
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic alkene skeletal modification for the construction of fluorinated tertiary stereocenters†
Liyin
Jiang
 a,
Pau
Sarró
ab,
Wei Jie
Teo
a,
Jordi
Llop
c and
Marcos G.
Suero
*a
a,
Pau
Sarró
ab,
Wei Jie
Teo
a,
Jordi
Llop
c and
Marcos G.
Suero
*a
aInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology. Av. Països Catalans, 16, 43007 Tarragona, Spain. E-mail: mgsuero@iciq.es
bDepartament de Química Analítica I Química Orgànica, Universitat Rovira I Virgili, C. Marcel·lí Domingo, 1, 43007 Tarragona, Spain
cCIC BiomaGUNE, Basque Research and Technology Alliance, 20014 San Sebastián, Guipuzcoa, Spain
First published on 15th March 2022
Abstract
Herein we describe the first construction of fluorinated tertiary stereocenters based on an alkene C(sp2)–C(sp2) bond cleavage. The new process, that takes advantage of a Rh-catalyzed carbyne transfer, relies on a branched-selective fluorination of tertiary allyl cations and is distinguished by a wide scope including natural products and drug molecule derivatives as well as adaptability to radiofluorination.
Introduction
The growing number of approved fluorinated small-molecule pharmaceuticals is a testimony of the tremendous research efforts in synthetic organofluorine chemistry1 and their application in fluorine-based drug design.2 The most prevalent chemotypes found in fluoro-pharmaceuticals feature monofluorinated moieties (Ar–F, Het–F, and alkyl–CH2F) and trifluoromethyl groups (Ar–CF3, Het–CF3, and alkyl–CF3).2d However, the appearance of fluorinated tertiary stereocenters is very rare (Fig. 1A), despite this fluorinated motif being an ideal bioisostere of tertiary stereocenters – a prevalent motif in drug molecules – and being found in fludrocortisone – the first approved fluorine-containing drug. The main reason for the lack of fluorinated tertiary stereocenters in drug molecules can be attributed to a less developed area of research and difficulties of adopting the known synthetic methods for drug molecule design.1d In this sense, it is of high contemporary interest to develop new synthetic concepts to such fluorinated motifs based on unconventional disconnection approaches that can expand and complement known synthetic protocols.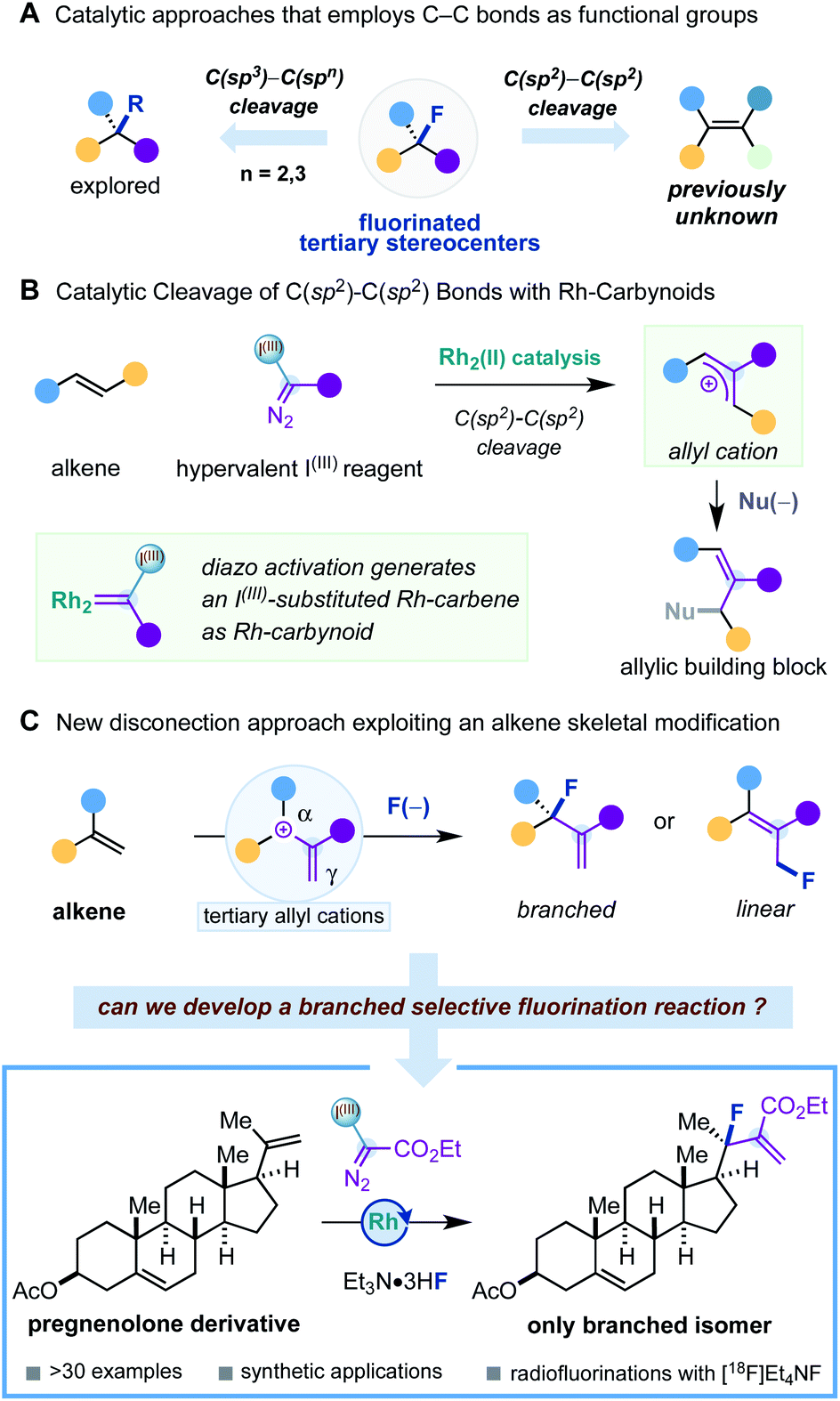 | ||
| Fig. 1 Fluorinated tertiary stereocenters. | ||
Catalytic methodologies based on (i) fluorination of alkenes, enol(ates), phenols and C–H bonds with electrophilic fluorinating reagents3 or in (ii) transformations of fluorine-containing starting materials,4 can be considered the most developed approaches for the regio- and enantioselective construction of fluorinated tertiary stereocenters. However, they respectively employ electrophilic fluorinating reagents – which are derived from fluorine gas – and use pre-functionalized fluorinated starting materials that could need multistep synthetic sequences. On the other hand, catalytic fluorination of alkenes, allylic electrophiles, and C–H bonds with nucleophilic fluoride sources represents in general an efficient option considering the availability of fluoride sources.5,6
Despite the variety of catalytic strategies developed, it is remarkable to observe that methodologies that employ C–C bonds as functional groups are scarce.7 Such processes rely on the skeletal modification of an organic molecule, offering new disconnection approaches.8 Examples of this class of fluorinations that reach fluorinated tertiary stereocenters are limited to the catalytic cleavage of C(sp3)–C(sp2) bonds of redox-active esters,9 carboxylic acids,10 and C(sp3)–C(sp3) bonds in cyclopropanes.11 However, to the best of our knowledge, synthesis of tertiary fluorinated stereocenters through catalytic alkene C(sp2)–C(sp2) bond cleavage is previously unknown (Fig. 1A).
As part of a research programme focused on the development of a carbyne transfer platform in organic synthesis, we reported a catalytic strategy that generates Rh-carbynoids as I(III)-substituted Rh-carbenes by selective diazo activation of bespoke hypervalent iodine reagents with a rhodium paddlewheel catalyst (Fig. 1B).12,13 We found that Rh-carbynoids provoked the skeletal modification of alkenes by formally inserting a cationic monovalent carbon unit (:+C–R) between both sp2-hybridized carbons. This constructive C(sp2)–C(sp2) bond cleavage process generated synthetically useful cation intermediates that converted to valuable chiral racemic allylic building blocks with a broad range of heteroatomic and carbon nucleophiles.
Recently, we wondered whether we could exploit our alkene skeletal modification platform for the catalytic conversion of 1,1-disubstituted alkenes into fluorinated tertiary stereocenters (Fig. 1C). We hypothesized that the fluoride nucleophilic attack would proceed with high branched selectivity considering that both the charge and the highest LUMO coefficient of the allyl cation may be centered at the α position, due to the double substitution with two stabilizing groups (alkyl and aromatic groups).14,15 However, we recognized that constructing a sterically demanding tertiary allylic fluoride could bring some problems associated with (i) parasitic proton eliminations promoted by fluoride and (ii) generation of undesirable branched/linear mixtures due to lack of regiocontrol in the nucleophilic fluoride attack.16
The successful development of such nucleophilic branched-selective fluorination of tertiary allyl cations would unlock a novel access to fluorinated tertiary stereocenters using readily or commercially available 1,1-disubstituted alkenes and nucleophilic fluoride sources. In addition, our strategy would represent also a novel approach to a class of allylic fluorides difficult to obtain with traditional bimolecular nucleophilic substitutions or transition metal-catalyzed platforms.1a,f,17
Herein, we would like to present the successful execution of this goal for a previously unknown disconnection approach to valuable fluorinated tertiary stereocenters based on the skeletal modification of 1,1-disubstituted alkenes. The synthetic protocol is amenable to a broad range of 1,1-disubstituted alkenes and permits the installation of a fluorinated tertiary stereocenter in natural products and drug molecule derivatives. Notably, the fluorination of the allyl cation intermediates occurred with excellent branched selectivity. Follow-up alkene transformations of the products and adaptability to radiofluorination were demonstrated.
Results and discussion
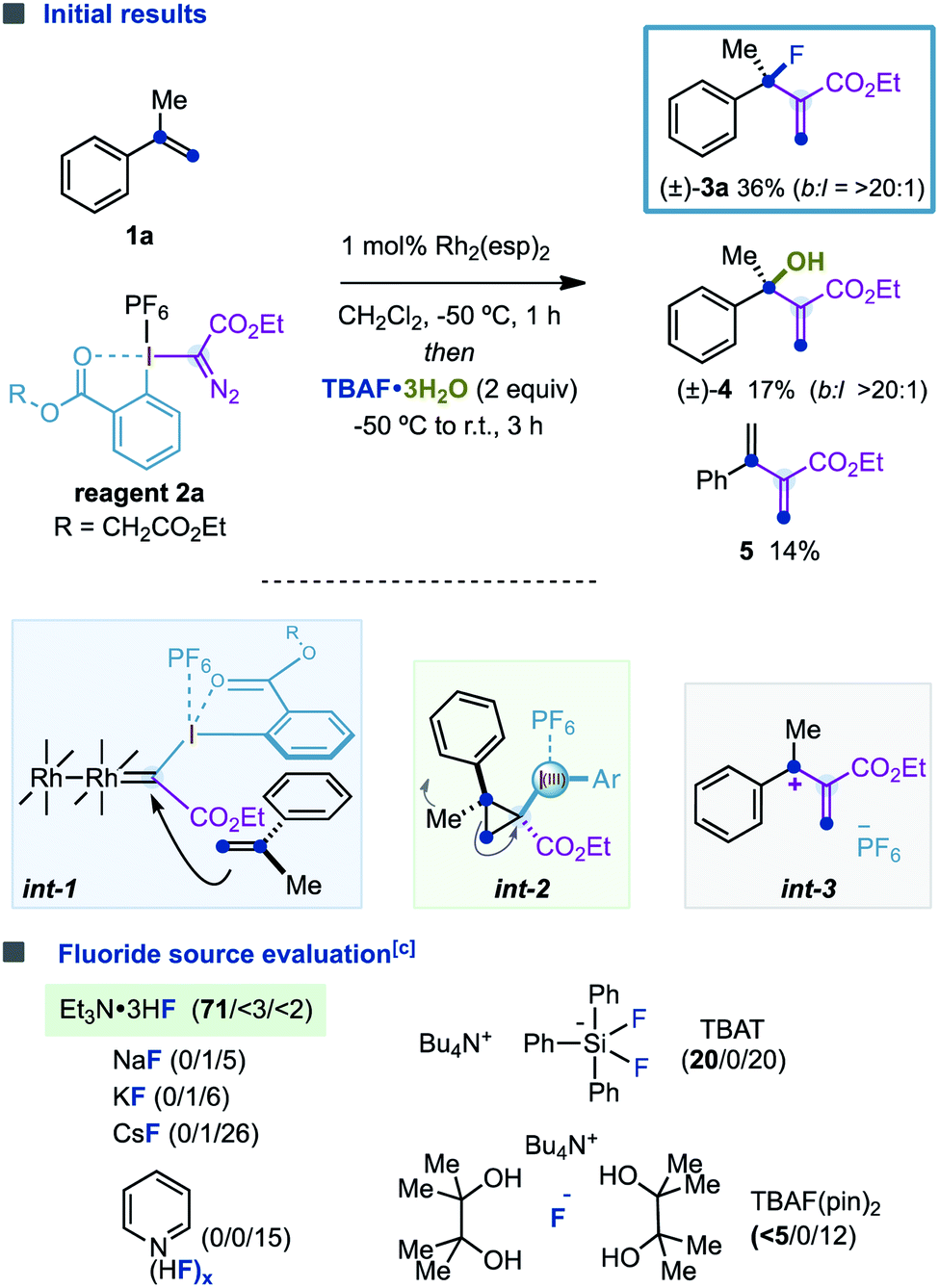
The initial experiments were performed using α-methylstyrene (1a, 2 equiv.), hypervalent iodine reagent 2a (1 equiv.), tetrabutylammonium fluoride trihydrate Bu4NF·3H2O (3 equiv.) and the Du Bois catalyst Rh2(esp)2 (1 mol%)18 in dichloromethane (Table 1). We were pleased to find that branched allylic fluoride (±)-3a was formed in a promising 36% isolated yield with excellent branched/linear selectivity (b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :l = >20:1). In addition to (±)-3a, alcohol (±)-4 and diene 5 were also formed as subproducts of the reaction in low yields. The formation of (±)-3a was proposed to proceed via (i) catalytic generation of a Rh-carbynoid by diazo activation of 2a; (ii) stereoselective cyclopropanation (int-1) to deliver a transient cyclopropyl–I(III) intermediate int-2; (iii) disrotatory ring-opening (the Ph ring rotates inwardly, and Me rotates outwardly) to give allyl cation int-3; and (iv) regioselective fluoride attack. Finally, alcohol (±)-4 could be formed by hydrolysis of (±)-3a19 or by a branched-selective water attack to int-319,20 and diene 5 by proton elimination.
:l = >20:1). In addition to (±)-3a, alcohol (±)-4 and diene 5 were also formed as subproducts of the reaction in low yields. The formation of (±)-3a was proposed to proceed via (i) catalytic generation of a Rh-carbynoid by diazo activation of 2a; (ii) stereoselective cyclopropanation (int-1) to deliver a transient cyclopropyl–I(III) intermediate int-2; (iii) disrotatory ring-opening (the Ph ring rotates inwardly, and Me rotates outwardly) to give allyl cation int-3; and (iv) regioselective fluoride attack. Finally, alcohol (±)-4 could be formed by hydrolysis of (±)-3a19 or by a branched-selective water attack to int-319,20 and diene 5 by proton elimination.
| a Performed with 1a (0.2 mmol, 2 equiv.), 2a (0.1 mmol, 1 equiv.), Rh2(esp)2 (0.002 mmol, 1 mol%), and a fluoride source (3 equiv.) in CH2Cl2 (0.1 M). b Yields are reported on the basis of 1H-NMR analysis using anisole as the internal standard; branched/linear ratio was determined by 19F NMR analysis. c Yields in parentheses are of (±)-3a/(±)-4/5. esp = α,α,α′,α′-tetramethyl-1,3-benzenedipropanoate. |
|---|
|
With these promising results, we were encouraged to optimize this novel fluorination reaction by evaluating a diverse variety of commercially available fluoride sources, hoping to improve the efficiency of the C–F bond-forming process while minimizing the parasitic proton elimination or water attack. We were pleased to find that Et3N·3HF led to (±)-3a in 71% isolated yield, and the formation of (±)-4 and 5 was certainly suppressed (<5% yield).21 However, while metallic fluorides MF (M = Na, K, Cs) and Olah's reagent [Py(HF)x] promoted the conversion to diene 5, tetrabutylammonium difluorotriphenylsilicate (TBAT) or the tetrabutylammonium fluoride bispinacol complex (TBAF(pin)2) provided (±)-3a in poor yields.22,23
With the optimized reaction conditions in hand, we next investigated the scope of this fluorination reaction by examining a broad range of α-substituted styrenes (Table 2). We were delighted to observe that substrates substituted in the para position of the aromatic ring with halogens [(±)-3b-e], trifluoromethyl [(±)-3f], ester [(±)-3g], trifluoromethoxy [(±)-3h], acyloxy [(±)-3i-j] and alkyne [(±)-3k] were well tolerated. However, methyl or methoxy substituents provided low levels of efficiency [(±)-3l] or no product [(±)-3m], as notable polymerizations were noticed with full starting material consumption. This observation might suggest that the corresponding allyl carbocation species are generated, before Et3N·3HF is added to the reaction, due to a significant acceleration in the ring-opening of cyclopropyl–I(III) intermediates (int-2) caused by the electron-rich aromatic rings. We later hypothesized that these electron-donating groups may not provoke such significant acceleration in the ring-opening step when placed in a meta position. As predicted, the reactions carried out with meta-MeO- and meta-Me-substituted α-methylstyrenes provided (±)-3n-o with satisfactory yields and excellent branched/linear ratios. Moreover, para- and meta-disubstituted aromatic rings or naphthalene provided the desired products with high level of efficiencies [(±)-3p,q].
| a Performed with 1 (0.4 mmol, 2 equiv.), 2a (0.2 mmol, 1 equiv.), Rh2(esp)2 (0.002 mmol, 1 mol%), and Et3N·3HF (3 equiv.) in CH2Cl2 (0.1 M). Yields are reported on the basis of the isolated pure product. b Branched/linear ratio, indicated in parentheses, and the diastereoselectivity ratio, indicated in brackets, were determined by 19F NMR or 1H NMR analysis of the crude reaction mixture. c Yield of the isolated product using 1.7 grams of 1a and 4.2 grams of 2a. |
|---|

|
In contrast to the exquisite branched selectivity obtained for para- and meta-substituted α-methylstyrenes, a different situation was observed for ortho-substituted derivatives. Equimolecular mixtures of branched/linear fluorides were obtained (3r,s) when using substrates substituted with a methyl or a chlorine group. Although this is a clear limitation of our method, it underlines a potential subtle effect of the ortho substituent in preventing the aromatic ring from stabilizing the charge at the α position of the tertiary allyl cation.
After this, we decided to extend the scope by varying the substituent in the α position of the styrene (Table 2). We observed that styrenes substituted with alkyl groups such as propyl, phenethyl, cyclohexyl, or iso-butyl [(±)-3t-w] worked well; however, a general decrease in the branched/linear ratio was observed. Fluoromethyl and fluoro substituents were well tolerated and provided access to difluoromethyl and 1,2-difluoroethyl compounds (±)-3x,y with high efficiency. The latter results highlight an added-value of our methodology in accessing an interesting and useful subset of organofluorine compounds increasingly observed in newly approved drug molecules.24 Phenyl or alkyne groups did not provide the expected branched fluorides, and instead, the corresponding linear derivatives were obtained (3z,aa). These results indicate that these substituents may provoke a delocalization of the charge in the allylic π system; however, we do not have a clear explanation for the excellent linear selectivity observed. The latter examples led us to question the behavior of allyl carbocations doubly-substituted in the α position with alkyl groups, which are generated from 1,1-dialkyl substituted alkenes. We were delighted to find that exocyclic and acyclic aliphatic olefins were well tolerated and provide tertiary allylic fluorides (3ab-ag) with good to excellent branched/linear ratios.
Further demonstration of the potential of our methodology was validated in the fluorination of a selection of natural products and drug molecule derivatives (3ah-an) (Table 2). It is worth highlighting the excellent degree of chemoselectivity observed in substrates containing more than one alkene. The results indicate that highly substituted alkenes are less reactive; however, the excellent selectivity observed for β-elemene (3al) highlights that the initial alkene cyclopropanation to form a cyclopropyl–I(III) intermediate is sensitive to steric effects.
We next aimed to transform the alkenyl-carboxylate moiety of (±)-3a into useful functionalities without compromising the integrity of the fluorinated tertiary stereocenter (Table 3). Hydrogenation (6), dihydroxylation (7), epoxidation (8) 1,3-cycloaddition reactions (9) and oxidation (10) provided a series of fluorinated derivatives that would be otherwise difficult to obtain by other means. To demonstrate the synthetic utility of our methodology in providing access to fluorinated analogues of medically relevant agents containing a tertiary stereocenter, we sought to synthesize (±)-F-flurbiprofen 12. Although this fluorinated analogue is known, it was synthesized using electrophilic reagents.25 Initially, we performed our fluorination reaction using a readily available styrene and obtained branched tertiary fluoride (±)-11 with high efficiency. Finally, oxidation with OsO4 transformed (±)-11 into the desired (±)-F-flurbiprofen 12 (Table 3).
| a Reaction conditions: (i) TsNHNH2, NaOEt, EtOH, 1 hour, and 80 °C; (ii) OsO4 (1 mol%), Oxone, DMF/H2O, 1 hour, and rt; (iii) m-CPBA, CH2Cl2, 14 hours, and reflux; (iv) chlorobenzaldoxime, Et3N, 5 hours, and 0 °C; (v) OsO4 (1 mol%), Oxone, DMF, 14 hours, and rt; then H2O2, 2M NaOH, and THF. b (i) 2-fluoro-4-(prop-1-en-2-yl)-1,1′-biphenyl, Rh2(esp)2 (1 mol%), and Et3N·3HF; (ii) OsO4 (1 mol%), Oxone, DMF, 14 hours, and rt; then H2O2, 2M NaOH, and THF. c 1a or 1j (20 μmol), 2a (20 μmol), Rh2(esp)2 (1 mol%), CH2Cl2, 1 hour, and −50 °C; then [18F]TEAF (∼0.2 GBq) in CH2Cl2 (100 μL), 20 min, and −50 °C → −30 °C. RCC was calculated with radio-HPLC with the number of replicates noted. |
|---|

|
18Fluorine [18F] is emerging as one of the most prominent radionuclides in the application of positron emission tomography (PET), a fundamental technology in precision medicine for (pre)clinical imaging.26 The preparation of radioactive molecules containing a [18F] fluorinated tertiary stereocenter remains a critical challenge. These motifs cannot be synthesized by nucleophilic substitution of alkylsulfonates with [18F]KF-K222, and current synthetic methodologies are limited in scope.9,27 We wondered whether our fluorination reaction could be a suitable methodology for the synthesis of [18F] tertiary allylic fluorides. The initial experiments were performed with α-methylstyrene, reagent 2a, Rh2(esp)2, and [18F]KF-K222 (∼0.2 GBq). The latter [18F]-fluoride source was added at −50 °C, and the reaction mixture was warmed until −30 °C during 20 min. Unfortunately, the radiolabelled product [18F](±)-3a was not detected, probably due to the low solubility of [18F]KF-K222. However, when using [18F]tetraethylammonium fluoride [18F]TEAF (∼0.2 GBq),28 labelled product [18F](±)-3a was formed in 10% ± 2% (n = 3) of radiochemical conversion (RCC). In addition, we observed that the radiolabelling could also work with other substrates ([18F](±)-3j, 9% ± 2% (n = 3) RCC). However, attempts to improve the RCC were unsuccessful (Table 3).29
Conclusions
In summary, we have developed a new synthetic methodology for the construction of fluorinated tertiary stereocenters from 1,1-disubstituted alkenes. The process relies on the generation of tertiary allyl cations, mediated by a catalytically-generated Rh-carbynoid, that undergoes nucleophilic fluorination with an excellent branched/selectivity ratio. Notable features of this process are the broad scope of 1,1-disubstituted alkenes, including natural products and drug molecule derivatives, applications in the synthesis of a fluorinated drug molecule – (±)-F-flurbiprofen – and its translation to radiofluorination with [18F]TEAF. The generality of our methodology and synthetic applications, based on a Rh-catalyzed carbyne transfer with alkenes, stands as a testament of its potential utility to expand the chemical space in fluorine-based drug design.Data availability
The data for this work, including optimization tables, general experimental procedures, characterization data for all new compounds and X-ray data are provided in the ESI.†Author contributions
L. J. discovered the fluorination reaction. L. J., P. S. & W. J. T. performed the experiments, and all authors contributed to the analysis and interpretation of the data. P. S. carried out the radiofluorination under the supervision and guidance of J. L. M. G. S. directed the project and wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The European Research Council (ERC-CoG 2019, 865554), Agencia Estatal de Investigación (AEI) of the Ministerio de Ciencia e Innovación (EUR2019-103814, PID2019-104101GB-I00, PID2020-117656RB-100, Severo Ochoa Excellence Accreditation 2020–2023 -CEX2019-000925-S, and Maria de Maeztu Excellence Accreditation MDM-2017-0720), ICIQ Foundation, and the CERCA Programme (Generalitat de Catalunya) are gratefully acknowledged for financial support. We thank the European Union for Marie Skłodowska-Curie Individual Fellowships (794815 to L. J.; 101028657 to W. J. T.), as well as AEI for a FPI pre-doctoral fellowship (BES-2017-080163) (to P. S.). Oscar Moreno and personnel at the Radiochemistry Platform of CIC biomaGUNE are gratefully acknowledged for experimental support.Notes and references
- (a) M. C. Pacheco, S. Purser and V. Gouverneur, Chem. Rev., 2008, 108, 1943 CrossRef CAS PubMed; (b) D. O’hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (d) Y. Zhu, J. Han, J. Wang, N. Shibata, M. Sodeoka, V. A. Soloshonok, J. A. S. Coelho and F. D. Toste, Chem. Rev., 2018, 118, 3887 CrossRef CAS PubMed; (e) R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824 CrossRef CAS PubMed; (f) A. M. Sorlin, F. O. Usman, C. K. English and H. M. Nguyen, ACS Catal., 2020, 10, 11980 CrossRef CAS.
- (a) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed; (d) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed; (e) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633 CrossRef CAS PubMed.
- For recent selected examples: (a) B. Greedy, J. M. Paris, T. Vidal and V. Gouverneur, Angew. Chem., Int. Ed., 2003, 42, 3291 CrossRef CAS PubMed; (b) R. J. Phipps, K. Hiramatsu and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 8376 CrossRef CAS PubMed; (c) J. Wu, Y. M. Wang, A. Drljevic, V. Rauniyar, R. J. Phipps and F. Dean Toste, Proc. Natl. Acad. Sci., 2013, 110, 13729 CrossRef CAS PubMed; (d) X. Yang, R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5225 CrossRef CAS PubMed; (e) W. Yuan and K. J. Szabõ, Angew. Chem., Int. Ed., 2015, 54, 8533 CrossRef CAS PubMed; (f) R. Guo, J. Huang and X. Zhao, ACS Catal., 2018, 8, 926 CrossRef CAS; (g) Q. Wang, M. Lübcke, M. Biosca, M. Hedberg, L. Eriksson, F. Himo and K. J. Szabó, J. Am. Chem. Soc., 2020, 142, 20048 CrossRef CAS PubMed; (h) Z. Liu, L. J. Oxtoby, M. Liu, Z. Q. Li, V. T. Tran, Y. Gao and K. M. Engle, J. Am. Chem. Soc., 2021, 143, 8962 CrossRef CAS PubMed; (i) J. Cao, H. Wu, Q. Wang and J. Zhu, Nat. Chem., 2021, 13, 671 CrossRef CAS PubMed.
- For recent selected examples: (a) Y. Liang and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 5520 CrossRef CAS PubMed; (b) Z. Jiao, J. J. Beiger, Y. Jin, S. Ge, J. S. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15980 CrossRef CAS PubMed; (c) K. Balaraman and C. Wolf, Angew. Chem., Int. Ed., 2017, 56, 1390 CrossRef CAS PubMed; (d) T. W. Butcher and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 13125 CrossRef CAS PubMed; (e) Z. T. He, X. Jiang and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 13066 CrossRef CAS PubMed; (f) J. Liu, Q. Yuan, F. D. Toste and M. S. Sigman, Nat. Chem., 2019, 11, 710 CrossRef CAS PubMed; (g) T. W. Butcher, J. L. Yang, W. M. Amberg, N. B. Watkins, N. D. Wilkinson and J. F. Hartwig, Nature, 2020, 583, 548 CrossRef CAS PubMed; (h) E. D. Kalkman and J. F. Hartwig, J. Am. Chem. Soc., 2021, 143, 11741 CrossRef CAS PubMed For a review:; (i) T. W. Butcher, W. M. Amberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2022, 61, 1 CrossRef PubMed.
- For recent selected examples: (a) M. H. Katcher, A. Sha and A. G. Doyle, J. Am. Chem. Soc., 2011, 133, 15902 CrossRef CAS PubMed; (b) J. J. Topczewski, T. J. Tewson and H. M. Nguyen, J. Am. Chem. Soc., 2011, 133, 19318 CrossRef CAS PubMed; (c) W. Liu, X. Huang, M. Cheng, R. J. Nielsen, W. a G. Iii and J. T. Groves, Science, 2012, 337, 1322 CrossRef CAS PubMed; (d) M. Braun and A. G. Doyle, J. Am. Chem. Soc., 2013, 4, 1 Search PubMed; (e) Z. Lu, X. Zeng, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2017, 139, 18202 CrossRef CAS PubMed; (f) X. Bertrand and J. F. Paquin, Org. Lett., 2019, 21, 9759 CrossRef CAS PubMed; (g) D. Bafaluy, Z. Georgieva and K. Muñiz, Angew. Chem., Int. Ed., 2020, 59, 14241 CrossRef CAS PubMed; (h) H. A. Sharma, K. M. Mennie, E. E. Kwan and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 16090 CrossRef CAS PubMed; (i) H. J. Tang, X. Zhang, Y. F. Zhang and C. Feng, Angew. Chem., Int. Ed., 2020, 59, 5242 CrossRef CAS PubMed; (j) H. J. Tang, B. Zhang, F. Xue and C. Feng, Org. Lett., 2021, 23, 4040 CrossRef CAS PubMed; (k) H. Qian, J. Chen, B. Zhang, Y. Cheng, W.-J. Xiao and J.-R. Chen, Org. Lett., 2021, 23, 6987 CrossRef CAS PubMed; (l) I. N.-M. Leibler, M. A. Tekle-Smith and A. G. Doyle, Nat. Commun., 2021, 12, 6950 CrossRef CAS PubMed; (m) Y. Zhang, A. N. Fitzpatrick, M. Das, I. P. Bedre, H. G. Yayla, M. S. Lall and P. Z. Musacchio, Chem. Catal., 2022, 2, 1 CrossRef.
- For alternative catalytic methods: (a) C. Sandford, R. Rasappan and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10100 CrossRef CAS PubMed; (b) E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744 CrossRef CAS PubMed; (c) G. H. Lovett, S. Chen, X. S. Xue, K. N. Houk and D. W. C. MacMillan, J. Am. Chem. Soc., 2019, 141, 20031 CrossRef CAS PubMed; (d) É. Vincent and J. Brioche, Eur. J. Org. Chem., 2021, 2021, 2421 CrossRef.
- For a review in catalytic C–C bond functionalizations: L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef CAS PubMed.
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed.
- E. W. Webb, J. B. Park, E. L. Cole, D. J. Donnelly, S. J. Bonacorsi, W. R. Ewing and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 9493 CrossRef CAS PubMed.
- (a) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401 CrossRef CAS PubMed; (b) J. Xiang, M. Shang, Y. Kawamata, H. Lundberg, S. H. Reisberg, M. Chen, P. Mykhailiuk, G. Beutner, M. R. Collins, A. Davies, M. Del Bel, G. M. Gallego, J. E. Spangler, J. Starr, S. Yang, D. G. Blackmond and P. S. Baran, Nature, 2019, 573, 398 CrossRef CAS PubMed; (c) Z. Wang, C. Y. Guo, C. Yang and J. P. Chen, J. Am. Chem. Soc., 2019, 141, 5617 CrossRef CAS PubMed.
- (a) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490 CrossRef CAS PubMed; (b) C. R. Pitts, B. Ling, J. A. Snyder, A. E. Bragg and T. Lectka, J. Am. Chem. Soc., 2016, 138, 6598 CrossRef CAS PubMed; (c) S. M. Banik, K. M. Mennie and E. N. Jacobsen, J. Am. Chem. Soc., 2017, 139, 9152 CrossRef CAS PubMed; (d) N. O. Ilchenko, M. Hedberg and K. J. Szabó, Chem. Sci., 2017, 8, 1056 RSC; (e) J. B. Roque, Y. Kuroda, L. T. Göttemann and R. Sarpong, Science, 2018, 361, 171 CrossRef CAS PubMed; (f) M. M. Wang and J. Waser, Angew. Chem., Int. Ed., 2020, 59, 16420 CrossRef CAS PubMed; (g) V. Lanke and I. Marek, J. Am. Chem. Soc., 2020, 142, 5543 CrossRef CAS PubMed; (h) J. Ren, F.-H. Du, M.-C. Jia, Z.-N. Hu, Z. Chen and C. Zhang, Angew. Chem., Int. Ed., 2021, 60, 24171 CrossRef CAS PubMed For recent examples involving cyclobutanol or bicyclic azaarene derivatives, see:; (i) Y. C. Lu and J. G. West, ACS Catal., 2021, 11, 12721 CrossRef CAS; (j) M. Komatsuda, A. Suto, H. Kondo, H. Takada, K. Kato, B. Saito and J. Yamaguchi, Chem. Sci., 2022, 13, 665 RSC.
- (a) Z. Wang, L. Jiang, P. Sarró and M. G. Suero, J. Am. Chem. Soc., 2019, 141, 15509 CrossRef CAS PubMed see also:; (b) Z. Wang, A. G. Herraiz, A. M. del Hoyo and M. G. Suero, Nature, 2018, 554, 86 CrossRef CAS PubMed; (c) L. Jiang, Z. Wang, M. Armstrong and M. G. Suero, Angew. Chem., Int. Ed., 2021, 60, 6177 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed; (b) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS PubMed; (c) H. M. L. Davies and K. Liao, Nat. Rev. Chem., 2019, 3, 347 CrossRef CAS PubMed.
- (a) I. Fleming, Molecular Orbitals and Organic Chemical Reactions, John Wiley & Sons, 2009, p. 142 CrossRef; (b) H. Mayr, W. Forner and P. von Ragué Schleyer, J. Am. Chem. Soc., 1980, 102, 3663 CrossRef.
- Previous reports proposing fluorination of allyl cations showed a preference of fluoride for the most substituted electrophilic carbon site: (a) M. Kirihara, T. Kambayashi and T. Momose, Chem. Commun., 1996, 1103 RSC; (b) J. C. Mixdorf, A. M. Sorlin, Q. Zhang and H. M. Nguyen, ACS Catal., 2018, 8, 790 CrossRef CAS See also ref 5l..
- We previously observed that bromide nucleophiles were converted selectively to the linear isomer. See ref. 12a.
- S. Takizawa, F. A. Arteaga, K. Kishi, S. Hirata and H. Sasai, Org. Lett., 2014, 16, 4162 CrossRef CAS.
- C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378 CrossRef CAS.
- A. Vasilopoulos, D. L. Golden, J. A. Buss and S. S. Stahl, Org. Lett., 2020, 22, 5753 CrossRef CAS PubMed.
- An experiment carried out under the same reaction conditions using water (10 equiv.) instead of TBAF·3H2O provided alcohol (±)-4 in 74% yield and with excellent branched selectivity (b:l > 20:1).
- Et3N·3HF was an effective fluorinating reagent for the synthesis of a secondary allyl fluoride derived from a reaction with cyclohexene, reagent 2a under Rh(II) catalysis, see ref. 12a.
- (a) K. M. Engle, L. Pfeifer, G. W. Pidgeon, G. T. Giuffredi, A. L. Thompson, R. S. Paton, J. M. Brown and V. Gouverneur, Chem. Sci., 2015, 6, 5293 RSC; (b) S. Liang, G. B. Hammond and B. Xu, Chem.–Eur. J., 2017, 23, 17850 CrossRef CAS PubMed.
- See the ESI† for further optimization.
- (a) C. F. Meyer, S. M. Hell, A. Misale, A. A. Trabanco and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 8829 CrossRef CAS PubMed; (b) I. G. Molnár and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 5004 CrossRef PubMed; (c) S. Meyer, J. Häfliger and R. Gilmour, Chem. Sci., 2021, 12, 10686 RSC.
- (a) H. Fujisawa, T. Fujiwara, Y. Takeuchi and K. Omata, Chem. Pharm. Bull., 2005, 53, 524 CrossRef CAS PubMed; (b) I. T. Schiefer, S. Abdul-Hay, H. Wang, M. Vanni, Z. Qin and G. R. J. Thatcher, J. Med. Chem., 2011, 54, 2293 CrossRef CAS PubMed.
- (a) I. T. Schiefer, S. Abdul-Hay, H. Wang, M. Vanni, Z. Qin and G. R. J. Thatcher, J. Med. Chem., 2011, 54, 2293 CrossRef CAS PubMed; (b) Z. Li and P. S. Conti, Adv. Drug Delivery Rev., 2010, 62, 1031 CrossRef CAS PubMed; (c) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS PubMed.
- (a) M. A. Cortés González, P. Nordeman, A. Bermejo Gómez, D. N. Meyer, G. Antoni, M. Schou and K. J. Szabó, Chem. Commun., 2018, 54, 4286 RSC; (b) W. Liu, X. Huang, M. S. Placzek, S. W. Krska, P. McQuade, J. M. Hooker and J. T. Groves, Chem. Sci., 2018, 9, 1168 RSC; (c) S. Verhoog, A. F. Brooks, W. P. Winton, B. L. Viglianti, M. S. Sanford and P. J. H. Scott, Chem. Commun., 2019, 55, 6361 RSC.
- (a) J. Zheng, R. Cheng, J. H. Lin, D. H. Yu, L. Ma, L. Jia, L. Zhang, L. Wang, J. C. Xiao and S. H. Liang, Angew. Chem., Int. Ed., 2017, 56, 3196 CrossRef CAS PubMed; (b) J. B. I. Sap, T. C. Wilson, C. W. Kee, N. J. W. Straathof, C. W. a. Ende, P. Mukherjee, L. Zhang, C. Genicot and V. Gouverneur, Chem. Sci., 2019, 10, 3237 RSC.
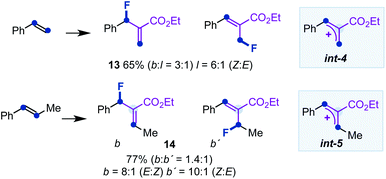
- To complement the study carried out with α-substituted styrenes, we performed experiments with styrene or β-methyl-styrene and 2a, under the optimized reaction conditions. The reactions provided a modest branched/linear ratio of allyl fluorides 13 or an equimolecular mixture of branched isomers 14, respectively (see below). The results clearly indicate that the substitution pattern of the allyl cations int-4 and int-5 is directly responsible for the charge distribution and reactivity towards the fluorination
 .
.

Footnote |
| † Electronic supplementary information (ESI) available. CCDC 20927292092730. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00968d |
| This journal is © The Royal Society of Chemistry 2022 |