
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssembling the carbon skeleton of A-74528†
Martin S.
Maier‡
 ab,
Andrej
Shemet‡
b and
Dirk
Trauner
*ab
ab,
Andrej
Shemet‡
b and
Dirk
Trauner
*ab
aDepartment of Chemistry, Ludwig Maximilian University of Munich, 81377 Munich, Germany
bDepartment of Chemistry, New York University, New York, New York 10003, USA. E-mail: dirktrauner@nyu.edu
First published on 5th July 2022
Abstract
The 2′-phosphodiesterase inhibitor A-74528, which combines an intriguing biosynthesis with unusual biological activity, is one of the most complex type II polyketides. As a synthetic target, it represents a significant challenge due to its size but also due to a unique carbon skeleton that features a hexacarbocyclic core with an appended pyrone. Here we report our efforts toward the synthesis of A-74528, which culminated in the construction of the full carbon skeleton and the correct installation of all but one stereocenter. Our strategy employs a molybdenum-catalyzed branched allylation to establish the central quaternary carbon and relies on establishing the remaining stereocenters in a substrate-controlled manner. Carbocycles were established using a spiro epoxide annulation, a 1,3-dipolar cycloaddition, followed by an aldol condensation, and a gold-catalyzed hydroarylation. The pyrone was appended to an aldehyde branching off the quaternary stereocenter by a one-carbon homologation and Mukaiyama aldol addition.
Introduction
In 2004, a research group at the Sankyo company reported their findings1 on a previously elusive 2′-phosphodiesterase (2′-PDE). They reported that the natural product A-74528, isolated from a streptomyces strain, acts as an inhibitor of 2′-PDE. Thereby, A-74528 modulates the activity of the 2′,5′-oligoadenylate (2-5A) system involved in anti-viral and anti-tumor immune responses in human cells. The 2-5A system triggers the enzyme RNase L, which degrades both viral and cellular RNAs and thus shuts down the protein biosynthesis in virus-infected cells. Remarkably, the natural product showed dose-dependent reduction of viral replication and no cytotoxic effects to the host cell. As such, it could serve as a lead compound for the development of mechanistically distinct antiviral and antitumor compounds. Structure elucidation based on extensive NMR studies2 showed that A-74528 is a highly fused type II polyketide with an unprecedented carbon skeleton and that it has a rather unusual structure compared to biosynthetically related polyketides3a (Scheme 1). | ||
| Scheme 1 (A) Structure and retrosynthetic analysis of A-74528. (B) Structural model of A-74528. | ||
A-74528 consists of a pyrone side-chain that is connected via a methylene bridge to a hexacyclic core, which adopts an L-shaped conformation and consist of two resorcinols that are flanking four annealed six-membered rings. These non-aromatic rings feature a benzylic carbonyl and an enolized 1,3-diketone as well as a sequence of six contiguous stereocenters, one of which is a quaternary carbon and one of which bears secondary alcohol. Biosynthetically, the positioning of the secondary alcohol is intriguing, as it does not follow the standard oxidation pattern of a nascent polyketide chain. Detailed investigations of the biosynthetic pathway were performed by the group of Khosla,3 who identified and characterized the gene cluster responsible for the production of A-74528. Consequently, several sequences that could transform a C30 polyketide precursor into A-74528 were suggested and it was proposed that the secondary alcohol would result from the opening of an epoxide. Furthermore, Khosla also pointed out the biosynthetic connections between A-74528 and fredericamycin, which are the two largest known compounds within the type II polyketides.
Despite its intriguing structure, A-74528 has received relatively little attention from the synthetic community. In 2011 our group reported a biomimetic approach based on a putative dearomatizing double Michael-addition sequence.4 Although this strategy featured novel condensation chemistry to furnish highly functionalized biaryls, it did not result in the establishment of the hexacyclic core. To the best of our knowledge there have been no further attempts to synthesize A-74528, apart from initial exploratory work by the Pronin group5 and A-74285 being mentioned as a potential target for applying a new photoenolization Diels–Alder methodology.6 We now disclose an approach toward A-74528 that is not based on biosynthetic speculations and their biomimetic execution, but rather on transition metal catalyzed reactions, which has resulted in the successful establishment of the complex carbon skeleton of our target molecule.
Results and discussion
Our retrosynthetic analysis of A-74528 (Scheme 1A) centered on establishing the quaternary carbon early in the synthesis and then approach the remaining stereochemical challenges in a substrate-controlled fashion. We presumed that it would be prudent to both establish the C-ring ketone by a benzylic oxidation and attach the pyrone moiety at a late stage of the synthesis, while the sensitive 1,3-diketone would best be masked as an isoxazole. These considerations led us aldehyde 1, which we further simplified to lactone 2, based on the expectation that the A-ring could be closed by an electrophilic aromatic substitution and the F-ring would be formed in an aldol condensation. Lactone 2 would be derived in a multi-step sequence from ketoester 3, with a 1,3-dipolar cycloaddition as the key reaction. Ketoesters similar to 3 have been prepared enantio-, diastereo- and regioselectively in high yield through iridium-catalyzed allylation by the group of Stoltz.7 However, we were also aware of the possibility to use other metals, such as molybdenum, as introduced by Trost and coworkers.8 Consequently, we prepared the tetralone ester 4 and the known cinnamyl carbonate 5, the optimized synthesis of which is given in the ESI.†The coupling of these two fragments is shown in Scheme 2. Employing a catalyst preparation method by Palucki9 and an electron-rich bis-pyridine ligand (L1) reported by Moberg,10 we found that ketoester 3 was formed with high diastereo- and regioselectivity by reaction of a slight excess (1.3 eq.) of the sodium enolate of tetralone 4 with carbonate 5. Thus, we established the quaternary stereocenter and adjacent tertiary stereocenter of A-74528 in a single operation. The desired product 3 was obtained in good yield (84%) after trituration with ethanol, which proved suitable to remove most of the minor regioisomeric and diastereomeric side-products. Regarding the enantioselectivity of this reaction, we performed initial tests without further optimization of the reaction and already observed an enantiomeric excess of 68%. However, as the more easily available racemic material was suitable to explore the following steps of the synthesis, the enantioselective allylation was not further examined.
 | ||
| Scheme 2 Fragment coupling. | ||
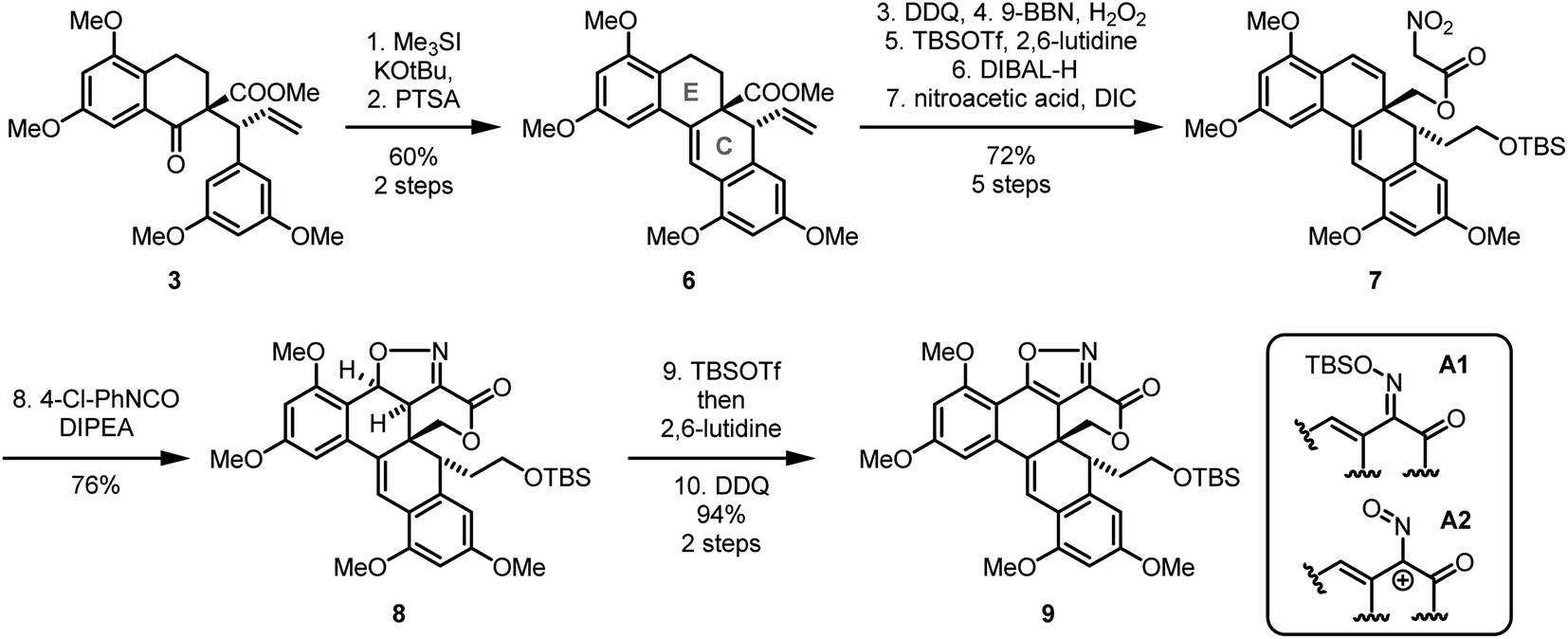
Having established a practical approach to ketoester 3, we then turned towards the C-ring of A-74528 (Scheme 3). To forge this ring, it was necessary to introduce one missing carbon atom, which we achieved via a Corey–Chaykovsky epoxidation.11 Treatment of ketoester 3 with trimethylsulfonium iodide and potassium tert-butoxide was followed directly by treatment with p-toluenesulfonic acid, which resulted in an electrophilic aromatic substitution and subsequent elimination of water to afforded stilbene 6 in decent yield (60% over two steps). We speculate that this reaction proceeds via formation of a spiro epoxide (see ESI, structure ESI-10†), followed by acid catalyzed Meinwald rearrangement, electrophilic aromatic substitution, and elimination. However, a direct nucleophilic attack of the electron rich arene into the spiro epoxide cannot be ruled out.
 | ||
| Scheme 3 C-Ring formation and isoxazole installation. | ||
With tetracycle 6 at hand, we next installed the isoxazole via intramolecular 1,3-dipolar cycloaddition. To prepare for this step, we needed to install a double bond in the E-ring, which could be achieved in high yield by treatment with DDQ. A highly chemo- and regioselective hydroboration gave a primary alcohol that was protected as a TBS ether. Reduction of the ester and acylation then gave nitroacetic acid ester 7 in good overall yield (72% for 5 steps).
The subsequent cycloaddition was enabled by using conditions similar to Mukaiyama's original work.12 Thus, isoxazoline 8 was formed by dehydration with p-chlorophenyl isocyanate in the presence of catalytic amounts of Hünig's base in hot toluene. In contrast to the previous benzylic unsaturation, the subsequent aromatization of 8 to yield isoxazole 9 proved quite challenging and suffered from sluggish conversion. However, in an attempt to increase the reactivity of a quinone oxidant we observed that isoxazolines like 8 are highly prone to acid-mediated ring-opening to the corresponding unsaturated oxime. This finding enabled us to develop a two-step method for this challenging isoxazoline oxidation: first, the isoxazoline was exposed to tert-butyldimethylsilyl trifluoromethanesulfonate, converting it into a O-silyl oxime (see Scheme 3, substructure A1). Second, this O-silyl oxime was oxidized with DDQ, which directly transformed it to the desired isoxazole.13 We presume that this sequence, which afforded isoxazole 9 with a yield of 94%, involves a nitrosoallyl cation (see Scheme 3, substructure A2) that undergoes a Nazarov-type 4π-electrocyclization.
With isoxazole 9 at hand, we proceeded with installing the next stereocenter through a hydrogenation of the stilbene moiety (Scheme 4). Using a modified version of Shenvi's hydrogen atom transfer (HAT) hydrogenation method,14 the desired product 2 was obtained in very good yield (86%) and as a single diastereomer without affecting the rather sensitive isoxazole N–O bond. At this point, we suspected that transforming the C-ring methylene to a carbonyl group could result in reduced stability and would limit our future choices of reaction conditions. Thus, we decided to defer this benzylic oxidation to a later stage in the synthesis. Instead, we set the stage for closing the F-ring. Reduction of the lactone moiety of 2, followed by protecting group manipulations, afforded diol 10 and oxidation with Dess–Martin periodinane,15 followed by treatment with potassium carbonate in methanol, led to an aldol condensation. This smoothly formed the F-ring and gave enal 11 (82% yield). Now, only two carbon atoms of the hexacyclic core of A-74528 were missing, which we delivered in the form of ethynylmagnesium bromide. Reaction of enal 11 with this Grignard reagent exhibited excellent diastereoselectivity and we exclusively observed the desired diastereomer 12. The last ring of the core was then established in an intramolecular gold-catalyzed hydroarylation, as treatment of propargylic alcohol 12 with triphenylphosphine gold triflimide gave bis-allyl alcohol 13 in very good yield. Considering the sensitive nature of the hydroarylation product 13, this reaction exemplifies the power of gold-catalyzed reactions under mild conditions.16
 | ||
| Scheme 4 Formation of A-ring and F-ring. | ||
The installation of the remaining stereocenters would require diastereoselective hydrogenation of the two alkenes in 13. However, this proved to be more challenging than anticipated and we were not able to reduce the exo-methylene moiety with satisfactory stereoselectivity. Molecular modeling suggested that the F-ring of 13 adopts a twist-boat conformation (Scheme 5B), which leads to preferential reductions from the undesired face. Thus, despite extensive experimentation (e.g. Crabtree's catalyst, Wilkinson's catalyst, palladium on carbon, Adam's catalyst, HAT hydrogenation), we were only able to achieve a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 mixture of 14a:14b in a combined yield of 80% by reaction with diimide (generated from o-nitrobenzenesulfonylhydrazide).17 After separation by silica gel chromatography, we initially continued with minor diastereomer 14a, but found that further reduction of the remaining trisubstituted alkene also proved problematic. The HAT hydrogen, which had worked very well at an earlier stage, now only proceeded to 15a in disappointing yield, as a similar reactivity of the alkene and isoxazole gave undesired by-products (mainly by N–O cleavage). Fortunately, benzyl protection of the secondary alcohol 15a gave crystalline ether 16a, which was suitable for X-ray analysis. Its result unambigously confirmed all our stereochemical assignments (Scheme 5C).
:2.5 mixture of 14a:14b in a combined yield of 80% by reaction with diimide (generated from o-nitrobenzenesulfonylhydrazide).17 After separation by silica gel chromatography, we initially continued with minor diastereomer 14a, but found that further reduction of the remaining trisubstituted alkene also proved problematic. The HAT hydrogen, which had worked very well at an earlier stage, now only proceeded to 15a in disappointing yield, as a similar reactivity of the alkene and isoxazole gave undesired by-products (mainly by N–O cleavage). Fortunately, benzyl protection of the secondary alcohol 15a gave crystalline ether 16a, which was suitable for X-ray analysis. Its result unambigously confirmed all our stereochemical assignments (Scheme 5C).
 | ||
| Scheme 5 (A) Final modifications of the A-ring and F-ring. (B) Structural model of intermediate 13. (C) X-ray structure of ether 16a (disordered TBS group not shown). | ||
Due to the limited supply of material at this stage of the synthesis we then switched to using the major product of the diimide reduction for our further investigations (Scheme 6). HAT hydrogenation and protecting group manipulations converted 14b into neopentylic alcohol 17b, which was oxidized using Stahl's method18 and homologated to aldehyde 18b. For this homologation, Morken's boron–Wittig reaction19 proved uniquely efficient and reliable. Next, we introduced the pyrone moiety in a three-step sequence20 including a vinylogous Mukaiyama aldol addition of silyl dienolate A3 to aldehyde 18b and oxidation of the intermediary alcohol with Dess–Martin periodinane, followed by thermal cyclization to provide compound 19b.
 | ||
| Scheme 6 Finishing the carbon skeleton of A-74528. | ||
Compound 19b contains all the carbons of the natural product and five of six stereocenters in the correct configuration. Completion of the synthesis would require a satisfactory solution of the alkene reduction, C-ring benzylic oxidation, unmasking of the 1,3-dicarbonyl by cleavage of the isoxazole, and global demethylation. Model studies with an advanced intermediate indicated that the latter two transformations should be straightforward.
In our studies towards the double reduction of the pentadienol moiety in 13 with Adams' catalyst we had observed the formation of enaminone 20b albeit with undesired stereochemical outcome (Scheme 7). This shows that the isoxazole can be cleaved under these conditions and that the enaminone does not spontaneously hydrolyze to afford the 1,3-diketone. After some experimentation, however, we found that treatment of 20b with tert-butyl nitrite in DMSO in the presence of triflic acid21 gave the enaminone hydrolysis product 21b (61% yield). This reaction is believed to proceed via formation of diazonium intermediate A4 followed by attack of water and expulsion of nitrogen. Finally, treatment of 21b with BBr3 led to the deprotection of all four methyl ethers and cleavage of the silyl ether with an unoptimized yield of 36%. This demonstrates that the secondary hydroxy group and the enolized 1,3-diketone are compatible with such demethylation conditions.
 | ||
| Scheme 7 Enaminone hydrolysis and global demethylation studies. | ||
Conclusions and outlook
In sum, we have synthesized compounds that closely resemble the natural product A-74528, bear most of its functional groups and stereocenters, and could share its biological activity. The key steps of our pathway include a molybdenum-catalyzed allylic alkylation to establish the central quaternary stereocenter, the installation of a masked 1,3-diketone via a (3 + 2)-cycloaddition, the formation of the A-ring by gold-mediated alkyne hydroarylation, and a late-stage pyrone attachment via a vinylogous Mukaiyama aldol reaction. Our experiments have provided insights into chemical stabilities and chemoselectivities as well as diastereoselectivities, that will inform future synthetic work. While it may be possible to finish the synthesis from compound 16a with brute force, ignoring the unfortunate diastereoselectivity of the exo-methylene double bond reduction, our study also provides a plethora of readily accessible intermediates that could eventually be forwarded to A-75428.Data availability
Synthetic procedures, characterization and X-ray crystallography data and NMR spectra are provided in the ESI† accompanying this paper.Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Michelle Neary (Hunter College) for X-ray crystallographic analysis. A. S. thanks the Swiss National Science Foundation (SNSF) for a postdoctoral fellowship (P2EZP_181623).References
- K. Kubota, K. Nakahara, T. Ohtsuka, Y. Yoshida, J. Kawaguchi, Y. Fujita, Y. Ozeki, A. Hara, C. Yoshimura, H. Furukawa, H. Haruyama, K. Ichikawa, M. Yamashita, T. Matsuoka and I. Iijima, Identification of 2′-Phosphodiesterase, Which Plays a Role in the 2-5A System Regulated by Interferon, J. Biol. Chem., 2004, 279, 37832 CrossRef CAS PubMed.
- Y. Fujita, A. Kasuya, Y. Matsushita, M. Suga, M. Kizuka, Y. Iijima and T. Ogita, Structural elucidation of A-74528, an inhibitor for 2′,5′-phosphodiesterase isolated from Streptomyces sp, Bioorg. Med. Chem. Lett., 2005, 15, 4317 CrossRef CAS PubMed.
- (a) K. Zaleta-Rivera, L. K. Charkoudian, C. P. Ridley and C. Khosla, Cloning, Sequencing, Heterologous Expression, and Mechanistic Analysis of A-74528 Biosynthesis, J. Am. Chem. Soc., 2010, 132, 9122 CrossRef CAS PubMed; (b) J. T. Fitzgerald, L. K. Charkoudian, K. R. Watts and C. Khosla, Analysis and Refactoring of the A-74528 Biosynthetic Pathway, J. Am. Chem. Soc., 2013, 135, 3752 CrossRef CAS PubMed.
- A. Hager, D. Mazunin, P. Mayer and D. Trauner, Synthetic Studies toward A-74528, Org. Lett., 2011, 13, 1386 CrossRef CAS PubMed.
- M. Young, Towards the Total Synthesis of A-74528, 2019, https://www.escholarship.org/uc/item/2wd8z35d Search PubMed.
- B. Yang, K. Lin, Y. Shi and S. Gao, Ti(Oi-Pr)4-promoted photoenolization Diels–Alder reaction to construct polycyclic rings and its synthetic applications, Nat. Commun., 2017, 8, 622 CrossRef PubMed.
- W.-B. Liu, C. M. Reeves, S. C. Virgil and B. M. Stoltz, Construction of Vicinal Tertiary and All-Carbon Quaternary Stereocenters via Ir-Catalyzed Regio-, Diastereo-, and Enantioselective Allylic Alkylation and Applications in Sequential Pd Catalysis, J. Am. Chem. Soc., 2013, 135, 10626 CrossRef CAS PubMed.
- (a) B. M. Trost and M. Lautens, Molybdenum catalysts for allylic alkylation, J. Am. Chem. Soc., 1982, 104, 5543 CrossRef CAS; (b) B. M. Trost and M. Lautens, Regiochemical diversity in allylic alkylations via molybdenum catalysts, Tetrahedron, 1987, 43, 4817 CrossRef CAS; (c) B. M. Trost and I. Hachiya, Asymmetric Molybdenum-Catalyzed Alkylations, J. Am. Chem. Soc., 1998, 120, 1104 CrossRef CAS; (d) B. M. Trost, J. R. Miller and C. M. Hoffman Jr, A Highly Enantio- and Diastereoselective Molybdenum-Catalyzed Asymmetric Allylic Alkylation of Cyanoesters, J. Am. Chem. Soc., 2011, 133, 8165 CrossRef CAS PubMed; (e) B. M. Trost, M. Osipov, S. Krüger and Y. Zhanga, A catalytic asymmetric total synthesis of (−)-perophoramidine, Chem. Sci., 2015, 6, 349 RSC; (f) S. W. Krska, D. L. Hughes, R. A. Reamer, D. J. Mathre, Y. Sun and B. M. Trost, The Unusual Role of CO Transfer in Molybdenum-Catalyzed Asymmetric Alkylations, J. Am. Chem. Soc., 2002, 124, 12656 CrossRef CAS PubMed.
- M. Palucki, J. M. Um, D. A. Conlon, N. Yasuda, D. L. Hughes, B. Mao, J. Wang and P. J. Reider, Molybdenum-Catalyzed Asymmetric Allylic Alkylation Reactions Using Mo(CO)6 as Precatalyst, Adv. Synth. Catal., 2001, 343, 46 CrossRef CAS.
- (a) O. Belda, N.-F. Kaiser, U. Bremberg, M. Larhed, A. Hallberg and C. Moberg, Highly Stereo- and Regioselective Allylations Catalyzed by Mo–Pyridylamide Complexes: Electronic and Steric Effects of the Ligand, J. Org. Chem., 2000, 65, 5868 CrossRef CAS PubMed; (b) O. Belda and C. Moberg, Substituted Pyridylamide Ligands in Microwave-Accelerated Mo(0)-Catalysed Allylic Alkylations, Synthesis, 2002, 1601 CAS.
- (a) E. J. Corey and M. Chaykovsky, Dimethylsulfoxonium Methylide, J. Am. Chem. Soc., 1962, 84, 867 CrossRef CAS; (b) V. Franzen and H. E. Driessen, Methylenierung mit sulfonium-yliden, Tetrahedron Lett., 1962, 3, 661 CrossRef; (c) E. J. Corey and M. Chaykovsky, Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis, J. Am. Chem. Soc., 1965, 87, 1353 CrossRef CAS.
- T. Mukaiyama and T. Hoshino, The Reactions of Primary Nitroparaffins with Isocyanates, J. Am. Chem. Soc., 1960, 82, 5339 CrossRef CAS.
- (a) V. G. Desai and S. G. Tilve, A Novel and Convenient Method for the Synthesis of 3, 5-Diarylisoxazoles, Synth. Commun., 1999, 29, 3017 CrossRef CAS; (b) R. F. Kurangi, R. Kawthankar, S. Sawal, V. G. Desai and S. G. Tilve, Convenient Synthesis of 3,5-Disubstituted Isoxazoles, Synth. Commun., 2007, 37, 585 CrossRef CAS.
- (a) K. Iwasaki, K. K. Wan, A. Oppedisano, S. W. M. Crossley and R. A. Shenvi, Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol, J. Am. Chem. Soc., 2014, 136, 1300 CrossRef CAS PubMed; (b) C. Obradors, R. M. Martinez and R. A. Shenvi, Ph(i-PrO)SiH2: An Exceptional Reductant for Metal-Catalyzed Hydrogen Atom Transfers, J. Am. Chem. Soc., 2016, 138, 4962 CrossRef CAS PubMed.
- D. B. Dess and J. C. Martin, Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
- Z. Zhou, A. X. Gao and S. A. Snyder, Total Synthesis of (+)-Arborisidine, J. Am. Chem. Soc., 2019, 141, 7715 CrossRef CAS PubMed , and references therein..
- (a) A. G. Myers, B. Zheng and M. Movassaghi, Preparation of the Reagent o-Nitrobenzenesulfonylhydrozide, J. Org. Chem., 1997, 62, 7507 CrossRef CAS PubMed; (b) K. R. Buszek and N. Brown, Improved Method for the Diimide Reduction of Multiple Bonds on Solid-Supported Substrates, J. Org. Chem., 2007, 72, 3125 CrossRef CAS PubMed.
- (a) J. M. Hoover, J. E Steves and S. S. Stahl, Copper(I)/TEMPO-catalyzed aerobic oxidation of primary alcohols to aldehydes with ambient air, Nat. Protoc., 2012, 7, 1161 CrossRef CAS PubMed; (b) Y. Nakayama, M. R. Maser, T. Okita, A. V. Dubrovskiy, T. L. Campbell and S. E. Reisman, Total Synthesis of Ritterazine B, J. Am. Chem. Soc., 2021, 143, 4187 CrossRef CAS PubMed.
- J. R. Coombs, L. Zhang and J. P. Morken, Synthesis of Vinyl Boronates from Aldehydes by a Practical Boron-Wittig Reaction, Org. Lett., 2015, 17, 1708 CrossRef CAS PubMed.
- T. Bach and S. Krisch, Synthesis of 6-Substituted 4-Hydroxy-2-pyrones from Aldehydes by Addition of an Acetoacetate Equivalent, Dess–Martin Oxidation and Subsequent Cyclization, Synlett, 2001, 1974 CrossRef CAS.
- Y. Yamashita, Y. Hirano, A. Takada, H. Takikawa and K. Suzuki, Total Synthesis of Bis-anthraquinone Antibiotic BE-43472B, Synthesis, 2018, 50, 2490 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and analytical data, X-ray crystallography, NMR spectra. CCDC 2156714 and 2156715. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01366e |
| ‡ M. S. M. and A. S. contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |