
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sustainable approach for the synthesis of recyclable cyclic CO2-based polycarbonates†
Xi
Liao
a,
Feng-Chao
Cui
c,
Jiang-Hua
He
a,
Wei-Min
Ren
 b,
Xiao-Bing
Lu
b and
Yue-Tao
Zhang
*a
b,
Xiao-Bing
Lu
b and
Yue-Tao
Zhang
*a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, 130012, Changchun, P. R. China. E-mail: ytzhang2009@jlu.edu.cn
bState Key Laboratory of Fine Chemicals, Dalian University of Technology, 116024, Dalian, P. R. China
cFaculty of Chemistry, Northeast Normal University, 130024, Changchun, P. R. China
First published on 22nd April 2022
Abstract
It is highly desirable to reduce the environmental pollution related to the disposal of end-of-life plastics. Polycarbonates derived from the copolymerization of CO2 and epoxides have attracted much attention since they can enable CO2-fixation and furnish biorenewable and degradable polymeric materials. So far, only linear CO2-based polycarbonates have been reported and typically degraded to cyclic carbonates. Here we synthesize a homogeneous dinuclear methyl zinc catalyst ((BDI-ZnMe)2, 1) to rapidly copolymerize meso-CHO and CO2 into poly(cyclohexene carbonate) (PCHC) with an unprecedentedly cyclic structure. Moreover, in the presence of trace amounts of water, a heterogeneous multi-nuclear zinc catalyst ((BDI-(ZnMe2·xH2O))n, 2) is prepared and shows up to 99% selectivity towards the degradation of PCHC back to meso-CHO and CO2. This strategy not only achieves the first case of cyclic CO2-based polycarbonate but also realizes the complete chemical recycling of PCHC back to its monomers, representing closed-loop recycling of CO2-based polycarbonates.
Introduction
The rapid development of plastics is a double-edged sword: on the one hand, they have penetrated into every aspect of our daily life because of their useful and versatile applications.1,2 On the other hand, with the increasing production of plastics worldwide, properly disposing of end-of-life plastics has become a very challenging task facing our society,3,4 since plastics limit landfills, produce microplastic issues during degradation that seriously contaminate waterways and aquifers, or generate toxic gases causing air pollution during incineration of plastic wastes, thus significantly intensifying environmental pollution.4–9 In order to reduce or prevent this “white pollution”,10–14 various approaches have been developed such as synthesizing bio-derived plastics from renewable monomers15–20 or maximizing the degradation value by transforming plastic waste into value-added chemicals.21–25 Among them, chemical recycling of polymers back to monomers (CRM) is an attractive strategy showing great potential in solving issues related to plastic waste because it can completely transform polymeric waste back into monomers for virgin polymer reproduction, thus closing the loop and realizing the ideal circular polymer economy.26–30The utilization of CO2, one of the most abundant C1 carbon sources, as co-monomers to be copolymerized with epoxides,31–39 has attracted intense attention since Inoue's seminal work,40 which will not only achieve CO2-fixation, thus reducing the greenhouse gas level in the atmosphere, but also furnish biorenewable and degradable polymeric materials.41–44 It should be noted that ring-opening copolymerization of epoxides and CO2 is typically initiated by either intrinsic metal-bonded initiators45–50 or additional initiators in binary systems.51–53 Therefore, linear polycarbonates capped by initiation species chain-ends are obtained in general. As we know, cyclic polymers possessing a ring-like structure exhibit a unique set of properties compared with their linear analogues, thus attracting particular interest in the precision polymer synthesis.54–57 However, there have been no reports on the synthesis of cyclic, CO2-based polycarbonates yet. As far as the degradation of polycarbonates is concerned, cyclic carbonate was the most reported degradation product in comparison with epoxide and carbon dioxide co-monomers.58,59 Polycarbonate derived from common monomers (i.e. poly(cyclohexene carbonate) (PCHC) based on meso-cyclohexene oxide (meso-CHO)) typically produced trans-CHC rather than meso-CHO, probably because the energy barrier for meso-CHO formation is 1.0 kcal mol−1 higher than that for trans-CHC.58 So far, only a few examples of polycarbonate based on epoxide monomers with large steric hindrance were reported to have been completely recycled back to their epoxide monomers.60,61 So far, we are unaware of any complete chemical recycling of PCHC back to meso-CHO yet. Here, we designed and synthesized a dinuclear methyl zinc catalyst ((BDI-ZnMe)2, 1) by reacting a BDI ligand with ZnMe2 (Fig. 1). Without the initiator, catalyst 1 exhibits high activity and chem-/regioselectivity towards the copolymerization of meso-CHO and CO2, furnishing polycarbonates with an unprecedented cyclic topology. Both the Mark–Houwink plot and MALDI-TOF-MS spectra confirmed that these cyclic polycarbonates possessed an “end group” of two CO2 molecules. Moreover, a heterogeneous catalyst ((BDI(ZnMe2·xH2O))n, 2) can be prepared from the same reaction in the presence of trace amounts of water, which can rapidly degrade PCHC to the meso-CHO monomer and CO2 comonomer with up to 99% high selectivity, which represented the perfect CRM example from PCHC to meso-CHO.
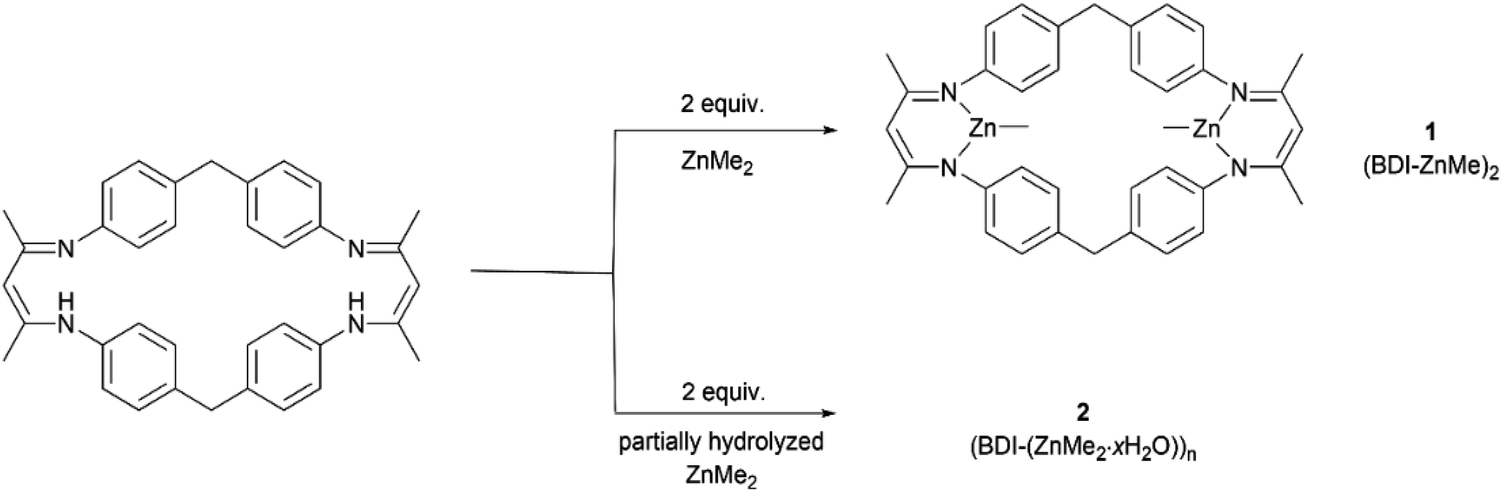 | ||
| Fig. 1 Preparation of catalyst 1 and 2 from the reaction of the BDI-ligand with ZnMe2. | ||
Results and discussion
Synthesis of catalyst 1 and its copolymerization activity
The β-diiminate ligand was prepared according to the literature,62 which was employed to react with pure ZnMe2 for the synthesis of (BDI-ZnMe)2, catalyst 1. It was structurally characterized by means of nuclear magnetic resonance (NMR) (Fig. S1†) and single-crystal X-ray diffraction (Fig. 2). Catalyst 1 is a symmetric structure bridged with two diphenyl methyl groups, while two β-diiminate methyl zinc planes were positioned face to face by bidentate zinc moieties such that both zinc centres bearing methyl groups shared the steric hindrance and there was an axial cavity between the zinc planes with a Zn⋯Zn distance of 6.507 Å. | ||
| Fig. 2 Crystal structure of catalyst 1 (hydrogen atoms are omitted for clarity and ellipsoids are set at 30% probability). | ||
Without requirement of initiators, catalyst 1 can rapidly polymerize 800 equiv. of meso-CHO into PCHC at 90 °C in 1 h under 40 bar CO2, thus affording copolymers with a turnover frequency (TOF) over 790 h−1 (entry 1, Table 1). This result contradicts previous literature findings that alkyl zinc complexes are inactive for epoxide/CO2 copolymerization in general and often act as precursors for alkoxide/acetate zinc derivatives.45,63,64 Moreover, we found that over 99% of the produced polymers are polycarbonates with a narrow molecular weight distribution (MWD) and there was no observation of polyether or cyclic carbonate (Fig. S2†). It is also noted that reducing CO2 pressure from 40 to 30, 20, and 10 bar, respectively, did not show a noticeable impact on the polymerization activity of catalyst 1, highly selectively producing polycarbonates with a similar molecular weight, narrow MWD and comparable TOF value (∼800 h−1) (entries 2 to 4 vs. 1, Table 1). Further reducing CO2 pressure to 1 bar led to the formation of both cis-cyclic carbonate and ether linkages, indicating that sufficient CO2 is required for high chemo- and region-selectivity (entry 5, Table 1 and Fig. S3†). The control experiment revealed that catalyst 1 is inactive for homopolymerization of meso-CHO up to 120 h (entry 6, Table 1), suggesting that CO2 is essentially important for the copolymerization of meso-CHO and CO2. We also evaluated the influence of catalyst loading on polymerization and were very glad to see that all polymerizations can proceed smoothly under 30 bar CO2 pressure (entries 7–10, Table 1). The corresponding TOF value increased from 396 to 792, 1164 and 1328 h−1 when the meso-CHO/1 ratio was changed from 400/1 to 800/1, 1200/1, and 1600/1, respectively (entries 7, 2, 8 and 9, Table 1). Even with a 2000/1 ratio, over 97% of the monomer can still be converted to PCHC with 99% selectivity in 2 h (entry 10, Table 1). Further investigation indicated that reaction temperature plays an important role in affecting polymerization activity. At room temperature (RT), only ∼30% of meso-CHO was converted to PCHC in 2 h for copolymerization performed in an 800/1 meso-CHO/1 ratio (entry 11, Table 1). In sharp contrast, it only took 0.5 h to reach 80% monomer conversion for polymerization heating at 110 °C, giving rise to a high TOF value of 3200 h−1 (entry 12, Table 1). It is worth noting that the produced PCHCs all exhibited comparable molecular weights (i.e. 400/1 31.4 kDa; 800/1 28.7 kDa; 1200/1 32.7 kDa, 1600/1 30.6 kDa, and 2000/1 33.5 kDa) regardless of the monomer feed ratios, while the dispersities remained narrow (Đ as low as 1.04) (Fig. S6†), thus strongly implying the occurrence of chain transfer during polymerization. This result aroused our interest since we did not add any chain transfer reagents to the system at all.
| Entry | [M]/[C] ratio | PCO2 (bar) | T (°C) | t (h) | Conv.b (%) | TOFb (h−1) | Polycarbonateb (%) | M w (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|---|
| a Carried out in toluene ([meso-CHO] = 2.0 M) in a 20 mL autoclave. b Conversion of meso-CHO and selectivity for polycarbonates determined by 1H NMR spectroscopy. Turnover frequency (TOF) = mol of product/mol of catalyst per hour. c Weight average molecular weight (Mw) was measured by GPC using a light scattering detector in THF. d [meso-CHO] = 1.0 M. e Using the mono-nuclear BDI-ZnMe complex as catalyst (Fig. S20). | |||||||||
| 1 | 800/1 | 40 | 90 | 1 | 99 | 792 | >99 | 28.6 | 1.11 |
| 2 | 800/1 | 30 | 90 | 1 | 99 | 792 | >99 | 28.7 | 1.09 |
| 3 | 800/1 | 20 | 90 | 1 | 98 | 784 | >99 | 27.9 | 1.11 |
| 4 | 800/1 | 10 | 90 | 1 | 99 | 792 | >99 | 32.2 | 1.02 |
| 5 | 800/1 | 1 | 90 | 1 | 18 | 144 | 67 | — | — |
| 6d | 400/1 | — | 90 | 120 | 0 | — | — | — | — |
| 7 | 400/1 | 30 | 90 | 1 | 99 | 396 | >99 | 31.4 | 1.04 |
| 8 | 1200/1 | 30 | 90 | 1 | 97 | 1164 | >99 | 32.7 | 1.06 |
| 9 | 1600/1 | 30 | 90 | 1 | 83 | 1328 | >99 | 30.6 | 1.11 |
| 10 | 2000/1 | 30 | 90 | 2 | 97 | 970 | >99 | 33.5 | 1.17 |
| 11 | 800/1 | 30 | 25 | 2 | 30 | 120 | >99 | ||
| 12 | 2000/1 | 20 | 110 | 0.5 | 80 | 3200 | >99 | 28.7 | 1.11 |
| 13 | 2000/1 | 20 | 110 | 1 | 95 | 1900 | >99 | 32.8 | 1.09 |
| 14e | 400/1 | 30 | 90 | 2 | 0 | — | — | — | — |
Characterization of cyclic polycarbonates produced by catalyst 1
The dinuclear zinc catalyst 1 not only exhibits unprecedentedly high activity and chemo/regio-selectivity towards meso-CHO/CO2 copolymerization in the absence of initiator, but also produces polycarbonates with a similar molecular weight (∼30 kDa). These intriguing features prompted us to detect more structural details about the produced polymers. At first, we analyzed the low molecular weight PCHCs produced by catalyst 1 with the matrix-assisted laser desorption/ionization time of flight mass spectroscopy (MALDI-TOF MS). As shown in Fig. 3A and S7,† only one series of molecular ion repeating in intervals of 142, corresponding to the sum of a repeat unit of meso-CHO and CO2 is clearly observed. An intercept of 127 obtained from the plot of m/z values of this series vs. the number of repeat units was commensurate with the sum of K+ and an unknown species with a molecular weight of 88. Obtaining such an unusual chain-end group ruled out that the polymerization was initiated by the following common species: (i) water; (ii) 1,2-cyclohexanediol generated in the early stage of polymerization; or (iii) meso-CHO rearrangement resulting in cyclopentyl, cyclohexyl, or cyclohexenyl, which excluded the corresponding chain transfer reactions related to these species. Moreover, the MALDI-TOF MS spectrum of the polymers produced by using BnOH as the initiator (Fig. S8†) also revealed that even though we observed a small portion of PCHC capped by BnOH, the majority of PCHC still possessed an end group with a molecular weight of 88, thus manifesting that the polymers obtained with a similar molecular weight under different conditions were not produced from the chain-transfer reaction initiated by BnOH.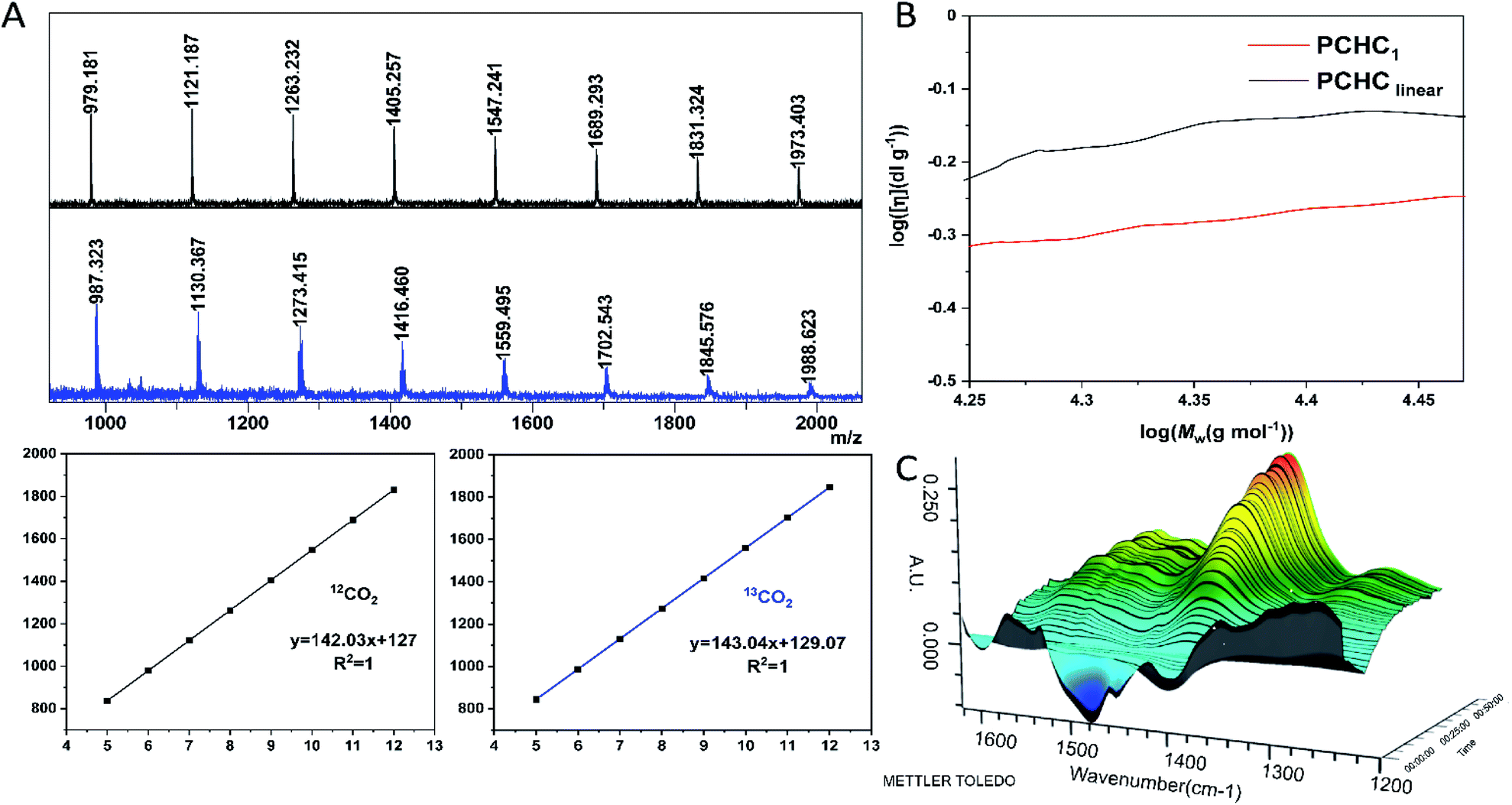 | ||
| Fig. 3 Characterization of the end-group and cyclic structure and in situ FTIR study of reaction between 1 and CO2. (A) The MALDI-TOF mass spectra of PCHC produced by 1 using 12CO2 (black) and 13CO2 (blue), respectively. CF3COOK was used as the ionizing agent. (B) Double logarithm (Mark–Houwink) plots of intrinsic viscosity [η] versus weight-average molecular weight of the PCHC produced by 1 (red line, entry 9, Table 1) and linear counterparts67 from the literature (black line). (C) Three-dimensional stack plot of the IR spectra during the reaction of CO2 and 1 in toluene at 90 °C, 40 bar pressure. | ||
Based on the above-mentioned results and the fact that the molecular weight for one molecule of CO2 is 44, we speculated that the unknown chain-end group with a molecular weight of 88 might be composed of two molecules of CO2. However, there have been no reports on the production of di(tri)carbonate linked polycarbonates so far, since it is believed to be enthalpically disfavored to consecutively insert two molecules of CO2 into polymer chains.32,65,66 To validate our assumption, we synthesized PCHC based on 13C-labeled CO2 and analyzed its MALDI-TOF MS spectrum (Fig. 3A). It turned out that the molecular weight of the repeat unit increased from 142 to 143. Meanwhile the molecular weight of the chain-end group changed from 88 to 90, indicating the formation of the chain-end group composed of two 13CO2 molecules. The 13C NMR spectrum of the 13CO2-labeled PCHC exhibited new peaks at 154.2 ppm and 154.4 ppm (Fig. S9 and S10†), which might be attributed to the tricarbonate linkage. These results clearly confirmed that the chain-end group is generated by the continuous insertion of two molecules of CO2. On the basis of the above-described results, it can be easily inferred that the produced polymers have a cyclic structure rather than a linear chain, because it would be unstable to consecutively insert two CO2 molecules into the end of a linear polymer chain, plus the molecular weight cannot be 88, since hydrogen or hydroxyl chain-end groups should be observed. To further validate our assumption, we heated the cyclic PCHC in toluene at 150 °C for 30 min. MALDI-TOF MS analysis did not show any difference between the low MW PCHC before and after heating (Fig. S11†), indicating that the cyclic PCHC is very stable. Next, in order to cleave such a tricarbonate linkage, low MW PCHC reacted with benzyl amine in DCM at room temperature for 30 min. The MALDI-TOF MS spectrum of the resulting products consisted of two series of mass ions. Beside the major series attributed to the cyclic PCHC, the minor series shows a similar molecular weight but capped by a chain end group with a molecular weight of 116 (155 = 116 + 39 (K+)), which corresponds to a linear polymer chain HO–(CHO–CO2)n–CHO–H resulting from the cleavage and release of tricarbonate linkage from the cyclic PCHC (Fig. S12†).
To the best of our knowledge, this is the first example of sequential carbonate linkage reported for a CO2-based polycarbonate. Furthermore, the formation of cyclic polymers would also help to explain that the produced polycarbonates all possessed a similar molecular weight since the ring-closure is a kind of chain-transfer reaction.
To confirm the formation of cyclic PCHC, we compared the intrinsic viscosity of the PCHC sample produced by catalyst 1 ([η]1) with that of its linear analogue with a comparable molecular weight ([η]linear).67 As shown in the Mark–Houwink plot (Fig. 3B), [η]1 is obviously lower than [η]linear ([η]1/[η]linear ratio ∼ 0.7). This result provides strong evidence to support the fact that the sample of PCHCs produced by catalyst 1 are cyclic polymers,68 which also further explained why the molecular weight of all the polymers are maintained around 30 kDa, regardless of the monomer feed ratio. That is to say, once reaching ∼30 kDa (∼200 repeating units) during propagation, the polymer chain would be closed to form a cyclic polymer while catalyst 1 was released from the polymer chain and entered into the next catalytic run. The fact that the initiation efficiency was greater than 100% suggested that more than one cyclic polymer chain is produced per catalyst molecule. This can be validated by the control experiment performed by sequential addition of two batches of 800 equiv. of meso-CHO. After complete consumption of the first batch of 800 equiv. meso-CHO under 20 bar CO2 and 90 °C in 1 h, a viscous aliquot was taken out for intrinsic viscosity measurement. Next, another 800 equiv. of meso-CHO was added to the above polymerization mixture and reacted for another 1.5 h to ensure full monomer conversion. GPC analyses revealed that there are no obvious molecular weight differences between the PCHCs produced by separate consumption of the first and second batch of 800 equiv. monomers, suggesting the formation of cyclic polymers (vide infra). The corresponding Mark–Houwink plots of both samples further confirmed the produced cyclic PCHCs have the same ring size (Fig. S13†). Considering that the repeat units are meso-CHO and CO2 plus the chain-end group is composed of two CO2 molecules, we presumed that after consecutive insertion of two CO2 molecules, the resultant dicarbonate anion would backbite the first carbonate in the polymer chain to close the ring and generate the cyclic polycarbonate containing a tricarbonate linkage (Fig. 4). The cyclic PCHC was amorphous showing a glass transition temperature of 113 °C and a decomposition temperature of 281 °C (Fig. S14 and S15†).
 | ||
| Fig. 4 Cyclic polycarbonates produced by catalyst 1. | ||
Mechanistic investigation
To gain more insights into the polymerization mechanism, we performed a series of NMR reactions and found that there are no obvious changes in chemical shifts between the 1H NMR spectra obtained before or after mixing catalyst 1 with meso-CHO at either RT for 24 h or 120 °C for 40 min (Fig. S16†), suggesting that catalyst 1 is incapable of initiating the polymerization of meso-CHO. Moreover, NMR spectroscopy also revealed that neither catalyst 1 reacted with CO2 under 1 bar at RT or under 20 bar at 90 °C, since all chemical shifts including the peak attributed to the most active zinc-methyl (−1.48 ppm) remained unchanged in 24 h (Fig. S17 and S18†). However, in situ FTIR studies revealed that in sharp contrast to the absorbance peak exhibited at 2430 cm−1 by CO2 dissolved in toluene (Fig. S19†), the reaction of catalyst 1 and CO2 carried out at 90 °C under 40 bar CO2 pressure showed the appearance of a gradually increased new νC–O stretch vibration peak69,70 at 1400 cm−1, indicating the weak activation of CO2 by catalyst 1 under high pressure (Fig. 3C). No new carbonyl formed but the free CO2 molecule was just restricted in the cavity of two zinc centres, causing the two oxygen atoms to become attached to the adjacent zinc atoms. Interestingly, in the presence of catalyst 1, the simultaneous addition of both meso-CHO and CO2 immediately initiated the copolymerization to produce PCHC. NMR spectroscopy revealed that catalyst 1 remained unchanged before and after copolymerization (Fig. S20†), indicating that the catalyst is released from the polymers after the ring-closure. These results can also be verified by successful sequential polymerization of the second feed of meso-CHO into new cyclic polymers by catalyst 1 (vide supra). In combination with control experiments where the mononuclear zinc complex (Fig. S21†) is inactive for meso-CHO/CO2 copolymerization under the same conditions (entry 14, Table 1), the dinuclear zinc catalyst 1 has demonstrated its essential importance on catalytic performance through the intramolecular cooperation effects between two zinc centres and meso-CHO or CO2.The above-mentioned experimental details, NMR reactions and in situ FTIR study coupled with the structural characterization of both catalyst 1 and polymers led to a proposed polymerization mechanism for meso-CHO/CO2 copolymerization by catalyst 1 (Fig. 5). With each oxygen weakly attached by the zinc centre, one CO2 molecule would be trapped while keeping its linear geometry still. The intramolecular cooperation effects between the dinuclear zinc catalyst 1 and meso-CHO or CO2 would enable two zinc centres to individually activate both monomers, respectively. The nucleophilic attack of the epoxide by the weakly activated CO2 initiated the ring-opening polymerization and produced an alkoxide. The metal alkoxide species subsequently underwent CO2 insertion to afford metal carbonate, which ring-opened another molecule of weak coordinated meso-CHO. The continuously alternative copolymerization of meso-CHO and CO2 led to the propagation. Once the molecular weight of the polycarbonates reached a certain level (ca. 30 kDa),55 the restricted CO2 between the two zinc centres would bridge the nucleophilic attack of the CO2 on the chain end by the last inserted CO2 on the propagation site, such that the polymer chain closed to form a cyclic polymer containing a tricarbonate linkage, while catalyst 1 was released from the polymer chain for the next catalytic cycle. Notably, the copolymerization can be ceased by releasing CO2 at a relatively low monomer conversion, which led to chain termination predominantly since the chain propagation was significantly suppressed as a result of the early release of CO2. Through such ways, we are able to prepare low-MW cyclic PCHC for MALDI-TOF MS analysis.
 | ||
| Fig. 5 The proposed copolymerization mechanism by catalyst 1. | ||
Synthesis of complex 2 and its degradation activity
During preparation of catalyst 1 from the reaction of the BDI ligand and ZnMe2, we observed the formation of a dark yellow precipitate (heterogeneous catalyst 2). In contrast to the literature findings that PCHC tends to be transformed into the thermodynamically favored cyclic carbonate rather than epoxide monomer, heterogeneous catalyst 2 can convert PCHC back to meso-CHO predominantly, which promoted us to further investigate the depolymerization of PCHC by catalyst 2 in detail. We noticed that the employment of ZnMe2 (1.0 M in toluene) stored for a long time would lead to the production of more dark yellow precipitates, indicating that heterogeneous catalyst 2 might be generated from the partial hydrolysis of ZnMe2. Therefore, by adjusting the amounts of water (0.02 to 0.5 equiv. relative to ZnMe2), we prepared a series of catalyst 2 ((BDI-(ZnMe2·xH2O))n, where x is the quantity of water relative to ZnMe2) with different hydrolysis degrees. The more water, the higher the yield of catalyst 2. Solid state NMR spectroscopy revealed that heterogeneous catalyst 2 is composed of the BDI framework (Fig. S22†) and the signals between 0 and 10 ppm are similar to that of catalyst 1, except the signals attributed to the methyl group (−1.90 ppm) on the zinc centre are partially replaced, thus indicating that catalyst 2 is multinuclear aggregates containing the BDI framework generated from the partial hydrolysis of the methyl group on the zinc centres.As shown in Table 2, the hydrolysis degree of ZnMe2 imposed an important impact on the degradation activity of catalyst 2. For degradation of PCHC performed in 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 repeat units (r.u.)/2 ratio, the catalysts prepared from the addition of 0.02 to 0.1 equiv. of water to ZnMe2 all exhibited comparable activity, furnishing meso-CHO in 99% selectivity. It only took 9 h for (BDI-(ZnMe2·0.1H2O))n to fully degrade PCHC to meso-CHO (Fig. S23†). After increasing the amount of water to 0.2 equiv., a longer reaction time was required and trace amounts of trans-CHC product was observed. Further increasing to 0.5 equiv. of water drastically slowed down the degradation; only 40% of PCHC was recycled back to meso-CHO in 15 h. Therefore, (BDI-(ZnMe2·0.1H2O))n was selected to screen the reaction parameters for PCHC depolymerization. For depolymerization performed in a 50:1 (r.u.)/2 ratio, PCHC can be near quantitatively converted back to meso-CHO in 20 h, whereas for depolymerization performed in a 100:1 (r.u.)/2 ratio, over 97% of PCHC was transformed into meso-CHO and trans-PCHC in 78% and 15% selectivity in 28 h. All these results demonstrated the high degradation activity and selectivity of catalyst 2 in the recycling of PCHC back to meso-CHO (Fig. 6). The remaining methyl group on the zinc centre is vital to the meso-CHO selectivity, while the synergistic action of the multinuclear zinc centres cannot be neglected as well. The high degradation activity and selectivity exhibited by catalyst 2 provides the possibility for complete chemical recycling of PCHC type plastics back to monomers to realize an ideal circular polymer economy.
:1 repeat units (r.u.)/2 ratio, the catalysts prepared from the addition of 0.02 to 0.1 equiv. of water to ZnMe2 all exhibited comparable activity, furnishing meso-CHO in 99% selectivity. It only took 9 h for (BDI-(ZnMe2·0.1H2O))n to fully degrade PCHC to meso-CHO (Fig. S23†). After increasing the amount of water to 0.2 equiv., a longer reaction time was required and trace amounts of trans-CHC product was observed. Further increasing to 0.5 equiv. of water drastically slowed down the degradation; only 40% of PCHC was recycled back to meso-CHO in 15 h. Therefore, (BDI-(ZnMe2·0.1H2O))n was selected to screen the reaction parameters for PCHC depolymerization. For depolymerization performed in a 50:1 (r.u.)/2 ratio, PCHC can be near quantitatively converted back to meso-CHO in 20 h, whereas for depolymerization performed in a 100:1 (r.u.)/2 ratio, over 97% of PCHC was transformed into meso-CHO and trans-PCHC in 78% and 15% selectivity in 28 h. All these results demonstrated the high degradation activity and selectivity of catalyst 2 in the recycling of PCHC back to meso-CHO (Fig. 6). The remaining methyl group on the zinc centre is vital to the meso-CHO selectivity, while the synergistic action of the multinuclear zinc centres cannot be neglected as well. The high degradation activity and selectivity exhibited by catalyst 2 provides the possibility for complete chemical recycling of PCHC type plastics back to monomers to realize an ideal circular polymer economy.
| Entry | Catalyst | Ratio | t (h) | PCHCb (%) | trans-CHCb (%) | PCHOb (%) | meso-CHOb (%) |
|---|---|---|---|---|---|---|---|
| a Carried out in toluene at 150 °C ([PCHC] = 7.1 mg mL−1). b Determined by comparison of the integrals of signals arising from the methylene protons in the 1H NMR spectra of reaction mixture aliquots without further purification due to PCHC (δ = 4.65 ppm), cis-CHC (δ = 4.63 ppm) and trans-CHC (δ = 4.00 ppm) against meso-CHO (δ = 3.20 ppm). | |||||||
| 1 | (BDI-(ZnMe2·0.02H2O))n | 25/1 | 15 | 0 | 0 | 0 | >99 |
| 2 | (BDI-(ZnMe2·0.04H2O))n | 25/1 | 15 | 0 | 0 | 0 | >99 |
| 3 | (BDI-(ZnMe2·0.08H2O))n | 25/1 | 15 | 0 | 0 | 0 | >99 |
| 4 | (BDI-(ZnMe2·0.1H2O))n | 25/1 | 9 | 0 | 0 | 0 | >99 |
| 5 | (BDI-(ZnMe2·0.2H2O))n | 25/1 | 15 | 0 | Trace | 0 | >99 |
| 6 | (BDI-(ZnMe2·0.5H2O))n | 25/1 | 15 | 60 | 0 | 0 | 40 |
| 7 | (BDI-(ZnMe2·0.1H2O))n | 50/1 | 20 | 0 | Trace | 0 | >99 |
| 8 | (BDI-(ZnMe2·0.1H2O))n | 100/1 | 28 | 7 | 15 | 0 | 78 |
 | ||
| Fig. 6 Recycling cyclic PCHC back to monomers by catalyst 2. | ||
Conclusions
In summary, we synthesized a homogeneous, zinc catalyst 1 (BDI-ZnMe)2 from the reaction of a BDI ligand and ZnMe2 and employed it to catalyze the highly efficient copolymerization of meso-CHO and CO2, furnishing PCHCs with comparable molecular weights (ca. 30 kDa) and narrow molecular weight distributions (Đ as low as 1.02). Both the MALDI-TOF MS spectra and Mark–Houwink plot confirmed the formation of the first example of cyclic PCHC containing a tricarbonate linkage. Systematic mechanistic studies including experimental details, NMR, in situ FTIR, and structural characterization of catalyst 1 and polymers led to a proposed polymerization mechanism for meso-CHO/CO2 copolymerization by catalyst 1. By adjusting the amount of water, we prepared a series of multinuclear zinc catalyst 2 containing a BDI framework generated from the partial hydrolysis of the methyl group on the zinc centres and found that catalyst 2 exhibited high activity and selectivity towards the degradation of PCHC to meso-CHO and CO2. To the best of our knowledge, it represents the perfect example of CRM of PCHC, thus providing the possibility to realize ideal circular polymer economy.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 22071077, 21774042, 21871107, and 21975102).Notes and references
- A. L. Andrady and M. A. Neal, Philos. Trans. R. Soc., B, 2009, 364, 1977–1984 CrossRef CAS PubMed.
- L. Shen, E. Worrell and M. Patel, Biofuels, Bioprod. Biorefin., 2010, 4, 25–40 CrossRef CAS.
- E. M. Foundation, The New Plastics Economy: Rethinking the future of plastics & catalysing action, 2017, https://www.ellenmacarthurfoundation.org/publications/the-new-plastics-economy-rethinking-the-future-of-plastics-catalysing-action Search PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- P. Europe, Plastics – the Facts 2020, an analysis of European plastics production, demand and waste data, 2020, https://www.plasticseurope.org/en/resources/publications/4312-plastics-facts-2020 Search PubMed.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- L. J. J. Meijer, T. van Emmerik, R. van der Ent, C. Schmidt and L. Lebreton, Sci. Adv., 2021, 7, eaaz5803 CrossRef PubMed.
- B. Nguyen, D. Claveau-Mallet, L. M. Hernandez, E. G. Xu, J. M. Farner and N. Tufenkji, Acc. Chem. Res., 2019, 52, 858–866 CrossRef CAS PubMed.
- K. Zhang, A. H. Hamidian, A. Tubic, Y. Zhang, J. K. H. Fang, C. Wu and P. K. S. Lam, Environ. Pollut., 2021, 274, 116554 CrossRef CAS PubMed.
- J. C. Worch and A. P. Dove, ACS Macro Lett., 2020, 9, 1494–1506 CrossRef CAS.
- X. Tang and E. Y. X. Chen, Chem, 2019, 5, 284–312 CAS.
- K. Khatami, M. Perez-Zabaleta, I. Owusu-Agyeman and Z. Cetecioglu, Nucl. Chem. Waste Manage., 2021, 119, 374–388 CrossRef CAS PubMed.
- P. K. Rai, J. Lee, R. J. C. Brown and K.-H. Kim, J. Cleaner Prod., 2021, 291, 125240 CrossRef CAS.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- F. B. Catia Bastioli, in Handbook of Biodegradable Polymers, ed. B. Catia, De Gruyter, 2020, ch. 6, pp. 147–182 Search PubMed.
- L. Hu, J. He, Y. Zhang and E. Y. X. Chen, Macromolecules, 2018, 51, 1296–1307 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- J. F. Wilson and E. Y. X. Chen, ACS Sustainable Chem. Eng., 2019, 7, 7035–7046 CrossRef CAS.
- A. Pellis, M. Malinconico, A. Guarneri and L. Gardossi, New Biotechnol., 2021, 60, 146–158 CrossRef CAS PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- A. Tennakoon, X. Wu, A. L. Paterson, S. Patnaik, Y. Pei, A. M. LaPointe, S. C. Ammal, R. A. Hackler, A. Heyden, I. I. Slowing, G. W. Coates, M. Delferro, B. Peters, W. Huang, A. D. Sadow and F. A. Perras, Nat. Catal., 2020, 3, 893–901 CrossRef CAS.
- J. M. Millican and S. Agarwal, Macromolecules, 2021, 54, 4455–4469 CrossRef CAS.
- X. Jie, W. Li, D. Slocombe, Y. Gao, I. Banerjee, S. Gonzalez-Cortes, B. Yao, H. AlMegren, S. Alshihri, J. Dilworth, J. Thomas, T. Xiao and P. Edwards, Nat. Catal., 2020, 3, 902–912 CrossRef CAS.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2021, 7, eabf8283 CrossRef CAS PubMed.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y. H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef CAS PubMed.
- C. Shi, M. L. McGraw, Z. C. Li, L. Cavallo, L. Falivene and E. Y. Chen, Sci. Adv., 2020, 6, eabc0495 CrossRef CAS PubMed.
- J. B. Zhu and E. Y. Chen, Angew. Chem., Int. Ed., 2019, 58, 1178–1182 CrossRef CAS PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- N. Vora, P. R. Christensen, J. Demarteau, N. R. Baral, J. D. Keasling, B. A. Helms and C. D. Scown, Sci. Adv., 2021, 7, eabf0187 CrossRef CAS PubMed.
- M. Hong and E. Y. X. Chen, Trends Chem., 2019, 1, 148–151 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- A. C. Deacy, A. F. R. Kilpatrick, A. Regoutz and C. K. Williams, Nat. Chem., 2020, 12, 372–380 CrossRef CAS PubMed.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed.
- G. W. Yang, C. K. Xu, R. Xie, Y. Y. Zhang, X. F. Zhu and G. P. Wu, J. Am. Chem. Soc., 2021, 143, 3455–3465 CrossRef CAS PubMed.
- A. Denk, S. Kernbichl, A. Schaffer, M. Kränzlein, T. Pehl and B. Rieger, ACS Macro Lett., 2020, 9, 571–575 CrossRef CAS.
- G. W. Yang, Y. Y. Zhang, R. Xie and G. P. Wu, J. Am. Chem. Soc., 2020, 142, 12245–12255 CrossRef CAS PubMed.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. Pena Carrodeguas, G. Trott, A. Santmarti, K. Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS PubMed.
- J. Furukawa, T. Saegusa, N. Mise and A. Kawasaki, Makromol. Chem., 1960, 39, 243–245 CrossRef CAS.
- Y.-Y. Zhang, G.-P. Wu and D. J. Darensbourg, Trends Chem., 2020, 2, 750–763 CrossRef CAS.
- G. A. Bhat, M. Luo and D. J. Darensbourg, Green Chem., 2020, 22, 7707–7724 RSC.
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed.
- A. Thevenon, A. Cyriac, D. Myers, A. J. P. White, C. B. Durr and C. K. Williams, J. Am. Chem. Soc., 2018, 140, 6893–6903 CrossRef CAS PubMed.
- G. Rosetto, A. C. Deacy and C. K. Williams, Chem. Sci., 2021, 12, 12315–12325 RSC.
- Y.-C. Su and B.-T. Ko, Inorg. Chem., 2021, 60, 852–865 CrossRef CAS PubMed.
- C.-H. Chang, C.-Y. Tsai, W.-J. Lin, Y.-C. Su, H.-J. Chuang, W.-L. Liu, C.-T. Chen, C.-K. Chen and B.-T. Ko, Polymer, 2018, 141, 1–11 CrossRef CAS.
- G.-L. Liu, H.-W. Wu, Z.-I. Lin, M.-G. Liao, Y.-C. Su, C.-K. Chen and B.-T. Ko, Polym. Chem., 2021, 12, 1244–1259 RSC.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers and A. L. Phelps, Inorg. Chem., 2004, 43, 1831–1833 CrossRef CAS PubMed.
- Y. Liu, W.-M. Ren, C. Liu, S. Fu, M. Wang, K.-K. He, R.-R. Li, R. Zhang and X.-B. Lu, Macromolecules, 2014, 47, 7775–7788 CrossRef CAS.
- W. T. Diment, T. Stößer, R. W. F. Kerr, A. Phanopoulos, C. B. Durr and C. K. Williams, Catal. Sci. Technol., 2021, 11, 1737–1745 RSC.
- Z. Jia and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2085–2097 CrossRef CAS.
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 CrossRef CAS PubMed.
- R. Liénard, J. De Winter and O. Coulembier, J. Polym. Sci., 2020, 58, 1481–1502 CrossRef.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 CrossRef CAS.
- D. J. Darensbourg, A. D. Yeung and S.-H. Wei, Green Chem., 2013, 15, 1578–1583 RSC.
- D. J. Darensbourg, Polym. Degrad. Stab., 2018, 149, 45–51 CrossRef CAS.
- Y. Liu, H. Zhou, J. Z. Guo, W. M. Ren and X. B. Lu, Angew. Chem., Int. Ed., 2017, 56, 4862–4866 CrossRef CAS PubMed.
- C. Li, R. J. Sablong, R. A. T. M. van Benthem and C. E. Koning, ACS Macro Lett., 2017, 6, 684–688 CrossRef CAS.
- M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A. K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821–9826 CrossRef CAS PubMed.
- C. Romain, M. S. Bennington, A. J. White, C. K. Williams and S. Brooker, Inorg. Chem., 2015, 54, 11842–11851 CrossRef CAS PubMed.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS PubMed.
- D. J. Darensbourg, J. C. Yarbrough, C. Ortiz and C. C. Fang, J. Am. Chem. Soc., 2003, 125, 7586–7591 CrossRef CAS PubMed.
- A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS.
- Y. Liu, W. M. Ren, J. Liu and X. B. Lu, Angew. Chem., Int. Ed., 2013, 52, 11594–11598 CrossRef CAS PubMed.
- J. Roovers, in Cyclic Polymers, ed. J. A. Semlyen, Kluwer Academic, 2nd edn, 2000, ch. 10, pp. 347–384 Search PubMed.
- U. Neugebauer, U. Schmid, K. Baumann, W. Ziebuhr, S. Kozitskaya, V. Deckert, M. Schmitt and J. Popp, ChemPhysChem, 2007, 8, 124–137 CrossRef CAS PubMed.
- K. Maquelin, C. Kirschner, L. P. Choo-Smith, N. van den Braak, H. P. Endtz, D. Naumann and G. J. Puppels, J. Microbiol. Methods, 2002, 51, 255–271 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, NMR spectra, MALDI-TOF MS spectra, GPC traces, double logarithm (Mark–Houwink) plots. CCDC 2092473. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01387h |
| This journal is © The Royal Society of Chemistry 2022 |