
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiohybrid photosynthetic charge accumulation detected by flavin semiquinone formation in ferredoxin-NADP+ reductase†
Lisa M.
Utschig
 *,
Udita
Brahmachari
,
Karen L.
Mulfort
,
Jens
Niklas
and
Oleg G.
Poluektov
*,
Udita
Brahmachari
,
Karen L.
Mulfort
,
Jens
Niklas
and
Oleg G.
Poluektov
Chemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL 60439, USA. E-mail: utschig@anl.gov
First published on 11th May 2022
Abstract
Flavin chemistry is ubiquitous in biological systems with flavoproteins engaged in important redox reactions. In photosynthesis, flavin cofactors are used as electron donors/acceptors to facilitate charge transfer and accumulation for ultimate use in carbon fixation. Following light-induced charge separation in the photosynthetic transmembrane reaction center photosystem I (PSI), an electron is transferred to one of two small soluble shuttle proteins, a ferredoxin (Fd) or a flavodoxin (Fld) (the latter in the condition of Fe-deficiency), followed by electron transfer to the ferredoxin-NADP+ reductase (FNR) enzyme. FNR accepts two of these sequential one electron transfers, with its flavin adenine dinucleotide (FAD) cofactor becoming doubly reduced, forming a hydride which is then passed onto the substrate NADP+ to form NADPH. The two one-electron potentials (oxidized/semiquinone and semiquinone/hydroquinone) are similar to each other with the FNR protein stabilizing the hydroquinone, making spectroscopic detection of the intermediate semiquinone state difficult. We employed a new biohybrid-based strategy that involved truncating the native three-protein electron transfer cascade PSI → Fd → FNR to a two-protein cascade by replacing PSI with a molecular Ru(II) photosensitizer (RuPS) which is covalently bound to Fd and Fld to form biohybrid complexes that successfully mimic PSI in light-driven NADPH formation. RuFd → FNR and RuFld → FNR electron transfer experiments revealed a notable distinction in photosynthetic charge accumulation that we attribute to the different protein cofactors [2Fe2S] and flavin. After freeze quenching the two-protein systems under illumination, an intermediate semiquinone state of FNR was readily observed with cw X-band EPR spectroscopy. The increased spectral resolution from selective deuteration allowed EPR detection of inter-flavoprotein electron transfer. This work establishes a biohybrid experimental approach for further studies of photosynthetic light-driven electron transfer chain that culminates at FNR and highlights nature's mechanisms that couple single electron transfer chemistry to charge accumulation, providing important insight for the development of photon-to-fuel schemes.
Introduction
Fundamental mechanisms of photosynthetic solar energy conversion provide a road map for renewable and clean energy strategies. Nature's mechanisms incorporate both large transmembrane protein complexes and small soluble redox proteins in a Z-scheme electron transport chain of oxygenic photosynthesis for photon capture and conversion, utilizing the resultant chemical energy for water splitting and carbon dioxide assimilation. The primary solar energy conversion reactions occur in the transmembrane reaction center proteins (RCs) and involve rapid sequential electron transfers between a chain of protein embedded electron donor and acceptor molecules.2 Following RC charge separation, the resultant electrochemical potential is converted to chemical energy by selective coupling to secondary reaction sequences. Electron paramagnetic resonance (EPR) spectroscopic studies have played a significant role in determining light-generated radical species and reactivities in RC electron transfer since they can selectively track the paramagnetic intermediates generated during the electron transfer events. Herein we build on these classic studies by using EPR methods to track and interrogate the photosynthetic electron transfer between the soluble redox proteins in the latter part of the Z-scheme (Fig. 1).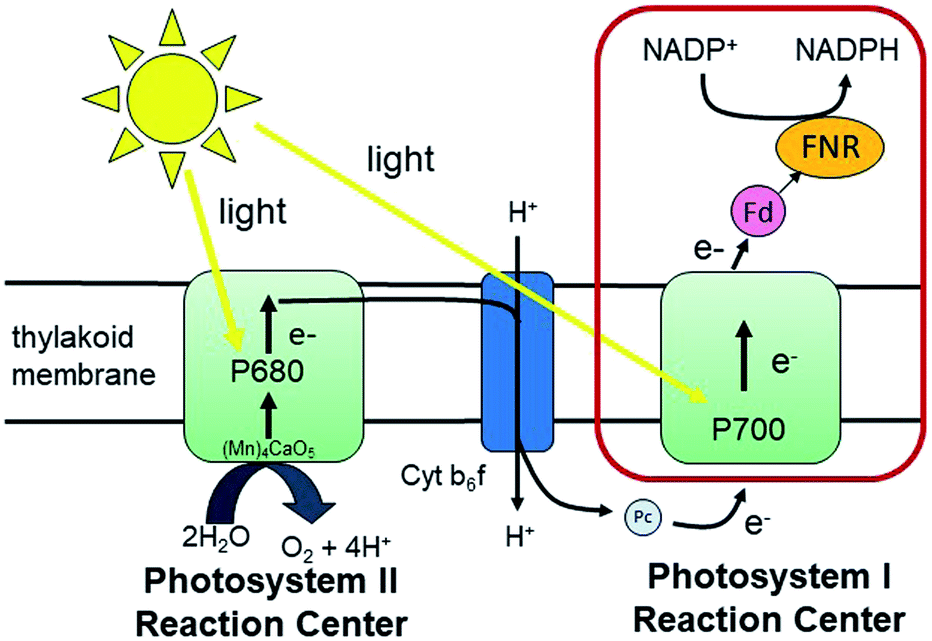 | ||
| Fig. 1 The Z-scheme of electron transport in oxygenic photosynthesis. Photosystem II (PSII) absorbs photons that are used to oxidize water. The extracted electrons are passed on to photosystem I (PSI) via the cytochrome b6f complex (Cyt b6f) and the electron transfer protein plastocyanin (Pc). PSI carries out light-driven transmembrane electron transfer from Pc to ferredoxin (Fd) (or flavodoxin (Fld) under conditions of Fe-deficiency). Fd (or Fld) then shuttles the electrons from PSI to ferredoxin NADP+-reductase (FNR) for the reduction of NADP+ to NADPH that is used in carbon fixation. The red box highlights the electron transfer reactions of interest in this study. | ||
In oxygenic photosynthesis of higher plants, cyanobacteria, and algae, the photosystem II (PSII) and photosystem I (PSI) RCs operate in series to transfer electrons from water to NADP+.2 PSII conducts light-driven water oxidation,3 whereas PSI catalyzes light-driven transmembrane electron transfer from plastocyanin to oxidized ferredoxin (Fd) (eqn (1)).4 The electrons from reduced Fd are used by the enzyme ferredoxin-NADP+ reductase (FNR) for the reduction of NADP+ to NADPH, the terminal step of the Z-scheme (eqn (2)). Under conditions of Fe-deficiency in cyanobacteria and some algae, flavodoxin (Fld) substitutes for Fd in the reduction of FNR.5
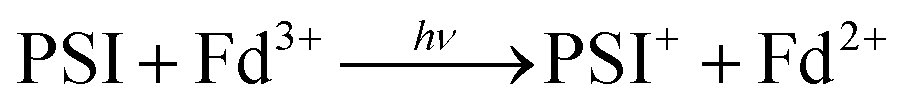 | (1) |
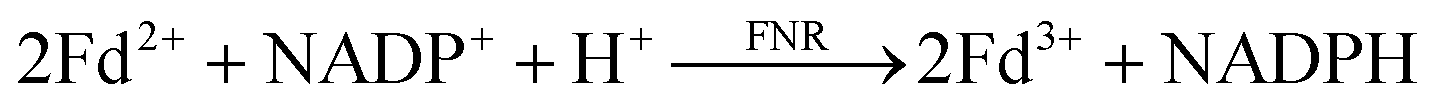 | (2) |
FNR contains a single flavin adenine dinucleotide (FAD) cofactor with an isoalloxazine ring which can stabilize three different oxidation states: fully oxidized, partially reduced by one electron (semiquinone), and fully reduced by two electrons (hydroquinone). FNR pairs single electrons from reduced Fd (or Fld) in a single hydride transfer step to protein bound substrate NADP+.6 Crystal structures of FNR reveal that the protein folds in two domains, one containing the noncovalently bound FAD molecule and the other NADP+ (Fig. 2).7,8 Fd and Fld can form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with FNR. The active site for Fd/Fld docking is at the interface of the two domains.9 Thus two reduced Fd or two reduced Fld proteins are needed for the formation of one NADPH molecule from a single FNR protein (eqn (2)). The two one-electron potentials of FAD are close to each other with FNR proteins stabilizing only 10–20% of the maximal amount of semiquinone.10,11 Nevertheless, the FAD semiquinone state certainly plays a role in the enzyme-catalyzed reduction of NADP+ as both Fd and Fld provide two successive one-electron reductions to FNR. To better understand photosynthetic coupling of one electron transfers to charge accumulation, we targeted generation of a stable semiquinone state of FNR for EPR characterization using the native electron transfer properties and interactions between the protein partners Fd and Fld.
:1 complexes with FNR. The active site for Fd/Fld docking is at the interface of the two domains.9 Thus two reduced Fd or two reduced Fld proteins are needed for the formation of one NADPH molecule from a single FNR protein (eqn (2)). The two one-electron potentials of FAD are close to each other with FNR proteins stabilizing only 10–20% of the maximal amount of semiquinone.10,11 Nevertheless, the FAD semiquinone state certainly plays a role in the enzyme-catalyzed reduction of NADP+ as both Fd and Fld provide two successive one-electron reductions to FNR. To better understand photosynthetic coupling of one electron transfers to charge accumulation, we targeted generation of a stable semiquinone state of FNR for EPR characterization using the native electron transfer properties and interactions between the protein partners Fd and Fld.
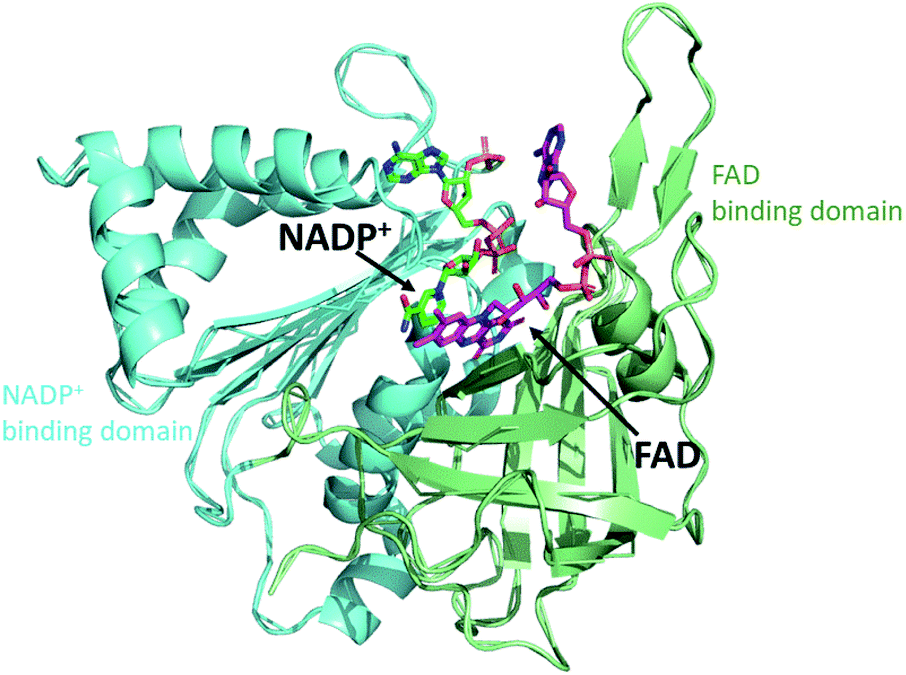 | ||
| Fig. 2 Crystal structure of FNR. The protein folds into two domains, one domain (green) contains the FAD cofactor (purple) and the other domain (blue) contains NADP+ (dark green). (2BSA) Fd/Fld dock at the interface of the two domains on the cofactor side (top of this view). | ||
Unlike Fld for which the semiquinone is exceptionally stable due to the relative midpoint potentials of the flavin mononucleotide (FMN) cofactor (EOX/SQ −195 mV; ESQ/HQ −390 mV), generation of a stable semiquinone species in FNR is challenging due to the similar midpoint potentials EOX/SQ −338 mV; ESQ/HQ −312 mV of the FAD cofactor and more positive potential of the hydroquinone.11–13 Two methods have been used to generate the semiquinone of FNR for EPR studies, chemical reduction with NADPH or steady-state photoreduction using diazariboflavin in the presence of EDTA.14–16 EPR/ENDOR spectroscopy has centered on a detailed examination of the localized molecular and electronic structural information of the flavin cofactor site in FNR.15,17,18 In this work we have extended these studies with directed photoreduction of FNR via light-driven interprotein electron transfer. This method will allow us to generate the semiquinone signal in high yield, avoiding the difficulties associated with chemical reduction like double reduction or addition of other radical species in solution such as diazariboflavin. In addition, we will observe the semiquinone state for electron transfer competent Fd-FNR and Fld–FNR complexes. To accomplish the spectroscopic detection of the semiquinone state, we used a bioinorganic approach, replacing the light-initiated chemistry of PSI with biohybrid complexes comprised of a photosensitizer molecule, ruthenium(II) tris(bipyridine) (RuPS), covalently bound to Fd and Fld (RuFd and RuFld). The resultant biohybrids add light-driven electron transfer capability to their electron shuttle function in photosynthetic electron transfer. Recently photosynthetic-based biohybrids were built with non-native functionality for photocatalytic H2 production.19–24 Herein we have applied biohybrid-driven chemistry to look at native photosynthetic function to better understand nature's mechanisms.
To aid in probing the two-protein electron transfer cascades RuFd → FNR and RuFld → FNR, we employed a method pioneered over 60 years ago, the deuteration of photosynthetic microorganisms by adaption and growth of bacteria, cyanobacteria and algae in heavy water (99.7% D2O).25–27 EPR spectroscopy of deuterated photosynthetic proteins is a classic approach that helped determine the cofactors involved with the electron transfer events in RCs.28–35 Deuteration often substantially increases EPR spectral resolution and provides the opportunity to directly probe flavin (in Fld) to flavin (in FNR) electron transfer via EPR for the first time. We targeted systematic study of Fd, a single electron carrier, Fld, a two electron carrier, and FNR, a two electron (hydride) transfer protein for insight on charge accumulation aspects of photosynthetic electron transfer in the latter part of nature's Z-scheme chemistry. The biohybrid methodologies developed inspire future study of ubiquitous flavin electron transfer chains in biology as well as inform artificial photosynthetic strategies for coupling single electron photochemistry to multielectron chemistry necessary for efficient photocatalytic solar fuel synthesis.
Results and discussion
Development of the two protein biohybrid system
Flavoproteins have the unique capacity of transferring either just one electron in each redox step or two electrons at once. Both FMN and FAD have an isoalloxazine ring, which is the redox active part of the flavin molecule (Fig. 3A). For this reason, nature incorporates flavoproteins in a wide variety of reaction schemes in addition to photosynthesis including energy transduction and biosynthesis.36 As such, spectroscopic methods for probing involvement of flavin semiquinone intermediates in flavoenzyme-catalyzed reactions is of wide interest, but experimentally challenging. To investigate the flavin reactions in the latter part of the photosynthetic Z-scheme we first targeted modification of the native proteins in two ways: insertion of deuterated flavin cofactor and specific binding of RuPS.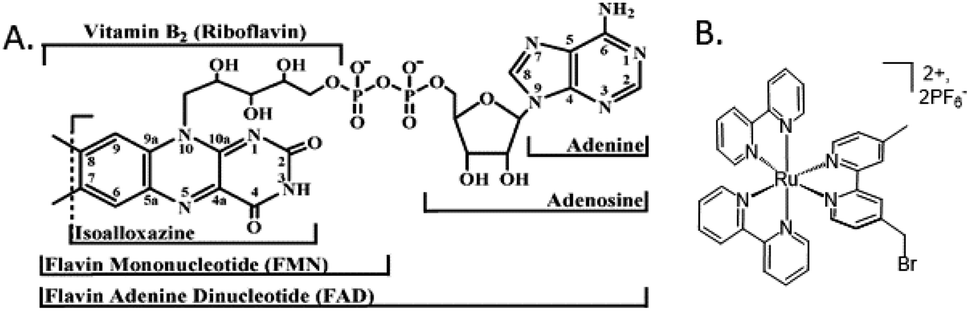 | ||
| Fig. 3 Molecular cofactors used in this study. (A) Chemical structures of flavin adenine dinucleotide, FAD, the native cofactor of ferredoxin-NADP+ reductase (FNR) and flavin mononucleotide, FMN, the native cofactor of flavodoxin (Fld).1 (B) The ruthenium photosensitizer (RuPS), [Ru(4-CH2Br-4′–CH3–2,2′-bpy)(bpy)2]·2PF6. | ||
Extracting significant quantities of deuterated FNR from cyanobacteria grown in 99.7% D2O media would be difficult due to low yielding protein purification protocols. Therefore, we isolated deuterated Fld protein, from which we extracted and purified the deuterated FMN cofactor.37 We confirmed successful deuterated flavin incorporation into FNR by UV-Vis spectroscopy (Fig. S1†). The 2H-FMN cofactor inserted into unreconstituted FNR “FNR(2H-FMN)” gave an acceptable A280/A456 = 7, where for overexpressed FNR with <5% FAD cofactor after purification, A280/A456 = 54. Crystal structures show that the FNR protein fold can productively use both FAD and FMN, as the adenosine moiety has no specific interactions with the protein.7,38 To affirm this, we tested the functionality of FMN substitution for FAD in FNR. PSI-driven NADP+ reduction activity is shown in Fig. S2.† Replacement of native FAD with 2H-FMN in FNR does not produce significant differences in activity under the reaction conditions. A slight slowing of enzymatic activity 77000 vs. 100400 mol NADPH (mol PSI)−1 h−1 and overall lower final quantity of NADPH formed was observed. This is most likely due to the FMN cofactor not positioning itself into the protein structure in exactly the same configuration as the native FAD cofactor, for which the protein was designed to hold. However, the same order of magnitude of activity indicates that selective deuteration of the flavin cofactor in FNR provides a highly functional system for EPR study.
RuPS (Fig. 3B) was covalently bound to free cysteine residues of Fd (Cys18) and Fld (Cys54) via a bromine substitution reaction to generate RuFd and RuFld.20 The ICP-AES analysis showed near stoichiometric ratios of 1.0 ± 0.1 Ru/Fd and 1.2 ± 0.2 Ru/Fld. Covalent binding was confirmed with 5,5′-dithiobi(2-nitrobenzoic acid) modification of cysteine residues.39 Upon illumination, RuFd and RuFld were able to drive NADP+ reduction from FNR as shown in Fig. 4 using a 10-fold excess of RuFd or RuFld compared to FNR. Interestingly, RuFld-driven NADP+ plateaued faster and achieved overall more turnovers per FNR than RuFd under the same experimental conditions. Initial rates were 8500 and 1050 mol NADPH (mol FNR)−1 h−1 for RuFld and RuFd respectively (Fig. S3†). This difference is interesting and may be related to the difference in 1 electron vs. 2 electron carrying abilities of Fd versus Fld, as discussed below. Note, employing the simplified 2-protein system averts a potential pitfall in EPR studies of using the native electron transfer cascades PSI → Fd → FNR and PSI → Fld → FNR to observe FNR semiquinone intermediates. In the native 3-protein system the prominent light-generated P700+ signal at g = 2.00 would significantly overlap with the semiquinone signal of FNR, making resolution of the semiquinone signal difficult. As the NADP+ reduction assays show, RuFd and RuFld are able to carry out light-driven NADPH formation and are thus suitable biohybrids to replace PSI chemistry in EPR studies of semiquinone formation in FNR.
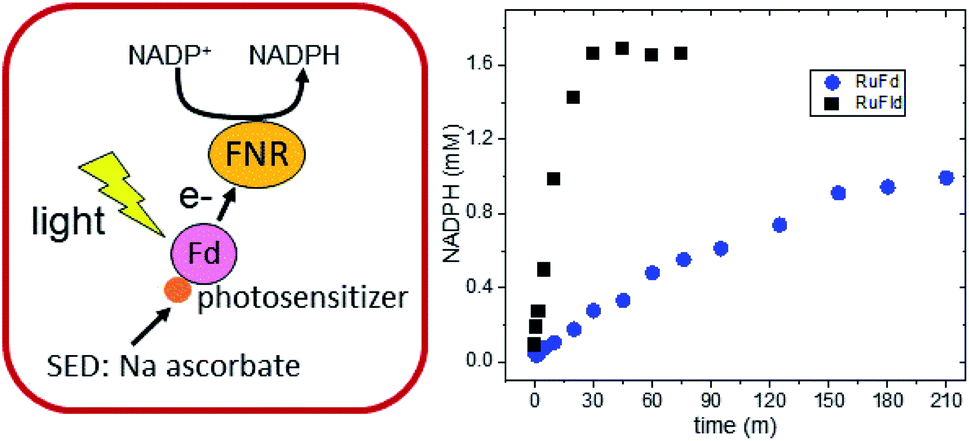 | ||
| Fig. 4 Biohybrid photosynthetic NADP+ reduction. (left) Schematic of the light-driven two protein cascade that replaces PSI with a ruthenium photosensitizer molecule bound to Fd. Fld can be substituted for Fd. (right) In vitro light-driven NADP+ reduction via RuFd (blue circles) and RuFld (black squares). | ||
EPR studies: electron transfer from ferredoxin to FNR
The light-driven chemistry of our two protein cascade biohybrid systems was examined with cw X-band EPR spectroscopy. The single flavin cofactor of FNR proceeds through three oxidation states in the transfer of electrons from Fd to NADP+, oxidized (FNROX), semiquinone (FNRSQ) and hydroquinone (FNRHQ) (Fig. S5†).5 Of these three, only the FNRSQ state has an unpaired electron that can be observed with EPR spectroscopy. We employed selective deuteration to aid in our detection of FNRSQ.We first examine the electron transfer RuFd → FNR. Fig. 5A shows the EPR spectra observed for FNR substituted with 1H-FAD, 1H-FMN, and 2H-FMN. The paramagnetic semiquinone state was generated by illumination with freeze trapping techniques of a sample containing both RuFd and FNR proteins in solution at a ratio of 2:1 with the sacrificial electron donor (SED) sodium ascorbate. In each case, a typical flavin semiquinone signal centered at g = 2.004 is observed. The spectrum of FNR(2H-FMN)SQ is different than the signals of the two FNR samples with protonated cofactors, FNR(1H-FMN)SQ and FNR(1H-FAD)SQ. Deuteration of FMN effectively decreases the inhomogeneous line width of the EPR signal that is largely due to unresolved proton (also called 1H in the following) hyperfine interactions. The substitution of 1H by 2H at nonexchangeable hydrogen positions on the flavin molecule results in a decrease in the hydrogen hyperfine coupling constants (relative gyromagnetic ratio of 1H/2H ≈ 6.5) so that the large hyperfine coupling constants of nitrogens and exchangeable hydrogen can be observed (protonated buffer).37 Thus, substitution of the native 1H-FAD cofactor with a non-native 2H-FMN cofactor gives a distinctive EPR signal that can be readily studied. Comparison of the EPR signal observed in H2O to that in D2O (Fig. S6†) shows a line narrowing, consistent with a neutral semiquinone species vs. anionic species as the exchangeable proton has stronger hyperfine coupling compared to deuteron (Table S1 and Fig. S5†). Freeze trapping of samples illuminated at room temperature was necessary to generate a large semiquinone signal as only very small signals are observed for samples that were dark adapted prior to freezing in liquid nitrogen and illuminated at 20 K in the EPR cavity. Based on the intensity of the FNRSQ signal formed after freeze trapping upon room temperature illumination, less than 5% low temperature electron transfer occurs between RuFd and FNR for samples frozen in the dark and illuminated at 20 K.
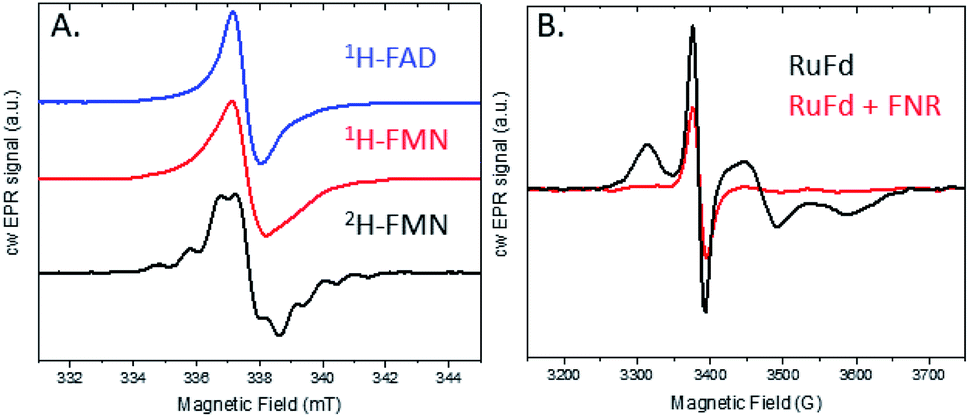 | ||
| Fig. 5 cw X-band EPR spectra of the two protein systems RuFd and FNR (both 1H). (A) Semiquinone signal for FNR reconstituted with 1H-FAD (blue), 1H-FMN (red), and 2H-FMN (black), all three samples containing RuFd:FNR in a 2:1 ratio. (B) Light-induced reduction of the [2Fe2S] cluster of RuFd in the presence (red) and absence of FNR (black). Samples were illuminated at room temperature for 5 s followed by immersion in liquid N2 while continuously illuminating. The 1H-FMN sample contained 486 μM RuFd, 270 μM FNR. The 1H-FAD sample contained 620 μM RuFd and 330 μM FNR. The 2H-FMN sample contained 620 μM RuFd and 287 μM FNR. | ||
EPR can also shed light on the biohybrid-driven interprotein electron transfer event that proceeds FNRSQ formation. In the RuFd hybrid, RuPS can transfer electrons to the [2Fe2S] cluster of Fd via either an oxidative or reductive quenching mechanism in the presence of sodium ascorbate. Upon illumination of the RuFd hybrid in the presence or absence of FNR with freeze trapping techniques, we do not observe formation of a Ru(III) species which would be indicative of an oxidative quenching mechanism for RuPS*.22 A reductive quenching pathway is probable due to the presence of a high concentration of the SED ascorbate.40 However we were unable to detect a ligand centered reduction of RuPS for a [Ru(bpy)3]+ species by EPR so the mechanism remains uncertain.
In the native three-protein electron transfer cascade PSI → Fd → FNR, the [2Fe2S] cofactor of Fd (Em −420 mV vs. NHE) shuttles a single electron from PSI to FNR. Does the RuFd → FNR electron transfer proceed through the [2Fe2S] cluster as well? Illumination of RuFd in the presence of sodium ascorbate with freeze trapping techniques leads to the formation of an EPR signal consistent with reduced [2Fe2S] cluster, gx = 2.05, gy = 1.96, gz = 1.89 (Fig. 5B).22,41 Thus, RuFd is capable of photoinduced electron transfer from the covalently bound RuPS to the [2Fe2S] cluster. In the presence of FNR, however, the signal corresponding to reduced [2Fe2S] is absent upon freeze trapping under illumination. This suggests that none of the light-generated electrons reside on the [2Fe2S] cluster; all electrons are transferred to FNR. However, whether or not electron transfer from RuPS to FNR in the RuFd-FNR complex proceeds through the [2Fe2S] cluster cannot be determined from EPR alone. For this reason, we prepared the Ru-apoFd analogue in which the [2Fe2S] cluster has been removed.42 NADP+ reduction experiments using photoexcitation of Ru-apoFd show a slightly faster rate of NADPH formation (1400 mol NADPH (mol FNR)−1 h−1) vs. the biohybrid prepared with native Fd (1050 mol NADPH (mol FNR)−1 h−1) (Fig. 6A). Based on these results, the [2Fe2S] cluster is not essential for NADPH formation. RuPS is a one electron donor. Therefore, in the case of apo-Fd, one electron is transferred directly from RuPS to FAD of FNR.43 This direct electron transfer must occur two times, to produce a hydride that yields NADPH. For native Fd, the electron transfer occurs step-wise: first from RuPS to [2Fe2S], then from [2Fe2S] to FAD of FNR. This step-wise electron transfer occurs two times to produce the hydride that yields NADPH (Fig. 7). A hypothesis that is consistent with redox potentials and EPR and explains the difference in NADPH production between RuFd and Ru-apoFd (Fig. 6A) is that one ET step is faster than two steps depending on the relative orientations of the two proteins as they dock together.
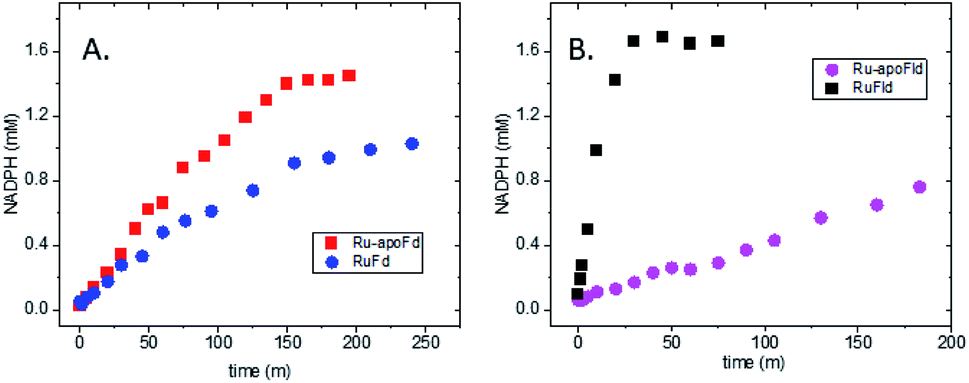 | ||
| Fig. 6 Importance of the intermediate electron acceptor cofactors in biohybrid photosynthetic NADP+ reduction. (A) In vitro light-driven NADP+ reduction for RuFd (blue circle) and Ru-apoFd (red square). (B) In vitro light-driven NADP+ reduction for RuFld (black square) and Ru-apoFld (magenta circle). | ||
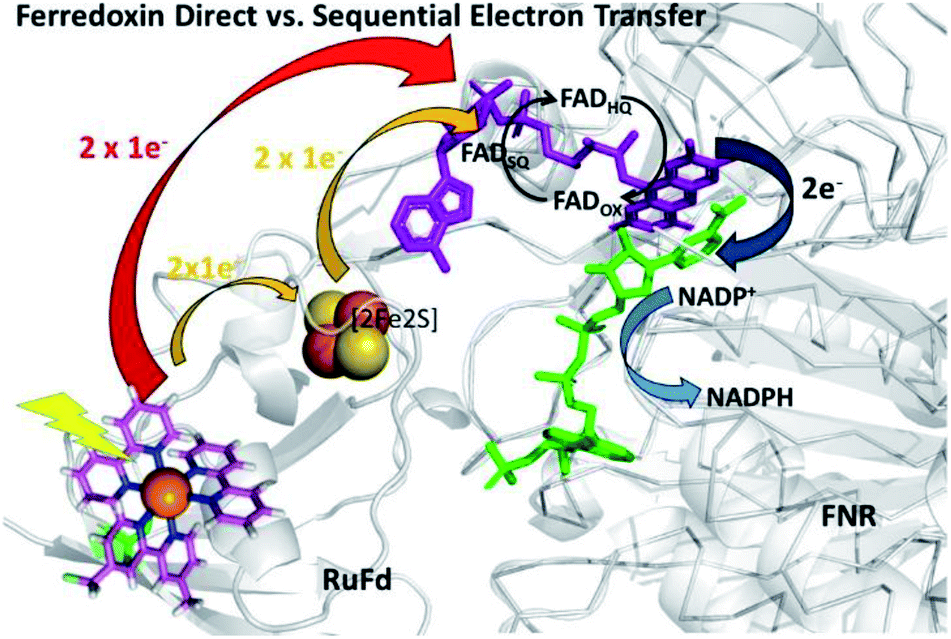 | ||
| Fig. 7 Scheme of photosynthetic electron transfer between RuFd and FNR showing two potential interprotein electron transfer pathways: a direct route between RuPS and FNR (red arrow) or a sequential transfer through the [2Fe2S] cluster of Fd to FNR. (Orange arrows). The [2Fe2S] cluster can accept one electron at a time, and donate one at a time. After receiving 2 single electron transfers from photoexcited RuFd, FNRHQ then donates 2 electrons in one step via a hydride to the bound substrate NADP+ to form NADPH. Reduced Fd and FNRSQ are observable via EPR. (PDB ID: 1A70, 2BSA). | ||
EPR studies: electron transfer from flavodoxin to FNR
We next examined the RuFld → FNR reaction, which provides a unique, yet challenging, opportunity to examine flavoprotein to flavoprotein electron transfer with EPR spectroscopy. Fld contains a FMN cofactor for shuttling a single electron from PSI to FNR. Like Fd, Fld forms a 1:1 complex with FNR. The FMN semiquinone in Fld is highly stable, so that close to 100% of the flavin is in this form after addition of one electron.44 This is a consequence of the relative midpoint potential for the FldOX/FldSQ couple (−195 mV) and the FldSQ/FldHQ couple (−390 mV).13 Note, the FldSQ/FldHQ pair is believed to shuttle electrons between PSI and FNR for the formation of NADPH.6 The study of the electron transfer between Fld and FNR by means of optical transient absorption spectroscopy is difficult because it is hard to detect the one electron transfer: one semiquinone flavin species gets oxidized (FMN in Fld) and a new semiquinone flavin species gets generated (FAD in FNR); both flavins exhibit optical spectral similarities if they are in the same redox state.13 Likewise, previous EPR studies of the Fld–FNR complex for fully protonated systems achieved formation of a semiquinone signal, but the EPR signals could not be distinguished between FldSQ and FNRSQ.15
We investigated this process using a selective deuteration approach. RuFld biohybrids were made using both fully deuterated Fld and protonated Fld (both in protonated buffer). Freeze quenching of these illuminated biohybrids in the presence of SED ascorbate resulted in a signal of the Fld semiquinone with g-value of 2.004 (Fig. 8A). The hyperfine splitting pattern of 2H-Fld is similar to 2H-FMN in FNR (black spectrum in Fig. 5A), indicating that the same paramagnetic flavin species is trapped. A good spectral simulation (Fig. 8A, green) was obtained assuming one strongly coupled proton in addition to the two prominent nitrogen atoms in the isoalloxazine moiety (see Table S1† for simulation parameters). Since the flavin is fully deuterated, this must be due to an exchangeable proton. Indeed, very good agreement is found with published values45 for neutral flavin semiquinones protonated at position 5 (Fig. 3A, Table S2†). Importantly, the 2H-Fld and 1H-Fld have identifying spectral features which can be used to examine inter flavoprotein electron transfer.
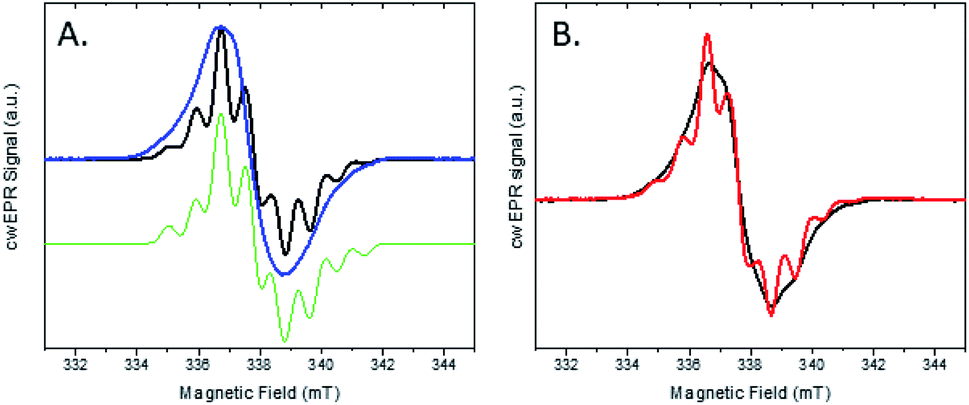 | ||
| Fig. 8 cw X-band EPR spectra of the two protein systems RuFld and FNR. (A) Semiquinone signal for the RuFld hybrid for 1H-Fld (blue) and 2H-Fld (black). Samples contained 260 μM Ru(1H-Fld) and 360 μM Ru(2H-Fld). Simulated spectrum of 2H-Fld (green), parameters Table S1.† (B) Mixed isotope strategy for defining flavin to flavin electron transfer, Ru(1H-Fld) + FNR(2H-FMN) (black) and Ru(2H-Fld) + FNR(1H-FMN) (red). Protein concentrations were: 400 μM Ru(1H-Fld), 280 μM FNR(2H-FMN), 230 μM Ru(2H-Fld), 200 μM FNR(1H-FMN). | ||
Matched samples were prepared, 2H-Fld + FNR(1H-FMN) and 1H-Fld + FNR(2H-FMN), with the goal of using the differences in isotopes between the flavin cofactors to track the RuFld → FNR electron transfer event. Interflavin electron transfer is spectroscopically complicated due to the multiple redox and protonation states of flavins,13 and only the semiquinone state is observed by EPR (Fig. S5†). Note, FldHQ delivers one electron at a time to FNROX, forming first the intermediate FNRSQ, then FNRHQ. After electron transfer, FldSQ is formed.
Fig. 8B shows the cw X-band EPR spectra of the selectively deuterated samples, Ru(2H-Fld) + FNR(1H-FMN) and Ru(1H-Fld) + FNR(2H-FMN), measured after freeze trapping under illumination to generate semiquinone signals. The samples contained both RuFld and FNR proteins in solution at a ratio of 1.4:1 as well as SED ascorbate. Comparison with 1H-Fld and 2H-Fld spectra in Fig. 8A unambiguously shows that both spectra in Fig. 8B are dominated by the signal of FldSQ. However, spectral differences due to the presence of a smaller contribution of FNRSQ are observed. To determine the relative contribution from these two radicals both spectra were simulated as a sum of FldSQ and FNRSQ spectra with different weight factors, normalized for the second integral. Using this conventional deconvolution procedure, we determine that 13% of the total EPR signal is due to FNRSQ generated via interprotein electron transfer. As observed in the NADP+ reduction assay, RuFld is capable of driving photocatalytic NADPH formation from FNR (Fig. 6B). The EPR spectral results are consistent with efficient light-driven electron transfer between RuFld and FNR. A hypothesis supported by the EPR results is that the flavin cofactor of Fld is reduced first by a reductive quenching mechanism of RuPS* by ascorbate, followed by rapid one electron transfers to the flavin cofactor of FNR, producing its semiquinone form (FNRSQ) which is in equilibrium with the EPR silent hydroquinone state, FNRHQ. The 13% FNRSQ observed by our selective deuteration scheme is in accord with the reported 10–20% FNR protein stabilization of the maximal amount of semiquinone and consistent with flavoproteins that transfer 2 electrons at a time.9,10
To further examine flavin cofactor involvement in RuFld → FNR interprotein electron transfer, we removed the FMN cofactor from Fld and prepared the Ru-apoFld biohybrid. The capability of Ru-apoFld to transfer light-generated electrons to FNR was measured in the NADP+ reduction assay (Fig. S4†) As Fig. 6B shows, removal of the flavin cofactor from Fld severely reduced the capability of the Ru-apoFld hybrid to efficiently drive NADP+ reduction upon illumination. The 630 mol NADPH (mol FNR−1) h−1 rate is 13-fold slower than that observed for the biohybrid made with native Fld, which confirms that the FMN cofactor in Fld is involved in the light-driven biohybrid RuFld → FNR electron transfer process. We assert that the multielectron capability of the flavin cofactor facilitates the two electron NADP+ reaction; providing an excellent example of photosynthetic charge accumulation. Fig. 9 depicts the scheme for light driven RuFld → FNR electron transfer NADPH formation. RuPS is a one electron donor, so to initiate the process RuPS must donate one electron two times to the FMN cofactor of Fld to generate FldHQ. FldHQ donates an electron to FAD cofactor of FNR. The Fld protein then cycles between the FldSQ and FldHQ states as it accepts one electron from RuPS and donates one electron to FAD cofactor of FNR. After FAD cofactor of FNR receives two electrons, it produces the hydride that yields NADPH.
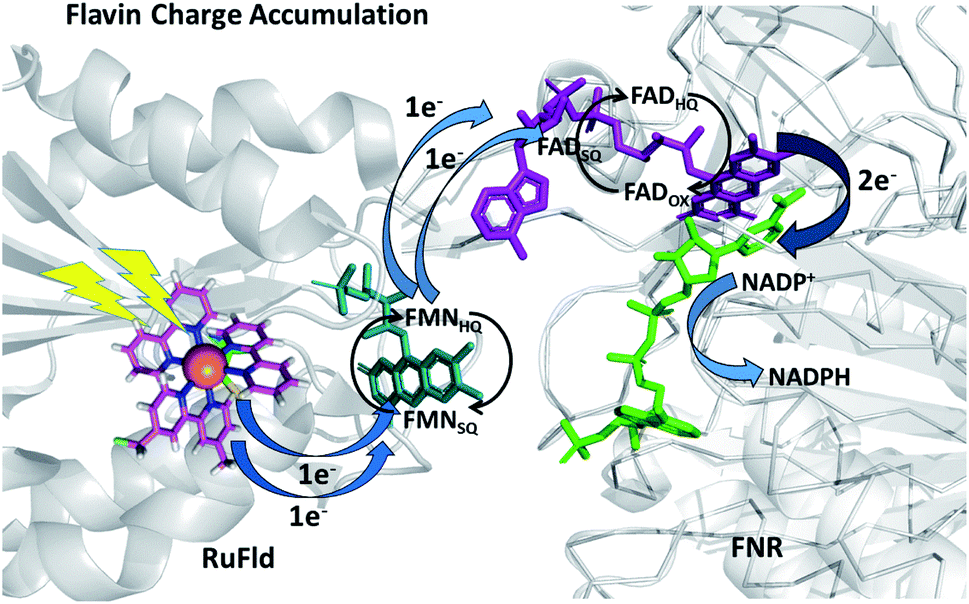 | ||
| Fig. 9 Scheme of photosynthetic electron transfer between RuFld and FNR highlighting the multielectron capabilities of the flavin cofactors. To initiate the interprotein electron transfer, Fldox must obtain two electrons from single electron transfers from RuPS covalently bound to Cys54 (RuPS is rereduced via the sacrificial electron donor sodium ascorbate). The Fld cofactor then cycles between the FldSQ and FldHQ transferring electrons one at a time to FNR. FNRHQ then donates 2 electrons in one step via a hydride to bound NADP+ to form NADPH. FldSQ and FNRSQ are observable via EPR. (PDB ID: 1CZN, 2BSA). | ||
Nature's mechanisms for coupling single electron transfers to charge accumulation
Nature has optimized the photosynthetic molecular machinery that couples single electron transfers, generated at the PSI RC protein, to charge accumulation at the FNR protein. Fd and Fld proteins shuttle light-generated electrons between PSI and FNR. Both are similarly capable of doing so, as demonstrated in PSI-driven in vitro NADP+ reduction experiments, with observed rates of 92500 mol NADPH (mol FNR−1) h−1 and 100400 mol NADPH (mol FNR−1) h−1 for Fld and Fd respectively (Fig. S7†). A surprising difference was revealed when we examined our biohybrid systems in which PSI was replaced with a molecular RuPS covalently bound to Fd and Fld. The RuFld biohybrid system exhibited an 8-fold higher rate of NADP+ reduction and nearly double the number of turnovers than our RuFd system (Fig. 4). The Em values for Fd (−420 mV vs. NHE) and Fld (−390 mV vs. NHE) are similar, ruling out driving force as the cause of the reactivity differences.
What is the molecular basis for this difference? With interprotein electron transfer, multiple factors are at play which make it experimentally difficult to address this question, but we can propose several likely rationale. One possibility is that the RuPS molecule bound to the protein surface interferes with protein–protein interactions. This could alter distances between the protein cofactors involved in electron transfer (most likely increasing them), thus impacting electron transfer rates. Fd (10.5 kDa) is smaller than Fld (17 kDa) so RuPS is a larger relative surface area of Fd than Fld, and more likely to disrupt structure and be a steric hindrance for RuFd- FNR complexation compared to RuFld–FNR complexation. A prior study, however, showed that covalent linkage of RuPS to the Fd protein did not limit formation of a competent RuFd–FNR complex for interprotein electron transfer and reported a rate of 6500 s−1 for the RuFd to FNR light-driven electron transfer reaction.43 Multiple orientations are available for Fd docking if in fact RuPS interferes with a particular interaction as a range of binding interactions for Fd have been observed in Fd-FNR crystal and NMR structures, with inter flavin isoalloxazine and [2Fe2S] distances < 8 Å.46–49 Crystal structures of Fld–FNR remain elusive, and it has been postulated that the interaction of Fld with FNR is less specific that of Fd.5 Perhaps this added flexibility in binding orientations aids in productive RuFld-FNR formation, facilitating the interprotein electron transfer. Quantum yields of electron transfer and intra and interprotein electron transfer rates may affect the overall reaction rates for NADP+ reduction.
Another possibility is that the RuPS molecule itself is not the sole cause of altered interprotein electron transfer, but rather brings to light, by way of the biohybrid system, a feature of nature's photosynthetic mechanism not observed before. Since a notable difference between Fd and Fld is the distinct type of cofactors, a [2Fe2S] cluster and a flavin molecule, we hypothesize that the higher rate of NADP+ reduction observed for RuFld → FNR versus RuFd → FNR light-driven electron transfer is due to the nature of the flavin cofactor; that is its ability to cycle between redox states of the semiquinone and hydroquinone (Fig. 9) compared to the less versatile single electron shuttling character of an Fe–S cluster (Fig. 7). We believe the difference between Fd and Fld is not observed in the PSI-driven system due to evolutionary optimization. Even in vitro, the observed rates of NADP+ reduction are 2 orders of magnitude faster than the rates we observe for our Ru biohybrid driven systems. Any differences due to the nature of the cofactors of Fd and Fld are obscured by the overall efficiency of the PSI-driven system. Thus, by using the biohybrid system we have uncovered an interesting observation about photosynthetic coupling of one electron transfers to charge accumulation.
Further investigation of flavin-based biohybrids can inform purely artificial systems about mechanisms and design strategies for light-induced charge accumulation. Artificial photosynthetic systems have been constructed that mimic structural and functional components of nature's light driven chemistry, yet it remains difficult to accomplish charge accumulation in purely synthetic systems.50,51 Photosynthetic biohybrid and artificial systems that accomplish charge accumulation are of interest in solar fuel schemes where efficient delivery of multiple electrons is needed. Understanding general mechanisms of charge accumulation is thus important for development of strategies for solar fuel development. Biohybrids experimentally bridge the gap between artificial and natural systems, combining beneficial features of both systems, which can provide new chemical insight.52
Conclusions
PSI-driven electron transfer events result in the charge accumulation of two successive electrons at the FAD cofactor site of FNR protein. A combined biohybrid and selective deuteration approach was used to generate and spectroscopically detect with EPR the semiquinone state of FNR formed following light-driven single interprotein electron transfer from the electron shuttle proteins Fd and Fld. In NADP+ reduction assays, a notable difference in RuFd versus RuFld reactivity was observed. The biohybrid technique revealed differentiated chemistry between the electron shuttle proteins, providing an example of the utility of incorporating synthetic molecules to creatively probe nature's mechanisms. We hypothesize that the observed differences are related to the versatility of flavin cofactors having three oxidation states readily available, and theorize that this intrinsic characteristic aids in photosynthetic charge accumulation at the FAD cofactor site of FNR. As such, this system provides a reminder of the evolutionary advantage of the functional inclusion of flavins in biological reaction schemes. Even though nature has mastered charge accumulation via photosynthetic schemes, it is still complicated to experimentally identify the individual mechanistic steps because of the multiple redox states of flavoproteins. Selective deuteration will enable application of advanced EPR techniques, such as high-field Mims-type pulsed electron-nuclear double resonance spectroscopy (ENDOR) or “matrix” ENDOR,53–56 to interrogate the local flavin site protein environmental responses to electron transfer events in future experiments. We expect using biohybrids to explore integral functional components of solar energy conversion will provide important insight about fundamental mechanisms of photochemical energy conversion.Experimental
Preparation of Fd and Fld biohybrids
The ruthenium photosensitizer (RuPS), [Ru(4-CH2Br-4′-CH3-2,2′-bpy)(bpy)2]·2PF6, was synthesized and characterized according to published methods.57,58 The RuFd hybrid was prepared using Spinacia oleracea ferredoxin (Fd) from Sigma-Aldrich.22 Spinach Fd (1.2 mM stock in 20 mM Hepes pH 7.9) was diluted to 32 μM in 10 mM MES buffer pH 6.0 and incubated with 8 mol equiv. RuPS (9.0 mM stock solution in DMSO) overnight at 4 °C. Samples were concentrated with Amicon 3000 MWCO filtration devices and repeatedly diluted (4 times) with 10 mM MES pH 6.0 to remove the unbound RuPS.Ru-2H-Fld hybrid was prepared using deuterated Fld isolated from Thermosynechococcus lividus (PCC6717) grown in deuterated medium and purified with protonated buffers.59 Ru-1H-Fld hybrid was prepared using Fld overexpressed in E. coli by standard procedures. UV-Vis spectroscopy of the purified 1H-Fld showed low incorporation of the native FMN cofactor. Fld was reconstituted by addition of 4 mol equiv. FMN followed by removal of excess unbound FMN by extensive washing with 3000 MWCO filtration devices until the filtrate was colorless. RuPS binding to 2H-Fld and 1H-Fld was performed by adding 4 mol equiv. RuPS/Fld (150 μM Fld) in 20 mM Hepes pH 8.0. The sample was tumbled (LabQuake) overnight at 4 °C in the dark. Unbound RuPS was removed by microfiltration (Amicon 3000 MWCO) using 20 mM Hepes pH 8.0.
Apo-Fd and Apo-Fld were prepared by cofactor removal as reported previously by a trichloroacetic acid precipitation in the presence of dithiothreitol.2,20,42 RuPS was bound to the apo-proteins using the same methods as for the native holoproteins.
Inductively coupled plasma atomic emission spectroscopic (ICP-AES) measurements with a Thermo Scientific iCAP6000 spectrometer were used to calculate metal-to-protein ratios using comparison to known metal standards. Fd protein concentration was determined using UV-visible absorption and a molar extinction coefficient of 9600 M−1 cm−1 at 422 nm or direct Fe content by ICP-AES.60 Fld protein content was determined by the Bradford protein assay method.61 Holo-Fld content was also verified with an extinction coefficient of 8300 M−1 cm−1 at 465 nm.59 RuPS driven NADP+ reduction activity was measured to verify successful interprotein electron transfer between the biohybrids and FNR protein. 4.8 μM biohybrid (RuFd, Ru-apoFd, RuFld, Ru-apoFld) was mixed with 500 nM FNR in a 20 mM Hepes pH 8.0 buffer containing 100 mM sodium ascorbate and 2 mM NADP+ (Sigma Aldrich) as substrate. Photoreduction was assayed by sample illumination with 455 nm LED (Thorlabs) in 2 mm cuvette and UV-Vis spectra obtained at time points after illumination (Fig. S3 and S4†). NADPH formed was determined using the extinction coefficient 6.22 mM−1 cm−1 at 340 nm.62
Preparation of protonated FNR
FNR was overexpressed from a synthetic gene encoding a domain from Anabaena PCC7119 (Uniprot P21890, amino acids 137-440) and purified as previously detailed.42 UV-Vis spectroscopy of the purified FNR protein showed low incorporation of the native FAD cofactor. To prepare protonated FNR EPR samples, the native FAD was reconstituted into purified FNR protein as verified by UV-Vis spectroscopy. FNR concentration was determined by extinction coefficient at 456 nm of 10740 M−1 cm−1 and verified separately by Bradford protein assays.42
Preparation of FNR (2H-FMN)
2H-FMN cofactor was extracted from deuterated Fld by trichloroacetic acid precipitation in the presence of dithiothreitol. The solution was neutralized with addition of NaOH to pH 8.0 and rotovapped to 1 ml. The sample was passed through a Sephadex G25 column equilibrated with MilliQ water, heart cut fractions combined, rotovapped to dryness, and stored at −20 °C. Stock solution of 2H-FMN was prepared by dilution with 20 mM Hepes pH 8.0 (in H2O) and concentration determined with extinction coefficient at 450 nm 12500 M−1 cm−1. ApoFNR (overexpressed FNR with <10% FAD cofactor) was reconstituted with 2H-FMN by addition of 5.8 mol equiv. FMN/FNR and overnight incubation. Unbound FMN cofactor was removed by microfiltration and washing with 20 mM Hepes pH 8.0 (protonated buffer). 2H-FMN incorporation into FNR was verified by UV-Vis spectroscopy. To verify function of FMN substituted FNR, NADP+ reduction was measured via PSI light-driven chemistry. Protein concentrations in the assay were 60 nM PSI purified from Synechococcus leopoliensis (UTEX625), 500 nM FNR, 4 μM Fd, and 10 μM cyt c6 purified from Thermosynechococcus lividus. The reaction mixture contained 20 mM Hepes pH 8.0, 10 mM sodium ascorbate, 3 mM MgCl2, 60 μM DCPIP, 0.03% n-dodecyl-β-maltopyranoside with 2 mM NADP+ substrate (Sigma Aldrich). UV Vis spectra were taken prior to and at specific time points of sample illumination with a white light LED (Solis-3C, Thorlabs).
EPR experiments
Ru biohybrids and FNR samples were prepared as described above and concentrated to 300–600 μM in 20 mM Hepes pH 7.97 (all EPR samples in protonated buffer, except for sample specifically exchanged into deuterated buffer, spectrum shown in Fig. S6†). For interprotein electron transfer experiments, the RuFd or RuFld was combined in appropriate ratio with FNR(1H-FAD), FNR(1H-FMN) or FNR(2H-FMN) in 100 mM sodium ascorbate and 120 mM NaCl. Samples were placed in quartz EPR tubes in a nitrogen box. A Bruker ELEXSYS II E500 EPR spectrometer (Bruker Biospin, Rheinstetten, Germany) equipped with a TE102 rectangular resonator (Bruker ER4102ST) and a helium gas-flow cryostat (ICE Oxford, UK) was used for cw X-band EPR measurements. An ITC (Oxford Instruments, UK) was used for temperature control. Dark spectra were obtained for all samples prior to illumination experiments. Samples were dark adapted in a nitrogen box prior to freezing in liquid nitrogen. Illumination during EPR measurements were performed using a white light LED (Solis-3C, Thorlabs). Samples were illuminated at room temperature for 5 s, followed by immersion in liquid N2 while continuously illuminating, and then placed in a precooled EPR resonator. All EPR spectra were obtained at 20 K. Data processing was performed using Xepr (Bruker Biospin, Rheinstetten, Germany) and spectral simulation using the EasySpin program63 in Matlab™ R2018b (MathWorks, Natick) environment.Conflicts of interest
There are no conflicts to declare.Data availability
Data are available upon request from the authors.Author contributions
L. M. U. conceived the project. U. B. prepared proteins. L. M. U. prepared the biohybrid complexes and conducted the biohybrid experiments. K. L. M. performed molecular synthesis. J. N. and O. G. P. performed the EPR spectroscopy and spectral data analysis. L. U. wrote the manuscript. All authors discussed the results and contributed to manuscript editing.Acknowledgements
The authors thank A. Wagner for growth of the cyanobacteria and P. Pokkuluri for overexpression of Fld and FNR. This work is supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, under Contract No. DE-AC02-06CH11357.Notes and references
- Y.-T. Kao, C. Saxena, T.-F. He, L. Guo, L. Wang, A. Sancar and D. Zhong, J. Am. Chem. Soc., 2008, 130, 13132–13139 CrossRef CAS PubMed.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell Science Ltd, Malden, USA, 2002 Search PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–U65 CrossRef CAS PubMed.
- P. Jordan, P. Fromme, H. T. Witt, O. Klukas, W. Saenger and N. Krauss, Nature, 2001, 411, 909–917 CrossRef CAS PubMed.
- M. Medina, FEBS J., 2009, 276, 3942–3958 CrossRef CAS PubMed.
- J. K. Hurley, G. Tollin, M. Medina and C. Gomez-Morena, in Photosystem I: the light-driven plastocyanin: ferredoxin oxidoreductase, Springer, Dordrecht, 2006, pp. 455–476 Search PubMed.
- C. M. Bruns and P. A. Karplus, J. Mol. Biol., 1995, 247, 125–145 CrossRef CAS PubMed.
- L. Serre, F. M. D. Vellieux, M. Medina, C. Gomez-Morena, J. C. Fontecilla-Camps and M. Frey, J. Mol. Biol., 1996, 263, 20–39 CrossRef CAS PubMed.
- P. Mulo and M. Medina, Photosynth. Res., 2017, 134, 265–280 CrossRef CAS PubMed.
- M. E. Corrando, A. Aliverti, G. Zanetti and S. G. Mayhew, Eur. J. Biochem., 1996, 239, 662–667 CrossRef PubMed.
- M. Faro, C. Gomez-Morena, M. Stankovich and M. Medina, Eur. J. Biochem., 2002, 269, 2656–2661 CrossRef CAS PubMed.
- M. Martinez-Julvez, J. Hermoso, J. K. Hurley, T. Mayoral, J. Sanz-Aparicio, G. Tollin, C. Gomez-Morena and M. Medina, Biochemistry, 1998, 37, 17680–17691 CrossRef CAS PubMed.
- M. C. Walker, J. J. Pueyo, C. Gomez-Morena and G. Tollin, Arch. Biochem. Biophys., 1990, 281, 76–83 CrossRef CAS PubMed.
- K. Huang, S.-I. Tu and J. H. Wang, Biochem. Biophys. Res. Commun., 1969, 34, 48–52 CrossRef CAS PubMed.
- M. Medina, C. Gomez-Morena and R. Cammack, Eur. J. Biochem., 1995, 227, 529–536 CrossRef CAS PubMed.
- A. Serrano, J. Rivas and M. Losada, FEBS Lett., 1984, 170, 85–88 CrossRef CAS.
- J. I. Martinez, P. J. Alonso, C. Gomez-Morena and M. Medina, Biochemistry, 1997, 36, 15526–15537 CrossRef CAS PubMed.
- M. Medina and R. Cammack, Perkin Trans. 2, 1996, 2, 633–638 RSC.
- S. C. Silver, J. Niklas, P. Du, O. G. Poluektov, D. M. Tiede and L. M. Utschig, J. Am. Chem. Soc., 2013, 135, 13246–13249 CrossRef CAS PubMed.
- S. R. Soltau, P. D. Dahlberg, J. Niklas, O. Poluektov, K. L. Mulfort and L. M. Utschig, Chem. Sci., 2016, 7, 7068–7078 RSC.
- S. R. Soltau, J. Niklas, P. D. Dahlberg, K. L. Mulfort, O. G. Poluektov and L. M. Utschig, ACS Energy Lett., 2017, 2, 230–237 CrossRef CAS.
- S. R. Soltau, J. Niklas, P. D. Dahlberg, O. G. Poluektov, D. M. Tiede, K. L. Mulfort and L. M. Utschig, Chem. Commun., 2015, 51, 10628–10631 RSC.
- L. M. Utschig, N. M. Dimitrijevic, O. G. Poluektov, S. D. Chemerisov, K. L. Mulfort and D. M. Tiede, J. Phys. Chem. Lett., 2011, 2, 236–241 CrossRef CAS.
- L. M. Utschig, S. C. Silver, K. L. Mulfort and D. M. Tiede, J. Am. Chem. Soc., 2011, 133, 16334–16337 CrossRef CAS PubMed.
- W. Chorney, N. Scully, H. L. Crespi and J. J. Katz, Biochim. Biophys. Acta, 1960, 37, 280–287 CrossRef CAS.
- H. L. Crespi, S. Archer and J. J. Katz, Nature, 1959, 184, 729–730 CrossRef CAS PubMed.
- H. L. Crespi, S. Conrad, R. A. Uphaus and J. J. Katz, Ann. N. Y. Acad. Sci., 1960, 84, 648–666 CrossRef CAS PubMed.
- G. Link, T. Berthold, M. Bechtold, J. U. Weidner, E. Ohmes, J. Tang, O. Poluektov, L. Utschig, S. L. Schlesselman, M. C. Thurnauer and G. Kothe, J. Am. Chem. Soc., 2001, 123, 4211–4222 CrossRef CAS PubMed.
- A. L. Morris, S. W. Snyder, Y. Zhang, J. Tang, M. C. Thurnauer, P. L. Dutton, D. E. Robertson and M. R. Gunner, J. Phys. Chem., 1995, 99, 3854–3866 CrossRef CAS.
- J. R. Norris, R. A. Uphaus, H. L. Crespi and J. J. Katz, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 625–628 CrossRef CAS PubMed.
- O. G. Poluektov, L. M. Utschig, S. L. Schlesselman, K. V. Lakshmi, G. W. Brudvig, G. Kothe and M. C. Thurnauer, J. Phys. Chem. B, 2002, 106, 8911–8916 CrossRef CAS.
- R. R. Rustandi, S. W. Snyder, L. L. Feezel, T. J. Michalski, J. R. Norris, M. C. Thurnauer and J. Biggins, Biochemistry, 1990, 29, 8030–8032 CrossRef CAS PubMed.
- J. Tang, L. M. Utschig, O. Poluektov and M. C. Thurnauer, J. Phys. Chem. B, 1999, 103, 5145–5150 CrossRef CAS.
- M. C. Thurnauer and P. Gast, Photobiochem. Photobiophys., 1985, 9, 29–38 CAS.
- L. M. Utschig, S. R. Greenfield, J. Tang, P. D. Laible and M. C. Thurnauer, Biochemistry, 1997, 36, 8548–8558 CrossRef CAS PubMed.
- V. Massey, Biochem. Soc. Trans., 2000, 28, 283–296 CrossRef CAS PubMed.
- H. L. Crespi, J. R. Norris and J. J. Katz, Biochim. Biophys. Acta, 1971, 253, 509–513 CrossRef CAS.
- C. C. Correll, M. L. Ludwig, C. M. Bruns and P. A. Karplus, Protein Sci., 1993, 2, 2112–2133 CrossRef CAS PubMed.
- P. W. Riddles, R. L. Blakeley and B. Zerner, Methods Enzymol., 1983, 91, 49–60 CAS.
- B. Shan, T. Baine, X. A. N. Ma, X. Zhao and R. H. Schmehl, Inorg. Chem., 2013, 52, 4853–4859 CrossRef CAS PubMed.
- R. Malkin and A. J. Bearden, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 16–19 CrossRef CAS PubMed.
- U. Brahmachari, P. R. Pokkuluri, D. M. Tiede, J. Niklas, O. G. Poluektov, K. L. Mulfort and L. M. Utschig, Photosynth. Res., 2020, 143, 183–192 CrossRef CAS PubMed.
- A. Quaranta, B. Lagoutte, J. Frey and P. Setif, J. Photochem. Photobiol., B, 2016, 160, 347–354 CrossRef CAS PubMed.
- J. J. Pueyo, C. Gomez-Morena and S. G. Mayhew, Eur. J. Biochem., 1991, 202, 1065–1071 CrossRef CAS PubMed.
- A. Okafuji, A. Schnegg, E. Schleicher, K. Möbius and S. Weber, J. Phys. Chem. B, 2008, 112, 3568–3574 CrossRef CAS PubMed.
- G. Kurisu, M. Kusunoki, E. Katoh, T. Yamazaki, K. Teshima, Y. Onda, Y. Kimata-Ariga and T. Hase, Nat. Struct. Biol., 2001, 8, 117–121 CrossRef CAS PubMed.
- R. Morales, M. H. Charon, G. Kachalova, L. Serre, M. Medina, C. Gomez-Moreno and M. Frey, EMBO Rep., 2000, 1, 271–276 CrossRef CAS PubMed.
- G. T. Hanke, G. Kurisu, M. Kusunoki and T. Hase, Photosynth. Res., 2004, 81, 317–327 CrossRef CAS PubMed.
- P. N. Palma, B. Lagoutte, L. Krippahl, J. J. G. Moura and F. Guerlesquin, FEBS Lett., 2005, 579, 4585–4590 CrossRef CAS PubMed.
- L. Hammarstrom, Acc. Chem. Res., 2015, 48, 840–850 CrossRef PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1993 CrossRef CAS PubMed.
- L. M. Utschig, Chem, 2022, 8, 5–6 CAS.
- O. G. Poluektov and L. M. Utschig, J. Phys. Chem. B, 2021, 125, 4025–4030 CrossRef CAS PubMed.
- O. G. Poluektov, L. M. Utschig, A. A. Dubinskij and M. C. Thurnauer, J. Am. Chem. Soc., 2004, 126, 1644–1645 CrossRef CAS PubMed.
- O. G. Poluektov, L. M. Utschig, A. A. Dubinskij and M. C. Thurnauer, J. Am. Chem. Soc., 2005, 127, 4049–4059 CrossRef CAS PubMed.
- L. M. Utschig, M. C. Thurnauer, D. M. Tiede and O. G. Poluektov, Biochemistry, 2005, 44, 14131–14142 CrossRef CAS PubMed.
- S. Gould, G. F. Strouse, T. J. Meyer and B. P. Sullivan, Inorg. Chem., 1991, 30, 2942–2949 CrossRef CAS.
- L. C. Sun, H. Berglund, R. Davydov, T. Norrby, L. Hammarstrom, P. Korall, A. Borje, C. Philouze, K. Berg, A. Tran, M. Andersson, G. Stenhagen, J. Martensson, M. Almgren, S. Styring and B. Akermark, J. Am. Chem. Soc., 1997, 119, 6996–7004 CrossRef CAS.
- H. L. Crespi, U. Smith, L. Gajda, T. Tisue and R. M. Ammeraal, Biochim. Biophys. Acta, 1972, 256, 611–618 CrossRef CAS.
- K. Tagawa and D. I. Arnon, Biochim. Biophys. Acta, 1968, 153, 602–613 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- R. Masaki, K. Wada and H. Matsubara, J. Biochem., 1979, 86, 951–962 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Flavin oxidation and protonation states, additional EPR spectra, EPR simulation parameters, and NADP+ reduction experiments. See https://doi.org/10.1039/d2sc01546c |
| This journal is © The Royal Society of Chemistry 2022 |