DOI:
10.1039/D2SC01394K
(Edge Article)
Chem. Sci., 2022,
13, 4922-4929
Nickel-catalyzed skeletal transformation of tropone derivatives via C–C bond activation: catalyst-controlled access to diverse ring systems†
Received
9th March 2022
, Accepted 3rd April 2022
First published on 4th April 2022
Abstract
We report herein on nickel-catalyzed carbon–carbon bond cleavage reactions of 2,4,6-cycloheptatrien-1-one (tropone) derivatives. When a Ni/N-heterocyclic carbene catalyst is used, decarbonylation proceeds with the formation of a benzene ring, while the use of bidentate ligands in conjunction with an alcohol additive results in a two-carbon ring contraction with the generation of cyclopentadiene derivatives. The latter reaction involves a nickel–ketene complex as an intermediate, which was characterized by X-ray crystallography. The choice of an appropriate ligand allows for selective synthesis of four different products via the cleavage of a seven-membered carbocyclic skeleton. Reaction mechanisms and ligand-controlled selectivity for both types of ring contraction reactions were also investigated computationally.
Introduction
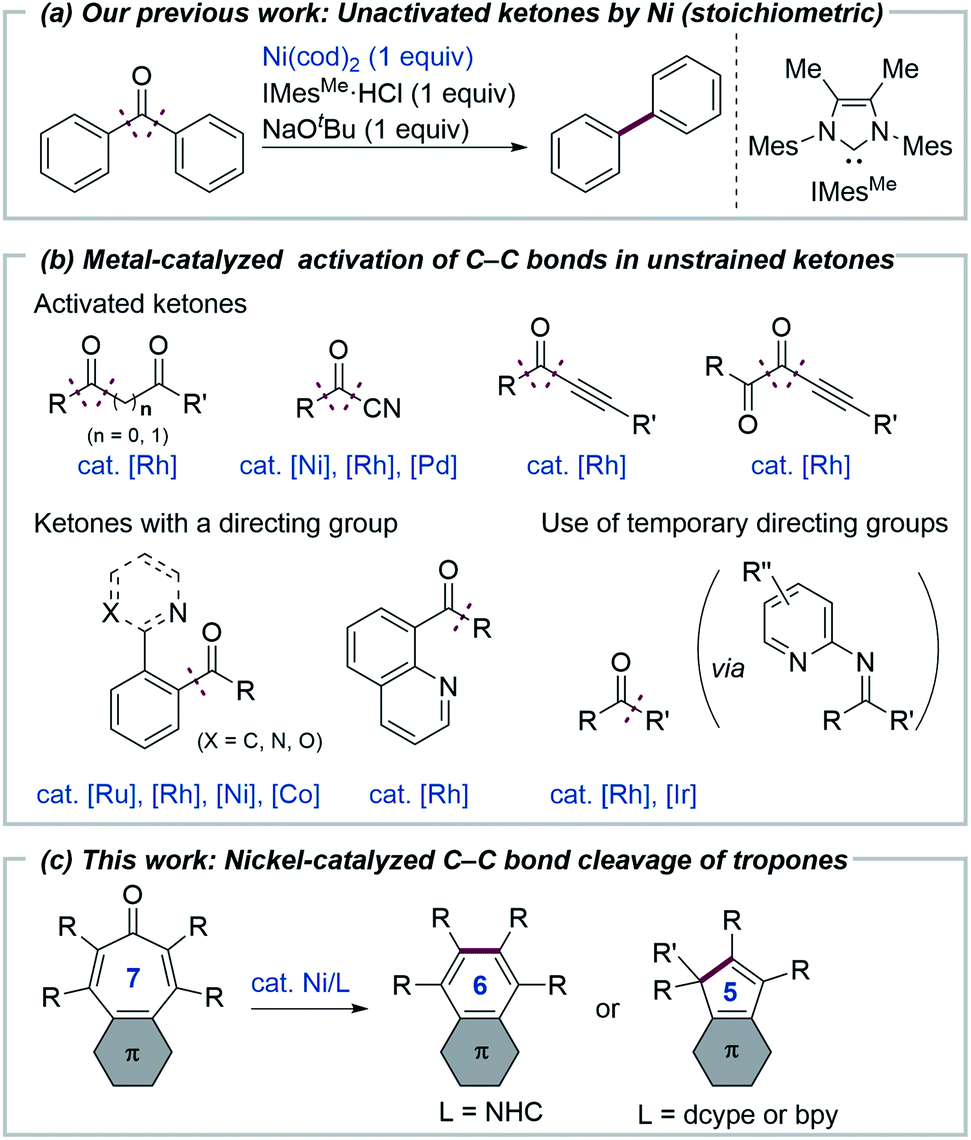
Transition metal-mediated selective cleavage of carbon–carbon (C–C) bonds has attracted the interest of researchers, since it would allow for the direct transformation of one of the most ubiquitous bonds in organic molecules.1 However, both the kinetic and thermodynamic stability of C–C bonds renders their cleavage a daunting challenge. To overcome this difficulty, several strategies, including the use of angle strain,2 a directing group,1,3 aromatization,4 and β-carbon elimination,5 have been successfully utilized to date. In contrast to nonpolar C–C bonds, polar C–C bonds, such as those in ketones, esters and nitriles, can be cleaved more readily by transition metals, although their bond dissociation energies are comparable to those for nonpolar C–C bonds.6 Regarding the metal-mediated cleavage of C(acyl)–C bonds in ketones, although most of the reported reactions continue to depend upon the use of angle strain2 or a directing group,1 several notable reactions that do not involve the use of such strategies have been reported. Murakami reported on a pioneering example of the metal-mediated activation of simple, unstrained ketones by developing the decarbonylation of cyclopentanones,7 while Brookhart later reported on rhodium-mediated decarbonylation of diaryl ketones.8 Our group previously reported that the decarbonylation of simple, unstrained ketones can also be mediated by a nickel/N-heterocyclic carbene (NHC) complex (Fig. 1a).9 Although these reactions are notable, in that neither ring strain nor a directing group is required to activate C–C bonds, it is necessary to use a stoichiometric amount of a metal complex. Catalytic C–C bond cleavage reactions of unstrained ketones have been reported to proceed when electronically activated ketones, such as diketones,10 acyl cyanides,11 alkynyl ketones,12 or ketones bearing a directing group13 are used (Fig. 1b). The catalytic C–C bond activation of simple ketones that proceed with the aid of a 2-aminopyridine cocatalyst, which serves as a temporary or removable directing group has been reported.3c,14 Although these reactions demonstrate the power of transition metals for activating otherwise unreactive C–C bonds, the scope of the catalytic transformation of C–C bonds in ketones remains limited, compared with other unreactive bonds, including C–H bonds. We envisioned that 2,4,6-cycloheptatrien-1-one (tropone)15 derivatives would be suitable substrates for catalytic decarbonylation because the process involves the formation of a benzene ring which would be expected to drive the otherwise difficult C–C bond cleavage reaction. It should be noted that the decarbonylation of tropone was reported to require heating at over 600 °C (ref. 16a) or photoirradiation conditions (∼1% yield).16b Herein, we report on the development of a nickel/NHC complex that can catalyze the decarbonylation of tropone derivatives with the formation of a benzene ring (one-carbon ring contraction). In addition, the use of a bidentate ligand was found to allow for an unprecedented catalytic two-carbon ring contraction reaction of tropones, leading to the formation of cyclopentadiene derivatives (Fig. 1c).
 |
| | Fig. 1 Transition metal-catalyzed C–C bond activation of ketones. | |
Results and discussion
Catalytic decarbonylation reactions
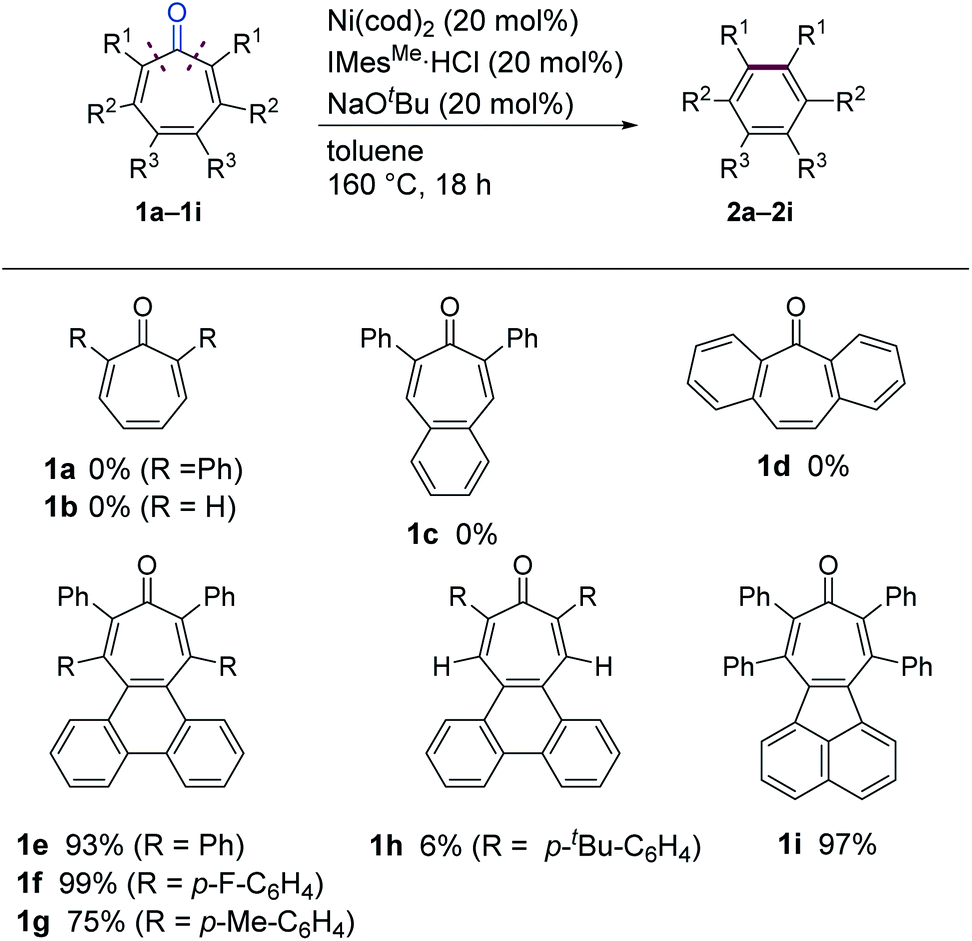
We initiated our study by examining the decarbonylation of a series of tropone derivatives 1a–1d using Ni(cod)2 (20 mol%) and IMesMe (20 mol%), which was used as an optimal catalyst in our previously reported nickel-mediated decarbonylation of simple ketones,9a in toluene at 160 °C for 18 h (Chart 1). No reaction occurred, however, and the starting ketones were recovered quantitatively under these conditions. Interestingly, the expected decarbonylation, in fact, proceeded, when a tropone bearing a fused phenanthrene ring (i.e., 1e) was used as a substrate, providing 2e in 93% yield. The 4,5-phenanthrene-fused tropones 1f and 1g bearing aryl substituents at α- and β-positions afforded the desired decarbonylated products 2f and 2g in a similar manner, whereas a tropone derivative lacking β-substituents, i.e., 1h, gave only a 6% yield. The acenaphtylene-fused analog 1i was decarbonylated successfully to furnish 2i in 97% yield.
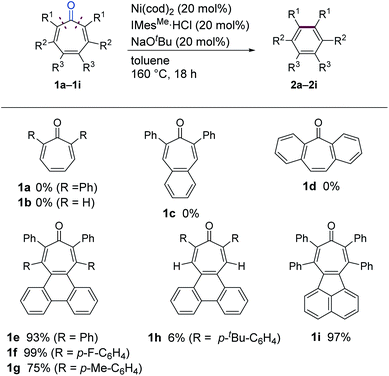 |
| | Chart 1 Scope of Ni/IMesMe-catalyzed decarbonylation of tropones. Reaction conditions: 1a (0.20 mmol), Ni(cod)2 (0.040 mmol), IMesMe·HCl (0.040 mmol), NaOtBu (0.040 mmol) toluene (1.0 mL), 160 °C for 18 h; run on a 0.30 mmol scale for 1b–1d and on a 0.20 mmol scale for 1e–1i. | |
Although tropones 1a–1i would all be expected to gain aromatic stabilization energy by decarbonylation, their reactivities toward the Ni/IMesMe catalyst were completely different. To obtain insights into structure/reactivity relationships, several structural and spectroscopic properties of the examined tropone derivatives were compared (Table 1). X-ray crystallographic analysis revealed that the Cα–CC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths of the successfully decarbonylated substrates 1e17a and 1i17b were relatively longer, and the bent angles of the tropone rings (θ and φ in Table 1) were larger than those for less reactive substrates 1c and 1h. All of these data suggest that the carbonyl groups in 1e and 1i are not conjugated with the π-system of the tropone ring in the solid state. 13C-NMR chemical shifts of the carbonyl carbons of 1e and 1i indicated that they were more deshielded than those of 1c and 1h, suggesting that 1e and 1i also behave as the non-conjugated ketones in solution. These bent structures found in 1e and 1i can be attributed to van der Waals (vdW) repulsion between the β-substituents and a large π-system fused to the tropone core. These analyses suggest that ground state destabilization by vdW strain18 is the key factor in promoting the C–C bond cleavage involved in this catalytic decarbonylation reaction of tropone derivatives.
O bond lengths of the successfully decarbonylated substrates 1e17a and 1i17b were relatively longer, and the bent angles of the tropone rings (θ and φ in Table 1) were larger than those for less reactive substrates 1c and 1h. All of these data suggest that the carbonyl groups in 1e and 1i are not conjugated with the π-system of the tropone ring in the solid state. 13C-NMR chemical shifts of the carbonyl carbons of 1e and 1i indicated that they were more deshielded than those of 1c and 1h, suggesting that 1e and 1i also behave as the non-conjugated ketones in solution. These bent structures found in 1e and 1i can be attributed to van der Waals (vdW) repulsion between the β-substituents and a large π-system fused to the tropone core. These analyses suggest that ground state destabilization by vdW strain18 is the key factor in promoting the C–C bond cleavage involved in this catalytic decarbonylation reaction of tropone derivatives.
Table 1 ORTEP drawings, C–C and C–O bond lengths, and bent angles of tropone scaffoldsa
To examine the catalyst turnover process, we next investigated the catalytic activity of Ni(CO)3(IMesMe), which would be expected to be formed as the decarbonylation reaction proceeds (Scheme 1a). It was found that Ni(CO)3(IMesMe) successfully catalyzed the decarbonylation of tropone 1e (94% yield). This result is in sharp contrast to the fact that this carbonyl complex does not catalyze the decarbonylation of simple ketones, which is the major reason for unsuccessful catalytic reactions.9a Therefore, the C(acyl)–C bond in 1e is more reactive than a corresponding bond of simple ketones because of vdW strain and can be activated by a less reactive CO-bound nickel species, allowing for a catalyst turnover. Indeed, the decarbonylation of 1e proceeded even at ambient temperature in 61% yield when a stoichiometric amount of a nickel complex was used (Scheme 1b), suggesting that a high temperature of 160 °C is required to release a CO ligand to regenerate an active Ni(0) species.
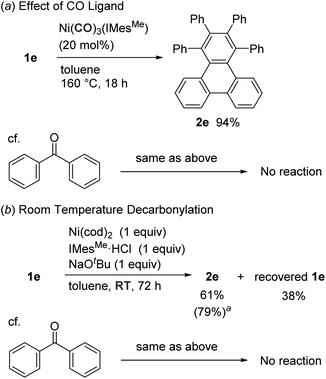 |
| | Scheme 1 Comparison between fused tropone and benzophenone. aRun at 40 °C for 18 h. | |
Catalytic two-carbon ring contraction reactions
During our optimization study of the decarbonylation of 1e, we envisioned that the use of a bidentate ligand would help to release a CO ligand from Ni(0) species, thereby improving the catalyst turnover step.9b,c Although these trials did not give satisfactory results for this decarbonylation, we noticed that a cyclopentadiene 3e was also formed in 8% yield, which appeared to be produced via the formal loss of CO and PhC units, when dcype was used as the ligand (Table 2, entry 1; Table S1, entry 6 in the ESI† for details), whereas no 3e was observed when NHCs or monodentate phosphine ligands, such as PCy3, were used (Table S1,† entries 1–5 and 7). The unexpected formation of 3e led us to re-examine the reaction conditions with respect to the selective formation of 3e (Table 2). The generation of the decarbonylated product 2e was suppressed when the reaction was performed at 120 °C (entry 2). A brief screening of ligands and additives revealed that the use of other bidentate phosphines also gave the desired two-carbon elimination product 3e, although the highest yield was only 15% (entries 3–6). Considering that a hydrogen atom is incorporated during the formation of 3e, we subsequently examined the addition of a hydrogen source to the reaction mixture (entries 7–9). As a result, 3e was selectively formed quantitatively, when the nickel/dcype-catalyzed reaction of 1e was carried out in the presence of MeOH (10 equiv.) (entry 7). Importantly, under these conditions, PhCH2CO2Me was also formed (80% yield), suggesting that the rest of the fragment of 1e was eliminated as 2-phenylethen-1-one (phenylketene), which was trapped by the added MeOH. To the best of our knowledge, this reaction represents the first catalytic conversion of a tropone ring system into a five-membered ring skeleton.
Table 2 Nickel-catalyzed two-carbon ring contraction of tropone 1ea
|
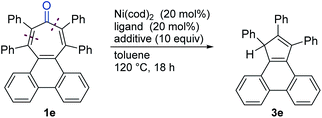
|
| Entry |
Ligand |
Additive |
Isolated yields (%) |
|
3e
|
2e
|
Recovered 1e |
|
Reaction condition: 1e (0.1 mmol), Ni(cod)2 (0.020 mmol), ligand (0.020 mmol), NaOtBu (0.020 mmol), toluene (1.0 mL), 120 °C for 18 h.
Run at 160 °C. NMR yields.
|
| 1b |
dcype |
None |
8 |
20 |
67 |
| 2 |
dcype |
None |
6 |
8 |
59 |
| 3 |
dcypm |
None |
11 |
18 |
55 |
| 4 |
dcypt |
None |
9 |
7 |
65 |
| 5 |
Dppf |
None |
15 |
9 |
76 |
| 6 |
Xantphos |
None |
12 |
2 |
84 |
| 7 |
dcype |
MeOH |
>99 |
0 |
0 |
| 8 |
dcype |
EtOH |
53 |
0 |
21 |
| 9 |
dcype |
2-Propanol |
24 |
0 |
10 |
|
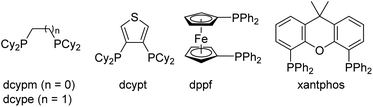
To obtain insights into the mechanism responsible for the generation of 3e, a stoichiometric reaction of 1e, Ni(cod)2 and dcype was performed in toluene at 100 °C, which led us to isolate the nickel–ketene complex 4e19,20 in 34% yield, along with 5e (56%) (Scheme 2a). The isolated ketene complex 4e was converted into the cyclopentadiene 3e in 84% yield by heating in toluene at 120 °C in the presence of EtOH (10 equiv.) (Scheme 2b). In this reaction, PhCH2CO2Et was also formed in 76% yield, indicating that the ketene fragment in 4e is reductively cleaved21 to give the cyclopentadiene 3e and a phenylketene, the latter of which was trapped by EtOH to form PhCH2CO2Et.
 |
| | Scheme 2 Intermediacy of a nickel–ketene complex. aORTEP drawings depicted at the 50% probability level. Hydrogen atoms were omitted for clarity. Phenyl and cyclohexyl groups were depicted as wireframe model. | |
To collect additional mechanistic details for the conversion of the ketene complex 4e to cyclopentadiene 3e, we examined the Ni/dcype-catalyzed reaction of 1e with EtOH at lower temperature (Scheme 3a). As a result, when the reaction was carried out at 80 °C, the cyclobutanone derivative 6e was formed in 75% yield. This transformation is not a simple skeletal isomerization, but two additional hydrogen atoms were incorporated into 1e. The generation of 6e could be explained as follows. The C–C bond that connects a cyclopentadiene scaffold and a ketene moiety in complex 4e is reductively cleaved4a,21,22 in the presence of EtOH to afford phenylketene (PhCHCO) and 3e, which subsequently undergoes a formal hetero [2 + 2] cycloaddition with the formation of the cyclobutanone 6e (Scheme 3a). Labelling experiments using EtOH-d6 confirmed that the two hydrogen atoms that are incorporated into 6e are derived from EtOH (Scheme 3b). An independently synthesized nickel-free ketene 7e could be converted to 3e in a quantitative yield when a Ni/dcype catalyst was used (Scheme 3c). In contrast, 6e did not undergo C–C bond fission under thermal conditions, but rather the addition of EtOH to a ketene moiety occurred to form 8e in 33% yield (see Scheme 3f for the structure of 8e), suggesting that the nickel catalyst plays an active role in the C–C bond cleavage step. The reaction between PhCHCO and 3e proceeded thermally (in toluene at 80 °C for 72 h) to afford 6e in 63% yield (Scheme 3d). It is noteworthy that a thermal reaction between ketene and cyclopentadiene was reported to form cyclobutanone derivatives via a [4 + 2] cycloaddtion/[3,3]-sigmatropic rearrangement sequence, resulting in a net [2 + 2] cycloaddition.20 Cyclobutanone 6e could be converted into 3e in 84% yield via a thermally-induced formal retro-[2 + 2] cycloaddition reaction at 120 °C (Scheme 3e). Additional studies revealed that the use of 2,2′-bipyridine allows for the addition of EtOH to the ketene moiety of 7e, providing a new cyclopentadiene derivative 8e (Scheme 3f). This result further highlights the profound impact of the ligand on the course of these types of reactions.
On the basis of the mechanistic findings shown in Schemes 2 and 3, the nickel/dcype-mediated reaction of 1e can be summarized as depicted in Scheme 4. The isomerization of tropone 1e to the ketene complex 4e initially occurs at 80 °C. Subsequent nickel-mediated C–C bond cleavage occurs at 80 °C in the presence of EtOH with the formation of the cyclopentadiene 3e and phenylketene, which then undergo a thermally-induced formal [2 + 2] cycloaddition to form cyclobutanone 6e. At higher temperatures, the reverse process from 6e to 3e begins to occur, leading to these compounds being in equilibrium. The phenylketene slowly and irreversibly reacts with EtOH at 120 °C (ref. 23) to form an ester (PhCH2CO2Et), thereby allowing for a selective formation of 3e at this temperature. Indeed, density functional theory (DFT) calculations at the M06-2X/6-31G(d,p) level of theory suggested that the sum of Gibbs free energies of 3e and PhCH2CO2Et is thermodynamically more stable than the sum of those for 6e and EtOH (ΔG = −25.9 kcal mol−1).
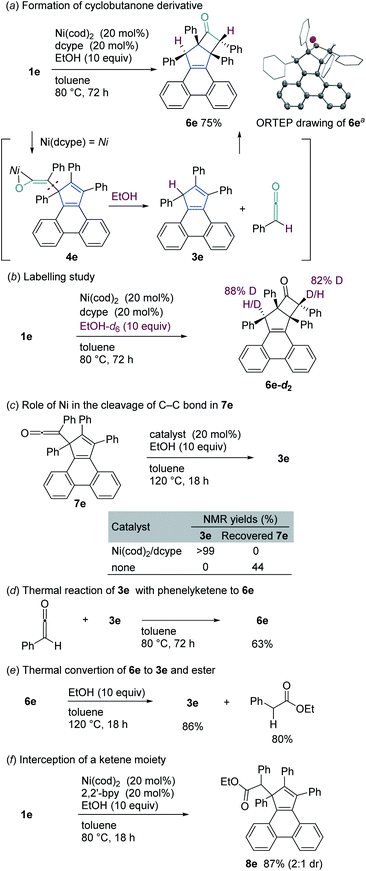 |
| | Scheme 3 Additional mechanistic studies. aORTEP drawing of 6e depicted at the 50% probability level. Hydrogen atoms were omitted for clarity. Phenyl and cyclohexyl groups were depicted as wireframe model 2,2′-bpy = 2,2′-bipyridine. | |
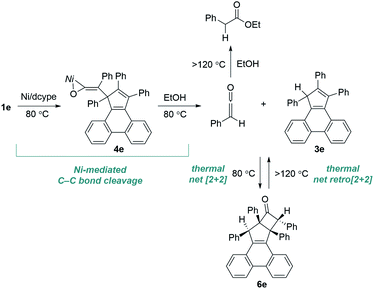 |
| | Scheme 4 Pathways involved in the conversion of tropone 1e to cyclopentadiene 3e. | |
Computational studies
Reaction mechanisms and ligand-controlled selectivity for both types of ring contraction reactions were also investigated by DFT calculations at the M06-2X/6-31G(d,p)-LanL2DZ level of theory (see ESI† in details). The mechanism for the oxidative addition of a C–C bond was initially investigated when IMesMe was used as a ligand. The coordination of 1e to Ni(IMesMe) forms Int I, in which the C1′–C2′ bond of a phenyl group at the α-position coordinates in a η2 manner, which is followed by the oxidative addition of a C1–C2 bond proceed via three-centred transition state TS I to give Int II. The activation barrier for this process is lower for 1e (11.7 kcal mol−1) than that for unreactive substrates such as 1c (28.2 kcal mol−1) and 1h (21.6 kcal mol−1) (Fig. 2a). The results of a distortion/interaction analysis24 indicated that the distortion energy (ΔE‡strain) of Ni–IMesMe and 1e for TS I is smaller compared with that for 1c or 1h (see Table S2 in ESI†). In contrast, the difference in the interaction energy (ΔE‡int) for these fragments for TS I is relatively small. The smaller ΔE‡strain of Ni–IMesMe and 1e indicates that 1e possesses a larger strain that originates from vdW repulsion between the bulky substituents compared to 1c or 1h, thereby leading to a lower activation barrier for this oxidative addition process. In contrast, when bidentate dcype is used as the ligand, oxidative addition occurs from the η2-(CO) coordinated Ni(0)-dcype intermediate (i.e., Int I′) via three-centred transition state TS I′ to generate INT II′. The activation barrier for this oxidative addition is higher than that for Ni–IMesMe (15.0 kcal mol−1) (Fig. 2b). The calculated activation barriers for oxidative addition of 1e are low for both IMesMe and dcype ligands, suggesting that the initial oxidative addition is not the rate-determining step of the decarbonylation reaction and high temperature is required for the dissociation of CO from Ni(0).
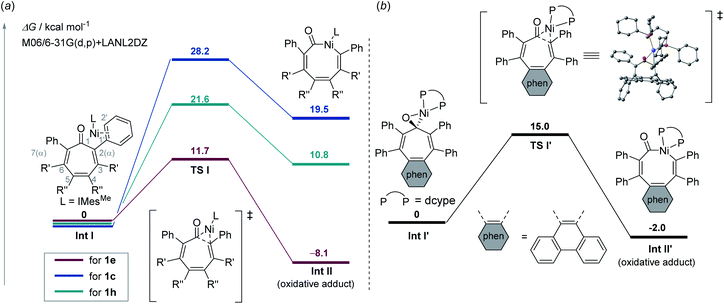 |
| | Fig. 2 (a) Substrate dependence on the oxidative addition of 1e (red), 1c (blue), and 1h (green) to Ni–IMesMe. For 1h, an α-phenyl analogue was used as a model for clarity. (b) Oxidative addition of 1e to Ni–dcype at the at the M06-2X/6-31G(d,p)-LanL2DZ level of theory. | |
The ligand-controlled selectivity was next examined by exploring the reaction pathways after the oxidative addition complexes Int II/Int II′ had been formed (Fig. 3). Using IMesMe as the ligand, Int II undergoes decarbonylation viaInt III, in which the C6–C7 double bond of the nickelacycle coordinates to the nickel centre, with a reasonable activation barrier of 10.2 kcal mol−1. The activation barrier for the competitive intramolecular insertion (ketene formation) process25 is higher by 3.9 kcal mol−1 compared to that for decarbonylation, thus rendering the decarbonylation pathway kinetically more favoured. When dcype is used as the ligand, Int II′ is converted to Int III′ by the coordination of an intramolecular alkene with one of the phosphorus atoms of dcype being dissociated. Although the difference in the activation barriers for the subsequent decarbonylation (i.e., the formation of Int IV′) and ketene formation (i.e., the formation of Int V′)25 steps is less than 1 kcal mol−1, the formation of Int V′ is exergonic, which explains the selective formation of cyclopentadiene in the case of a dcype ligand (Fig. 3).
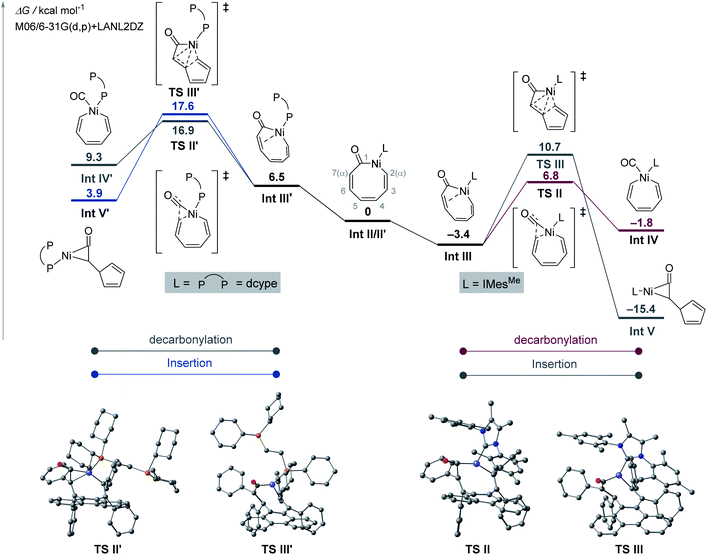 |
| | Fig. 3 Energy diagram for ligand-controlled selective ring contraction reactions of 1e at the at the M06-2X/6-31G(d,p)-LanL2DZ level of the theory. | |
Conclusions
In conclusion, we report on C–C bond cleavage reactions of tropone derivatives by nickel catalysis. When NHC is used as a ligand, the decarbonylation reaction proceeds catalytically to form a benzene ring (one-carbon ring contraction). In contrast, when dcype is used as the ligand, the tropone derivatives undergo a two-carbon ring contraction with the formation of cyclopentadiene derivatives. The addition of alcohol is essential for an efficient reaction, and the dissociated ketene fragment can be trapped by the cyclopentadiene moiety to generate polycyclic cyclobutanone derivatives. X-ray crystallography studies revealed that the formation of these compounds involves a nickel-ketene intermediate, which can be trapped by EtOH catalytically by changing the ligand from dcype to 2,2′-bypyridine. The judicious selection of the ligand and reaction temperature enables four different products, including three different ring systems, to be produced catalytically from a single substrate (Scheme 5).
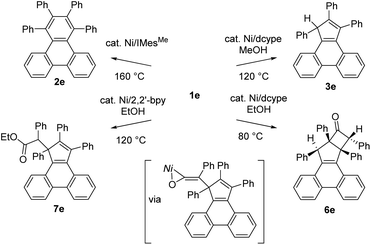 |
| | Scheme 5 Catalyst controlled-conversion of 1e to diverse ring systems. | |
Data availability
All experimental and characterization data are available in the ESI.† Crystallographic data for compounds c1, 1i, 4e, 5e, 6e, 8e, and Ni(CO)3(IMesMe) have been deposited in the Cambridge Crystallographic Data Centre under accession numbers CCDC 2115589–2115595.
Author contributions
T. K. and M. T. conceived the project. T. K. and K. S. performed the experimental work. T. K. and K. S. collected and analysed the spectroscopic data. T. K. and M. T. wrote the manuscript. All of the authors discussed the results and contributed to the preparation of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Grant-in-Aid for Early-Career Scientists (JP19K15564), Grant-in-Aid for Scientific Research (A) (JP21H04682) from MEXT, Japan and TOBE MAKI Scholarship Foundation (20-JA-013). We thank the Instrumental Analysis Center, Faculty of Engineering, Osaka University, for their assistance with HRMS.
Notes and references
- Selected reviews:
(a)
M. Murakami, Cleavage of Carbon—Carbon Single Bonds by Transition Metals, in Activation of Unreactive Bonds and Organic Synthesis, Springer-Verlag Berlin Heidelberg, 1999, pp. 97–129 Search PubMed
 ;
(b) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed ;
(c)
C–C Bond Activation, ed. G. Dong, Springer-Verlag Berlin Heidelberg, 2014 Search PubMed ;
(d)
Cleavage of Carbon-Carbon Single Bonds by Transition Metals, ed. M. Murakami and N. Chatani, Wiley-VCH Verlag GmbH & Co. KGaA, 2015 Search PubMed ;
(e) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed ;
(f) M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed ;
(g) P.-H. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340–1360 CrossRef CAS PubMed ;
(h) Z. Nairoukh, M. Cormier and I. Marek, Nat. Rev. Chem., 2017, 1, 0035 CrossRef ;
(i) F. Song, T. Gou, B.-Q. Wang and Z.-J. Shi, Chem. Soc. Rev., 2018, 47, 7078–7115 RSC ;
(j) L. Deng and G. Dong, Trends Chem., 2020, 2, 183–198 CrossRef CAS ;
(k) H. Lu, T.-Y. Yu, P. F. Xu and H. Wei, Chem. Rev., 2021, 121, 365–411 CrossRef CAS PubMed .
;
(b) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed ;
(c)
C–C Bond Activation, ed. G. Dong, Springer-Verlag Berlin Heidelberg, 2014 Search PubMed ;
(d)
Cleavage of Carbon-Carbon Single Bonds by Transition Metals, ed. M. Murakami and N. Chatani, Wiley-VCH Verlag GmbH & Co. KGaA, 2015 Search PubMed ;
(e) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed ;
(f) M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed ;
(g) P.-H. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340–1360 CrossRef CAS PubMed ;
(h) Z. Nairoukh, M. Cormier and I. Marek, Nat. Rev. Chem., 2017, 1, 0035 CrossRef ;
(i) F. Song, T. Gou, B.-Q. Wang and Z.-J. Shi, Chem. Soc. Rev., 2018, 47, 7078–7115 RSC ;
(j) L. Deng and G. Dong, Trends Chem., 2020, 2, 183–198 CrossRef CAS ;
(k) H. Lu, T.-Y. Yu, P. F. Xu and H. Wei, Chem. Rev., 2021, 121, 365–411 CrossRef CAS PubMed .
- Selected reviews:
(a) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed ;
(b) L. Jiao and Z.-X. Yu, J. Org. Chem., 2013, 78, 6842–6848 CrossRef CAS PubMed ;
(c) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740–7752 CrossRef CAS PubMed ;
(d) P.-H. Chen and G. Dong, Chem.–Eur. J., 2016, 22, 18290–18315 CrossRef CAS PubMed ;
(e) D. J. Mack and J. T. Njardarson, ACS Catal., 2013, 3, 272–286 CrossRef CAS ;
(f) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed ;
(g) R. Vicente, Chem. Rev., 2021, 121, 162–226 CrossRef CAS PubMed .
- Selected reviews:
(a) Y. J. Park, J.-W. Parka and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222–234 CrossRef CAS PubMed ;
(b) D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS PubMed ;
(c) Y. Xia and G. Dong, Nat. Rev. Chem., 2020, 4, 600–614 CrossRef CAS PubMed ; Selected examples:;
(d) M. Gozln, A. Weisman, Y. Ben-David and D. Milstein, Nature, 1993, 364, 699–701 CrossRef ;
(e) J. Zhu, J. Wang and G. Dong, Nat. Chem., 2019, 11, 45–51 CrossRef CAS PubMed ;
(f) S. Sakurai and M. Tobisu, Organometallics, 2019, 38, 2834–2838 CrossRef CAS ;
(g) J. Zhu, R. Zhang and G. Dong, Nat. Chem., 2021, 13, 836–842 CrossRef CAS PubMed .
-
(a) R. B. King and A. Efraty, J. Am. Chem. Soc., 1972, 94, 3773–3779 CrossRef CAS ;
(b) R. H. Crabtree, R. P. Dion, D. J. Gibboni, D. V. Mcgrath and E. M. Holt, J. Am. Chem. Soc., 1986, 108, 7222–7227 CrossRef CAS ;
(c) M. A. Halcrow, F. Urbanos and B. Chaudret, Organometallics, 1993, 12, 955–957 CrossRef CAS ;
(d) S. W. Youn, B. S. Kim and A. R. Jagdale, J. Am. Chem. Soc., 2012, 134, 11308–11311 CrossRef CAS PubMed ;
(e) G. Smits, B. Audic, M. D. Wodrich, C. Corminboeuf and N. Cramer, Chem. Sci., 2017, 8, 7174–7179 RSC ;
(f) Y. Xu, X. Qi, P. Zheng, C. C. Berti, P. Liu and G. Dong, Nature, 2019, 567, 373–378 CrossRef CAS PubMed ; Review: ;
(g) F. Hu, L. Wang, L. Xu and S.-S. Li, Org. Chem. Front., 2020, 7, 1570–1575 RSC .
- Selected reviews:
(a) M. D. R. Lutz and B. Morandi, Chem. Rev., 2021, 121, 300–326 CrossRef CAS PubMed ;
(b) K. Nogi and H. Yorimitsu, Chem. Rev., 2021, 121, 345–364 CrossRef CAS PubMed .
- Selected reviews:
(a) M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300–307 RSC ;
(b)
M. Tobisu, Reactions via Cleavage of Carbon–Carbon Bonds of Ketones and Nitriles, in Cleavage of Carbon-Carbon Single Bonds by Transition Metals, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 193–220 Search PubMed ;
(c) Y. Nakao, Chem. Rev., 2021, 121, 327–344 CrossRef CAS PubMed .
- M. Murakami, H. Amii and Y. Ito, Nature, 1994, 370, 540–541 CrossRef CAS .
- O. Daugulis and M. Brookhart, Organometallics, 2004, 23, 527–534 CrossRef CAS .
-
(a) T. Morioka, A. Nishizawa, T. Furukawa, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2017, 139, 1416–1419 CrossRef CAS PubMed ; Our group also reported on nickel-catalyzed decarbonylation of amides and acylsilanes via C–C bond cleavage:;
(b) T. Morioka, S. Nakatani, Y. Sakamoto, T. Kodama, S. Ogoshi, N. Chatani and M. Tobisu, Chem. Sci., 2019, 10, 6666–6671 RSC ;
(c) S. Nakatani, Y. Ito, S. Sakurai, T. Kodama and M. Tobisu, J. Org. Chem., 2020, 85, 7588–7594 CrossRef CAS PubMed .
- K. Kaneda, H. Azuma, M. Wayaku and S. Tehanishi, Chem. Lett., 1974, 3, 215–216 CrossRef .
-
(a) J. Blum, E. Oppenheimer and E. D. Bergmann, J. Am. Chem. Soc., 1967, 89, 2338–2341 CrossRef CAS ;
(b) S. Murahashi, T. Naota and N. Nakajima, J. Org. Chem., 1986, 51, 898–901 CrossRef CAS ;
(c) K. Nozaki, N. Sato and H. Takaya, J. Org. Chem., 1994, 59, 2679–2681 CrossRef CAS ;
(d) Y. Nishihara, Y. Inoue, M. Itazaki and K. Takagi, Org. Lett., 2005, 7, 2639–2641 CrossRef CAS PubMed ;
(e) Y. Kobayashi, H. Kamisaki, R. Yanada and Y. Takemoto, Org. Lett., 2006, 8, 2711–2713 CrossRef CAS PubMed ;
(f) Y. Nakao, Y. Hirata and T. Hiyama, J. Am. Chem. Soc., 2006, 128, 7420–7421 CrossRef CAS PubMed .
-
(a) A. Dermenci, R. E. Whittaker and G. Dong, Org. Lett., 2013, 15, 2242–2245 CrossRef CAS PubMed ;
(b) A. Dermenci, R. E. Whittaker, Y. Gao, F. A. Cruz, Z.-X. Yu and G. Dong, Chem. Sci., 2015, 6, 3201–3210 RSC .
- Selected examples:
(a) N. Chatani, Y. Ie, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 1999, 121, 8645–8646 CrossRef CAS ;
(b) Z.-Q. Lei, H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang, J. Sun and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 2690–2694 CrossRef CAS PubMed ;
(c) R. Zeng and G. Dong, J. Am. Chem. Soc., 2015, 137, 1408–1411 CrossRef CAS PubMed ;
(d) R. Zeng, P. H. Chen and G. Dong, ACS Catal., 2016, 6, 969–973 CrossRef CAS PubMed ;
(e) Z.-Q. Lei, F. Pan, H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang, Y.-X. Li, J. Sun and Z.-J. Shi, J. Am. Chem. Soc., 2015, 137, 5012–5020 CrossRef CAS PubMed ;
(f) C. Jiang, W.-Q. Wu, H. Lu, T.-Y. Yu, W.-H. Xu and H. Wei, Asian J. Org. Chem., 2019, 8, 1358–1362 CrossRef CAS ;
(g) C. Jiang, Z.-J. Zheng, T.-Y. Yu and H. Wei, Org. Biomol. Chem., 2018, 16, 7174–7177 RSC ;
(h) T.-T. Zhao, W.-H. Xu, Z.-J. Zheng, P.-F. Xu and H. Wei, J. Am. Chem. Soc., 2018, 140, 586–589 CrossRef CAS PubMed ;
(i) T.-Y. Yu, W.-H. Xu, H. Lu and H. Wei, Chem. Sci., 2020, 11, 12336–12340 RSC .
- Selected examples:
(a) C.-H. Jun and H. Lee, J. Am. Chem. Soc., 1999, 121, 880–881 CrossRef CAS ;
(b) Y. Xia, G. Lu, P. Liu and G. Dong, Nature, 2016, 539, 546–550 CrossRef CAS PubMed ;
(c) J. Zhong, W. Zhou, X. Yan, Y. Xia, H. Xiang and X. Zhou, Org. Lett., 2022, 24, 1372–1377 CrossRef CAS PubMed .
- Selected reviews on tropones, see:
(a) P. L. Pauson, Chem. Rev., 1955, 55, 9–136 CrossRef CAS ;
(b) F. Pietra, Chem. Rev., 1973, 73, 293–364 CrossRef CAS ;
(c) F. Pietra, Acc. Chem. Res., 1979, 12, 132–138 CrossRef CAS .
-
(a) T. Murai, T. Nakazawa and T. Shishido, Tetrahedron Lett., 1967, 8, 2465–2469 CrossRef ;
(b) T. Murai and T. Sato, Bull. Chem. Soc. Jpn., 1968, 41, 2819 CrossRef .
-
(a) T. Kodama, Y. Kawashima, Z. Deng and M. Tobisu, Inorg. Chem., 2021, 60, 4332–4336 CrossRef CAS PubMed ;
(b) T. Kodama, Y. Kawashima, K. Uchida, Z. Deng and M. Tobisu, J. Org. Chem., 2021, 86, 13800–13807 CrossRef CAS PubMed ; A similar Xray crystal structure of 1c was reported; see:;
(c) K. Ibata, H. Shimanouchi and Y. Sasada, Acta Cryst, 1977, B33, 1129–1138 CrossRef CAS .
- A similar ground state destabilization by vdW strain is frequently utilized in the C–N bond activation of amides. A leading review: G. Meng, J. Zhang and M. Szostak, Chem. Rev., 2021, 121, 12746–12783 CrossRef CAS PubMed .
-
(a) The structure of 4e was unambiguously determined by X-ray crystallography (Fig. S8 in ESI†);;
(b) Related Ni/ketene complexes: N. D. Staudaher, A. M. Arif and J. Louie, J. Am. Chem. Soc., 2016, 138, 14083–14091 CrossRef CAS PubMed .
-
(a) S. Yamabe, T. Dai, T. Minato, T. Machiguchi and T. Hasegawa, J. Am. Chem. Soc., 1996, 118, 6518–6519 CrossRef CAS ;
(b) T. Machiguchi, T. Hasegawa, A. Ishiwata, S. Terashima, S. Yamabe and T. Minato, J. Am. Chem. Soc., 1999, 121, 4771–4786 CrossRef CAS .
- Although the detailed mechanism responsible for this process is unclear, C−C bond cleavage is likely to be driven by aromatization (i.e., formation of cyclopentadienyl anion). See ref. 4a for a related reaction in this context..
- The overall process is a reductive C−C bond cleavage, in which two hydrogen atoms are incorporated. The results of labelling studies confirmed that an external alcohol serves as the hydride source (Scheme 3b). β-Hydrogen elimination of metal–alkoxide species is presumably involved. For example, see: K. Yasui, M. Higashino, N. Chatani and M. Tobisu, Synlett, 2017, 28, 2569–2572 CrossRef CAS .
- The thermal reaction of PhCHCO with EtOH in toluene was slower (19% at 80 °C; 59% at 120 °C for 72 h) than that with the cyclopentadiene 3e (see ESI†), which is also consistent with the mechanistic scheme shown in Scheme 4.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed .
- The isomerization to a cyclopentadiene derivative is a concerted process that proceeds via TSIII/III′ in which C2 and C6 carbons are located close to facilitate the bond formation between these carbons.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Kanako
Saito
a and
Mamoru
Tobisu
ab,
Kanako
Saito
a and
Mamoru
Tobisu