
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable and highly controlled aryl couplings revealed by systematic assessment of photoactivatable linkers†
Veit G.
Haensch
 a,
Toni
Neuwirth
a,
Alexander
Bergner
a,
Jakob
Bruhnke
a,
Florian
Kloss
b and
Christian
Hertweck
*ac
a,
Toni
Neuwirth
a,
Alexander
Bergner
a,
Jakob
Bruhnke
a,
Florian
Kloss
b and
Christian
Hertweck
*ac
aDepartment of Biomolecular Chemistry, Leibniz Institute for Natural Product Research and Infection Biology (HKI), Beutenbergstr. 11a, 07745 Jena, Germany. E-mail: christian.hertweck@leibniz-hki.de
bTransfer Group Anti-infectives, Leibniz Institute for Natural Product Research and Infection Biology (HKI), 07745 Jena, Germany
cFaculty of Biological Sciences, Friedrich Schiller University Jena, 07743 Jena, Germany
First published on 13th April 2022
Abstract
The controlled synthesis of biphenyls, which play a prominent role in pharmaceuticals, agrochemicals, and liquid crystals, typically requires hazardous organometallic reagents, aryl halides, and heavy metal catalysts. We recently reported a metal-free, photochemical alternative (“photosplicing”) for the selective preparation of a wide range of pharmaceutically important biphenyls. Whereas the traceless sulfonamide linker enables and controls the aryl coupling, unwanted toxic byproducts are released. Therefore, we designed over 25 different temporary linkers and tested them for their suitability for the photosplicing reaction in a flow reactor. We found that a surprisingly high number of functional groups enable light-induced aryl fusion and identified a number of linkers for environmentally friendly procedures. We also report that a thiol-ene (click) – photosplicing sequence enables a convenient route to biaryls such as liquid crystals. This work sheds light on thus far neglected photochemistry of temporary linkers, reduces toxic byproducts, and expands the available starting materials for metal-free biphenyl synthesis.
1 Introduction
The biaryl motif is an important functional group that is widely used in dyes, liquid crystals, ligands for metal catalysis, agrochemicals, polymers, and pharmaceuticals.1–3 To gain access to this prominent substructure, a large variety of synthetic protocols have been developed.4–6 The commonly used avenue to biaryls is the metal-catalyzed cross-coupling of an organometallic reagent to an aryl halide.7 While this process benefits from high efficacy and regioselectivity of the coupling, it requires energy-intensive, hazardous organometallic reagents and toxic redox-active heavy metals.8 Although metal-free alternatives can overcome some of these drawbacks, they often suffer from lack of selectivity or a narrow substrate scope.1–3,9,10Recently, we discovered a novel, metal-free aryl cross-coupling reaction that is highly selective and provides access to a broad range of substituted biaryls in good yields. We found that sulfonamide precursors (e.g.1a or 1b) undergo a photochemical rearrangement to biphenyls (e.g.2a in 92% or 2b in 76%) with extrusion of the linker, termed photosplicing (Scheme 1A).11 However, precursor synthesis has so far required the use of corrosive sulfonic acid chlorides and harmful benzylamines, with the additional disadvantage of low atomic efficiency. Specifically, the aryl moieties are tethered via a linker by base-mediated nucleophilic substitution in a non-protic solvent (e.g. dichloromethane (DCM)) together with a base (Scheme 1A). For aryl coupling, the sulfonamide precursor is dissolved in a suitable solvent (e.g. acetonitrile (MeCN)) and irradiated with UV-C light to enable the photosplicing reaction. The major drawback of the reaction is the release of toxic gases (sulfur dioxide, ammonia and formaldehyde) from the fragmented linker.12 Furthermore, H2 and CO can result from subsequent reactions of formaldehyde with UV-C light.13
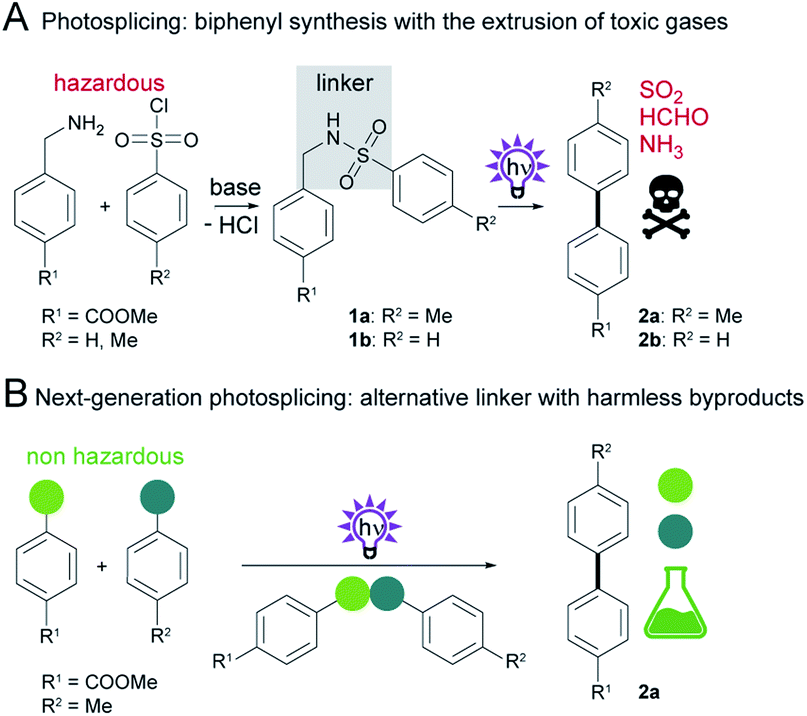 | ||
| Scheme 1 Synthesis of biphenyls. (A) Two-step photosplicing reaction starting from a sulfonic acid chloride and benzylamine. (B) This work describes the selective and additive-free photochemically induced aryl coupling from non-hazardous educts without the formation of toxic byproducts. | ||
Although the photosplicing reaction could be employed as an alternative to the metal-catalyzed aryl cross-coupling, the formation of toxic byproducts and the use of some hazardous reagents and solvents are suboptimal and do not meet the requirements for “green” syntheses.14
Here, we report a systematic examination of the impact of linker composition on the formation of both the desired products and byproducts in photosplicing reactions (Scheme 1B). In this way we identified various modifications to the photosplicing reaction that enable biphenyl synthesis in a more environmentally friendly and sustainable manner. The optimized process relies on thioether linkers and has several benefits: the use of harmless reactants, additive-free precursor synthesis without solvent or purification, irradiation at less harmful wavelength in ethanol and avoiding the formation of toxic byproducts.
2 Results and discussion
We reasoned that the sulfonamide linker (SO2–NH–CH2) (Scheme 1A) used for the photosplicing reaction has potential for modification. Our goal was to find alternative linkers that allow a broader range of reactants while making the photosplicing reaction more sustainable. Thus, we systematically exchanged individual atoms in the linker and monitored the potential production of the desired biphenyl and the – ideally non-toxic – byproducts of the reaction.2.1 Substitution of the NH group in the linker
First, we evaluated the impact of NH substitutions on biphenyl yields and byproduct formation (Scheme 2A). According to the proposed reaction mechanism,12 the irradiation of an N-methyl derivative would form methylamine instead of ammonia. To test this, we prepared sulfonamide 1c,12 irradiated a MeCN solution in an NMR tube, and treated the crude solution with trityl chloride (TrCl). The mixture was analyzed by GC-MS, showing a new compound with a mass that is consistent with TrNHMe. The identity of the product was verified by comparison of the EI fragmentation pattern and retention times with a synthetic TrNHMe reference (Scheme 2B).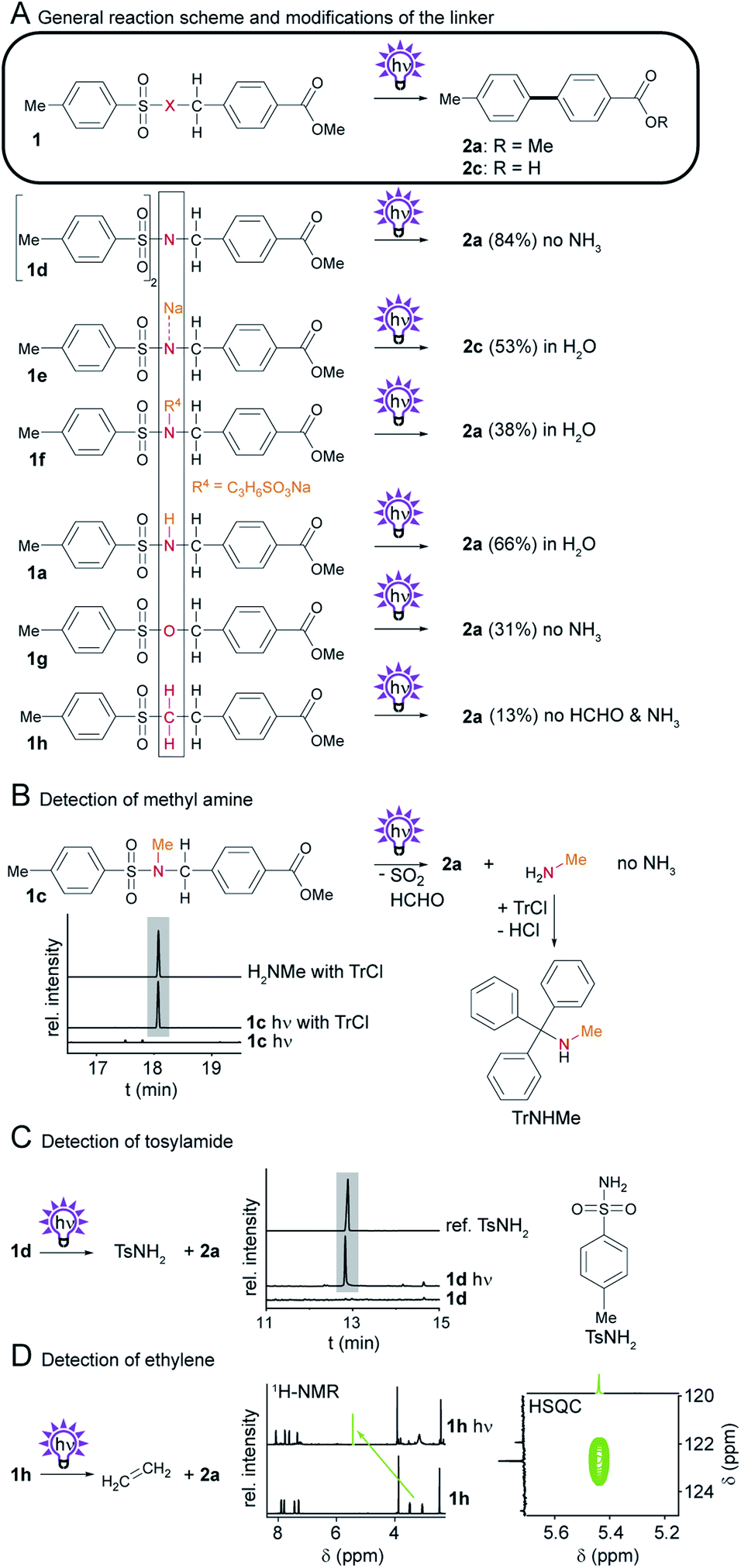 | ||
| Scheme 2 Investigation of the photochemistry of compounds with modifications to X, the NH group in the sulfonamide 1a. (A) General reaction scheme, and structures of derivatives. Isolated yields obtained by photosplicing are given in brackets. (B) Irradiation of 1c and detection of MeNH2 by GC-MS. (C) Irradiation of 1d and detection of TsNH2 by GC-MS. (D) Irradiation of 1h and detection of ethylene with 1H-NMR and HSQC experiments. | ||
Since N-substitutions do not hamper the biaryl formation, we next considered a reaction involving aromatic sulfonamides that are not basic or volatile as byproducts. According to the reaction mechanism, tosylamide (TsNH2) would result as a byproduct of the irradiation of ditolylsulfimides. To test this, we synthesized ditolylsulfimide 1d by treating 1a with 4-dimethylaminopyridine (DMAP) and tosyl chloride (TsCl). After irradiation, GC-MS analysis of the crude photoproducts indicated the formation of biphenyl 2a along with TsNH2, which was verified by comparison with an authentic reference and its characteristic EI-MS fragmentation pattern (Scheme 2C and Fig. S7†).
Furthermore, the amine functionality turned out to be a useful handle to modify the properties of the sulfonamide, such as increased water solubility. Initially, the use of water as a solvent for the photoreaction was hampered by the very low solubility of sulfonamide 1a. By NaOMe-mediated deprotonation of the acidic N–H proton15 of sulfonamide 1a we generated a water-soluble anion 1e. Irradiation of 1e in water, however, gave hydrolyzed biphenyl 2c. Since this approach is not applicable for base-labile functional groups and the use of an inert photoreactor is required (see ESI†), we tested an alternative approach by means of a water solubility-conferring moiety.
The concept of a water-soluble sulfonamide could be realized with compound 1f, which was formed from 1e and 1,3-propane sultone. Irradiation of an aqueous solution of 1f yielded biphenyl 2a, albeit with decreased yield compared to the model reaction with 1a. Since the photoreaction also takes place in solid state on TLC plates,11 we reasoned that a suspension of 1a in water could also give the desired results. To produce a suspension of small crystals, we precipitated a concentrated solution of 1a in MeCN with an excess of water (see ESI†). This suspension in water was irradiated to form 2a in up to 66% yield.
Beyond N-substitutions, we explored the possibility of replacing the amine with either oxygen or a methylene group. Therefore, we prepared the highly labile tosylbenzyl ester 1g from the corresponding benzyl alcohol and TsCl, whereas sulfone 1h was prepared from sodium benzenesulfinate and 2-phenylethyl bromide (see ESI†). Remarkably, both the sulfonate ester 1g and the sulfone 1h provide 2a upon UV irradiation. Advantageously, neither ammonia nor formaldehyde should be formed in the photoreactions of sulfone 1h. To identify the byproduct from the photoreaction of sulfone 1h, we irradiated a solution of 1h in a fused silica NMR tube and observed the formation of a new singlet representing a proton resonating at δ = 5.41 ppm (Scheme 2D). The suggestion that ethylene is formed as a byproduct is further supported by 13C-NMR and HSQC spectra, which show the characteristic chemical shifts and cross peak of ethylene.16
2.2 Substitution of the CH2 group in the linker
We also sought to determine the impact of CH2-group substitutions on biphenyl yields and byproduct formation (Scheme 3A). Sulfonamide 1i12 with two methyl groups at the methylene group was predicted to form acetone in lieu of harmful formaldehyde. We tested this by adding 2,4-dinitrophenylhydrazine (DNHP) to an irradiated solution of 1i. Indeed, the acetone-derived hydrazone could be detected by HLPC-HRMS (Scheme 3B), and the identity of the acetone adduct was verified by comparison of the UV-vis spectra, HRMS data (see ESI†) and retention time with an analytical reference.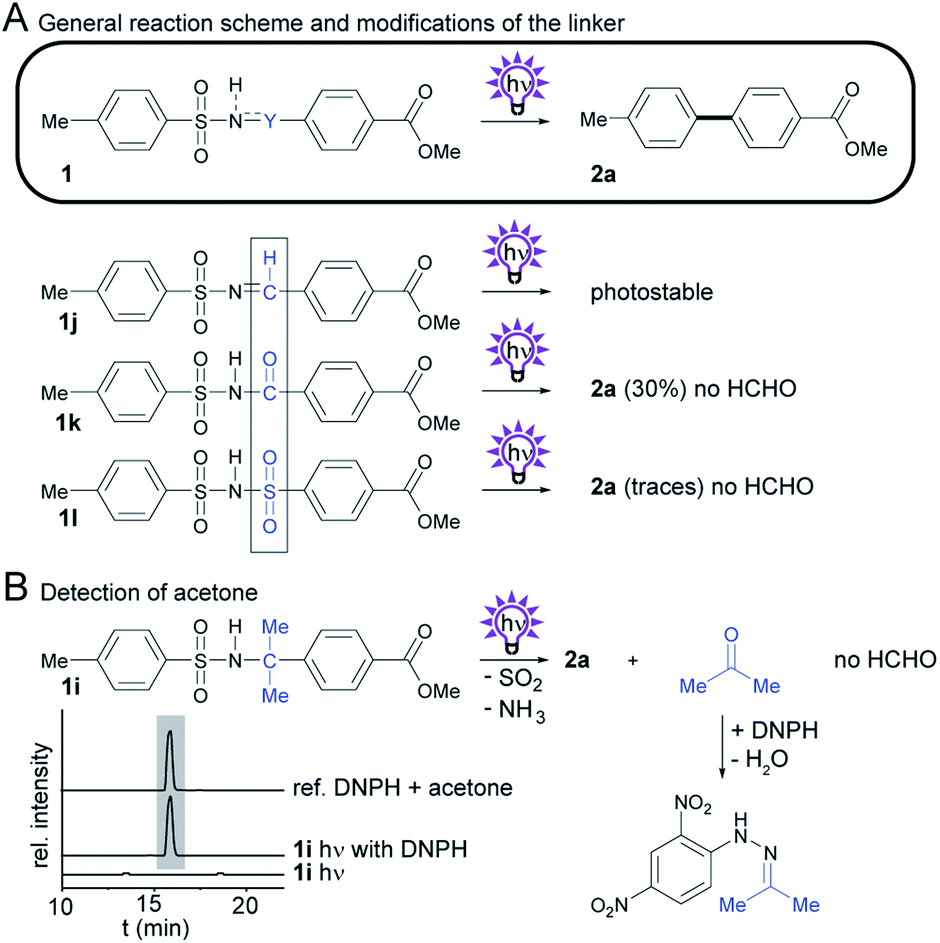 | ||
| Scheme 3 Investigation of the photochemistry of compounds with modifications to Y, the CH2 group in the sulfonamide 1a. (A) General reaction scheme, and structures of derivatives. Isolated yields obtained by photosplicing are given in brackets. (B) Detection of acetone by HPLC-HRMS. | ||
To learn more about the effect of the methylene group in the linker, we next altered its oxidation state to +1 and +3 and tested the dehydrogenated and oxygenated derivatives (1j and 1k) in the photosplicing reaction. We reasoned that a methine group could produce formic acid, whereas the amide carbon could form carbon dioxide after hydrolysis. To test this, we prepared tosylimine 1j by condensing TsNH2 with the corresponding benzaldehyde in the presence of P4O10. This compound proved to be surprisingly photostable, and no biphenyl 2a was detected upon irradiation. In contrast, tosylimide 1k forms biphenyl 2a under same conditions, but with low selectivity compared to 1a. Recently, a similar method using N-methyl-aroylsulfonamides has been reported, but this procedure is less attractive from an environmental point of view because toxic methyl isocyanate is formed as the byproduct.17
We have also replaced the carbonyl in 1k for a sulfone group to give bis-sulfonimide 1l, which has a linker containing two sulfone groups. In analogy to sulfonamides, this functional group also undergoes rearrangement upon exposure to UV light to form biphenyl 2a, yet only in trace amounts.
2.3 Variation of quantity and position of CH2 groups in the linker
As another way to modify the byproducts of the photosplicing reaction, we synthesized and tested linker derivatives with different positions and numbers of methylene groups (Scheme 4). These derivatives are particularly interesting because they stress the theory of the required horseshoe conformation, in which both aryl groups are superimposed prior to C–C coupling.12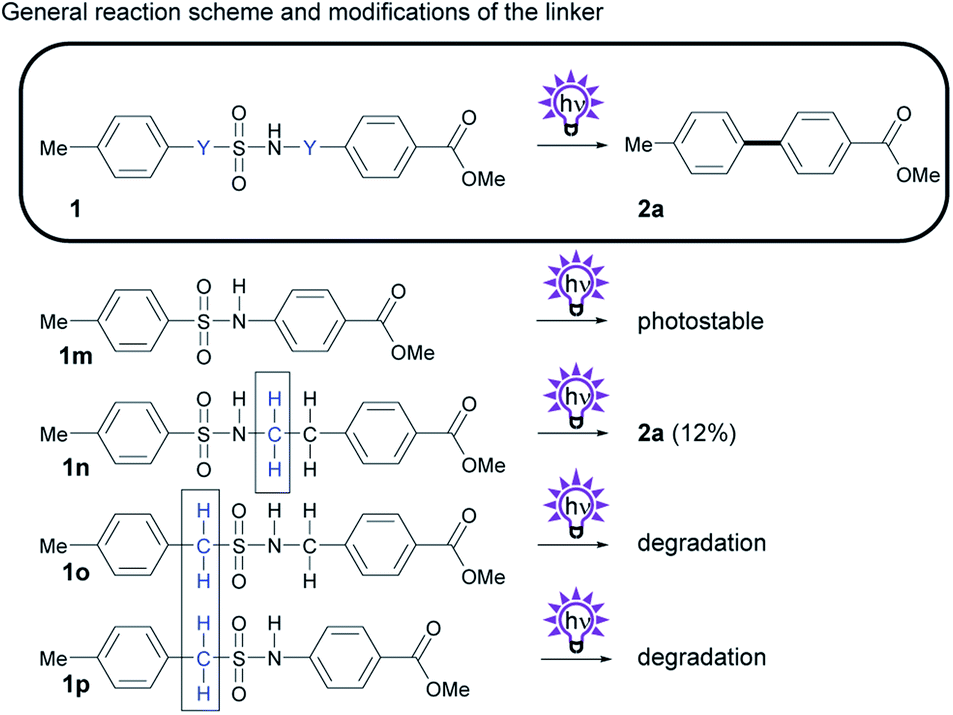 | ||
| Scheme 4 Investigation of the photochemistry of compounds with modifications of the number and position of Y, the CH2 group in the sulfonamide 1a. General reaction scheme, and structures of derivatives. Isolated yields obtained by photosplicing are given in brackets. | ||
We synthesized a shorter version of the sulfonamide linker without the methylene group by reacting TsCl with 4-aminobenzoic acid methyl ester to give sulfonamide 1m (Scheme 4). While removal of the methylene group would prevent the formation of formaldehyde, we suspected that this truncation would hinder the cyclic transition state.12,18 As we suspected, subsequent UV irradiation did not form biphenyl 2a.
We then attempted to add an additional methylene group to the sulfonamide linker to investigate whether an extended linker with four atoms could still facilitate biphenyl formation upon irradiation. The generation of 2a from 1n indicates that photosplicing is still possible, albeit with reduced yields. The fact that linkers with four atoms also rearrange to form biphenyls makes the occurrence of intramolecular excimers, in which one excited aryl residue directly interact with the other aryl residue in the ground state, unlikely because they are too far apart from each other.19
To evaluate whether the position of the methylene group plays an essential role in the aryl coupling, we synthesized two structural isomers of 1a and 1n (1p and 1o) with a methylene group between the aromatic ring and the sulfur of the sulfonamide moiety. These two compounds are unable to rearrange into biphenyl 2a upon irradiation, although they have three or four atoms in each linker. These results indicate that sulfur with a relatively weak carbon–sulfur bond must directly be attached to the aromatic core.
2.4 Substitution of the SO2 group in the linker
To avoid the formation of toxic sulfur dioxide, we sought to replace the SO2 group in the sulfonamide linker (Scheme 5). Therefore, we considered using phenylphosphonic acid derivatives as an alternative that would not generate volatile byproducts. It has been reported that phenyl phosphates generate biphenyls upon irradiation.20 This photoreaction proceeds via a singlet-excited intramolecular excimer21 and involves radicals.22 We first synthesized phosphinic amide 1q from PhPOCl2 and a benzyl amine. We found that the photoreaction with 1q yields small amounts of biphenyl 2b and, to our surprise, biphenyl 2d, which is a result of undesired homocoupling. Phosphonic amide 1r shows similar photochemistry to 1q and produces biphenyl 2b but not the homocoupling product 2d. Owing to the very low yields of biphenyl formed from phosphonic and phosphinic amides, however, phosphorous compounds are a poor alternative to the sulfonamide linker. Notably, substitution of phosphor for silicon or carbon (1s or 1t) yields no biphenyls after irradiation.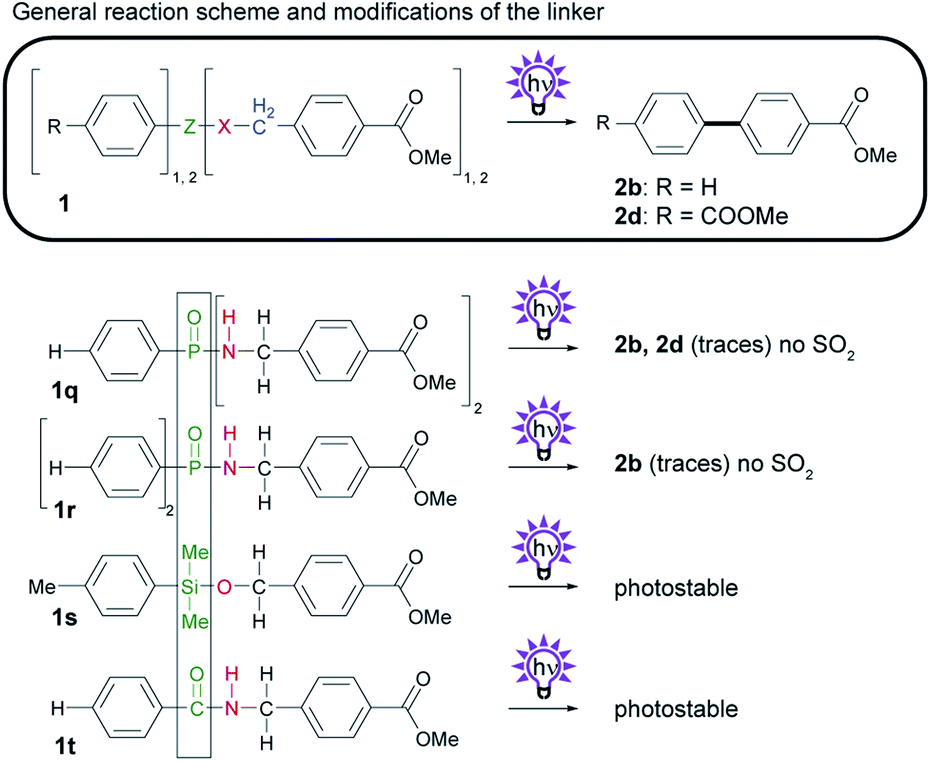 | ||
| Scheme 5 Investigation of the photochemistry of compounds with modifications to Z and X, the SO2 and NH group in sulfonamide 1a. General reaction scheme, and structures of derivatives. Isolated yields obtained by photosplicing are given in brackets. | ||
2.5 Further simplification of the linker system
To identify the essential components of the linker and to facilitate the synthetic access to biphenyl precursors we aimed to further simplify the linker system and probed sulfoxides and thioethers (Scheme 6A). Therefore, we prepared the regioisomeric phenylethyl phenyl thioethers (1v and 1w) from the corresponding thiophenols and phenylethyl bromides, and sulfoxide 1u by oxidation of 1v. We found that irradiation of the sulfoxide provides biphenyl 2a in 15% yield. To our surprise, even the substituted phenylethyl phenyl thioether 1v provides the desired photosplicing product upon irradiation, along with some side products from thioether cleavage and benzyl cleavage (Fig. S27†). By GC-MS of the bromine adduct we concluded that ethylene is formed as a byproduct, as during the photoreaction of 1h (Scheme 6B, Fig. S14 and S15†). Considering the overall oxidation state, elemental sulfur is the other byproduct, which was detected as diphenyl sulfide by GC-MS after the addition of phenylmagnesium bromide to irradiated thioether 1v (Scheme 6B, Fig. S17 and S18†). Notably, the isolated yields (49%) are better than those in the reaction with the corresponding sulfone (1h).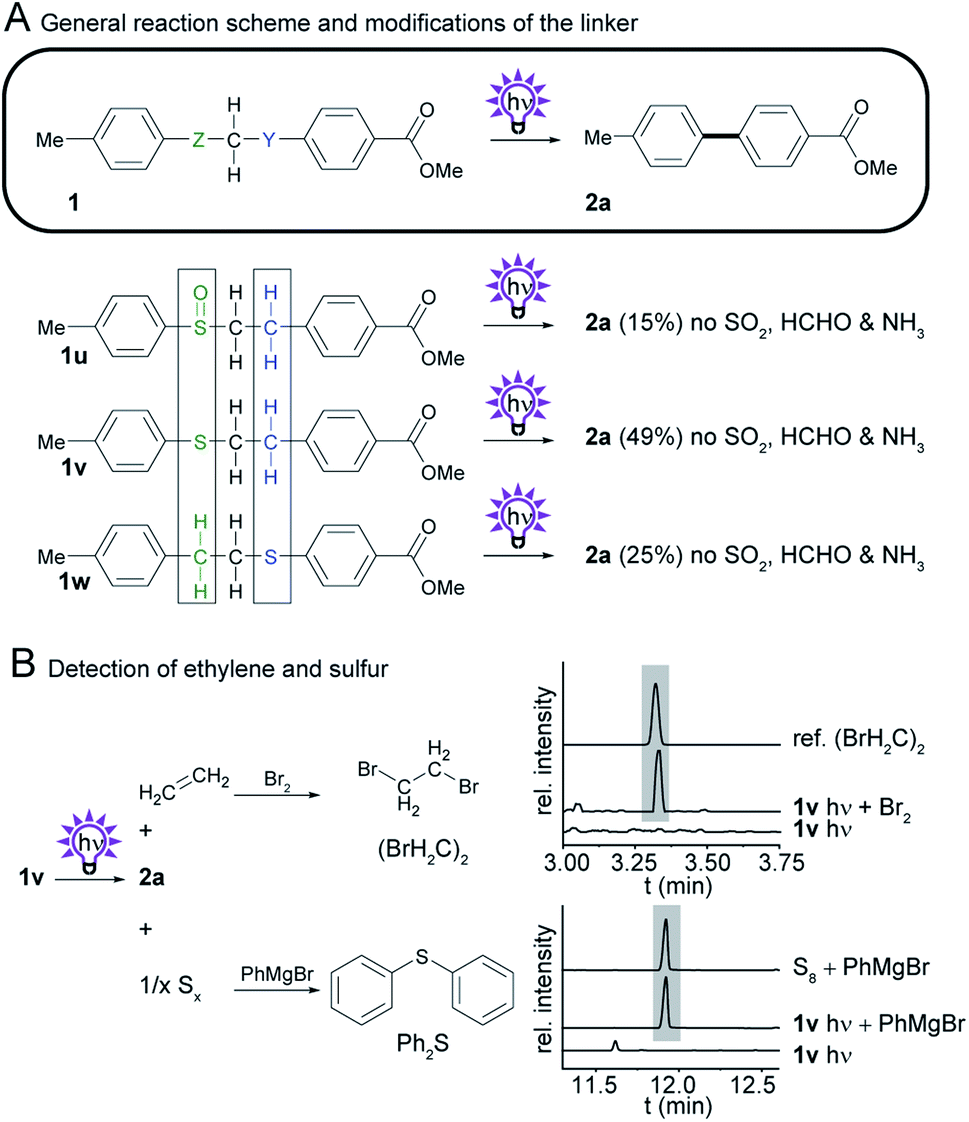 | ||
| Scheme 6 Investigation of the photochemistry of compounds with modifications to Z and Y, the SO2 and CH2 group in sulfone 1h. (A) General reaction scheme, and structures of derivatives. Isolated yields obtained by photosplicing are given in brackets. (B) Detection of ethylene and sulfur with GC-MS. | ||
We found that thioether 1w, in which the linker is flipped, is also suitable for photosplicing but gives lower yields of 2a (25%) compared to 1v. Experiments at the analytical scale revealed that a broad range of substituents, such as methyl-, cyano-, chloro-, hydroxy-, carboxy-, amino-, and trifluoromethyl-, is tolerated by the method (see Fig. S21 and S22†). Thus, we expect a similar scope compared to the original photosplicing reaction. Limiting factors are certainly photolabile groups such as iodo- and nitro-groups. The findings that phenylethyl phenyl thioethers are useful precursors for the production of biphenyls is remarkable.
We also probed other precursors with two methylene carbons and selenium,23 oxygen, nitrogen or sulfur in different positions (1x to 1ac) and noted that biphenyl 2a is not formed by irradiation of these compounds (Fig. S19†). The observation that ethers are not suitable for photosplicing is noteworthy in the context of the reported intramolecular [2+2] photocycloaddition in naphthylethyl resorcinolethers.24 O–B–O, O–S–O, O–Si–O, and O–C–O linkers do not form biphenyls after irradiation, either.25,26 Thioether-based linkers in photosplicing reactions benefit from acceptable yields of biphenyl as well as the formation of non-toxic byproducts, making them the ideal alternative to sulfonamides.
2.6 Combination of thiol-ene-reaction and photosplicing
The use of the phenylethyl phenyl thioether system as in 1v is by far the most environmentally friendly linker system with good yield of the corresponding biphenyl. Instead of ammonia, sulfur dioxide and formaldehyde, non-toxic ethylene, and elemental sulfur are formed as byproducts. Initially, we prepared the precursor thioether 1v by nucleophilic substitution of a thiophenol and phenylethyl bromide in DCM using N,N-diisopropylethylamine (DIPEA) as a base. Since these conditions for precursor synthesis using alkylhalides are not ideal, we explored alternative reaction conditions.A thiol-ene-click reaction of thiophenol and a styrene appeared to be ideally suited, since no base is needed and the reaction proceeds with perfect atom economy.27 Since this thiol-ene-reaction could also potentially be initiated by UV irradiation, we mixed the corresponding thiol and styrene and irradiated the solution with UV light. In the first attempts of this cascade reaction, however, the yield of the biphenyl was very low because of inefficient coupling of the thiol and styrene. We serendipitously found that the thioether forms in high yields when the two reactants are mixed without solvent at room temperature.28 As purification of the product is not necessary, the thioether could be directly irradiated. After optimization of the dwell time of the educts and the flow rate we achieved a yield of 60%. We subsequently substituted the UV-C light source for UV-B lamps and used ethanol as a “green” solvent instead of acetonitrile.
To demonstrate the utility of this reaction sequence, we synthesized a biphenyl that was inaccessible by conventional photosplicing (Scheme 6A) unless protecting groups are involved.29 Using 4-hydroxythiophenol and 4-cyanostyrene as substrates, we formed biphenyl 2e in 60% yield in one step by mixing followed by irradiation in EtOH with UV-B light (Scheme 7C). This yield is comparable with the protective group-free Suzuki reaction (Scheme 7B), but does not require expensive and scarce raw materials and offers a better atom efficiency.30 This example is relevant because biphenyl 2e represents an important precursor of liquid crystals.31
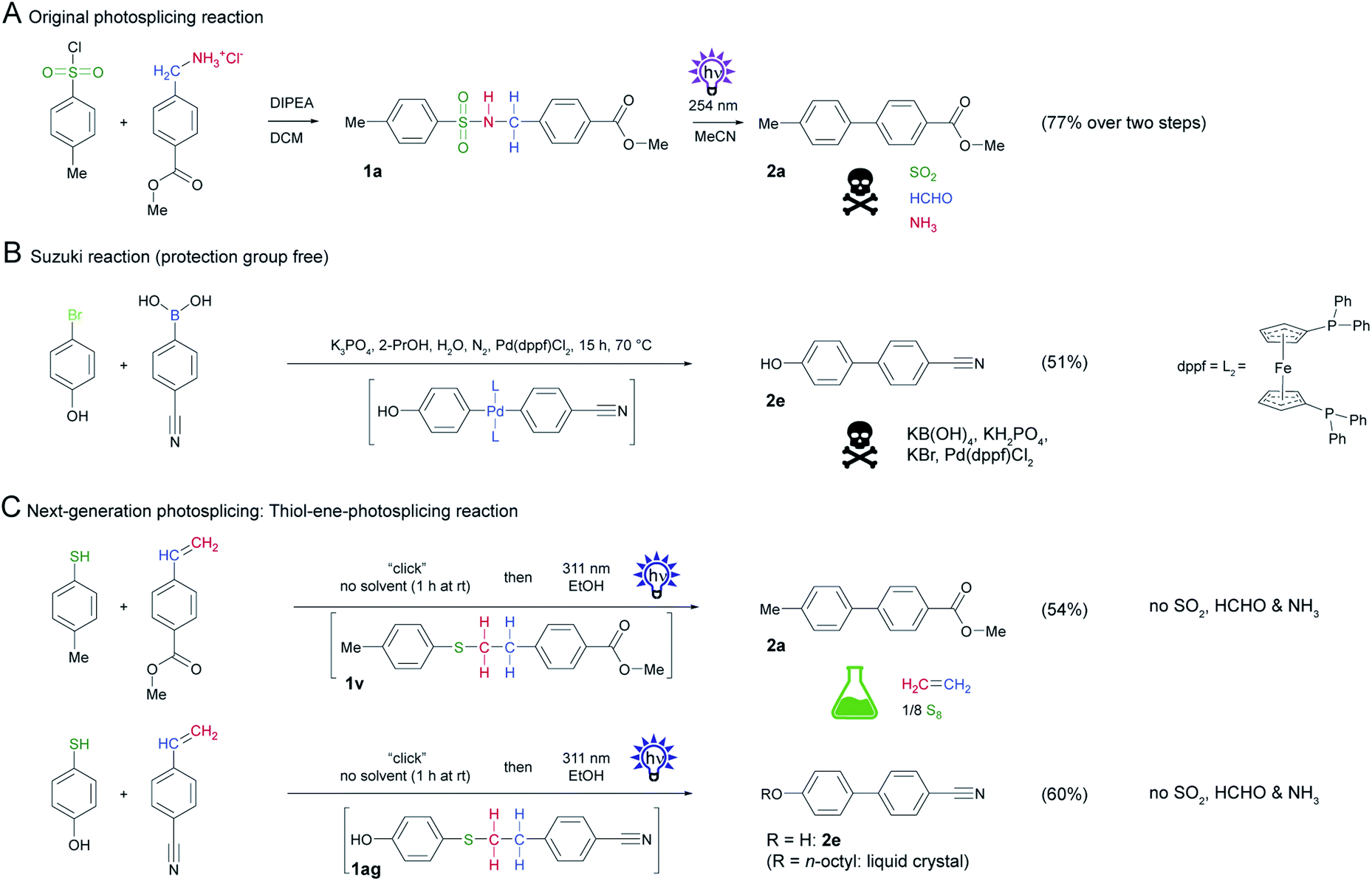 | ||
| Scheme 7 Comparison of original photosplicing reaction with the thiol-ene-photosplicing reaction and Suzuki reaction. (A) Original photosplicing reaction in two steps starting from TsCl and benzylamine. (B) Suzuki reaction with critical raw materials for comparison. (C) Thiol-ene-photosplicing starting from phenylthiol and styrene. Two different examples to give biphenyl 2a and biphenyl 2e. | ||
3 Conclusion
In this systematic linker investigation, we have substantially expanded the scope of the photosplicing reaction. A prerequisite for this light-driven aryl coupling is a flexible three-membered linker. Truncated or elongated homologues, however, give either no or only reduced yields. Terminal groups comprising a π-system are essential, yet many substituents in the linker are permitted, except for photolabile groups (see Fig. S20†). Remarkably, the importance of one sulfur atom as the linking atom (not as the central element, see Fig. S19†) to one aryl substituent was highlighted (see Scheme 8A). This structural feature seems to provide the perfect balance of a labile bond and facilitates the formation of the newly formed C–C bond while it ensures sufficient stability against unselective photochemical decomposition. While phosphorous containing linkers afforded traces of the corresponding biphenyl products, replacements against other elements were not giving the desired products (see Scheme 8B).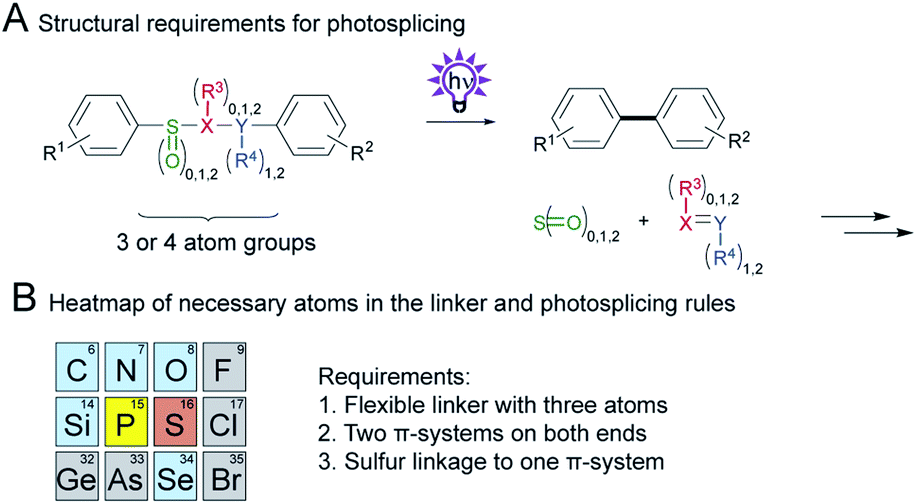 | ||
| Scheme 8 Summary of necessary parts in the linker. (A) Structure of the linker with required parts. (B) Section of the periodic table with the essential elements (grey: not possible or not tested, blue: no biphenyl formation, yellow: traces of biphenyl, red: successful biphenyl formation) and the general rules for photosplicing. | ||
We have shown that the nature of the temporary linkers is crucial for the selective synthesis of biaryls by photosplicing. We designed and synthesized a broad range of substrates with over 25 different linkers and tested their ability to regioselectively form biphenyls upon irradiation. Through systematic interrogation of the structure and reaction conditions we succeeded in identifying a variety of linkers that are viable alternatives to the previously used sulfonamide-based linkers. As a result, we were able to deduce general criteria for the photosplicing reaction. This study not only broadens the scope of the photosplicing reaction and the range of precursors, but also allows for a substantially more sustainable biphenyl production using more environmentally friendly educts and solvents. Importantly, the new protocol prevents the formation of problematic byproducts. The combination of the thiol-ene-reaction and photosplicing is particularly useful, as it allows for the synthesis of an important liquid crystal precursor with high atom efficiency using UV-B light in the presence of oxygen and water. The reaction proceeds robustly and rapidly at room temperature, without critical raw materials. To our knowledge, this example represents one of the most environmentally friendly biphenyl syntheses currently known.
4 Experimental
General procedure of the photosplicing reaction: a potential photosplicing precursor derivative (prepared by any method available) is dissolved in methanol, acetonitrile or ethanol to give a concentration between 1 and 2 mg mL−1. The solution is loaded on a continuous-flow thin-film photoreactor and irradiated with a 254 nm or 311 nm UV lamp. Average dwell times should be set in the range of 5–30 min, depending on the individual reaction speed. A typical silica window size of a photoreactor is 17 × 32 cm and suitable UV sources are six low-pressure metal vapor UV-C or UV-B lamps (15 W power consumption each). The solvent feed of the photoreactor should be operated using an adjustable pump with appropriate flow rates. Reactor temperatures should be in the range of 10 to 20 °C. Throughout the reaction proper shielding of the setup must be warranted to protect the user from UV irradiation. The fractions containing the crude photoproducts are collected, the solvents are removed under reduced pressure and the residues are purified by silica gel filtration, flash chromatography or crystallization to yield the desired biaryls. Full experimental details, methods, detailed synthetic procedures and physicochemical characterization of new compounds including NMR spectra are available in the ESI.†Data availability
The datasets supporting this article (1D and selected 2D NMR spectra with complete assignment of the signals, GC-MS and HPLC-HRMS data of calculated and observed exact masses) have been uploaded as part of the ESI.†Author contributions
V. H. and C. H. conceived the presented idea. V. H., T. N., A. B. and J. B. carried out the experiments. V. H. did the project administration. V. H. and C. H. wrote the manuscript with support from T. N., A. B., J. B. and F. K,. F. K. and C. H. acquired funding. C. H. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Andrea Perner for MS analyses and Heike Heinecke for NMR measurements. We thank Prof. Ivan Vilotijević, FSU Jena, for valuable discussions and comments. We thank Lisa Sachse, Lisa Birkner, Jennifer Marie Quast and Vivian Schraps for synthetic support. We thank Evelyn M. Molloy for accurate proofreading. We thank the Deutsche Forschungsgemeinschaft (Leibniz Prize, for C. H.), the Federal Ministry of Education and Research (BMBF) of Germany within the program InfectControl 2020 (FKZ 03ZZ0803 and FKZ 03ZZ0835 for F. K.).Notes and references
- J.-A. García-López and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766–6798 RSC.
- F. R. Leroux, A. Berthelot, L. Bonnafoux, A. Panossian and F. Colobert, Chem.–Eur. J., 2012, 18, 14232–14236 CrossRef CAS PubMed.
- F. Fringuelli, R. Girotti, O. Piermatti, F. Pizzo and L. Vaccaro, Org. Lett., 2006, 8, 5741–5744 CrossRef CAS PubMed.
- R. Shang and L. Liu, Sci. China: Chem., 2011, 54, 1670–1687 CrossRef CAS.
- W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC.
- C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS.
- Y.-F. Zhang and Z.-J. Shi, Acc. Chem. Res., 2018, 52, 161–169 CrossRef PubMed.
- V. Dichiarante, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2007, 46, 6495–6498 CrossRef CAS PubMed.
- C. M. Holden, S. M. Sohel and M. F. Greaney, Angew. Chem., Int. Ed., 2016, 128, 2496–2499 CrossRef.
- F. Kloss, T. Neuwirth, V. G. Haensch and C. Hertweck, Angew. Chem., Int. Ed., 2018, 57, 14476–14481 CrossRef CAS PubMed.
- V. G. Haensch, T. Neuwirth, J. Steinmetzer, F. Kloss, R. Beckert, S. Gräfe, S. Kupfer and C. Hertweck, Chem.–Eur. J., 2019, 25, 16068–16073 CrossRef CAS PubMed.
- M. Cooke, S. Utembe, P. G. Carbajo, A. Archibald, A. Orr-Ewing, M. Jenkin, R. Derwent, D. Lary and D. Shallcross, Atmos. Ocean. Sci. Lett., 2010, 11, 33–38 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- E. Krylov and L. Virzum, Russ. Chem. Bull., 2019, 68, 527–531 CrossRef CAS.
- G. R. Fulmer, A. J. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- P. Wessig and S. Krebs, Eur. J. Org. Chem., 2021, 2021, 6367–6374 CrossRef CAS.
- J. Steinmetzer, S. Kupfer and S. Gräfe, Int. J. Quantum Chem., 2021, 121, e26390 CrossRef CAS.
- F. Hirayama, J. Chem. Phys., 1965, 42, 3163–3171 CrossRef CAS.
- H. Qrareya, L. Meazza, S. Protti and M. Fagnoni, Beilstein J. Org. Chem., 2020, 16, 3008–3014 CrossRef CAS PubMed.
- Y. Okamoto, T. Tatsuno and S. Takamuku, Heteroat. Chem., 1996, 7, 257–261 CrossRef CAS.
- Y. Okamoto, M. Nakamura, M. S. And and S. Takamuku, Photochem. Photobiol., 1992, 56, 403–407 CrossRef CAS.
- P. J. Kropp, G. E. Fryxell, M. W. Tubergen, M. W. Hager, G. D. Harris Jr, T. P. McDermott Jr and R. Tornero-Velez, J. Am. Chem. Soc., 1991, 113, 7300–7310 CrossRef CAS.
- N. Hoffmann, Tetrahedron, 2002, 58, 7933–7941 CrossRef CAS.
- M. Nakamura, Y. Okamoto and S. Takamuku, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 106, 137–144 CrossRef CAS.
- M. Shi, K. Yamamoto, Y. Okamoto and S. Takamuku, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 60, 1–14 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- K. Choudhuri, A. Mandal and P. Mal, Chem. Commun., 2018, 54, 3759–3762 RSC.
- J. I. Levin, M. T. Du and K. Park, Synth. Commun., 2004, 34, 2773–2781 CrossRef CAS.
- T. Dutronc, E. Terazzi, L. Guénée, K.-L. Buchwalder, A. Spoerri, D. Emery, J. Mareda, S. Floquet and C. Piguet, Chem.–Eur. J., 2013, 19, 8447–8456 CrossRef CAS PubMed.
- A. Selevou, G. Papamokos, M. Steinhart and G. Floudas, J. Phys. Chem. B, 2017, 121, 7382–7394 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc00886f |
| This journal is © The Royal Society of Chemistry 2022 |