR data revealed that Aβ40Os adopt the structural motif of β-loop-β but the chemical shifts manifested that they may be structurally different from low-MW AβOs and mature fibrils. From the thioflavin-T results, we found that high-MW Aβ42Os can accelerate the fibrillization of Aβ40 monomers. Our protocol allows performing cross-seeding experiments among oligomeric species. By comparing the chemical shifts of Aβ40Os cross seeded by Aβ42Os and those of Aβ40Os prepared in the absence of Aβ42Os, we observed that the chemical states of E11, K16, and E22 were altered, whereas the backbone conformation of the β-sheet region near the C-terminus was structurally invariant. The use of reverse micelles allows hitherto the most detailed characterization of the structural variability of Aβ40Os.
Introduction
Aggregation of amyloid beta peptides (Aβ) is closely associated with Alzheimer's disease (AD), where the 40-residue (Aβ40) and 42-residue (Aβ42) peptides are two major Aβ species derived from amyloid precursor protein (APP).1–3 Their primary sequences are identical from residues 1–40 and the two additional residues at the C-terminus of Aβ42 are I41 and A42. Although the severity of dementia correlates only weakly to the density or number of fibrillar amyloid plaques, there is a robust correlation between soluble Aβ levels and the extent of synaptic loss.4–6 Since the discovery of Aβ oligomers (AβOs) as potent neurotoxins,7 it is increasingly accepted that AβOs are the most pathogenic form of Aβ.8–11 There is a continuous distribution of soluble Aβ species from monomeric form up to oligomers in excess of 100 kDa.5 At the molecular level, a chemical equilibrium is being re-established among the large with smaller species and monomers. Therefore, it is very difficult to obtain a homogeneous preparation of a particular oligomeric species,9 rendering the comparison of the biochemical results of AβOs in the literature very difficult.12 Oligomers of low molecular weight (low-MW) refer to AβOs of MW ≤ 30 kDa, whereas high-MW AβOs have MW > 100 kDa.5 There are many different ways to prepare low-MW AβOs.13 Dimers of Aβ peptides can be prepared by the use chemical linkers.14–16 The technique of photo induced cross-linking of unmodified proteins has been used to demonstrate that the Aβ40 tetramers (n = 4) and other low-n are in rapid equilibrium, whereas the stable oligomers of Aβ42 (n = 5 and 6) have a strong propensity to assemble further to form superstructures.17–19 Very recently, homogenous and stable Aβ42 tetramers and octamers were successfully prepared in detergent micelles.20,21 On the other hand, high-MW AβOs are usually prepared by incubating the monomer solution of Aβ in vitro at relatively low temperature or in a short period of time.22–28 Other chemical approaches such as binding with metal ions,29,30 antibody,31 small molecules,32,33 and polymer-nanodiscs34 have also been developed to prepare high-MW AβOs. Both the low-MW and high-MW Aβ aggregates have different impacts on synapses.10 In fact, high-MW AβOs may dissociate into low-MW AβOs in mildly alkaline aqueous buffer.35 Currently, it remains unsettled which forms of AβOs are the true culprits of AD.6,11The aggregation of Aβ40 and Aβ42 have different dependence on the peptide concentration or other environmental factors.36 The kinetic profiles of the Aβ aggregation process usually comprise the lag phase, elongation, and eventually the plateau phase. Adding fragments of preformed fibrils (fibril seeds) of the same protein can significantly shorten the period of lag phase. This phenomenon, known as self-seeding, is consistent with the nucleation-growth mechanism.37,38 While the self-seeding effect has been well established for Aβ40 and Aβ42,39–43 the situation is more complicated for the cross seeding between Aβ40 and Aβ42. Earlier reports suggested that the aggregation of Aβ40 monomers can be facilitated by the presence of Aβ42 fibril seeds.40,42,44 By contrast, more recent studies reported that the seeding effect of Aβ42 fibrillar seeds on Aβ40 monomer is insignificant.45,46 Indeed, solid-state NMR studies revealed that the backbone conformation of the β-strands of Aβ40 and Aβ42 fibrils are different.47–49 The formation of a sufficiently large peptide cluster, which is coined as the “nucleus” according to the classical nucleation theory for crystal growth, is the most critical event of Aβ aggregation. As proposed by Knowles and co-workers,50–53 the formation of Aβ nucleus can occur via the processes of primary nucleation, fibril fragmentation, and the fibril-assisted mechanism (secondary nucleation). The existence of multiple nucleation pathways might well be the underlying physical reason for the structural polymorphism commonly observed for Aβ aggregates prepared in bulk solution.54 Furthermore, it has been argued that the interaction between Aβ40 and Aβ42 was restricted at the step of primary nucleation.45 The aforementioned studies indicated that it is highly desirable to develop a method to control the aggregation pathway of Aβ peptides so that the interaction between Aβ40 and Aβ42 could be characterized at the molecular level.
Reverse micelles (RMs), which correspond to water droplets in an oil phase, have been extensively used as microreactors for a large variety of materials.55,56 In this chemical system, the RMs undergo coalescence and separation at a rate constant of 106–108 dm3 mol−1 s−1.57 The material transfer among RMs has been convincingly demonstrated in many studies of enzymatic activities, where substrate molecules were delivered to the enzyme via the continuous fusion and fission of RMs.55,58,59 Consequently, we hypothesize that Aβ peptides could aggregate via a single nucleation process in a space confined by the physical dimension of RMs. This concept has been proved feasible in our earlier attempt to prepare protofibrils (curvilinear fibrils) of Aβ40 peptides.60 In this work, we have prepared high-MW oligomeric aggregates of Aβ40 with a single dominant structure (monomorphic) based on the RMs formed by the non-ionic surfactants poly(oxyethylene) nonylphenyl ether (Igepal CO520). For the first time in the literature, we were able to prepare Aβ40Os cross seeded by Aβ42Os. The solid-state NMR results showed that the chemical states of E11, K16, and E22 of Aβ40 are altered upon the interaction between high-MW Aβ42Os and Aβ40 peptides.
Results
Molecular weight of RMCO520Aβ40
The purity of the Aβ peptides were >95% (Fig. S1†). Size exclusion chromatography (SEC) and dynamic light scattering (DLS) were used to confirm that the freshly prepared Aβ40 peptides were in monomeric state (Fig. S2 and S3†). Aβ40 monomers (100 μM) were incubated in the RMs formed by the ternary system of CO520/cyclohexane/NH4OAc(aq) for 7 d (Fig. S4†).61 DLS measurements indicated that the RMs were stable for at least 10 days (Fig. S5†), where the RMs had a size of ca. 23 nm with polydispersity index (PdI) less than 0.10. To back extract the Aβ peptides from the RM solution, the stripping buffer (ZnCl2, 50 μM) was added to the RM solution. The ZnCl2 solution was used to suppress any further aggregation of the AβOs.62 The aqueous phase was then lyophilized to remove ammonium acetate and water. The obtained powder sample is henceforth referred to as RMCO520Aβ40. To estimate the amount of residual CO520, we carried out elemental analysis (EA) for RMCO520Aβ40 (Fig. S6 and Table S1†). CO520 and Aβ40 had different contributions to the atomic percentage of C, N, S, H of RMCO520Aβ40. As shown in Fig. S6,† the molar ratio of CO520 to Aβ40 was estimated to be 9.0 ± 0.7. In addition, the mass percentage of Aβ40 was ca. 24.9%, from which the efficiency of the back extraction of Aβ peptides was calculated to be ca. 66%. Although analytical size exclusion chromatography (SEC) could be used to estimate the MW of protein aggregates, it has been pointed out that the results for AβOs are unreliable because of the possible interaction between Aβ peptides and the SEC column material.28 On the other hand, the mass-per-length of amyloid fibrils can be accurately determined by scanning transmission microscopy (STEM) or tilted-beam TEM using tobacco mosaic virus (TMV) rods as reference objects.63Fig. 1 shows the STEM image of RMCO520Aβ40 and TMV, where the size of RMCO520Aβ40 particles was 23.1 ± 5.3 nm (Fig. S7†). Following the procedure described in the ESI,† the MW of RMCO520Aβ40, which contained CO520 and Aβ40, was determined as 4600 kDa, from which the AβO in RMCO520Aβ40 was estimated to have an MW of 2400 kDa (∼554 monomers). In comparison, the amyloid intermediate (Iβ) of Aβ40, with a size of 10–35 nm, was estimated to have MW ≥ 650 kDa by a glycerol gradient sedimentation assay.23 Using a similar method, the patient derived amylospheroids (ASPDs, 10–15 nm) were reported to have MW in the range of 158–669 kDa.64,65 It is well known that Aβ oligomers are poorly defined in size and a distribution of 150–1000 kDa has been reported for Aβ peptides in bulk solution by multiangle laser light scattering detection.66 While RMCO520Aβ40 is a non-fibrillar aggregate of Aβ40, its sizable MW suggested that RMCO520Aβ40 might possess the structural features required for the onset of fibrillar growth. We note in passing that micelle-like aggregates of Aβ40 peptides of 7 nm in diameter were identified as the nucleation center for Aβ40 fibrillization.67,68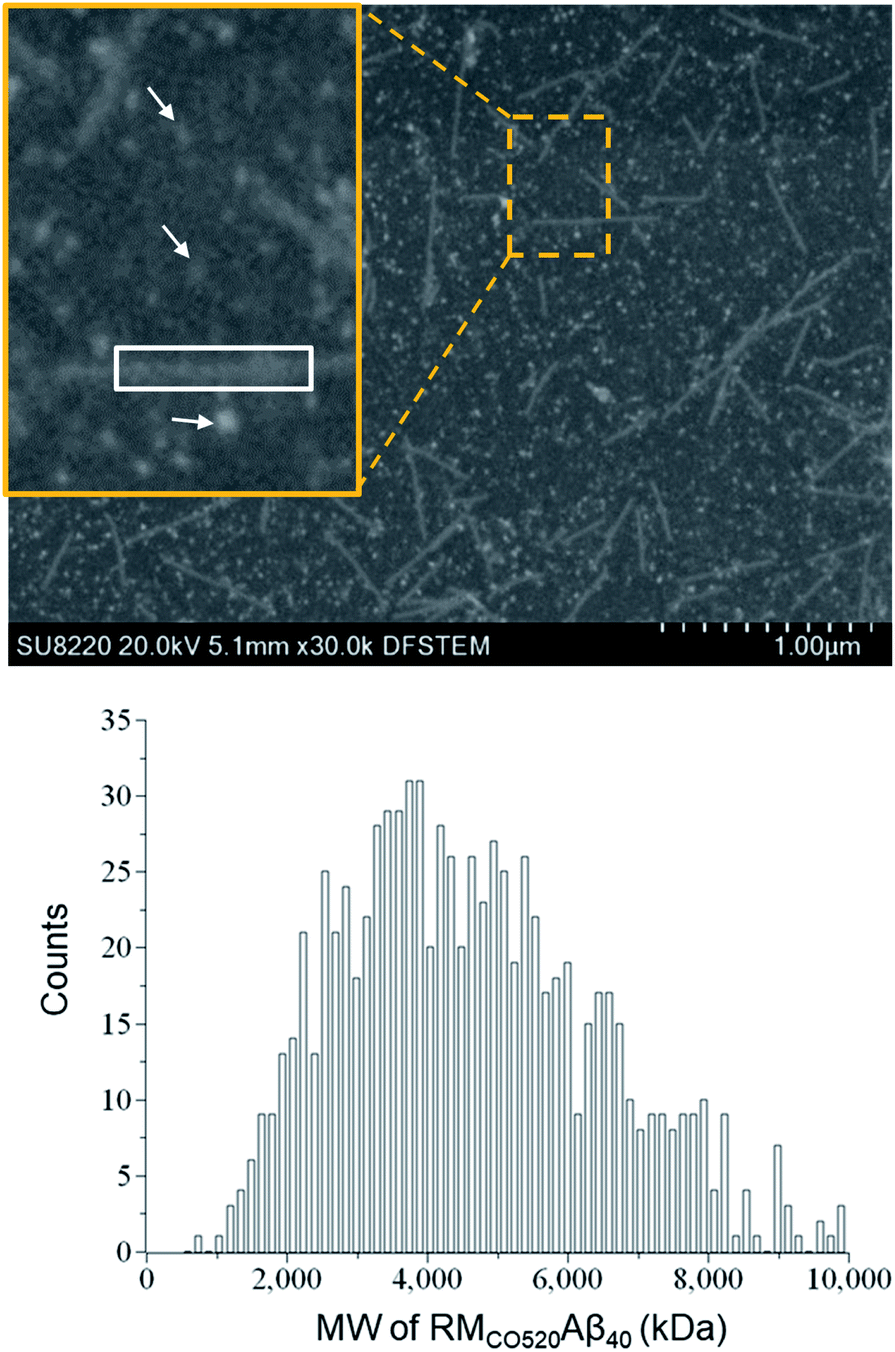 | ||
| Fig. 1 STEM image of a mixture of RMCO520Aβ40 and TMV. Typical oligomers of RMCO520Aβ40 are indicated by arrows. The rectangular box in white shows a fragment of TMV. The mean value of the MW of RMCO520Aβ40 was estimated to be 4600 kDa, which was obtained by the bootstrapping resampling method. | ||
Seeding and self-aggregating effects of RMCO520Aβ
The seeding effects of RMCO520Aβ40 on Aβ40 monomers (25 μM) was examined by the thioflavin T (ThT) assay, where EDTA solution was added to chelate Zn2+ ions. The control sample was prepared in the absence of Aβ peptides (RMCO520). As expected, the seeding effect of RMCO520Aβ40 on Aβ40 monomers was revealed by the shortening of the lag time (Fig. 2). Remarkably, the ThT results revealed that RMCO520Aβ42 was also able to promote the aggregation of Aβ40 monomers. Recently, it has been demonstrated that the oligomeric aggregates of a chimeric peptide can completely eliminate the lag period of Aβ40, resulting in immediate fibril elongation.16 Although the lag phase of Aβ40 was not completely eliminated by RMCO520Aβ42, the extent of the shortening of the lag period was comparable to what reported for the cross-seeding effect between Aβ40 and Aβ42.69 Similar ThT results were observed when the amount of seeds was increased from 4% to 10% (Fig. S8†). Amyloid fibrils were also formed by the self-aggregation of RMCO520Aβ (Fig. S9†). Goto and co-workers reported that protein-detergent complex would become an off-pathway aggregate when the mixed micelles of detergent and protein are thermodynamically and kinetically more stable than the amyloid fibrils.70 Because CO520 is a non-ionic surfactant (Fig. S4†), its interaction with Aβ peptides are expected to be small. The self-aggregating behaviors of RMCO520Aβ indicated that CO520 did not form any stable complex with Aβ peptides.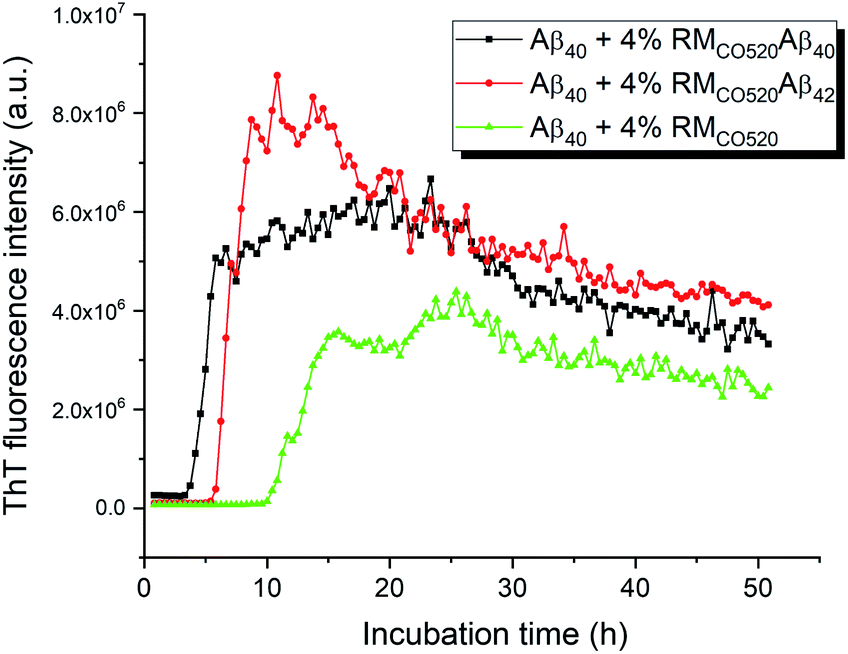 | ||
| Fig. 2 ThT Fluorescence results for probing the seeding effects of RMCO520Aβ. For all the measurements, the concentration of Aβ40 monomer was 25 μM. The amount of the seeds was adjusted to 4 mole% of the monomers. RMCO520 was prepared in the absence of Aβ peptides. | ||
Interactions between Aβ alloforms could modulate their aggregation behaviors in vitro.71 Previous studies suggested that there is a significant molecular cross-talk between Aβ40 monomers and various aggregation states of Aβ42.40,42,44 In particular, Aβ40 monomers have strong binding affinity for Aβ42 aggregates.72,73 Because the aggregation kinetics of Aβ42 is much faster than Aβ40,53 we attempted to investigate the effect of Aβ42Os on the aggregation process of Aβ40 monomers through our RM system. Briefly, the Aβ42 peptides were first incubated for three days and Aβ40 monomers were subsequently added in such a way that the final molar ratio of Aβ42 and Aβ40 was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. We henceforth refer the sample to as RMCO520Aβ42/40, which also exhibited seeding effects on Aβ40 monomers and the self-aggregating behavior (Fig. S10 and S11†). The size of RMCO520Aβ42/40 particles was 22.5 ± 6.1 nm (Fig. S12†), indicating that the size of both AβOs were constrained by the physical dimension of the RMs. The ThT fluorescence excitation spectrum revealed that ThT molecules can bind to RMCO520Aβ42/40, RMCO520Aβ42, and RMCO520Aβ40 (Fig. S13†), which verified that these high-MW Aβ oligomers had fibril-like structures.
:1. We henceforth refer the sample to as RMCO520Aβ42/40, which also exhibited seeding effects on Aβ40 monomers and the self-aggregating behavior (Fig. S10 and S11†). The size of RMCO520Aβ42/40 particles was 22.5 ± 6.1 nm (Fig. S12†), indicating that the size of both AβOs were constrained by the physical dimension of the RMs. The ThT fluorescence excitation spectrum revealed that ThT molecules can bind to RMCO520Aβ42/40, RMCO520Aβ42, and RMCO520Aβ40 (Fig. S13†), which verified that these high-MW Aβ oligomers had fibril-like structures.
Structural motif of RMCO520Aβ40
Four 13C enriched samples of RMCO520Aβ40 were prepared, covering 23 residues in the region from E11 to V40 (Table S2†). The enrichment level was 60%. A typical 13C–13C correlation spectrum acquired for RMCO520Aβ40 is shown in Fig. 3. While structural polymorphism was commonly observed for the Aβ40Os or Aβ fibrils prepared in bulk solution,26,54 the structure of RMCO520Aβ40 was monomorphic as evidenced by the fact that the NMR spectra were basically dominated by a single set of signals (Fig. S14–S16†). We attributed this favorable result to the scenario that the nucleation process of Aβ40 in a reverse micelle (23 nm in diameter) was dominated by a single pathway. The TEM images of the samples after the solid-state NMR measurements confirmed that the samples remained in the oligomeric state (Fig. S17†). The full width at half maximum (FWHM) data, with an average of 3.4 ppm (Fig. 4), were larger than the fibrillar aggregates.74 We did not rehydrate the lyophilized oligomer samples for NMR measurements, in order to prevent fibrillization from occurring. Although oligomeric aggregates of Aβ are transient species, the FWHM data supported that the molecular structure of RMCO520Aβ40 was not completely disordered. All the chemical shift data are summarized in Table S3,† from which the backbone torsion angles were estimated by TALOS-N.75 Fig. S18† illustrates the backbone torsion angles, indicating that the structural motif adopted by RMCO520Aβ40 was β1-loop-β2, which is the same as that of the spherical amyloid assembly (SPA) of Aβ42.76 The similar structural motif for Aβ40Os and Aβ42Os is consistent with the observation that both RMCO520Aβ40 and RMCO520Aβ42 could seed the fibrillization of Aβ40 monomers.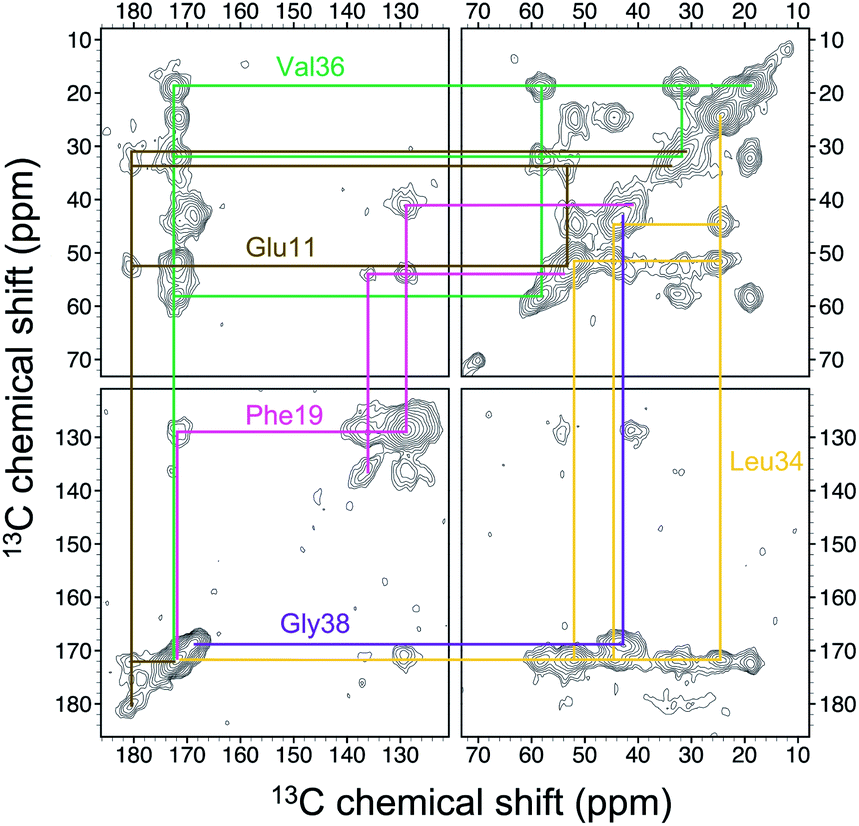 | ||
| Fig. 3 Spectral assignment for the 13C homonuclear correlation spectrum of RMCO520Aβ40 with uniformly 13C and 15N labeling at E11, F19, L34, V36, and G38 (sample S1). The contour levels were increased by a factor of 1.4 successively, where the base levels were set to 4× root-mean-square noise. | ||
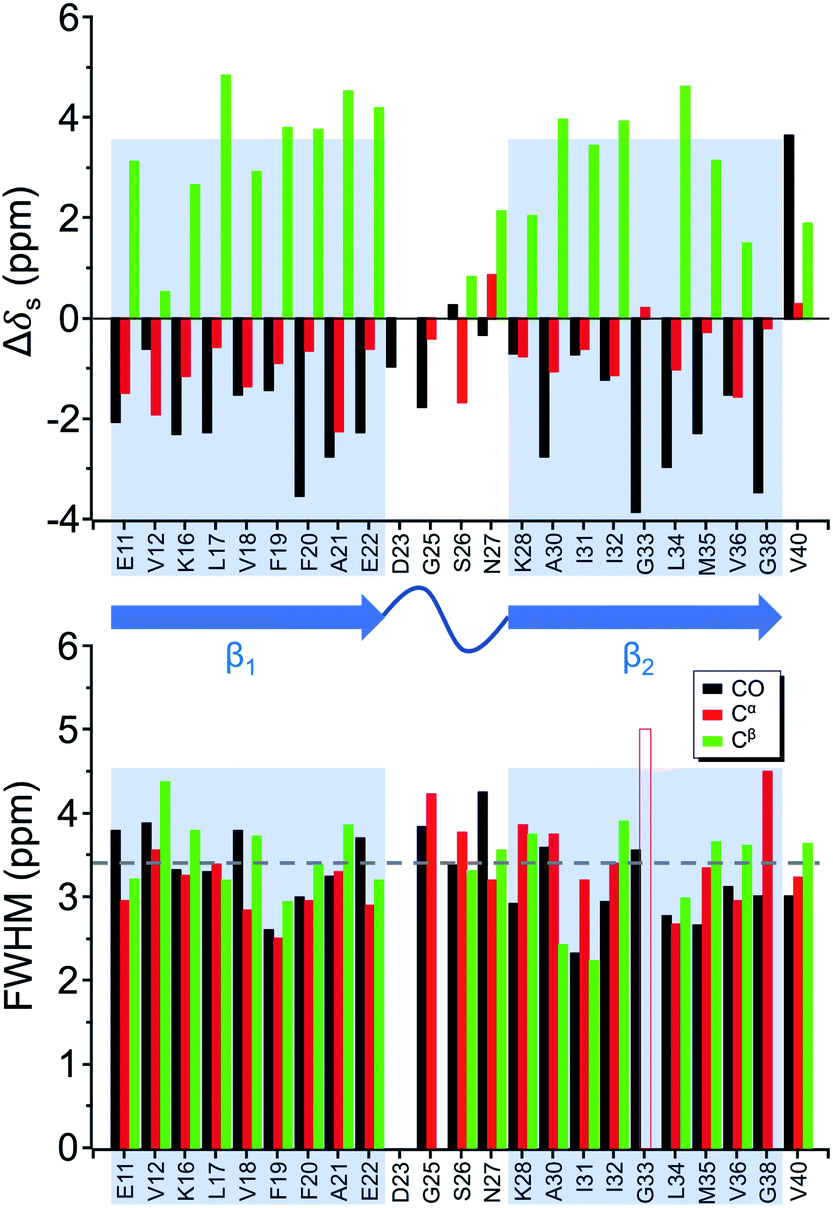 | ||
| Fig. 4 Secondary chemical shift (upper trace) and full width at half maximum (FWHM) of the NMR signals (lower trace) extracted from the 13C–13C correlation spectra of RMCO520Aβ40. The β-sheet region was highlighted in shaded rectangular blocks. The dashed line indicates the average FWHM. The open bar denotes FWHM > 5 ppm. | ||
The chemical shift data revealed some interesting structural features of RMCO520Aβ40. The backbone chemical shifts of RMCO520Aβ40 were very different from those reported for Aβ40 fibrils (Fig. 5), where 13C chemical shifts of mature fibrils were taken from wild-type Aβ40 fibrils incubated under quiescent conditions and agitated conditions,77 and brain-derived Aβ40 fibrils.78–80 The data for Aβ oligomers were taken from low-MW Aβ40Os,26 SPA,76 ASPD,27 and Iβ.23 Overall, a large chemical shift difference (>1.5 ppm) was found at the loop region (D23 to N27). Glycine residues are important structural markers for sharp loops or turns in amyloid structures because of their structural flexibility.49,81 Accordingly, the FWHM of all the 13C enriched glycines of RMCO520Aβ40 were >4 ppm. Comparing the glycine chemical shifts of RMCO520Aβ40 and those reported for in vitro fibrillar aggregates of Aβ40, a large discrepancy (>2 ppm) was observed at G25 and G33, whereas a reasonable agreement was at G38 (Fig. S19†). We surmised that a major conformational change between high-MW Aβ40Os and Aβ40 fibrils occurred at G25 and G33. While the structure of Aβ oligomers could be fibrillar like,82 a recent solid-state NMR study suggested that there is a significant difference in supramolecular organization between protofibrils and fibrils.28 Thus, it is possible that the supramolecular structure of RMCO520Aβ40 was different from mature fibrils. On the other hand, the backbone chemical shifts of RMCO520Aβ40, albeit very similar to those reported for Iβ (high-MW Aβ40Os), exhibit a significant difference from those reported for low-MW Aβ40Os (Fig. S20†).26 In other words, there could be a continuous structural evolution from low-MW Aβ40Os, to high-MW Aβ40Os, and then to mature fibrils.28 But this hypothesis has to be validated by more experimental evidences. In fact, it is not yet clear whether such difference in chemical shift is owing to the difference in incubation conditions or the size effect of the oligomers. We also note that the long-range contact between F19 and L34 were not observed in this work. While the relatively low level of 13C enrichment (60%) might have rendered the contact unobservable, it is equally plausible that such contact was absent in our samples.83
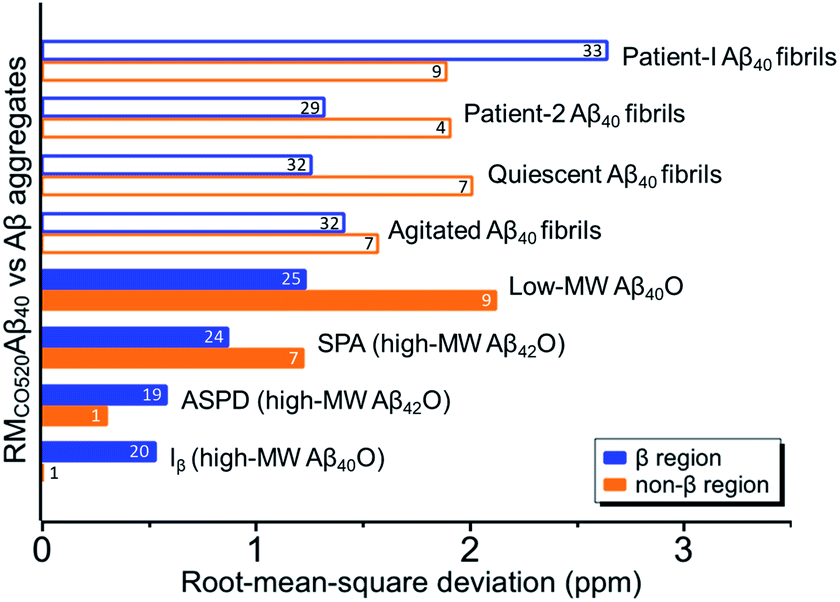 | ||
| Fig. 5 Comparison of the Cα and Cβ chemical shifts of RMCO520Aβ40 and the data of other Aβ40 aggregates in the literature. Solid bars indicate the results obtained for oligomeric species and open bars for fibrillar aggregates. The number of the data points for calculating the rms deviations are shown on the bars, which were largely limited by the availability of the literature data. The β-sheet and non-β-sheet regions were defined with respect to the RMCO520Aβ40 sample. | ||
Structural motif of oligomeric RMCO520Aβ42/40
In the foregoing section, we have demonstrated that RMs could be used to investigate the effect of Aβ42Os on the aggregation process of Aβ40. We carried out solid-state NMR measurements of RMCO520Aβ42/40 with the same isotopic labeling scheme as RMCO520Aβ40 (Fig. S21–24 and Table S4†). To facilitate the comparison of the data of RMCO520Aβ40 and RMCO520Aβ42/40, only the Aβ40 peptides were 13C enriched for the latter. Fig. 6 shows the selected excerpts of the spectra of RMCO520Aβ40 and RMCO520Aβ42/40. In addition to Aβ40Os and Aβ42Os, RMCO520Aβ42/40 might, in principle, contain the oligomeric aggregates of a mixture of Aβ40 and Aβ42 because of the continuous attachment and detachment of Aβ peptides from AβOs. Were it the case that the Aβ40 peptides of RMCO520Aβ42/40 had different local environments, multiple sets of NMR signals would have been observed for all the residues. Interestingly, the NMR spectra of RMCO520Aβ42/40 were again dominated by a single set of signals for the β2 region, suggesting that RMCO520Aβ42/40 did not contain a random oligomeric mixture of Aβ40 and Aβ42. That is, Aβ42Os were relatively stable and would not dissociate during the aggregation of Aβ40 peptides. Overall, the region of the β1-loop-β2 motif is the same for RMCO520Aβ40 and RMCO520Aβ42/40, as indicated by the secondary chemical shifts (Fig. S25†). With reference to the data shown in Fig. 7, we found that the backbone 13C chemical shift difference between RMCO520Aβ42/40 and RMCO520Aβ40 was immaterial in the β2 region (K28 to G38), where the only exception was A30-Cα. Thus, it is legitimate to infer that their backbone conformations were largely the same. On the other hand, relatively large chemical shift deviations were observed for some residues, viz., E11, K16, E22, G25, and S26. In addition, the FWHM data shown in Fig. S26† revealed that N27, K28, and G33 of RMCO520Aβ42/40 might have considerable difference in structural disorder and/or motional dynamics from RMCO520Aβ40. For other residues with minor chemical shift deviation, a careful scrutiny of the cross-peak patterns revealed that there were multiple cross peaks for the region of LVFFA (Fig. S27†). That is, the intermolecular packing of Aβ40 peptides in that region was rather heterogeneous, but their backbone and sidechain conformations were largely the same.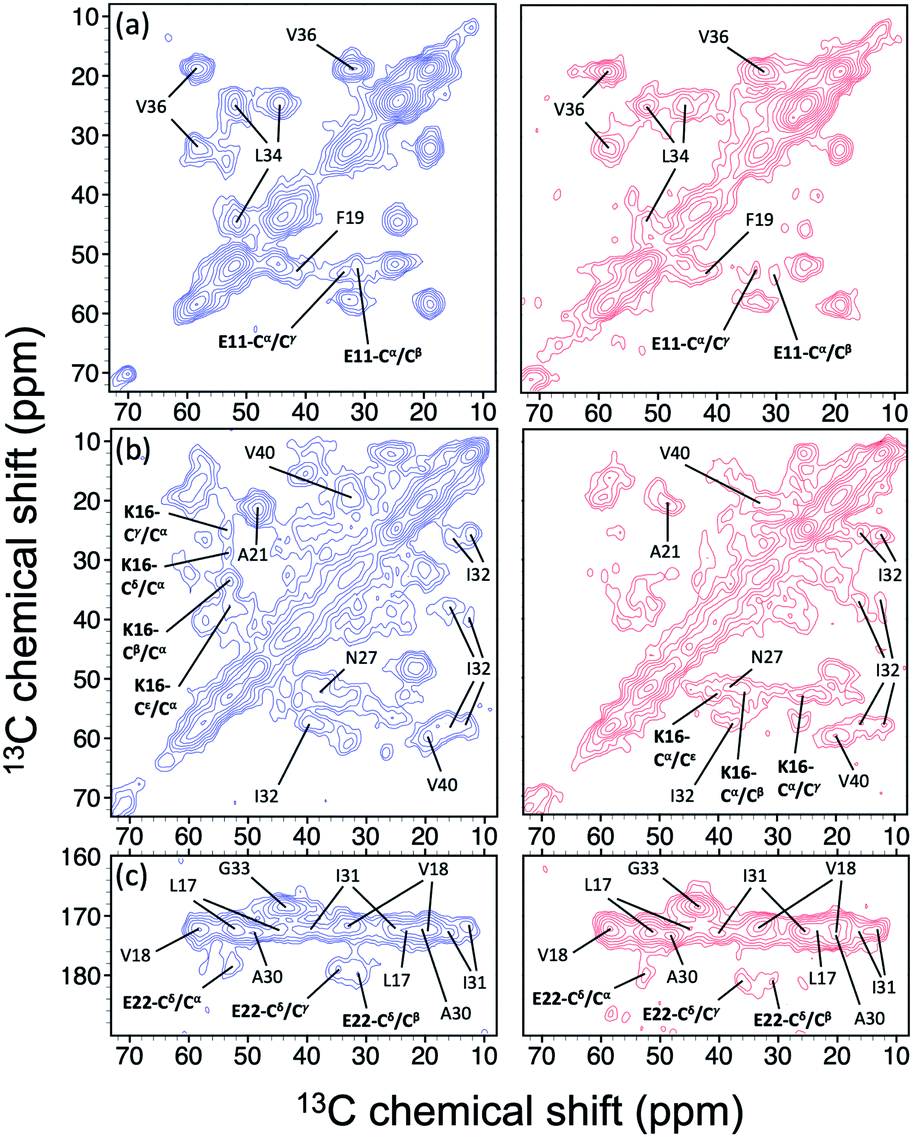 | ||
| Fig. 6 Comparison of the excerpts of the 13C–13C correlation spectra of RMCO520Aβ40 (blue) and RMCO520Aβ42/40 (red). The spectral assignments in bold type were specifically given for (a) E11, (b) K16, and (c) E22. For brevity, brief assignments were given for other peaks. The peak positions of the weak signals had been confirmed by other relay cross peaks. The contour levels of all spectra were set as described in Fig. 3. | ||
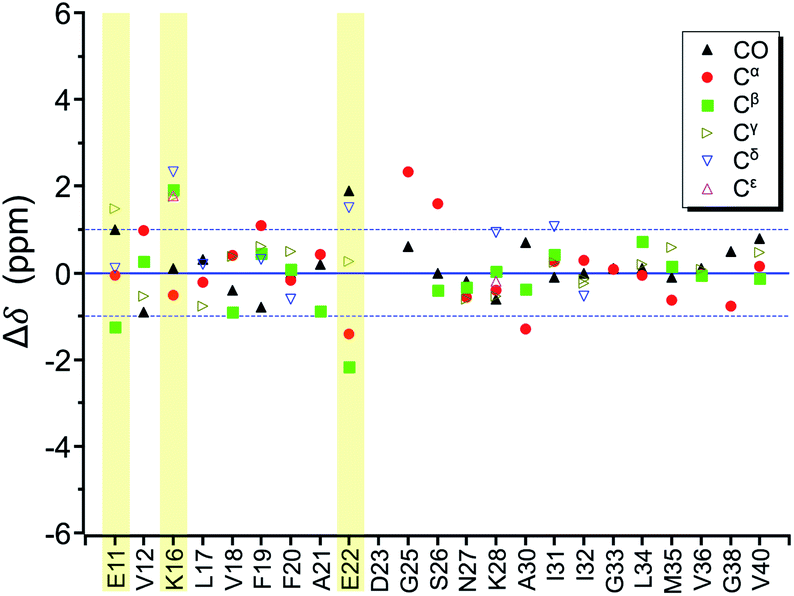 | ||
| Fig. 7 Difference of chemical shifts (Δδ) between RMCO520Aβ42/40 and RMCO520Aβ40. The residues with the most notable chemical shift differences are highlighted with yellow bars. The two Cγ signals of I32 were resolved. | ||
Fibrillar aggregates of RMCO520Aβ42/40
By incubating RMCO520Aβ42/40 in phosphate buffer, we obtained the corresponding fibrillar aggregates (Fig. S28†). The NMR spectra of the wet pellets of the fibril samples were largely dominated by a single set of signals, except the signals of CO and Cα of I32 (Fig. S29–S32 and Table S5†). The average FWHM was 2.3 ppm (Fig. S33†), which was substantially sharper than that of the lyophilized oligomeric aggregates. However, such reduction in FWHM for the hydrated fibril samples were not unexpected because of the conformational averaging effect. From the secondary chemical shifts and the backbone torsion angles estimated by TALOS-N (Fig. S34†), fibrillar RMCO520Aβ42/40 also exhibited the structural motif of β1-loop-β2 motif. While there were multiple weak signals of D23 for oligomeric RMCO520Aβ42/40, there was only one dominant set of D23 signals for fibrillar RMCO520Aβ42/40. The intensities of K16-Cδ/Cε, D23-Cγ/CO, and I32-Cγ/Cβ were stronger than those of the corresponding cross peaks of the oligomer spectra. By contrast, the cross peaks of E11-Cδ/Cα, E11-Cδ/Cγ, V12-Cγ/Cβ, F20-Cβ/Cδ, E22-Cδ/Cα, and V36-Cβ/Cα of the fibril spectra were much weaker than those of the oligomer spectra. This remarkable observation revealed that the peptide conformation of RMCO520Aβ42/40 oligomers and fibrils might be similar, but their sidechain packings could be very different.Fig. 8a shows the deviation between the chemical shift data of oligomeric and fibrillar RMCO520Aβ42/40. Interestingly, the top three largest deviations were again observed for the charged residues, viz., E11, K16, and E22. We had envisioned two possible interpretations. First, the charged residues near the β1 region of Aβ40, together with D23, might play the key role in the structural rearrangement from high-MW oligomeric state to the fibrillar state. On the other hand, Fig. 8b illustrates that the chemical shift difference between RMCO520Aβ42/40 fibrils and RMCO520Aβ40 oligomers only exhibited a slightly larger root-mean-square deviation than the results shown in Fig. 7 and 8a. Thus, as an alternative interpretation, the chemical shift variation of these charged residues might merely reflect the fact that chemical shifts are sensitive to the charge state of these residues. Upon a change in intermolecular packing, the solvent exposure of the charged residues could have altered, leading to a variation of their electrostatic charges. However, this change in the chemical state is not necessarily accompanied with a major conformational change. This rationalization could also apply to the effect of Aβ42Os on the aggregation of Aβ40. An unequivocal interpretation of our chemical shift data may require the justifications by extensive theoretical calculations.
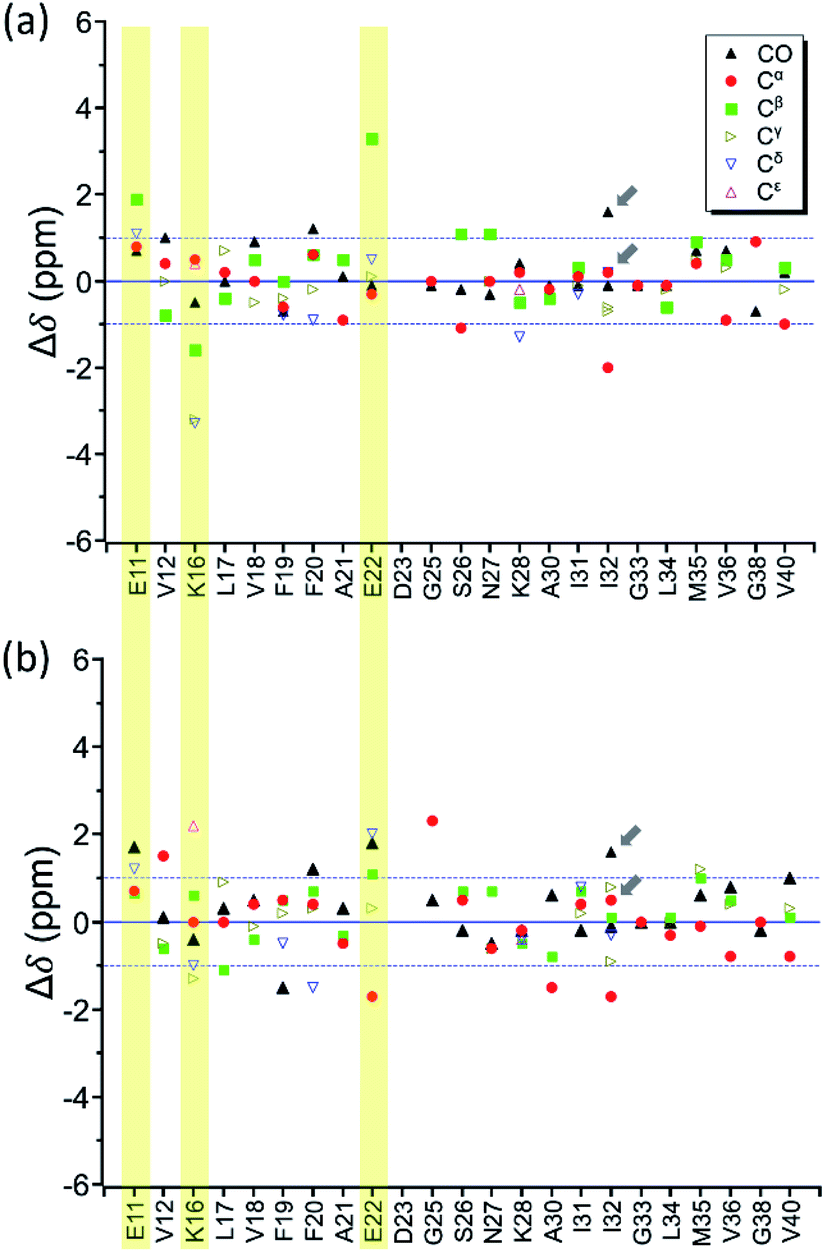 | ||
| Fig. 8 Difference of chemical shifts (Δδ) (a) between RMCO520Aβ42/40 fibrils and RMCO520Aβ42/40 oligomers; (b) between RMCO520Aβ42/40 fibrils and RMCO520Aβ40 oligomers. The residues with the most notable chemical shift differences in (a) are highlighted with yellow bars. The two Cγ signals of I32 were resolved. The arrows indicate there were two sets of CO and Cα signals for I32. | ||
Discussion
Use of reverse micelles as nano-incubator for amyloidogenic peptides
There are several different approaches developed to prepare high-MW AβOs.84 All the methods reported thus far in the literature can be categorized as the passive approach, where the size distribution of the oligomers is primarily determined by the aggregation kinetics. In this work, we reported the use of RMs to prepare AβOs. A detailed discussion of the technical issues associated with the encapsulation of Aβ peptides within RMs is given in ESI.† The success of this protocol, as validated by the solid-state NMR data, has several important implications. First, it is well known that the toxicity of AβOs depends on its size.8 Therefore, it is highly desirable to probe the structures of AβOs of different size. Because the chemistry of RMs is rather mature,85 a judicious choice of surfactants and/or the oil phase can be used to control the size of reverse micelles, which in turn can allow an active control of the size of AβOs. To the best of our knowledge, such a control of the oligomeric state is not possible for other “passive” methods. Second, RM is an ideal device for the study of the protein–protein interactions associated with amyloidogenic peptides. On the one hand, the fusion and fission of RMs allow the material transfer among RMs. Thus, two different types of amyloidogenic peptides can be well mixed in RMs. On the other hand, the fusion-fission process has a tiny energy barrier.57 Thus, the peptides encapsulated in a RM have significantly longer interaction time than in bulk solution. This may better mimic the crowded environment in vivo. On a more general note, the RM approach can be used as a platform to study protein–protein interactions for other amyloidogenic proteins such as α-synuclein, prion, TDP-43, tau, etc. Third, RMs could be used to amplify a minute quantity of the oligomeric species extracted from the brain tissues of AD patients, provided that the seeding kinetics are much faster than the homogeneous nucleation rate of the Aβ monomers. While the idea of seeding has been successfully adapted to amplify the fibrillar aggregates of brain extracts,78 seeding in bulk solution cannot be used to amplify any oligomeric species because further transformation to mature fibrils cannot be avoided. Overall, our RM approach has provided an exciting possibility for a systematic study of oligomeric aggregates of amyloidogenic peptides and their more aggregation-prone mutants, prepared in vitro or derived from brain tissues.Aβ40 aggregation seeded by Aβ42Os
Aβ peptides can be found in many body fluids, such as cerebrospinal fluid, plasma, and brain. Under normal physiological conditions, Aβ40 (80–90%) is more abundantly produced than Aβ42 (5–10%).86,87 For AD brains, however, Aβ42 is the major component in the parenchymal plaques, whereas Aβ40 is usually deposited in the cerebral vessels.39,88 Many efforts have been dedicated to study the cross-seeding effect between them under in vitro conditions. In most of these studies the seeds were prepared by fragmentation of the mature fibrils. However, it is debatable whether it is biological relevant to study the interaction between the monomeric form of one Aβ form and the short fibril fragments of another form, because there is no in vivo evidence showing that the fibrillization of one Aβ form must precede another. Furthermore, it is very difficult to compare the cross-seeding data published by different laboratories if fibril-assisted secondary nucleation is an effective Aβ aggregation pathway. The notion of fibril-assisted nucleation is appealing because it can rationalize the observation that Aβ42 fibrils can nucleate the fibrillogenesis of Aβ40 but Aβ42 protofibrils have no seeding effects on Aβ40 monomers.40 Thus, it is very likely that the aggregation kinetics depend on the surface structure of the fibrils, which is in turn strongly influenced by the incubation conditions.89 Consequently, it may be a futile exercise to compare the vast body of literature in Aβ cross-seeding experiments because the molecular structure of the seeds were usually uncharacterized. In this work, we showed that high-MW Aβ42Os could seed the aggregation of Aβ40 monomers. That is, both Aβ40Os and Aβ42Os could serve as “template” for the aggregation of Aβ40 monomers. As a partial support to this conjecture, it has been reported that the spherical amyloid assembly (SPA) of Aβ42 exhibited a β-loop-β motif,76 similar to what we observed for Aβ40Os. Altogether, our results suggest that the cross-seeding of Aβ in vivo does not necessarily require the formation of amyloid fibrils. Furthermore, the seeding effect of Aβ42Os on Aβ40 might be dominated by electrostatic interactions via the charged residues. This inference is consistent with the observations that liposomes formed by charged lipids can accelerate the aggregation kinetics of Aβ.90,91On-pathway or off-pathway?
Recently, Hoyer and co-workers used a flexible (Gly4Ser)4 linker to covalently connect two Aβ40 peptides in a head-to-tail fashion to form the so-called dimAβ molecules.16 Surprisingly, the globular oligomeric aggregates of dimAβ, with an estimated size of 62 kDa,92 would inhibit the nucleation and growth of Aβ fibrils. Consequently, the authors claimed that AβOs of sizes >50 kDa are not intermediates on the pathway to amyloid fibrils, i.e., off-pathway.16,92 While dimAβ may be considered as a reasonable mimic of two Aβ molecules in close proximity, it remains a single chimeric peptide. It is also unclear whether the same results would be obtained for the head-to-head or tail-to-tail fashions. Even though dimAβ can form fibrillar assembly, it does not imply that the oligomeric aggregates of dimAβ are structurally the same as AβOs. After all, neither the molecular structure of the oligomeric aggregates of dimAβ nor Aβ are known in the literature. The 3D reconstruction by cryoEM for the oligomeric aggregates of dimAβ had a spatial resolution of 17 Å only.92 In this work, our seeding experiments provide a direct evidence that high-MW AβOs did not inhibit the fibrillization of Aβ40 monomers. It remains an open question whether there is any structural similarity between the AβOs and the seeded fibrils. If they are structurally distinctive, the seeding phenomenon could be driven by non-specific molecular interactions. Thus, further investigation of the fibrillar structures of the seeded fibrils by cryoEM are warranted. In any case, the critical role of AβOs in AD pathogenesis is a consensus,5,6,10–12,93,94 should it be on-pathway or off-pathway.Implications of the NMR data of RMCO520Aβ42/40
Although the solid-state NMR data unequivocally showed that Aβ42Os can modulate the peptide conformation of Aβ40Os, it is not straightforward to unravel the underlying molecular events. While the chemical shift data of low-MW Aβ40Os and high-MW Aβ40Os are different,23,26 it is well known that multiple distinct structures could be obtained for fibrils incubated under different conditions.54 Yet, we cannot rule out the possibility that structural rearrangement could occur for Aβ40Os at different sizes. The NMR spectra of RMCO520Aβ40 and RMCO520Aβ42/40, which were of similar size and were incubated under the same conditions, might unravel the effects of Aβ42Os on the nucleation process of Aβ40Os. As revealed in the chemical shift data, the residues of the β2 region were less affected by the seeding effect. The significant perturbation of the 13C chemical shifts of E11, K16, E22, G25, and S26 of Aβ40 implies that their chemical states were altered during the nucleation process. The chemical shift perturbations at G25 and S26 seemed consistent with the observation that G25 and S26 of brain-derived Aβ40 fibrils are solvent exposed.78 However, the 13C chemical shifts of CO and Cβ of S26 were almost the same for RMCO520Aβ42/40 and RMCO520Aβ40. Thus, the large deviation of Cα alone may not guarantee a major conformational change at S26. In addition, the hydrogen-sidechain of glycine should have very limited sidechain–sidechain interactions. Hence, we inferred that the sidechains of E11, K16, and E22 of Aβ40 played an important role when its monomers aggregated on the surface of Aβ42Os. This process should resemble the event of secondary nucleation. More experiments are required to verify whether or not other residues near the N-terminus were involved.The central fragment of Aβ such as LVFF, KLVFF, KLVFFA, and KLVFFAE have been identified as the template for the development of Aβ inhibitors,95–100 peptide homologs to attenuate fibril formation and moderately dissemble preformed fibrils,101,102 and β-sheet-breaker peptides to reduce cerebral damage in APP transgenic mouse model.103 Residues of F19 and F20 were considered to favor the nucleation of Aβ aggregation.104 Mutations at residues of A21, E22, and D23 would affect the kinetics of nucleation in the fibrillization process of Aβ.105 These studies reflect the consensus that the region of K16 to E22 is of great importance for Aβ aggregation. The solid-state NMR data for RMCO520Aβ42/40 shed additional insights into this widely accepted notion. Upon the interaction between Aβ40 monomers and Aβ42Os, there were chemical changes at K16 and E22. However, those residues in the hydrophobic region of LVFFA were not structurally altered as indicated by their relatively minor variation in chemical shifts. Yet, the presence of multiple peaks implies that this region might be susceptible to non-specific intermolecular interaction. The observation that E22 and K16 are involved in governing the interaction between Aβ40 peptides and Aβ42Os may look counterintuitive in the light of the genetics of familial AD, viz., E22K (Italian), E22Q (Dutch), E22G (Arctic), and E22Δ (Osaka),106 and the fact that the K16N mutation of APP produces highly toxic heteromeric Aβ oligomers.107 However, we make no claim that electrostatic interactions are the only viable mechanism governing the interaction between Aβ40 and Aβ42. Thus, our results do not rule out the possibility that mutations at E22 might enhance the interactions between Aβ40 and Aβ42.
Conclusions
The structure of AβOs prepared in bulk solution are intrinsically heterogeneous because AβOs are transient species and they would readily transform to fibrillar aggregates. We demonstrated that RMs could be exploited to provide an active control of the size of the AβOs. Our approach also opens a new avenue to study the interaction of monomer peptides with the oligomeric aggregates of other amyloidogenic peptides. This important advantage had been illustrated by our results that there were considerable differences for the chemical shifts of Aβ40Os incubated with and without the influences of Aβ42Os.The cross-seeding effect of Aβ42 aggregates on Aβ40 is an important issue in Aβ research because it is a widespread notion that Aβ42 could trigger amyloid plaque deposition. While early results on the cross-seeding effect are consistent with the notion, many recent studies argued that the fibrillar fragments of Aβ42 have negligible seeding effect on Aβ40. Herein, we reported that oligomeric aggregates of Aβ42 could indeed accelerate the fibrillization process of Aβ40. Our results indicated that a careful characterization of the aggregation state of the seeds is necessary to warrant a meaningful interpretation of any cross-seeding experiments. This may help resolve the controversy over the cross-seeding effect between Aβ40 and Aβ42.
Data availability
All relevant data are presented in the main text and ESI.†Author contributions
J. C. C. C. designed the research, H. W. C., H. I. M., Y. S. W., and M. C. L. performed Aβ preparation and biophysical assays. E. C. Y. Y. and H. W. C. performed EA analysis and analyzed data. M. H. W., L. H. T., and M. C. L. prepared isotope-labeled Aβ peptide. H. W. C., H. I. M., and S. J. H. performed and analyzed ssNMR data. J. C. C. C. wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Ministry of Science and Technology (MOST 108-2113-M-002-001). The TMV samples were kindly provided by Dr Hsin-Hung Yeh and Dr Yi-Shu Chiu (Agricultural Biotechnological Research Center, Academia Sinica). The NMR, TEM, and STEM measurements were carried out at the Instrumentation Center of National Taiwan University, supported by the Ministry of Science and Technology. The assistance of Ya-Yun Yang in STEM measurements is gratefully acknowledged.Notes and references
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- D. M. Walsh and D. J. Selkoe, J. Neurochem., 2007, 101, 1172–1184 CrossRef CAS PubMed.
- K. L. Viola and W. L. Klein, Acta Neuropathol., 2015, 129, 183–206 CrossRef CAS PubMed.
- M. P. Lambert, A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, C. E. Finch, G. A. Krafft and W. L. Klein, Proc. Natl. Acad. Sci., 1998, 95, 6448–6453 CrossRef CAS PubMed.
- F. Bemporad and F. Chiti, Chem. Biol., 2012, 19, 315–327 CrossRef CAS PubMed.
- E. Y. Hayden and D. B. Teplow, Alzheimer's Res. Ther., 2013, 5, 60 CrossRef PubMed.
- S. T. Ferreira, M. V. Lourenco, M. M. Oliveira and F. G. De Felice, Front. Cell. Neurosci., 2015, 9, 191 Search PubMed.
- E. N. Cline, M. A. Bicca, K. L. Viola and W. L. Klein, J. Alzheimers Dis., 2018, 64, S567–S610 CAS.
- I. Benilova, E. Karran and B. De Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- T. Härd, FEBS J., 2011, 278, 3884–3892 CrossRef PubMed.
- T. Yamaguchi, H. Yagi, Y. Goto, K. Matsuzaki and M. Hoshino, Biochemistry, 2010, 49, 7100–7107 CrossRef CAS PubMed.
- B. O'Nuallain, D. B. Freir, A. J. Nicoll, E. Risse, N. Ferguson, C. E. Herron, J. Collinge and D. M. Walsh, J. Neurosci., 2010, 30, 14411–14419 CrossRef PubMed.
- F. Hasecke, T. Miti, C. Perez, J. Barton, D. Schölzel, L. Gremer, C. S. R. Grüning, G. Matthews, G. Meisl, T. P. J. Knowles, D. Willbold, P. Neudecker, H. Heise, G. Ullah, W. Hoyer and M. Muschol, Chem. Sci., 2018, 9, 5937–5948 RSC.
- G. Bitan, A. Lomakin and D. B. Teplow, J. Biol. Chem., 2001, 276, 35176–35184 CrossRef CAS PubMed.
- G. Bitan, M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek and D. B. Teplow, Proc. Natl. Acad. Sci., 2003, 100, 330–335 CrossRef CAS PubMed.
- D. B. Teplow, N. D. Lazo, G. Bitan, S. Bernstein, T. Wyttenbach, M. T. Bowers, A. Baumketner, J. E. Shea, B. Urbanc, L. Cruz, J. Borreguero and H. E. Stanley, Acc. Chem. Res., 2006, 39, 635–645 CrossRef CAS PubMed.
- M. Serra-Batiste, M. Ninot-Pedrosa, M. Bayoumi, M. Gairí, G. Maglia and N. Carulla, Proc. Natl. Acad. Sci., 2016, 113, 10866–10871 CrossRef CAS PubMed.
- S. Ciudad, E. Puig, T. Botzanowski, M. Meigooni, A. S. Arango, J. Do, M. Mayzel, M. Bayoumi, S. Chaignepain, G. Maglia, S. Cianferani, V. Orekhov, E. Tajkhorshid, B. Bardiaux and N. Carulla, Nat. Commun., 2020, 11, 3014 CrossRef CAS PubMed.
- S. Chimon and Y. Ishii, J. Am. Chem. Soc., 2005, 127, 13472–13473 CrossRef CAS PubMed.
- S. Chimon, M. A. Shaibat, C. R. Jones, D. C. Calero, B. Aizezi and Y. Ishii, Nat. Struct. Mol. Biol., 2007, 14, 1157–1164 CrossRef CAS PubMed.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostrand and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561–568 CrossRef CAS PubMed.
- W. B. Stine, L. Jungbauer, C. Yu and M. J. LaDu, in Alzheimer's Disease and Frontotemporal Dementia: Methods and Protocols, ed. E. D. Roberson, Humana Press, Totowa, NJ, 2011, pp. 13–32 Search PubMed.
- B. Sarkar, V. S. Mithu, B. Chandra, A. Mandal, M. Chandrakesan, D. Bhowmik, P. K. Madhu and S. Maiti, Angew. Chem., Int. Ed., 2014, 53, 6888–6892 CrossRef CAS PubMed.
- S. Parthasarathy, M. Inoue, Y. Xiao, Y. Matsumura, Y. Nabeshima, M. Hoshi and Y. Ishii, J. Am. Chem. Soc., 2015, 137, 6480–6483 CrossRef CAS PubMed.
- A. Potapov, W.-M. Yau, R. Ghirlando, K. R. Thurber and R. Tycko, J. Am. Chem. Soc., 2015, 137, 8294–8307 CrossRef CAS PubMed.
- Y. Miller, B. Ma and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9490–9495 CrossRef CAS PubMed.
- T. M. Ryan, N. Kirby, H. D. T. Mertens, B. Roberts, K. J. Barnham, R. Cappai, C. L. L. Pham, C. L. Masters and C. C. Curtain, Metallomics, 2015, 7, 536–543 CrossRef CAS PubMed.
- H. A. Scheidt, I. Morgado and D. Huster, J. Biol. Chem., 2012, 287, 22822–22826 CrossRef CAS PubMed.
- J. M. Lopez del Amo, U. Fink, M. Dasari, G. Grelle, E. E. Wanker, J. Bieschke and B. Reif, J. Mol. Biol., 2012, 421, 517–524 CrossRef CAS PubMed.
- J. Wang, T. Yamamoto, J. Bai, S. J. Cox, K. J. Korshavn, M. Monette and A. Ramamoorthy, Chem. Commun., 2018, 54, 2000–2003 RSC.
- B. R. Sahoo, T. Genjo, M. Bekier, S. J. Cox, A. K. Stoddard, M. Ivanova, K. Yasuhara, C. A. Fierke, Y. Wang and A. Ramamoorthy, Chem. Commun., 2018, 54, 12883–12886 RSC.
- T. Yang, S. Li, H. Xu, D. M. Walsh and D. J. Selkoe, J. Neurosci., 2017, 37, 152–163 CrossRef CAS PubMed.
- D. A. White, A. K. Buell, T. P. J. Knowles, M. E. Welland and C. M. Dobson, J. Am. Chem. Soc., 2010, 132, 5170–5175 CrossRef CAS PubMed.
- J. T. Jarrett and P. T. Lansbury, Cell, 1993, 73, 1055–1058 CrossRef CAS PubMed.
- A. Lomakin, D. S. Chung, G. B. Benedek, D. A. Kirschner and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1125–1129 CrossRef CAS PubMed.
- J. T. Jarrett, E. P. Berger and P. T. Lansbury, Biochemistry, 1993, 32, 4693–4697 CrossRef CAS PubMed.
- A. Jan, O. Gokce, R. Luthi-Carter and H. A. Lashuel, J. Biol. Chem., 2008, 283, 28176–28189 CrossRef CAS PubMed.
- K. Hasegawa, I. Yamaguchi, S. Omata, F. Gejyo and H. Naiki, Biochemistry, 1999, 38, 15514–15521 CrossRef CAS PubMed.
- S. W. Snyder, U. S. Ladror, W. S. Wade, G. T. Wang, L. W. Barrett, E. D. Matayoshi, H. J. Huffaker, G. A. Krafft and T. F. Holzman, Biophys. J., 1994, 67, 1216–1228 CrossRef CAS PubMed.
- K. Ono, R. Takahashi, T. Ikeda and M. Yamada, J. Neurochem., 2012, 122, 883–890 CrossRef CAS PubMed.
- D. Frost, P. M. Gorman, C. M. Yip and A. Chakrabartty, Eur. J. Biochem., 2003, 270, 654–663 CrossRef CAS PubMed.
- R. Cukalevski, X. Yang, G. Meisl, U. Weininger, K. Bernfur, B. Frohm, T. P. J. Knowles and S. Linse, Chem. Sci., 2015, 6, 4215–4233 RSC.
- B. K. Yoo, Y. Xiao, D. McElheny and Y. Ishii, J. Am. Chem. Soc., 2018, 140, 2781–2784 CrossRef CAS PubMed.
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Nat. Struct. Mol. Biol., 2015, 22, 499–505 CrossRef CAS PubMed.
- M. T. Colvin, R. Silvers, Q. Z. Ni, T. V. Can, I. Sergeyev, M. Rosay, K. J. Donovan, B. Michael, J. Wall, S. Linse and R. G. Griffin, J. Am. Chem. Soc., 2016, 138, 9663–9674 CrossRef CAS PubMed.
- M. A. Wälti, F. Ravotti, H. Arai, C. G. Glabe, J. S. Wall, A. Böckmann, P. Güntert, B. H. Meier and R. Riek, Proc. Natl. Acad. Sci., 2016, 113, E4976–E4984 CrossRef PubMed.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci., 2013, 110, 9758–9763 CrossRef CAS PubMed.
- G. A. Garcia, S. I. A. Cohen, C. M. Dobson and T. P. J. Knowles, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 89, 032712 CrossRef PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- G. Meisl, X. Yang, E. Hellstrand, B. Frohm, J. B. Kirkegaard, S. I. A. Cohen, C. M. Dobson, S. Linse and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9384–9389 CrossRef CAS PubMed.
- R. Tycko, Neuron, 2015, 86, 632–645 CrossRef CAS PubMed.
- P. L. Luisi, M. Giomini, M. P. Pileni and B. H. Robinson, Biochim. Biophys. Acta, Biomembr., 1988, 947, 209–246 CrossRef CAS.
- M. P. Pileni, J. Phys. Chem., 1993, 97, 6961–6973 CrossRef CAS.
- P. D. I. Fletcher, A. M. Howe and B. H. Robinson, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 985–1006 RSC.
- E. Ruckenstein and P. Karpe, J. Phys. Chem., 1991, 95, 4869–4882 CrossRef CAS.
- Y. Miyake, Colloids Surf., A, 1996, 109, 255–262 CrossRef CAS.
- Y.-L. Lin, Y.-S. Cheng, C.-I. Ho, Z.-H. Guo, S.-J. Huang, M.-L. Org, A. Oss, A. Samoson and J. C. C. Chan, Chem. Commun., 2018, 54, 10459–10462 RSC.
- J.-L. Lemyre, S. Lamarre, A. Beaupré and A. M. Ritcey, Langmuir, 2010, 26, 10524–10531 CrossRef CAS PubMed.
- M.-C. Lee, W.-C. Yu, Y.-H. Shih, C.-Y. Chen, Z.-H. Guo, S.-J. Huang, J. C. C. Chan and Y.-R. Chen, Sci. Rep., 2018, 8, 4772 CrossRef PubMed.
- B. Chen, K. R. Thurber, F. Shewmaker, R. B. Wickner and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14339–14344 CrossRef CAS PubMed.
- M. Hoshi, M. Sato, S. Matsumoto, A. Noguchi, K. Yasutake, N. Yoshida and K. Sato, Proc. Natl. Acad. Sci., 2003, 100, 6370–6375 CrossRef CAS PubMed.
- A. Noguchi, S. Matsumura, M. Dezawa, M. Tada, M. Yanazawa, A. Ito, M. Akioka, S. Kikuchi, M. Sato, S. Ideno, M. Noda, A. Fukunari, S. Muramatsu, Y. Itokazu, K. Sato, H. Takahashi, D. B. Teplow, Y. Nabeshima, A. Kakita, K. Imahori and M. Hoshi, J. Biol. Chem., 2009, 284, 32895–32905 CrossRef CAS PubMed.
- R. W. Hepler, K. M. Grimm, D. D. Nahas, R. Breese, E. C. Dodson, P. Acton, P. M. Keller, M. Yeager, H. Wang, P. Shughrue, G. Kinney and J. G. Joyce, Biochemistry, 2006, 45, 15157–15167 CrossRef CAS PubMed.
- W. Yong, A. Lomakin, M. D. Kirkitadze, D. B. Teplow, S.-H. Chen and G. B. Benedek, Proc. Natl. Acad. Sci., 2002, 99, 150–154 CrossRef CAS PubMed.
- R. Sabaté and J. Estelrich, J. Phys. Chem. B, 2005, 109, 11027–11032 CrossRef PubMed.
- J. Tran, D. Chang, F. Hsu, H. Wang and Z. Guo, FEBS Lett., 2017, 591, 177–185 CrossRef CAS PubMed.
- M. So, A. Ishii, Y. Hata, H. Yagi, H. Naiki and Y. Goto, Langmuir, 2015, 31, 9973–9982 CrossRef CAS PubMed.
- G. A. Braun, A. J. Dear, K. Sanagavarapu, H. Zetterberg and S. Linse, Chem. Sci., 2022, 13, 2423–2439 RSC.
- Y. Yan and C. Wang, J. Mol. Biol., 2007, 369, 909–916 CrossRef CAS PubMed.
- T. Yamaguchi, K. Matsuzaki and M. Hoshino, FEBS Lett., 2013, 587, 620–624 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci., 2002, 99, 16742–16747 CrossRef CAS PubMed.
- Y. Shen and A. Bax, in Artificial Neural Networks, ed. H. Cartwright, Springer, New York, NY, 2015, pp. 17–32 Search PubMed.
- Y. Xiao, I. Matsuda, M. Inoue, T. Sasahara, M. Hohi, s and Y. Ishii, J. Biol. Chem., 2020, 295, 458–467 CrossRef CAS PubMed.
- A. T. Petkova, R. D. Leapman, Z. H. Guo, W. M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262–265 CrossRef CAS PubMed.
- J.-X. Lu, W. Qiang, W.-M. Yau, C. D. Schwieters, S. C. Meredith and R. Tycko, Cell, 2013, 154, 1257–1268 CrossRef CAS PubMed.
- W. Qiang, W.-M. Yau, J.-X. Lu, J. Collinge and R. Tycko, Nature, 2017, 541, 217–221 CrossRef CAS PubMed.
- U. Ghosh, K. R. Thurber, W.-M. Yau and R. Tycko, Proc. Natl. Acad. Sci., 2021, 118, e2023089118 CrossRef CAS PubMed.
- A. Daskalov, M. Gantner, M. A. Wälti, T. Schmidlin, C. N. Chi, C. Wasmer, A. Schütz, J. Ceschin, C. Clavé, S. Cescau, B. Meier, R. Riek and S. J. Saupe, PLoS Pathog., 2014, 10, e1004158 CrossRef PubMed.
- J. C. Stroud, C. Liu, P. K. Teng and D. Eisenberg, Proc. Natl. Acad. Sci., 2012, 109, 7717–7722 CrossRef CAS PubMed.
- M. Kollmer, W. Close, L. Funk, J. Rasmussen, A. Bsoul, A. Schierhorn, M. Schmidt, C. J. Sigurdson, M. Jucker and M. Fändrich, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- B. R. Sahoo, S. J. Cox and A. Ramamoorthy, Chem. Commun., 2020, 56, 4627–4639 RSC.
- J. Eastoe, M. J. Hollamby and L. Hudson, Adv. Colloid Interface Sci., 2006, 128–130, 5–15 CrossRef CAS PubMed.
- D. J. Selkoe, Physiol. Rev., 2001, 81, 741–766 CrossRef CAS PubMed.
- M. P. Murphy and H. LeVine, J. Alzheimer's Dis., 2010, 19, 311–323 Search PubMed.
- A. Charidimou, G. Boulouis, M. E. Gurol, C. Ayata, B. J. Bacskai, M. P. Frosch, A. Viswanathan and S. M. Greenberg, Brain, 2017, 140, 1829–1850 CrossRef PubMed.
- A. K. Paravastu, R. D. Leapman, W. M. Yau and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18349–18354 CrossRef CAS PubMed.
- M. Bokvist, F. Lindström, A. Watts and G. Gröbner, J. Mol. Biol., 2004, 335, 1039–1049 CrossRef CAS PubMed.
- K. Matsuzaki, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 1935–1942 CrossRef CAS PubMed.
- M. P. Schützmann, F. Hasecke, S. Bachmann, M. Zielinski, S. Hänsch, G. F. Schröder, H. Zempel and W. Hoyer, Nat. Commun., 2021, 12, 4634 CrossRef PubMed.
- W. M. Berhanu and U. H. E. Hansmann, Adv. Protein Chem. Struct. Biol., 2014, 96, 113–141 CAS.
- L. Nagel-Steger, M. C. Owen and B. Strodel, ChemBioChem, 2016, 17, 657–676 CrossRef CAS PubMed.
- L. O. Tjernberg, J. Näslund, F. Lindqvist, J. Johansson, A. R. Karlström, J. Thyberg, L. Terenius and C. Nordstedt, J. Biol. Chem., 1996, 271, 8545–8548 CrossRef CAS PubMed.
- M. A. Findeis, G. M. Musso, C. C. Arico-Muendel, H. W. Benjamin, A. M. Hundal, J.-J. Lee, J. Chin, M. Kelley, J. Wakefield, N. J. Hayward and S. M. Molineaux, Biochemistry, 1999, 38, 6791–6800 CrossRef CAS PubMed.
- D. J. Gordon, K. L. Sciarretta and S. C. Meredith, Biochemistry, 2001, 40, 8237–8245 CrossRef CAS PubMed.
- R. J. Chalifour, R. W. McLaughlin, L. Lavoie, C. Morissette, N. Tremblay, M. Boulé, P. Sarazin, D. Stéa, D. Lacombe, P. Tremblay and F. Gervais, J. Biol. Chem., 2003, 278, 34874–34881 CrossRef CAS PubMed.
- T. Takahashi and H. Mihara, Acc. Chem. Res., 2008, 41, 1309–1318 CrossRef CAS PubMed.
- M. Ouberai, P. Dumy, S. Chierici and J. Garcia, Bioconjugate Chem., 2009, 20, 2123–2132 CrossRef CAS PubMed.
- C. Soto, M. S. Kindy, M. Baumann and B. Frangione, Biochem. Biophys. Res. Commun., 1996, 226, 672–680 CrossRef CAS PubMed.
- M. A. Chacón, M. I. Barría, C. Soto and N. C. Inestrosa, Mol. Psychiatry, 2004, 9, 953–961 CrossRef PubMed.
- B. Permanne, C. Adessi, G. P. Saborio, S. Fraga, M.-J. Frossard, J. V. Dorpe, I. Dewachter, W. A. Banks, F. V. Leuven and C. Soto, FASEB J., 2002, 16, 860–862 CrossRef CAS PubMed.
- R. Cukalevski, B. Boland, B. Frohm, E. Thulin, D. Walsh and S. Linse, ACS Chem. Neurosci., 2012, 3, 1008–1016 CrossRef CAS PubMed.
- C.-L. Ni, H.-P. Shi, H.-M. Yu, Y.-C. Chang and Y.-R. Chen, FASEB J., 2011, 25, 1390–1401 CrossRef CAS PubMed.
- J. Nasica-Labouze, P. H. Nguyen, F. Sterpone, O. Berthoumieu, N.-V. Buchete, S. Coté, A. De Simone, A. J. Doig, P. Faller, A. Garcia, A. Laio, M. S. Li, S. Melchionna, N. Mousseau, Y. Mu, A. Paravastu, S. Pasquali, D. J. Rosenman, B. Strodel, B. Tarus, J. H. Viles, T. Zhang, C. Wang and P. Derreumaux, Chem. Rev., 2015, 115, 3518–3563 CrossRef CAS PubMed.
- D. Kaden, A. Harmeier, C. Weise, L. M. Munter, V. Althoff, B. R. Rost, P. W. Hildebrand, D. Schmitz, M. Schaefer, R. Lurz, S. Skodda, R. Yamamoto, S. Arlt, U. Finckh and G. Multhaup, EMBO Mol. Med., 2012, 4, 647–659 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01555b |
| This journal is © The Royal Society of Chemistry 2022 |